

Abstract
Background
McLeod syndrome (MLS) is an X-linked multisystemic progressive disorder caused by loss of function mutations in the XK gene. The rare blood group phenotype of MLS patients with absent Kx antigen requires the support of specialized transfusion institutions because of the risk of transfusion complications. Acanthocytosis of red blood cells occurs in almost all patients. Nonhematological manifestations of MLS are very similar to those of VPS13A disease (chorea-acanthocytosis), an autosomal-recessive condition. Their shared phenotype apart from acanthocytosis includes movement disorders such as chorea and dystonia, epilepsy, peripheral neuropathy, and muscle involvement, typically with creatine kinase (CK) elevation, cardiomyopathy included.
Summary
In this review, we describe the nonhematological manifestations of MLS in comparison with those of VPS13A disease. While there are many similarities, differences such as mode of inheritance, sex distribution, age at manifestation, severity of heart involvement, frequency of feeding dystonia or of involuntary head drops may help to distinguish these disorders in the clinic. Immunohematological demonstration of the McLeod-Kell phenotype or detection of pathogenic mutations of XK (or VPS13A, respectively) is the gold standard for distinction. “Neuroacanthocytosis” was often used as an overarching term, but is potentially misleading, as the term does not refer to a defined disease entity. Its use, if continued, must not prevent clinicians to seek a final diagnosis on the basis of molecular findings. The clinical similarity of MLS and VPS13A disease has long suggested some shared pathophysiology. Evidence for molecular interaction between XK, the McLeod protein, and chorein, the VPS13A gene product, has recently been put forward: XK forms a complex with chorein/VPS13A, a bulk lipid transporter located at various membrane contact sites. The exact role of XK in this complex needs to be further elucidated. Impairment of bulk lipid transport appears as the common denominator of both MLS and VPS13A disease. A variety of further conditions may in time be added to the “bulk lipid transport diseases,” such as the recently recognized disorders caused by mutations in the VPS13B, VPS13C, and VPS13D genes.
Key Messages
(1) Patients diagnosed with the rare red cell McLeod phenotype (McLeod syndrome, MLS) require interdisciplinary collaboration of transfusion medicine specialists, neurologists, and cardiologists for both their hematological and nonhematological disease manifestations. (2) The phenotypical similarity of MLS and VPS13A disease, often leading to either confusion or insufficient diagnostic depth (under the label of “neuroacanthocytosis”), is based on interaction of the respective proteins, XK and chorein, within the cellular machinery for bulk lipid transport. (3) Overall, the term “bulk lipid transport diseases” seems useful for further research on a group of conditions that may not only share pathophysiology, but may also share treatment approaches.
Keywords: Kx antigen, XK protein, Chorein, Neuroacanthocytosis, Bulk lipid transport diseases
Introduction
McLeod syndrome (OMIM #300842) is an ultra-rare progressive multisystemic disorder due to pathogenic variants in the XK gene with X-chromosomal inheritance [1]. Delineation of the syndrome began in the early 1960s with the description of a hitherto unknown Kell antigen profile in an otherwise healthy 25-year-old male blood donor, then a Harvard dental student, who developed neurological symptoms much later in life [1, 2, 3]. The “McLeod blood group phenotype” that necessitates specific transfusion precautions was later found associated with degeneration within the central and peripheral nervous system and with a wide spectrum of neurological signs and symptoms and led to naming of the multisystemic condition as McLeod syndrome (MLS) after the original propositus [1, 4, 5]. Because of the particular “neurohematological” coincidence, diagnosis and management of MLS patients clearly require efforts by multidisciplinary collaboration.
The “McLeod blood group phenotype” results from absence of the Kx antigen (located at an extracellular loop of the transmembrane protein XK) and an associated reduced expression of antigens of the Kell blood group system [5, 6]. MLS therefore affects both the XK (ISBT 019) and KEL (ISBT 06) systems [4, 7]. Kx+ transfusions should be strictly avoided for all individuals carrying the McLeod blood phenotype: this implicates the need for support from specialized transfusion institutions with immunohematological expertise. Because of the rarity of compatible units of stored blood, autologous donation prior to an anticipated need or blood donation for long-term cryopreservation, banking and interinstitutional, world-wide exchange of blood units are required. The impact on blood banking and cryopreservation of the presence of acanthocytic red cells and of subsequent chronic-compensated hemolysis in MLS [4] so far has not been systematically studied. Single case observations, however, indicate the absence of major issues with freezing and thawing of McLeod blood [8, 9].
Acanthocytosis of red blood cells occurs in almost all patients, but was first recognized as a feature of the McLeod propositus only years after the immunohematological definition [10]. Our recent chance observation of slowed erythrocyte sedimentation of acanthocytic blood was confirmed by systematic analysis that showed a clear inverse correlation of acanthocyte proportion and sedimentation rate [11].
Compensated, usually clinically asymptomatic hemolysis with decreased erythrocyte lifespan [12] probably underlies the hepatosplenomegaly seen in around half of the cases [1], and the commonly elevated levels of aspartate and alanine transaminases and of lactate dehydrogenase in MLS may relate to such liver involvement [13].
MLS and the clinically similar, yet autosomal-recessive VPS13A disease (also known as chorea-acanthocytosis; OMIM #200150), with an estimated prevalence of 1:10,000,000 and 1:1,000,000, respectively, may also be called “core neuroacanthocytosis syndromes” [14, 15, 16]. The term “neuroacanthocytosis,” however, is no longer recommended, as it blurs the distinction of genetically separate conditions [14]. In the past, the term was used in an even broader sense: neurological disorders associated with acanthocytosis comprise pantothenate kinase-associated neurodegeneration (OMIM #606157) as well as disorders of lipid absorption, with peripheral neuropathy and cerebellar signs, such as abetalipoproteinemia (Bassen-Kornzweig syndrome; OMIM #200100) or familial hypobetalipoproteinemia (FHBL1, OMIM #615558; FHBL2, OMIM #605019) [14].
In the present article, we review the nonhematological manifestations of MLS in comparison to VPS13A disease and point out the molecular evidence that may explain the similarity of the two conditions.
XK and VPS13A Genes, Mode of Inheritance
MLS is caused by pathogenic variants in the XK gene which is located at the p21.1 region of the X chromosome [17], in close proximity to the CYBB gene that codes for cytochrome b558, subunit β (also known as NADPH oxidase 2 and relevant for the microbicidal system of phagocytes). Deletions or partial deletions of the two genes thus lead to a contiguous gene deletion syndrome that combines MLS with chronic granulomatous disease (OMIM #306400). As the latter often requires blood transfusion [18], MLS ought to be ruled out in chronic granulomatous disease patients prior to allogenic transfusions in order to avoid alloimmunization and subsequent posttransfusion complications [19]. Larger deletions of the Xp21 region may additionally involve genes associated with Duchenne muscular dystrophy (OMIM #310200), retinitis pigmentosa (OMIM #300029) and ornithine transcarbamylase deficiency (OMIM #311250), respectively [1]. Partial inclusion of the PRRG1 gene that codes for transmembrane γ-carboxyglutamic acid protein 1 has recently been found included in an MLS patient´s deletion, without any as yet clinically tangible consequence [20]. Due to the X-chromosomal mode of inheritance, most individuals clinically affected by MLS are male. Exceptionally, heterozygous female gene carriers may develop symptoms, too [21, 22, 23, 24, 25], which is likely due to skewed X-chromosome inactivation [26]. Commonly, however, females with a single XK mutation will not be affected. Females homozygous for XK gene mutations have not yet been recognized.
VPS13A disease, also known as chorea-acanthocytosis [14], is caused by pathogenic variants in the VPS13A gene, located at the q21.2 region of chromosome 9. Initially, the gene was called CHAC and its product was called chorein [27, 28]. As expected with autosomal-recessive inheritance, females and males likewise are affected by VPS13A disease without an as yet obvious genotype-phenotype correlation [29].
VPS13A is part of a mammalian gene family of 4 paralogues [30]. Mutations in the other VPS13 family genes are also associated with neurodevelopmental or neurodegenerative diseases. VPS13B (COH1) variants underlie Cohen syndrome [31], and VPS13C variants have been observed in early-onset Parkinson's [32, 33] and in Lewy body disease [34]. VPS13D disease may start with movement disorders in early childhood and was diagnosed also in cases initially labeled as recessive spinocerebellar ataxia type 4 or spinocerebellar ataxia with saccadic intrusions [35].
Nonhematological Manifestations of MLS
Multiple systems are affected in MLS, mainly the blood, the central and peripheral nervous system, the skeletal muscle, and the heart [36]. The McLeod blood phenotype is present at birth in male XK mutation carriers, while the time of first occurrence of red cell acanthocytosis is still a matter of speculation. In contrast, signs and symptoms from nonhematological involvement usually develop after the age of 30 years, with a broad range of variability [1, 4, 20, 23]. Overall, these manifestations of MLS often cause severe disability and may reduce or abolish independent living and shorten life expectancy [37].
Central nervous system manifestation of MLS is typically with “huntingtonism,” a progressive triple disorder of movement, behavior, and cognition. To which extent each domain is affected, varies among MLS individuals, but symptoms seem to correlate with progressive degeneration of the basal ganglia. There is progressive widening of the anterior horn of the lateral ventricles due to caudate nucleus atrophy [38] and MRI volumetry shows an inverse correlation of basal ganglia volumes with duration of disease and, in particular, a decrease in caudate volume with disease progression [39, 40]. FDG-PET reveals bilaterally reduced striatal glucose uptake [41, 42] and post mortem studies show the atrophy of the striatum more pronounced than that of the globus pallidus [43, 44]. In the exceptional “L family” female mutation carrier [26], the substantia nigra was felt unaffected, as was her brainstem, subthalamic nucleus, thalamus, cerebral cortex, cerebellum, and spinal cord [45, 46]. A recent male case confirmed the normal findings in subthalamic nucleus, thalamus, and substantia nigra, and displayed no neuronal or glial inclusions with TDP 43, α-synuclein and p62 immunohistochemistry. Only few τ-positive neuronal and glial inclusions, compatible with aging, were detected [47].
Disorders of movement often are the presenting manifestations of MLS, most commonly hyperkinesia, i.e., chorea, which develops over time in 95% of the patients [36]. Chorea can affect all parts of the body [48]. Dystonia can also occur as can parkinsonism, the latter typically later into the disease [1, 36]. Involuntary facial and perioral contractions as well as unintended vocalizations may be present [1, 48]. Dysarthria impacts communication and social participation. Dysphagia may impair caloric intake and lead to complications such as aspiration and recurrent pneumonia [37]. In addition to these well-established features, less common manifestations occur in single MLS patients, such as involuntary tongue and cheek biting, feeding dystonia, and head drops [49, 50], findings that are much more typical in VPS13A disease.
Behavioral and cognitive manifestations of MLS, even if commonly observed, as yet await systematic prospective study. Both figurative and verbal memory is impaired, the latter with documented progression, and there is executive (“frontal”) dysfunction [51]. Changes of behavior and/or personality occur in about 80% of the patients [36], and depression, anxiety, psychosis, irritability, and obsessive-compulsive disorder have been reported [1, 39, 52, 53].
About 40% of patients are diagnosed with epilepsy, which in half of these is the very first presentation of MLS [36]. Seizures are commonly described as generalized, yet little is known about the exact semiology or pathogenesis.
Neuromuscular manifestations are obvious from the regular findings of diminished or absent deep tendon reflexes and of, even excessive, hyperCKemia [54, 55]. The high levels of transaminases (ALT/AST) and LDH, mentioned in relation to hepatomegaly, might alternatively originate from muscle affection. MLS may be considered a primary myopathy [56]. As sensory and motor axonal polyneuropathy is very common [1, 23, 39, 57], muscle atrophy and weakness have thus been interpreted predominantly due to motor neuropathy [3]. Muscle biopsies show both neurogenic and myopathic changes [1, 3, 57, 58]. MLS thus seems to be a mixed neuromuscular condition with an individually variable proportion of the two components. Clinically relevant impairment occurs in about half of MLS individuals [1], such as pronounced bilateral foot drop, but also axially predominant weakness [59]. Of interest are biopsy findings that suggest additional inflammatory changes in muscle [57, 59, 60] and correspond to comparable observations in the heart [61].
Heart involvement in MLS is potentially life-threatening and comprises organ failure, dilated cardiomyopathy, and arrhythmias such as ventricular tachycardia [58, 61, 62, 63, 64]. Heart muscle fibrosis may be detected on histology [58, 63]. Its presence can also be inferred from cardiac MRI [64].
MLS and VPS13A disease behave as phenocopies with respect to many nonhematological manifestations. Apart from their respective mode of inheritance, X-linked or autosomal-recessive (with resultant sex distribution), the only phenotypical feature to tell the two disorders apart with high certainty is the presence or absence of the McLeod-Kell phenotype, i.e., weak Kell antigen expression in combination with absent Kx antigen.
Some additional features may help in distinguishing MLS from VPS13A disease (Fig. 1). In MLS, nonhematological manifestations occur at a later age, and cardiac involvement is more severe. Episodes of sudden loss of muscle tone in the neck, trunk, or legs (leading to head drops, “clasping behavior,” or the peculiar “rubber-man-like appearance/gait”) as well as tongue protrusion/feeding dystonia seem more typical for VPS13A disease. Nevertheless, proper distinction of the two disorders requires molecular characterization, preferably on both the levels of their genes and the corresponding proteins, XK and chorein.
Fig. 1.
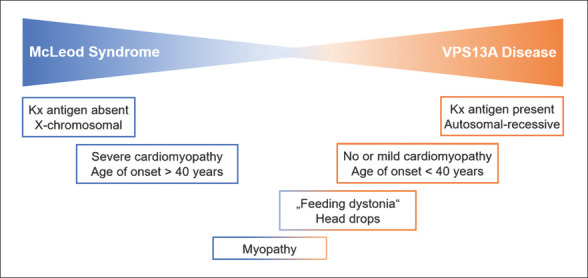
McLeod syndrome (MLS) and VPS13A disease (chorea-acanthocytosis) are phenotypically very similar neurohematological disorders. Certain features, such as cardiomyopathy or involuntary movements that interfere with chewing and swallowing (feeding dystonia), may be less common in one of the two, but presence of the Kell-McLeod phenotype (absence of Kx antigen) is the main distinguishing feature.
It is not yet clear how to interpret the novel observation of a 67-year-old male with a suggestive neurological syndrome but normal Kell phenotype in spite of a possibly pathogenic XK missense mutation [65]. This appears as the reverse of situations where the McLeod red cell phenotype is detected but nonhematological manifestations are not [66, 67]. For the latter cases one could argue that clinical follow-up was not sufficiently long to witness symptom development, yet both types of observations, if confirmed, would provide valuable insights into MLS disease mechanisms at the molecular level.
Diagnosis and Management
In adults presenting with the clinical triad of progressive disorders of movement, cognition, and behavior (huntingtonism), genetic testing for mutations in the Huntington´s disease gene is mandatory. If negative, further differential diagnosis is complex [68]. Low erythrocyte sedimentation rate, elevated levels of CK, ALT, AST, and LDH, as well as red cell acanthocytosis − that, however is difficult to determine and not even an obligatory finding under routine clinical conditions [69, 70] − may lead one to consider the two related conditions of MLS and VPS13A disease. While male sex, older age, and affection also of a brother or maternal uncle support an assumption of MLS, the two conditions cannot, however, be sufficiently distinguished on the basis of clinical phenotype alone. Several cases are on record where the diagnosis had to be changed to MLS after more extensive testing (cases of, e.g., Gandhi et al. [71] and the case of Faillace et al. [72] and Marsh [73]).
For an elderly male patient (>40 years of age) with clinical and laboratory features as detailed above, we thus first recommend an immunohematological search for the “McLeod blood group phenotype” and/or genetic testing to identify a pathogenic variant or a deletion involving the XK gene. Patient blood samples are first evaluated serologically for KEL and Kx antigen reactivity which is expected to be either negative or weakened (further details and flowchart in Frey et al. [74]). At the level of genetic analysis, a specific approach to overcome diagnostic difficulties with XK gene deletions has been proposed [75].
Similar to the diagnosis of MLS that is based on the absence of the XK protein (carrier of the Kx antigen) and/or presence of XK gene mutations, diagnosis of VPS13A disease should be based upon the absence of the chorein protein in erythrocyte membrane Western blot [76] and/or the identification of mutations in the VPS13A gene. VPS13A disease is clinically more suspicious in younger patients of both sexes, in particular in siblings without cases in the parents´ generation who might, nevertheless, share a common family or regional background. Based on the literature [65, 66, 67] and on our own experience [77], we propose that only analyses on both the gene and the protein levels should be considered as sufficient for full diagnosis of the two clinical conditions of MLS and of VPS13A disease.
Treatment of both diseases is currently purely symptomatic: a detailed review for the resultant manifestation-specific approach is available, but treatment decisions are highly individual [8]. Chorea, for example, may be addressed by dopamine-depleting drugs such as tetrabenazine. For seizures, conventional anticonvulsants are usually effective, yet there is no evidence for superiority of any particular drug. Most important in the management of every single MLS patient, because of the risk of sudden cardiac death [37], is cardiological surveillance on a regular (yearly) basis with subsequent individual treatment decisions that may even include implantation of a cardioverter-defibrillator [61] or a heart transplant [63].
The other highly relevant management topic relates to the rarity of compatible blood units worldwide and the question of their availability in cases of urgent need. In spite of regional and national differences with respect to the procedures involved, the banking of autologous blood units in specialized transfusion institutions is highly recommended.
Family studies are helpful to detect additional, perhaps subclinically affected members (patients´ brothers and maternal uncles, but in particular their mothers and sisters) and to offer genetic counseling [36] about risks for individuals (and their precautions, see above) and for family offspring (possibly including discussion of preimplantation genetic testing).
At the present date, no disease-modifying approach is known for MLS, even if first exploratory results are available for VPS13A disease [78, 79, 80]. There, inhibition of hyperactive Lyn kinase may have addressed a relevant disease mechanism, but so far this approach has not been extended to MLS where Lyn kinase may not even be a potential drug target.
XK and VPS13A Proteins, Localization and Interaction
XK and VPS13A are ubiquitously expressed proteins that interact with each other (as recently demonstrated by Park and Neiman [81]). Prior to this, the intriguing clinical similarities of MLS and VPS13A disease had already suggested some sharing of pathways [82].
XK protein is an integral membrane protein [17] and in red blood cells forms a heterodimer with the Kell glycoprotein [83], as part of a larger membrane multiprotein complex important for red cell membrane cytoskeleton stability [84]. The exact localization of XK in nonerythroid tissues remains elusive, but it was shown that it is not coexpressed with the Kell protein in brain tissue [85] and probably not in skeletal muscle either [86, 87].
As shown early in red blood cells, XK absence leads to a reduction of phosphatidylserine in the inner leaflet of the cell membrane [88]. In line with this, XK-related (Xkr) proteins were later found to translocate phosphatidylserine between the two membrane leaflets [89, 90]. This specific activity of a molecule is summarized by its designation as a “scramblase.” The structure of two human XKrs (Xkr8 and Xkr9) − with Xkr8 working in a complex with the chaperone basigin − was just successfully elucidated [91, 92]. For XK, foremost member of the Xkr family, however, the putative scramblase function remains to be proven.
VPS13 proteins localize to membrane contact sites and, according to a rapidly growing body of evidence, seem responsible for bulk lipid transport between the membranes of various organelle types [for reviews, see 93, 94]. Impaired interorganellar mobility of lipids impresses as the common denominator of this novel group of neurodegenerative/neurodevelopmental VPS13 diseases. As illustrated here (Fig. 2a), VPS13A/chorein localizes to membrane contact sites of the endoplasmic reticulum and mitochondria and, respectively, lipid droplets in human cells [95, 96]. Tethering of mitochondria and endosomes was also described [97]. In yeast, organelle-specific adaptor proteins recruit Vps13 to the various interorganellar contact sites that each mediates a distinct function [98].
Fig. 2.
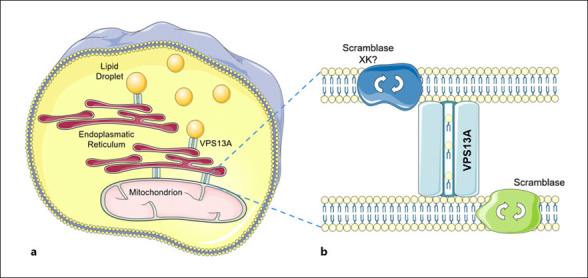
Subcellular localization and putative functions of VPS13A and XK.aThe VPS13A protein (chorein) localizes at membrane contact sites between the endoplasmic reticulum and mitochondria or lipid droplets, respectively.bVPS13A seems to function as bulk lipid transporter between membranes. Putative partner proteins include scramblases (possibly XK among them) that translocate phospholipids between the two sheets of the membranes tethered by VPS13A. Contains modified images from Servier Medical Art (https://smart.servier.com) licensed by a Creative Commons Attribution 3.0 Unported License.
Recent studies suggest that XK and VPS13A are partner proteins: their coimmunoprecipitation was shown in HEK293 cells [99], and they form complexes in human cells [81]. It was also shown that XK is involved in the relocalization of VPS13A from lipid droplets to endoplasmic reticulum subdomains when overexpressed [81]: XK seems to recruit VPS13A to probably the endoplasmic reticulum. Thus, at the beginning or the end of a VPS13 bulk lipid transport chain, Xkr family proteins may act as scramblases that exchange lipids between the leaflets of the membrane of origin or, respectively, destiny. This process would allow lipid equilibration within the membranes that were temporarily tethered for the purpose of bulk lipid transport [81, 93, 94] (Fig. 2b).
Overall, conditions caused by dysfunction of VPS13 proteins and of partners such as the McLeod protein XK might be lumped together under the common concept of “bulk lipid transport diseases.” This label stresses the presence of shared pathways, and, if investigated from such a more general point of view, therapeutic options that are focused on the commonalities of these diseases might become available faster than approaches that result from studying the single conditions in isolation.
A pressing question among the many that are still open is the question whether proteins of the VPS13 and of the Xkr families might, at least partially, compensate for deficiency of a member within the specific protein family or even across protein families. Such mechanisms would provide an explanation for both the clinical heterogeneity as well as the late onset observed in MLS patients and in individuals affected by VPS13 disease. For VPS13A disease and MLS it is of particular interest whether the presence of both chorein and XK in membranes of mature red blood cells (that lack subcellular organelles and thus contact sites) indicates interaction that is still ongoing or is just a trace of a pathway once shared, e.g., in erythropoiesis.
Conclusion
In this review, we focused on nonhematological manifestations of the rare neurohematological disorder MLS. Because of the multisystemic nature of their condition, MLS patients necessarily deserve interdisciplinary collaboration, with participation of transfusion medicine specialists, neurologists, and cardiologists.
MLS strongly resembles VPS13A disease. The basis for the phenotypical commonalities most likely is the interaction of the two proteins affected, XK and VPS13A/chorein. We propose the term “bulk lipid transport diseases” to place special emphasis on the putative main function of VPS13 proteins that unfolds in interaction with a number of partner proteins.
Bulk lipid transport is essential for the rapid availability of membrane constituents necessary for shrinkage or growth of organelles. Fission and fusion of mitochondria as well as the formation of autophagosomes are pertinent examples. XK appears to work as scramblase, equilibrating lipids within the bilayers tethered by the VPS13A molecular machinery. Consideration of this function and its involvement in a variety of processes at the subcellular level might be particularly relevant for a deeper understanding of McLeod syndrome pathophysiology.
Conflict of Interest Statement
The authors declare that they have no competing interest related to the paper.
Financial disclosures of all authors for the preceding 3 years: K.P. has received funding from the Else Kröner Clinician Scientist Program (TU Dresden, Germany), the Rostock Academy for Clinician Scientists (RACS, University of Rostock, Germany), the MeDDrive grant of the Technische Universität Dresden, and the FORUN grant of the University of Rostock. A.H. has received funding from the Federal Ministry of Education and Research (BMBF), the Helmholtz Association, the Hermann und Lilly Schilling-Stiftung für medizinische Forschung im Stifterverband, and the “Innovationsfond des gemeinsamen Bundesausschusses.” He has received honoraria for presentations, advisory boards, consultations from Biogen and Desitin. He has received royalties from Elsevier Press. A.D. has received honoraria for presentations from the Zürich Blood Bank, the Aarau and Schlössli Hospitals, all in Switzerland, and for expert testimony required by German courts of law as well as funding from the Advocacy for Neuroacanthocytosis Patients.
Funding Sources
There are no funding sources.
Author Contributions
Conceptualization − K.P., A.H., and A.D. Writing of the first draft of the paper − K.P. Critical review and editing of the paper − A.H. and A.D.
Acknowledgements
We are grateful to Glenn (†) and Ginger Irvine as the founders of the Advocacy for Neuroacanthocytosis Patients (www.naadvocacy.org) and to Susan Wagner and Joy Willard-Williford as representatives of the NA Advocacy USA (www.naadvocacyusa.org). K.P. is supported by the Rostock Academy for Clinician Scientists (RACS) and the FORUN program (University of Rostock, Germany). A.H. is supported by the “Hermann und Lilly Schilling-Stiftung für medizinische Forschung im Stifterverband.”
References
- 1.Danek A, Rubio JP, Rampoldi L, Ho M, Dobson-Stone C, Tison F, et al. McLeod neuroacanthocytosis: genotype and phenotype. Ann Neurol. 2001 Dec;50((6)):755–64. doi: 10.1002/ana.10035. [DOI] [PubMed] [Google Scholar]
- 2.Allen FH, Krabbe SM, Corcoran PA. A new phenotype (McLeod) in the Kell blood-group system. Vox Sang. 1961;6((5)):555–60. doi: 10.1111/j.1423-0410.1961.tb03203.x. [DOI] [PubMed] [Google Scholar]
- 3.Hewer E, Danek A, Schoser BG, Miranda M, Reichard R, Castiglioni C, et al. McLeod myopathy revisited: more neurogenic and less benign. Brain. 2007 Dec;130((Pt 12)):3285–96. doi: 10.1093/brain/awm269. [DOI] [PubMed] [Google Scholar]
- 4.Roulis E, Hyland C, Flower R, Gassner C, Jung HH, Frey BM. Molecular basis and clinical overview of McLeod syndrome compared with other neuroacanthocytosis syndromes: a review. JAMA Neurol. 2018;75((12)):1554–62. doi: 10.1001/jamaneurol.2018.2166. [DOI] [PubMed] [Google Scholar]
- 5.Redman CM, Russo D, Lee S. Kell, Kx and the McLeod syndrome. Baillieres Best Pract Res Clin Haematol. 1999;12((4)):621–35. doi: 10.1053/beha.1999.0045. [DOI] [PubMed] [Google Scholar]
- 6.Redman CM, Marsh WL. The Kell blood group system and the McLeod phenotype. Semin Hematol. 1993;30:209–18. [PubMed] [Google Scholar]
- 7.Denomme GA. Kell and Kx blood group systems. Immunohematology. 2015;31((1)):14–9. [PubMed] [Google Scholar]
- 8.Walker RH. Management of neuroacanthocytosis syndromes. Tremor Other Hyperkinet Mov (N Y) 2015;5:346. doi: 10.7916/D8W66K48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Turner TR, Clarke G, Denomme GA, Skeate R, Acker JP. Effect of cryopreservation on a rare McLeod donor red blood cell concentrate. Immunohematology. 2021 Jun;37((2)):78–83. doi: 10.21307/immunohematology-2021-012. [DOI] [PubMed] [Google Scholar]
- 10.Wimer BM, Marsh WL, Taswell HF, Galey WR. Haematological changes associated with the McLeod phenotype of the Kell blood group system. Br J Haematol. 1977;36((2)):219–24. doi: 10.1111/j.1365-2141.1977.tb00642.x. [DOI] [PubMed] [Google Scholar]
- 11.Darras A, Peikert K, Rabe A, Yaya F, Simionato G, John T, et al. Acanthocyte sedimentation rate as a diagnostic biomarker for neuroacanthocytosis syndromes: experimental evidence and physical justification. Cells. 2021 Apr;10((4)):788. doi: 10.3390/cells10040788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Symmans WA, Shepherd CS, Marsh WL, Oyen R, Shohet SB, Linehan BJ. Hereditary acanthocytosis associated with the McLeod phenotype of the Kell blood group system. Br J Haematol. 1979 Aug;42((4)):575–83. doi: 10.1111/j.1365-2141.1979.tb01170.x. [DOI] [PubMed] [Google Scholar]
- 13.Walker RH, Danek A, Jung HH, Davey R, Reid M. McLeod syndrome presenting with hepatic disease. Mov Disord. 2005;20((Suppl 10)):S6–7. [Google Scholar]
- 14.Walker RH, Danek A. “Neuroacanthocytosis” − overdue for a taxonomic update. Tremor Other Hyperkinet Mov (N Y) 2021 Jan;11((11)):1–6. doi: 10.5334/tohm.583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Walker RH, Gatto EM, Bustamante ML, Bernal-Pacheco O, Cardoso F, Castilhos RM, et al. Huntington's disease-like disorders in Latin America and the Caribbean. Parkinsonism Relat Disord. 2018 Aug;53:10–20. doi: 10.1016/j.parkreldis.2018.05.021. [DOI] [PubMed] [Google Scholar]
- 16.Peikert K, Danek A, Hermann A. Current state of knowledge in chorea-acanthocytosis as core neuroacanthocytosis syndrome. Eur J Med Genet. 2018 Nov;61((11)):699–705. doi: 10.1016/j.ejmg.2017.12.007. [DOI] [PubMed] [Google Scholar]
- 17.Ho M, Chelly J, Carter N, Danek A, Crocker P, Monaco AP. Isolation of the gene for McLeod syndrome that encodes a novel membrane transport protein. Cell. 1994 Jun;77((6)):869–80. doi: 10.1016/0092-8674(94)90136-8. [DOI] [PubMed] [Google Scholar]
- 18.Brzica SM, Pineda AA, Taswell HF, Rhodes KH. Chronic granulomatous disease and the McLeod phenotype. Successful treatment of infection with granulocyte transfusions resulting in subsequent hemolytic transfusion reaction. Mayo Clin Proc. 1977 Mar;52((3)):153–6. [PubMed] [Google Scholar]
- 19.Lhomme F, Peyrard T, Babinet J, Abou-Chahla W, Durieu I, Moshous D, et al. Chronic granulomatous disease with the McLeod phenotype: a French national retrospective case series. J Clin Immunol. 2020 Jul;40((5)):752–62. doi: 10.1007/s10875-020-00791-w. [DOI] [PubMed] [Google Scholar]
- 20.Peikert K, Schlotter-Weigel B, Montagnese F, Reilich P, Saft C, Marxreiter F, et al. Novel mutations and findings in a cohort of McLeod neuroacanthocytosis, an X-linked HD phenocopy. J Neurol Neurosurg Psychiatry. 2021 Sep;92((Suppl 1)):A30. [Google Scholar]
- 21.Kawakami T, Takiyama Y, Sakoe K, Ogawa T, Yoshioka T, Nishizawa M, et al. A case of McLeod syndrome with unusually severe myopathy. J Neurol Sci. 1999 Jun;166((1)):36–9. doi: 10.1016/s0022-510x(99)00108-2. [DOI] [PubMed] [Google Scholar]
- 22.Ueyama H, Kumamoto T, Nagao S, Masuda T, Sugihara R, Fujimoto S, et al. A novel mutation of the McLeod syndrome gene in a Japanese family. J Neurol Sci. 2000 Jun;176((2)):151–4. doi: 10.1016/s0022-510x(00)00307-5. [DOI] [PubMed] [Google Scholar]
- 23.Weaver J, Sarva H, Barone D, Bobker S, Bushara K, Hiller A, et al. McLeod syndrome: five new pedigrees with novel mutations. Parkinsonism Relat Disord. 2019 Jul;64:293–9. doi: 10.1016/j.parkreldis.2019.04.022. [DOI] [PubMed] [Google Scholar]
- 24.Balint B, Lang AE. Expert comment to: Novel Xp21.1 deletion associated with unusual features in large McLeod syndrome kindred. Parkinsonism Relat Disord. 2020 Oct;79:133–4. doi: 10.1016/j.parkreldis.2019.02.024. [DOI] [PubMed] [Google Scholar]
- 25.Sveinsson O, Udd B, Svenningsson P, Gassner C, Engström C, Laffita-Mesa J, et al. Involuntary movements, vocalizations and cognitive decline. Parkinsonism Relat Disord. 2020 Oct;79:135–7. doi: 10.1016/j.parkreldis.2019.05.029. [DOI] [PubMed] [Google Scholar]
- 26.Ho MF, Chalmers RM, Davis MB, Harding AE, Monaco AP. A novel point mutation in the McLeod syndrome gene in neuroacanthocytosis. Ann Neurol. 1996 May;39((5)):672–5. doi: 10.1002/ana.410390518. [DOI] [PubMed] [Google Scholar]
- 27.Rampoldi L, Dobson-Stone C, Rubio JP, Danek A, Chalmers RM, Wood NW, et al. A conserved sorting-associated protein is mutant in chorea-acanthocytosis. Nat Genet. 2001 Jun;28((2)):119–20. doi: 10.1038/88821. [DOI] [PubMed] [Google Scholar]
- 28.Ueno S, Maruki Y, Nakamura M, Tomemori Y, Kamae K, Tanabe H, et al. The gene encoding a newly discovered protein, chorein, is mutated in chorea-acanthocytosis. Nat Genet. 2001 Jun;28((2)):121–2. doi: 10.1038/88825. [DOI] [PubMed] [Google Scholar]
- 29.Bader B, Dobson-Stone C, Velayos-Baeza A, Monaco AP, Danek A. The genotype and phenotype of chorea-acanthocytosis. Eur J Neurol. 2008;15((Suppl 3)):16. [Google Scholar]
- 30.Velayos-Baeza A, Vettori A, Copley RR, Dobson-Stone C, Monaco AP. Analysis of the human VPS13 gene family. Genomics. 2004 Sep;84((3)):536–49. doi: 10.1016/j.ygeno.2004.04.012. [DOI] [PubMed] [Google Scholar]
- 31.Kolehmainen J, Black GCM, Saarinen A, Chandler K, Clayton-Smith J, Träskelin A-L, et al. Cohen syndrome is caused by mutations in a novel gene, COH1, encoding a transmembrane protein with a presumed role in vesicle-mediated sorting and intracellular protein transport. Am J Hum Genet. 2003 Jun;72((6)):1359–69. doi: 10.1086/375454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lesage S, Drouet V, Majounie E, Deramecourt V, Jacoupy M, Nicolas A, et al. Loss of VPS13C function in autosomal-recessive parkinsonism causes mitochondrial dysfunction and increases PINK1/parkin-dependent mitophagy. Am J Hum Genet. 2016 Mar;98((3)):500–13. doi: 10.1016/j.ajhg.2016.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schormair B, Kemlink D, Mollenhauer B, Fiala O, Machetanz G, Roth J, et al. Diagnostic exome sequencing in early-onset Parkinson's disease confirms VPS13C as a rare cause of autosomal-recessive Parkinson's disease. Clin Genet. 2018;93((3)):603–12. doi: 10.1111/cge.13124. [DOI] [PubMed] [Google Scholar]
- 34.Smolders S, Philtjens S, Crosiers D, Sieben A, Hens E, Heeman B, et al. Contribution of rare homozygous and compound heterozygous VPS13C missense mutations to dementia with Lewy bodies and Parkinson's disease. Acta Neuropathol Commun. 2021 Feb;9((1)):25. doi: 10.1186/s40478-021-01121-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Meijer IA. VPS13D movement disorder. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJ, Stephens K, et al., editors. GeneReviews®. Seattle: University of Washington; 2019. Available from: http://www.ncbi.nlm.nih.gov/books/NBK537720/ [PubMed] [Google Scholar]
- 36.Jung HH, Danek A, Walker RH, Frey BM, Peikert K. McLeod neuroacanthocytosis syndrome. GeneReviews®. Seattle: University of Washington;; 2021. Available from: http://www.ncbi.nlm.nih.gov/books/NBK1354/ [PubMed] [Google Scholar]
- 37.Walker RH, Miranda M, Jung HH, Danek A. Life expectancy and mortality in chorea-acanthocytosis and McLeod syndrome. Parkinsonism Relat Disord. 2019 Mar;60:158–61. doi: 10.1016/j.parkreldis.2018.09.003. [DOI] [PubMed] [Google Scholar]
- 38.Danek A, Uttner I, Vogl T, Tatsch K, Witt TN. Cerebral involvement in McLeod syndrome. Neurology. 1994;44((1)):117–20. doi: 10.1212/wnl.44.1.117. [DOI] [PubMed] [Google Scholar]
- 39.Jung HH, Hergersberg M, Kneifel S, Alkadhi H, Schiess R, Weigell-Weber M, et al. McLeod syndrome: a novel mutation, predominant psychiatric manifestations, and distinct striatal imaging findings. Ann Neurol. 2001 Mar;49((3)):384–92. [PubMed] [Google Scholar]
- 40.Valko PO, Hänggi J, Meyer M, Jung HH. Evolution of striatal degeneration in McLeod syndrome. Eur J Neurol. 2010;17((4)):612–8. doi: 10.1111/j.1468-1331.2009.02872.x. [DOI] [PubMed] [Google Scholar]
- 41.Ehrlich DJ, Walker RH. Functional neuroimaging and chorea: a systematic review. J Clin Mov Disord. 2017 Dec;4((1)):8. doi: 10.1186/s40734-017-0056-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ryu HS, Hong CM. Brain F-18 FDG and F-18 FP-CIT PET/CT findings of c.856_860delCTCTA mutation McLeod syndrome. Cogn Behav Neurol. 2021 Sep;34((3)):207–11. doi: 10.1097/WNN.0000000000000267. [DOI] [PubMed] [Google Scholar]
- 43.Danek A, Neumann M, Brin MF, Symmans WA, Hays AP. Cerebral involvement in McLeod syndrome: the first autopsy revisited. In: Walker RH, Saiki S, Danek A, editors. Neuroacanthocytosis syndromes II. Berlin: Springer; 2008. pp. p. 205–15. [Google Scholar]
- 44.Geser F, Tolnay M, Jung HH. The neuropathology of McLeod syndrome. In: Walker RH, Saiki S, Danek A, editors. Neuroacanthocytosis syndromes II. Berlin: Springer; 2008. pp. p. 197–203. [Google Scholar]
- 45.Rinne JO, Daniel SE, Scaravilli F, Pires M, Harding AE, Marsden CD. The neuropathological features of neuroacanthocytosis. Mov Disord. 1994;9((3)):297–304. doi: 10.1002/mds.870090303. [DOI] [PubMed] [Google Scholar]
- 46.Rinne JO, Daniel SE, Scaravilli F, Harding AE, Marsden CD. Nigral degeneration in neuroacanthocytosis. Neurology. 1994;44((9)):1629–32. doi: 10.1212/wnl.44.9.1629. [DOI] [PubMed] [Google Scholar]
- 47.Deutschländer AB, Dickson DW, Wszolek ZK. Neuropathology of McLeod syndrome. Mov Disord. 2022 doi: 10.1002/mds.28882. [DOI] [PubMed] [Google Scholar]
- 48.Danek A, Tison F, Rubio J, Oechsner M, Kalckreuth W, Monaco AP. The chorea of McLeod syndrome. Mov Disord. 2001 Sep;16((5)):882–9. doi: 10.1002/mds.1188. [DOI] [PubMed] [Google Scholar]
- 49.Gantenbein AR, Damon-Perrière N, Bohlender JE, Chauveau M, Latxague C, Miranda M, et al. Feeding dystonia in McLeod syndrome. Mov Disord. 2011 Sep;26((11)):2123–6. doi: 10.1002/mds.23843. [DOI] [PubMed] [Google Scholar]
- 50.Chauveau M, Damon-Perriere N, Latxague C, Spampinato U, Jung H, Burbaud P, et al. Head drops are also observed in McLeod syndrome. Mov Disord. 2011 Jul;26((8)):1562–3. doi: 10.1002/mds.23605. [DOI] [PubMed] [Google Scholar]
- 51.Danek A, Sheesley L, Tierney M, Uttner I, Grafman J. Cognitive and neuropsychiatric findings in McLeod syndrome and in chorea-acanthocytosis. In: Danek A, editor. Neuroacanthocytosis syndromes. Dordrecht: Springer; 2004. pp. p. 95–115. [Google Scholar]
- 52.Walterfang M, Evans A, Looi JCL, Jung HH, Danek A, Walker RH, et al. The neuropsychiatry of neuroacanthocytosis syndromes. Neurosci Biobehav Rev. 2011 Apr;35((5)):1275–83. doi: 10.1016/j.neubiorev.2011.01.001. [DOI] [PubMed] [Google Scholar]
- 53.Jung HH, Haker H. Schizophrenia as a manifestation of X-linked Mcleod-neuroacanthocytosis syndrome. J Clin Psychiatry. 2004 May;65((5)):722–3. doi: 10.4088/jcp.v65n0520c. [DOI] [PubMed] [Google Scholar]
- 54.Marsh WL, Marsh NJ, Moore A, Symmans WA, Johnson CL, Redman CM. Elevated serum creatine phosphokinase in subjects with McLeod syndrome. Vox Sang. 1981 Jun;40((6)):403–11. doi: 10.1111/j.1423-0410.1981.tb00728.x. [DOI] [PubMed] [Google Scholar]
- 55.Jung HH, Brandner S. Malignant McLeod myopathy. Muscle Nerve. 2002 Sep;26((3)):424–7. doi: 10.1002/mus.10199. [DOI] [PubMed] [Google Scholar]
- 56.Dotti MT, Malandrini A, Federico A. Neuromuscular findings in eight Italian families with neuroacanthocytosis. In: Danek A, editor. Neuroacanthocytosis syndromes. Dordrecht: Springer; 2004. pp. p. 127–38. [Google Scholar]
- 57.Vaisfeld A, Bruno G, Petracca M, Bentivoglio AR, Servidei S, Vita MG. Neuroacanthocytosis syndromes in an Italian cohort: clinical spectrum, high genetic variability and muscle involvement. Genes (Basel) 2021 Feb;12((3)):344. doi: 10.3390/genes12030344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Witt TN, Danek A, Reiter M, Heim MU, Dirschinger J, Olsen EG. McLeod syndrome: a distinct form of neuroacanthocytosis. Report of two cases and literature review with emphasis on neuromuscular manifestations. J Neurol. 1992;239((6)):302–6. doi: 10.1007/BF00867584. [DOI] [PubMed] [Google Scholar]
- 59.Díaz-Manera J, Sotoca-Fernández J, Alonso-Jiménez A, Marzo ME, Gallardo E, Segovia-Simón S, et al. McLeod syndrome is a new cause of axial muscle weakness: axial myopathy secondary to McLeod syndrome. Muscle Nerve. 2018 Jul;58((1)):E5–8. doi: 10.1002/mus.26086. [DOI] [PubMed] [Google Scholar]
- 60.Barnett MH, Yang F, Iland H, Pollard JD. Unusual muscle pathology in McLeod syndrome. J Neurol Neurosurg Psychiatry. 2000;69((5)):655–7. doi: 10.1136/jnnp.69.5.655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Montagnese F, Grabmaier U, Abicht A, Schoser B. Dilative cardiomyopathy displaying double trouble etiology: myocarditis and McLeod syndrome? Clin Neurol Neurosurg. 2020 Oct;197:106122. doi: 10.1016/j.clineuro.2020.106122. [DOI] [PubMed] [Google Scholar]
- 62.Oechslin E, Kaup D, Jenni R, Jung HH. Cardiac abnormalities in McLeod syndrome. Int J Cardiol. 2009 Feb;132((1)):130–2. doi: 10.1016/j.ijcard.2007.07.167. [DOI] [PubMed] [Google Scholar]
- 63.Laurencin C, Sebbag L, Jousserand G, Demontes M, Campean L, Thivolet-Bejui F, et al. Novel XK mutation in a McLeod patient diagnosed after heart transplant. Clin Neurol Neurosurg. 2018;168:64–6. doi: 10.1016/j.clineuro.2018.02.039. [DOI] [PubMed] [Google Scholar]
- 64.Quick S, Heidrich FM, Winkler M-V, Winkler AH, Ibrahim K, Linke A, et al. Cardiac manifestation is evident in chorea-acanthocytosis but different from McLeod syndrome. Parkinsonism Relat Disord. 2021 Jul;88:90–5. doi: 10.1016/j.parkreldis.2021.05.015. [DOI] [PubMed] [Google Scholar]
- 65.Floch A, Lomas-Francis C, Vege S, Westhoff CM. Three new XK alleles; two associated with a McLeod RBC phenotype. Transfusion. 2021 Oct;61((10)):E69–70. doi: 10.1111/trf.16650. [DOI] [PubMed] [Google Scholar]
- 66.Jung HH, Hergersberg M, Vogt M, Pahnke J, Treyer V, Röthlisberger B, et al. McLeod phenotype associated with a XK missense mutation without hematologic, neuromuscular, or cerebral involvement. Transfusion. 2003 Jul;43((7)):928–38. doi: 10.1046/j.1537-2995.2003.t01-1-00434.x. [DOI] [PubMed] [Google Scholar]
- 67.Walker RH, Danek A, Uttner I, Offner R, Reid M, Lee S. McLeod phenotype without the McLeod syndrome. Transfusion. 2007 Feb;47((2)):299–305. doi: 10.1111/j.1537-2995.2007.01106.x. [DOI] [PubMed] [Google Scholar]
- 68.Walker RH. The differential diagnosis of chorea. New York: Oxford University Press; 2010. [Google Scholar]
- 69.Storch A, Kornhass M, Schwarz J. Testing for acanthocytosis: a prospective reader-blinded study in movement disorder patients. J Neurol. 2005;252((1)):84–90. doi: 10.1007/s00415-005-0616-3. [DOI] [PubMed] [Google Scholar]
- 70.Klempíř J, Roth J, Zárubová K, Písačka M, Špačková N, Tilley L. The McLeod syndrome without acanthocytes. Parkinsonism Relat Disord. 2008 May;14((4)):364–6. doi: 10.1016/j.parkreldis.2007.07.011. [DOI] [PubMed] [Google Scholar]
- 71.Gandhi S, Hardie RJ, Lees AJ. An update on the Hardie neuroacanthocytosis series. In: Walker RH, Saiki S, Danek A, editors. Neuroacanthocytosis syndromes II. Berlin: Springer; 2008. pp. p. 43–51. [Google Scholar]
- 72.Faillace RT, Kingston WJ, Nanda NC, Griggs RC. Cardiomyopathy associated with the syndrome of amyotrophic chorea and acanthocytosis. Ann Intern Med. 1982;96:616–7. doi: 10.7326/0003-4819-96-5-616. [DOI] [PubMed] [Google Scholar]
- 73.Marsh WL. Deleted antigens of the rhesus and Kell blood groups: association with cell membrane defects. In: Garraty G, editor. Blood group antigens and disease. Arlington: American Association of Blood Banks; 1983. pp. p. 165–85. [Google Scholar]
- 74.Frey BM, Gassner C, Jung HH. Neurodegeneration in the elderly − when the blood type matters: an overview of the McLeod syndrome with focus on hematological features. Transfus Apher Sci. 2015 Jun;52((3)):277–84. doi: 10.1016/j.transci.2015.04.007. [DOI] [PubMed] [Google Scholar]
- 75.Gassner C, Brönnimann C, Merki Y, Mattle-Greminger MP, Sigurdardottir S, Meyer E, et al. Stepwise partitioning of Xp21: a profiling method for XK deletions causative of the McLeod syndrome. Transfusion. 2017;57((9)):2125–35. doi: 10.1111/trf.14172. [DOI] [PubMed] [Google Scholar]
- 76.Dobson-Stone C, Velayos-Baeza A, Filippone LA, Westbury S, Storch A, Erdmann T, et al. Chorein detection for the diagnosis of chorea-acanthocytosis. Ann Neurol. 2004 Aug;56((2)):299–302. doi: 10.1002/ana.20200. [DOI] [PubMed] [Google Scholar]
- 77.Miltenberger-Miltenyi G, Bader B, Velayos-Baeza A, Burghaus L, Goldsmith P, Abicht A, et al. Normal chorein signal on red cell membrane Western blotting in VPS13A disease. 2021. p. p. 1. [Google Scholar]
- 78.Peikert K, Glaß H, Federti E, Matte A, Pelzl L, Akgün K, et al. Targeting Lyn kinase in chorea-acanthocytosis: a translational treatment approach in a rare disease. J Pers Med. 2021 May;11((5)):392. doi: 10.3390/jpm11050392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Peikert K, Federti E, Matte A, Constantin G, Pietronigro EC, Fabene PF, et al. Therapeutic targeting of Lyn kinase to treat chorea-acanthocytosis. Acta Neuropathol Commun. 2021 May;9((1)):81. doi: 10.1186/s40478-021-01181-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Rabe A, Kihm A, Darras A, Peikert K, Simionato G, Dasanna AK, et al. The erythrocyte sedimentation rate and its relation to cell shape and rigidity of red blood cells from chorea-acanthocytosis patients in an off-label treatment with dasatinib. Biomolecules. 2021 May;11((5)):727. doi: 10.3390/biom11050727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Park J-S, Neiman AM. XK is a partner for VPS13A: a molecular link between chorea-acanthocytosis and McLeod syndrome. Mol Biol Cell. 2020 Oct;31((22)):2425–36. doi: 10.1091/mbc.E19-08-0439-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Danek A. Neuroacanthocytosis syndromes: what links red blood cells and neurons? In: Danek A, editor. Neuroacanthocytosis Syndromes. Dordrecht: Springer-Verlag; 2004. pp. p. 1–14. [Google Scholar]
- 83.Russo D, Redman C, Lee S. Association of XK and Kell blood group proteins. J Biol Chem. 1998 May;273((22)):13950–6. doi: 10.1074/jbc.273.22.13950. [DOI] [PubMed] [Google Scholar]
- 84.Lux SE. Anatomy of the red cell membrane skeleton: unanswered questions. Blood. 2016 Jan;127((2)):187–99. doi: 10.1182/blood-2014-12-512772. [DOI] [PubMed] [Google Scholar]
- 85.Clapéron A, Hattab C, Armand V, Trottier S, Bertrand O, Ouimet T. The Kell and XK proteins of the Kell blood group are not co-expressed in the central nervous system. Brain Res. 2007 May;1147:12–24. doi: 10.1016/j.brainres.2007.01.106. [DOI] [PubMed] [Google Scholar]
- 86.Russo D, Wu X, Redman CM, Lee S. Expression of Kell blood group protein in nonerythroid tissues. Blood. 2000 Jan;96((1)):340–6. [PubMed] [Google Scholar]
- 87.Jung HH, Russo D, Redman C, Brandner S. Kell and XK immunohistochemistry in McLeod myopathy. Muscle Nerve. 2001 Oct;24((10)):1346–51. doi: 10.1002/mus.1154. [DOI] [PubMed] [Google Scholar]
- 88.Redman CM, Huima T, Robbins E, Lee S, Marsh WL. Effect of phosphatidylserine on the shape of McLeod red cell acanthocytes. Blood. 1989 Oct;74((5)):1826–35. [PubMed] [Google Scholar]
- 89.Suzuki J, Denning DP, Imanishi E, Horvitz HR, Nagata S. Xk-related protein 8 and CED-8 promote phosphatidylserine exposure in apoptotic cells. Science. 2013 Jul;341((6144)):403–6. doi: 10.1126/science.1236758. [DOI] [PubMed] [Google Scholar]
- 90.Suzuki J, Imanishi E, Nagata S. Exposure of phosphatidylserine by Xk-related protein family members during apoptosis. J Biol Chem. 2014 Oct;289((44)):30257–67. doi: 10.1074/jbc.M114.583419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Sakuragi T, Kanai R, Tsutsumi A, Narita H, Onishi E, Nishino K, et al. The tertiary structure of the human Xkr8-Basigin complex that scrambles phospholipids at plasma membranes. Nat Struct Mol Biol. 2021 Oct;28((10)):825–34. doi: 10.1038/s41594-021-00665-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Straub MS, Alvadia C, Sawicka M, Dutzler R. Cryo-EM structures of the caspase-activated protein XKR9 involved in apoptotic lipid scrambling. eLife. 2021 Jul;10:e69800. doi: 10.7554/eLife.69800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Dziurdzik SK, Conibear E. The VPS13 family of lipid transporters and its role at membrane contact sites. Int J Mol Sci. 2021 Mar;22((6)):2905. doi: 10.3390/ijms22062905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Leonzino M, Reinisch KM, De Camilli P. Insights into VPS13 properties and function reveal a new mechanism of eukaryotic lipid transport. Biochim Biophys Acta Mol Cell Biol Lipids. 2021 Oct;1866((10)):159003. doi: 10.1016/j.bbalip.2021.159003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kumar N, Leonzino M, Hancock-Cerutti W, Horenkamp FA, Li P, Lees JA, et al. VPS13A and VPS13C are lipid transport proteins differentially localized at ER contact sites. J Cell Biol. 2018 Oct;217((10)):3625–39. doi: 10.1083/jcb.201807019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Yeshaw WM, van der Zwaag M, Pinto F, Lahaye LL, Faber AI, Gómez-Sánchez R, et al. Human VPS13A is associated with multiple organelles and influences mitochondrial morphology and lipid droplet motility. Elife. 2019 Feb;8:e43561. doi: 10.7554/eLife.43561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Muñoz-Braceras S, Tornero-Écija AR, Vincent O, Escalante R. *VPS13A is closely associated with mitochondria and is required for efficient lysosomal degradation. Dis Model Mech. 2019 Feb;12((2)):dmm036681. doi: 10.1242/dmm.036681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Bean BDM, Dziurdzik SK, Kolehmainen KL, Fowler CMS, Kwong WK, Grad LI, et al. Competitive organelle-specific adaptors recruit Vps13 to membrane contact sites. J Cell Biol. 2018 Oct;217((10)):3593–607. doi: 10.1083/jcb.201804111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Urata Y, Nakamura M, Sasaki N, Shiokawa N, Nishida Y, Arai K, et al. Novel pathogenic XK mutations in McLeod syndrome and interaction between XK protein and chorein. Neurol Genet. 2019 Jun;5((3)):e328. doi: 10.1212/NXG.0000000000000328. [DOI] [PMC free article] [PubMed] [Google Scholar]