

Abstract
Objective: RNA extraction and library preparation from formalin-fixed, paraffin-embedded (FFPE) samples are crucial pre-analytical steps towards achieving optimal downstream RNA sequencing (RNASeq) results. In this study, we assessed 2 Illumina library preparation methods for RNA-Seq analysis using archived FFPE samples from human cancer indications at 2 independent vendors. Methods: Twenty-five FFPE samples from 5 indications (non-small cell lung cancer, colorectal cancer, renal carcinoma, breast cancer, and hepatocellular carcinoma) were included, covering a wide range of sample storage durations (3-25 years-old), sample qualities, and specimen types (resection vs core needle biopsy). Each sample was processed independently by both vendors. Total RNA was isolated using the Qiagen miRNeasy FFPE kit followed by library construction using either TruSeq Stranded Total RNA library preparation kit with Ribo-Zero Gold, or TruSeq RNA Access library preparation kit. Libraries were normalized to 20 pM and sequenced on an Illumina HiSeq 2500 using V3 chemistry in paired-end mode with a read length of 2 × 50 bp. The data were processed through a standard RNASeq pipeline to produce counts and transcripts per millions for each gene in each sample to compare 2 library kits at 2 different vendors. Results: Our data showed that TruSeq RNA Access libraries yield over 80% exonic reads across different quality samples, indicating higher selectivity of the exome pull down by the capture approach compared to the random priming of the TruSeq Stranded Total kit. The overall QC data for FFPE RNA extraction, library preparation, and sequencing generated by the 2 vendors are comparable, and downstream gene expression quantification results show high concordance as well. With the TruSeq Stranded Total kit, the mean Spearman correlation between vendors was 0.87 and the mean Pearson correlation was 0.76. With the TruSeq RNA Access kit, the mean Spearman correlation between vendors was 0.89 and the mean Pearson correlation was 0.73. Interestingly, examination of the cross-vendor correlations compared to various common QC statistics suggested that library concentration is better correlated with consistency between vendors than is the RNA quantity. Conclusions: Our analyses provide evidence to guide selection of sequencing methods for FFPE samples in which the sample quality may be severely compromised.
Keywords: Gene expression, RNASeq, breast cancer, renal cancer, non-small cell lung cancer, colorectal cancer, hepatocellular carcinoma
Introduction
High analytical sensitivity and broad dynamic range render RNA sequencing (RNA-Seq) very appealing for mRNA expression analyses in clinical biomarker development and identification for enabling precision oncology. 1 However, the reliability and accuracy of RNA-Seq data is largely dependent on template RNA quality and input amount as well as the cDNA library preparation methods applied, especially in samples with suboptimal quality that is extracted from FFPE specimens. 2 Several next generation sequencing (NGS) protocols are currently available for the profiling of suboptimal RNA samples, including RNase H, Ribo-Zero, DSN-lite, NuGEN, SMART, and exome capture, each with its own strengths and weakness.3–5
Among these NGS protocols, Illumina offers 2 library preparation methods for samples with suboptimal quality: the TruSeq RNA Access library preparation method is based on RNA capture by targeting known exons with exon capture probes to enrich for coding RNAs; 4 the TruSeq Stranded Total RNA library kit with Ribo-Zero rRNA removal (TruSeq Stranded Total RNA) is a method that reduces the highly abundant ribosomal RNAs from total RNA samples using ribosomal capture probes. 3 The TruSeq RNA Access protocol is not only suitable for the profiling of samples of severely compromised quality, but also appropriate for very heterogeneous RNA samples including a wider range of low quantity and extremely low-quality samples. 6 The performance of the TruSeq Stranded Total RNA and TruSeq RNA Access library preparation kits has been evaluated on well-established human reference RNA samples from the Microarray/Sequencing Quality Control consortium. 7 It is essential to conduct a systemic comparison of these protocols using human samples across various cancer indications using different vendors to ensure clinical translatability.
In this study, we compared the performance of the Illumina TruSeq Stranded Total RNA and Illumina TruSeq RNA Access library preparation kits using 25 FFPE samples from patients with 5 cancers of various sample quality, age of samples, and sample type and between 2 vendors (Vendor A and Vendor B).
Materials and Methods
Clinical Samples
Twenty-five FFPE samples from 5 indications (non-small cell lung cancer, colorectal cancer, renal carcinoma, breast cancer, and hepatocellular carcinoma) of various sample quality, age of samples (collection year: 1993-2015) and sample type (22 resection vs 3 core needle biopsy) were procured from 3 suppliers (Asterand, Cureline and Conversant Biologics, Inc.). The written informed consent was received from all subjects used in this study and all samples were collected under the institutional review board approved protocols, as defined by the specimen providers. Ethical approval was obtained from Asterand, Cureline and Conversant Biologics, Inc. ethics committee, respectively. This study was conducted under the guidelines put forth into the Declaration of Helsinki. The same set of samples were processed at both vendors and with both library preparation kits by splitting the available FFPE slides. Both vendors used the same protocols from RNA extraction to sequencing and both are CLIA/CAP certified diagnostics laboratories (Vendor A and Vendor B).
RNA Extraction and Assessment of Quality
The RNA extraction of FFPE tumor specimens was performed on five 5 μm-deep tissue cuts using the Qiagen RNeasy Mini Kit (Qiagen), according to the manufacturer's recommendations. Total RNA concentration was measured using Qubit® RNA HS Assay Kit on a Qubit® 2.0 Fluorometer (Thermo Fisher Scientific Inc.). Integrity was assessed using Agilent RNA 6000 Nano Kit on a 2100 Bioanalyzer instrument (Agilent Technologies). The RNA integrity number (RIN) score and the percentages of fragments larger than 200 nucleotides (DV200) were calculated. According to Agilent 2100 bioanalyzer system assessment and Illumina library preparation input recommendation, the degraded RNA samples can be classified according to their size distribution DV200. FFPE RNA samples with DV200 >70% are high-quality samples, 50% to 70% are medium quality samples, 30% to 50% is defined as low-quality FFPE while DV200 <30% indicates the FFPE RNA is likely too degraded for RNASeq.
RNA Library Construction and Sequencing
Ribosomal RNA depleted strand-specific RNA libraries were generated with the TruSeq Stranded Total RNA sample preparation kit with Ribo-Zero Gold (#RS-122-2301 and #RS-122-2302, Illumina) and transcriptome capture based libraries were generated with the TruSeq RNA Access Library Prep Kit (#RS-301-2001, Illumina). All protocols were performed following the manufacturer's instructions. Each library was sequenced on an Illumina HiSeq 2500 (Illumina, Inc.) using V3 chemistry, in paired-end mode with a read length of 2 × 50 bp. Each library was normalized to 20 pM and subjected to cluster and pair read sequencing was performed for 50 cycles on a HiSeq2500 instrument, according to the manufacturer's instructions. As the data generated here was initially for other purposes and not for cross-vendor comparison, the vendors used different sequencing depths. Vendor B generated 2 × 50 bp paired end sequencing at minimum 50 million reads per sample from TruSeq Stranded Total RNA library construction whereas 100 million reads per sample from TruSeq RNA Access library preparation. Vendor A generated at minimum 100 million reads per sample from both library preparation. Image analysis, base calling, and base quality scoring of the run were processed on the HiSeq instrument by Real Time Analysis (RTA 1.17.21.3) and followed by generation of FASTQ sequence files by CASAVA 1.8 (Illumina, Inc.). Data are available in the repository NCBI Sequence Read Archive, accession number PRJNA660476.
Data Processing
All raw data of the samples were processed through a standard RNASeq pipeline to produce counts and transcripts per million (TPM) for each gene in each sample. Reads were aligned with STAR version 2.5.2b 8 against hg19 and the gencode gene annotations version 24. 9 RSEM version 1.2.29 was then used to quantify and compute TPMs. 10 All downstream processing of the TPM and counts was performed in R version 3.6.1. 11 QC was performed on the STAR output using Picard (http://broadinstitute.github.io/picard). Unless otherwise noted, we present output only using the 15 samples which passed QC for all 4 attempts (2 vendors times 2 protocols). We quantified data quality for a sample in terms of the number of genes detected (count greater than zero) and by the 90th percentile count in a sample (low RNA input often yields extremely high amplification of a few highly expressed genes and few reads at the vast majority of genes, leading to a low 90th percentile count). We assessed the results in terms of Spearman and Pearson correlation of TPM values between vendors for the same kit, and between the 2 kits at the same vendor. Spearman correlation measures whether 2 sets of values are in the same order, even if the relationship is nonlinear, while the Pearson correlation measures whether the relationship between 2 datasets is linear. For example, if the values in one dataset are the square of the values in the other dataset, they would have a high Spearman correlation but low Pearson correlation. Differences between groups were tested using t-test or analysis of variance, where appropriate.
Results
RNA and Library QC Measurements
To evaluate the performance of RNA-seq methods in profiling FFPE samples, we conducted a technical assessment of the 2 different RNA library preparation protocols on 25 FFPE samples (Figure 1). The samples were first sent to Vendor A. There, 25 FFPE samples were extracted and analyzed for RNA integrity and quality. A total of 23 FFPE specimens had sufficient yield (>100 ng, average DV200 is 28% with the range from 5% to 51%) to proceed to TruSeq Stranded Total RNA library preparation. Four sample libraries had a final concentration of less 2 nM and therefore did not proceed to sequencing. A total of 19 libraries had sufficient yield to proceed to sequencing. All sequenced samples generated adequate reads (100 M or greater). Since more RNA sample is required in the TruSeq Stranded Total RNA protocol, only 21 samples had sufficient RNA remaining (total yield >20 ng, average DV200 is 27 with the range from 5 to 51) for the TruSeq RNA Access library preparation kit. Two Access library final concentration were less than 2 nM and failed library QC, then the remaining 19 samples were sequenced.
Figure 1.

Study design & workflow. Schematic of the sample flow through the 2 vendors and 2 protocols. The number of samples processed at each step is noted.
Due to insufficient FFPE slides for 3 samples, Vendor B performed RNA extraction on the 22 remaining FFPE samples. From these 22 samples, all extracted RNA passed extraction QC (total yield >100 ng, average DV200 is 44% with the range from 12% to 72%) and then could proceed to TruSeq Stranded Total RNA library preparation. Following library preparation steps, all 22 samples had sufficient yield and could proceed to sequencing. After the TruSeq Stranded Total RNA library process, 2 samples did not have sufficient material to process in the TruSeq RNA access workflow. The remaining 20 RNA samples (total yield >20 ng, average DV200 is 44% with the range from 12% to 72%) were reprepared using the Illumina TruSeq RNA Access library kit and 16 samples passed the library QC for sequencing. From our original cohort of 25 samples, 15 samples were processed and sequenced at both vendors and with both kits. This cohort of 15 samples was thus used for further analysis.
For each vendor and kit, Table 1 shows the mean (and range or standard deviation) for process QC measures. The library preparation output is characterized by the average fragment size (measured by Bioanalyzer) and the library concentration. The sequencing output is characterized by the number of reads and a variety of metrics concerning the read alignment rates to exons and ribosomal regions. Supplemental Table 1 shows the sample annotations and presequencing QC results for each sample. Supplemental Figures 1-3 show the relationship between vendors for amount of RNA extracted and for the library concentrations.
Table 1.
Illumina TruSeq RNA Access Versus TruSeq Stranded Total RNA: Overall QC and Alignment Stats.
| Step | QC measure | Vendor A | Vendor B | ||
|---|---|---|---|---|---|
| Total Stranded
a
(n = 15) |
RNA Access
a
(n = 15) |
Total Stranded
b
(n = 15) |
RNA Access
a
(n = 15) |
||
| Library Prep | Average Fragment size: Mean (Range) | 318 (260-413) |
309.83 (280-326) |
296.63 (268-324) |
324.5 (280-394) |
| Concentration (nM): Mean (Range) | 57.43 (18.68-108.81) |
51.24 (3.89-150.63) |
224.57 (6.51-507.85) |
205.81 (9.76-600.62) |
|
| Sequencing | Total paired end reads: Mean (Range) | 137 M (117-157) |
141 M (104-169) |
64.7 M (22.5-191) |
184 M (114-294) |
| %Aligned reads rate: Mean ± SD | 89.6 ± 10.8 | 95.2 ± 1.0 | 86.3 ± 7.8 | 92.7 ± 1.5 | |
| %Exonic rate: Mean ± SD | 18.5 ± 4.7 | 81.0 ± 2.3 | 41.6 ± 13.2 | 84 ± 2.4 | |
| %Intragenic rate: Mean ± SD | 83.1 ± 6.8 | 89.1 ± 2.3 | 81.5 ± 17.6 | 92.1 ± 1.9 | |
| %rRNA rate: Mean ± SD | 2.0 ± 1.9 | 2.2 ± 1.6 | 9.9 ± 1.83 | 1.8 ± 2.2 | |
| % Correct strand reads rate: Mean ± SD | 94.1 ± 3.3 | 95.9 ± 2.3 | 97.5 ± 1.3 | 97.8 ± 1.5 | |
2 × 50 bp paired end sequencing at minimum 100 million reads per sample.
2 × 50 bp paired end sequencing at minimum 50 million reads per sample.
Figure 3.

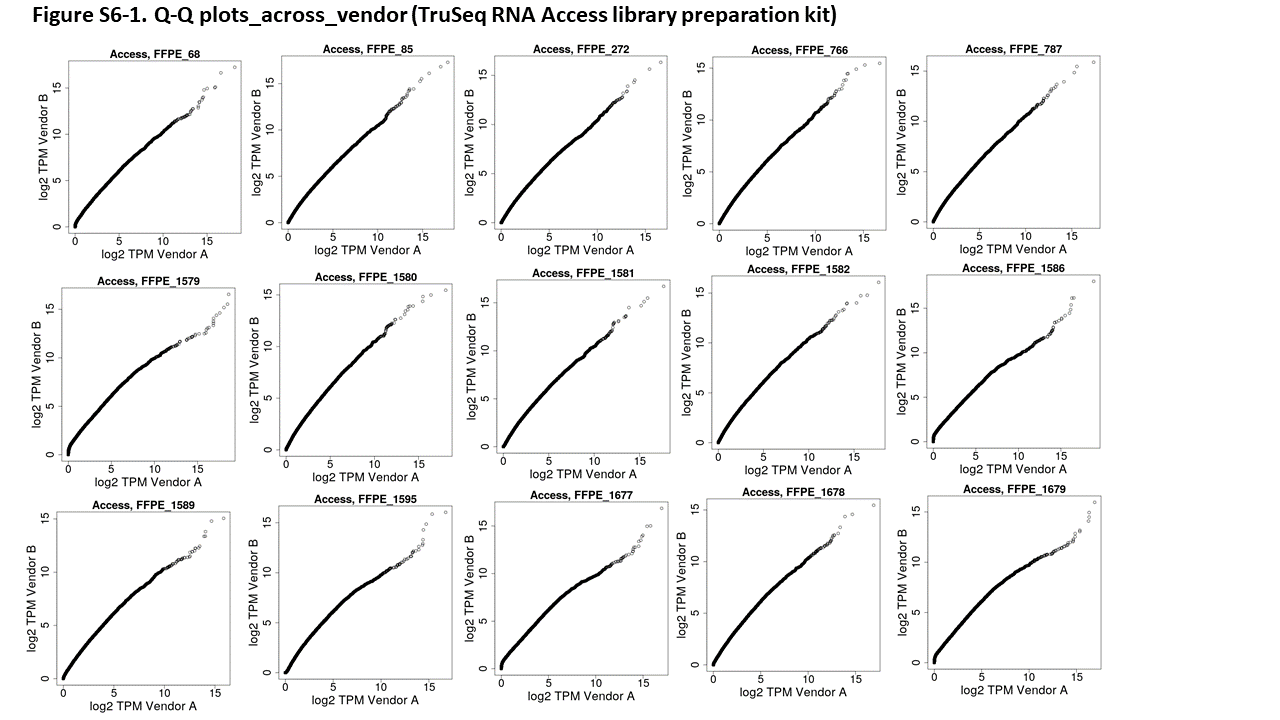
Q–Q plots. The Q–Q plots help visualize the shape of the correlation or distribution between the 2 kits. Here, the data for sample FFPE_766 is shown from both vendors. At both, the majority of the plot shows a straight diagonal line, indicating identical distribution of TPMs for most percentiles. However, the highest percentiles diverge and the TruSeq Stranded Total RNA kit shows higher levels than the TruSeq RNA Access kit. The plot should not be interpreted to mean that either kit is necessarily correct; only that the highest expressed genes in the TruSeq Stranded Total RNA kit yield higher TPM values than the highest expressed genes in the TruSeq RNA Access kit. The plots also show divergence at very low expression values, potentially genes which are not present in the Access probe set and thus generate no signal in the TruSeq RNA Access results while generating some signal in the TruSeq Stranded Total RNA kit.
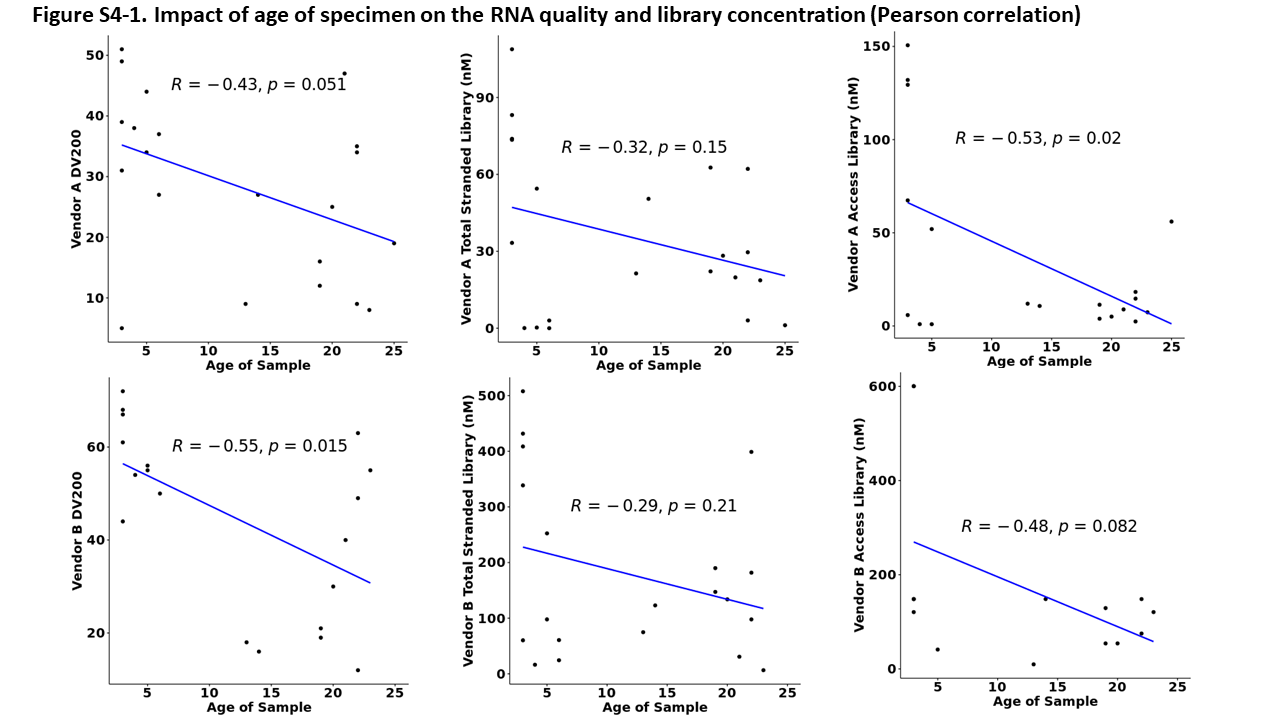
By correlating the extracted RNA quality (DV200) with the age of the specimen, we observed lower RNA quality in older samples (Vendor A: P = .051; Vendor B: P = .015). The concentration of libraries prepared by TruSeq RNA Access library preparation kit at Vendor A was significant decreased with the age of specimen (P = .02). However, there was no association for Vendor B (Supplemental Figure 4). The age of the specimen did not correlate with the concentration of libraries prepared by TruSeq Stranded Total RNA library kit at either vendor. The amount of tissue didn't affect the RNA yield—here was no significant difference in the total amount of RNA extraction between the resection samples (Vendor A: 3.72 ± 6.10; Vendor B: 1.21 ± 2.06) and core needle biopsy samples (Vendor A: 2.50 ± 0.58; Vendor B: 0.24 ± 0.17) at both vendors (Vendor A: p = .37; Vendor B: p = .06). The type of the cancer also didn't affect either the RNA yield or RNA quality (DV200) at either vendor (Vendor A DV200 P = .30; Vendor B DV200, P = .11; Vendor A RNA yield P = .35; Vendor B P = .44).
Alignment Statistics
Our results showed that the TruSeq RNA Access library preparation protocol produced higher alignment rates at both vendors (means 95% and 93% vs 83% and 78%; Table 1). Compared to the TruSeq Stranded Total RNA protocol, the TruSeq RNA Access protocol showed marked differences in the percent of reads aligned to exons, introns, and intergenic regions. For TruSeq RNA Access the percentages of exonic reads were over 80% across different quality samples at both vendors, reflecting the high efficiency of the exome pull down by the capture approach. The mean exonic percentages were 81% and 84% with the TruSeq RNA Access kit and 17% and 31% with the TruSeq Stranded Total kit. Supplemental Table 2 shows per-sample data, including output from Picard, genes detected (> = 1 read), and the 90th percentile gene count.
Agreement Between Vendors
The 2 kits showed similar correlation between vendors. With the TruSeq Stranded Total RNA kit, the mean per-sample Spearman correlation between vendors was 0.87 and the mean Pearson correlation was 0.76. With the TruSeq RNA Access kit, the mean per-sample Spearman correlation between vendors was 0.89 and the mean Pearson correlation was 0.73. Per-sample correlations across vendors are in Supplemental Table 3 and Supplemental Figure 5 contains the scatterplots. The relationship of logTPMs between vendors appears to be roughly linear for both kits, though the RNA Access kit shows a slight bend. Across individual samples, the correlation between vendors ranged from R = 0.94 to R = 0.01 (Figure 2).
Figure 2.

Example cross-vendor scatterplots. The overall correlation between vendors ranged from excellent (eg, A: FFPE_1582 in both TruSeq Stranded Total RNA [R = 0.873, rho = 0.927] and TruSeq RNA Access kit [R = 0.858, rho = 0.927]) to moderate (eg, B: FFPE_1579 in both TruSeq Stranded Total RNA [R = 0.012, rho = 0.760] and TruSeq RNA Access kit [R = 0.131, rho = 0.869]).
Agreement Between Protocols
QC data for FFPE RNA extraction, library preparation, and sequencing from both vendors are comparable. Both vendors achieved similar agreement between protocols. Among the 15 samples available in all 4 datasets, the mean Spearman correlation between protocols at Vendor A was 0.81 and at Vendor B it was 0.83. The mean Pearson correlation was 0.13 at Vendor A and 0.22 at Vendor B.
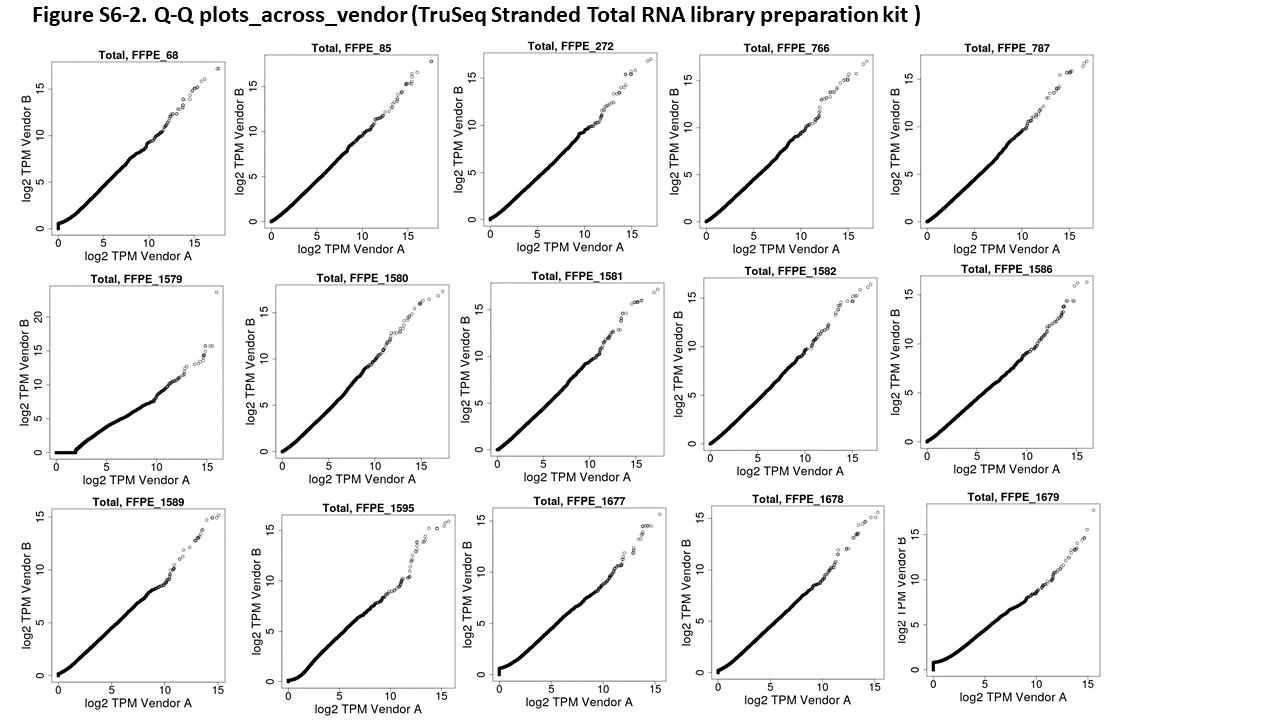
While the scatterplots for the individual samples (see Supplemental Figure 5 for the complete set) make it clear that the correlation between protocols is generally good, it is difficult to tell whether there is any systematic difference between protocols. For this, we used Q–Q plots; deviations from a straight line in these plots suggest systematic differences in the dynamics between the 2 protocols. A number of samples show off-diagonal behavior at the upper end of expression, suggesting that either the TruSeq Stranded Total RNA protocol is saturating or that the TruSeq RNA Access protocol is over-amplifying very highly expressed genes (see Figure 3 for one example; see Supplemental Figure 6 for full set).
We were interested in whether any QC factors (eg, RNA input, library concentration), especially those obtained before sequencing, might predict the correlation between vendors. Such a predictor could be used in future experiments to distinguish samples likely to produce high-quality output from those which may not. As there is no gold-standard data against which to compare results, we used the Spearman correlation between vendors as a measure of data quality for each sample. We thus examined the Spearman correlation between QC factors (the RNA quantity from each vendor and the library concentration from each vendor) and the Spearman correlation between the data from the 2 vendors. Table 2 summarizes the results, which suggest that library concentration is better correlated with consistency between vendors than is the RNA quantity. For example, for the TruSeq Stranded Total RNA data, the Spearman correlation of Vendor A's library concentration with the correlation between Vendor A's results and Vendor B's results is 0.96. Figure 4 shows the data in detail, plotting the cross-vendor Spearman correlation versus library concentration.
Table 2.
Spearman Correlations Between Library QC Factors (Total RNA Extracted, Library Concentration) and the Spearman Correlation Between Vendors of the Eventual Gene-Level Quantification.
| Kit | predictive_factor | Spearman's rho | P-value | Bonferroni adjusted_P-value |
|---|---|---|---|---|
| RNA Access | Vendor A, ug RNA | 0.421 | 1.192×10−1 | 3.577×10−1 |
| RNA Access | Vendor B, ug RNA | 0.481 | 6.965×10−2 | 2.786×10−1 |
| RNA Access | Vendor A, library conc. | 0.732 | 2.733×10−3 | 1.367×10−2 |
| RNA Access | Vendor B, library conc | 0.812 | 2.329×10−4 | 1.630×10−3 |
| TruSeq Total Stranded | Vendor A, ug RNA | 0.264 | 3.401×10−1 | 4.010×10−1 |
| TruSeq Total Stranded | Vendor B, ug RNA | 0.35 | 2.005×10−1 | 4.010×10−1 |
| TruSeq Total Stranded | Vendor A, library conc. | 0.964 | <1×10−10 | <1×10−10 |
| TruSeq Total Stranded | Vendor B, library conc | 0.821 | 2.578×10−4 | 1.630×10−3 |
This uses cross-vendor correlation as a proxy for the quality of the result and looks at which QC factors might predict that result quality. For each row, a P-value and a Bonferroni adjusted P-value (adjusted over all rows) is provided. While the quantity of RNA is never significantly associated with the eventual data quality, the library concentrations are significantly associated with downstream data quality for both kits and when using the library concentration from either vendor.
Figure 4.

Cross-vendor correlation versus library concentration. Examination of the cross-vendor correlations compared to various common QC statistics suggested that the library concentration was most informative in predicting the cross-vendor correlation. Plotted here are the cross-vendor correlation values versus library concentration. For the TruSeq Stranded Total RNA kit, there is a trend of increasing (though perhaps nonlinear) correlation as library concentration increases. For TruSeq RNA Access kit, there appears to be notably better results from library concentrations above 50 nM.
Discussion
RNA-seq is a powerful technology in transcriptome profiling. However, the challenge remains to choose suitable RNA-seq protocols for oncology FFPE specimens with degraded and low quantity RNA sample material. To guide the experimental design of clinical FFPE sample RNA-Seq, we conducted a comparison study using 2 Illumina library preparation protocols at 2 vendors for analyzing human RNA isolated from FFPE tissues.
Our results showed that both kits have the similar cross-vendor correlations, suggesting that both protocols offer reproducible results between different operators. However, the 2 library preparation kits yielded substantial differences in output consistent with the different approaches that the 2 kits take. Since more RNA sample is required in the TruSeq Stranded Total RNA protocol, only 20 samples had remaining RNA for TruSeq RNA Access library preparation. Thus, the TruSeq RNA Access protocol may be the preferred library prep for samples with limited quantity. The Illumina TruSeq RNA Access Library kit generated a higher fraction of reads from protein coding regions compared to other genomic regions; thus, it is a more efficient way to assay the expression of protein coding genes given a limited sequencing budget.
While the TruSeq RNA Access kit may be preferred for difficult samples, the resulting data may not be completely comparable to data from the TruSeq Stranded Total kit or to other kits based on ribosomal depletion and random priming. Further, the 2 library preparation kits yielded different dynamics of the output transcripts-per-million data at high expression levels where the TruSeq Stranded Total protocol tended to capture genes with higher expression and GC content. The probe-based selection of TruSeq RNA Access libraries may influence output differently than the random priming in other kits. Finally, the probe selection approach precludes certain downstream analyses, such as testing for viral or bacterial content that may be valuable in some settings. Thus, the lower sequencing costs should be weighed carefully against the anticipated uses for the data to decide which is appropriate for a given experiment. However, since the probe selection step in the RNA Access protocol may bias results compared to other platforms, further exploration may be needed.
In our study, we observed that extracted RNA quality decreases with the age of specimen. In addition, 2 samples (ages 14 years and 16 years) failed RNA extraction QC in Vendor A. Due to limited FFPE material, there was not enough sample to reextract failed samples. Thus, the main reason for failed library preparation was RNA sample depletion. This evidence suggested the influence of age on sample quality on RNAseq library preparation and sequencing. Supplemental Table 1 lists the detailed reasons for all failures in extraction and library preparation. Interestingly, library concentration appeared to be the best predictor of reproducibility across vendors and thus may be a preferred QC metric for future experiments on FFPE material. While this finding may be useful in avoiding sequencing samples with a low chance of providing quality data, it is not optimal as it can only be applied after the FFPE material is consumed, RNA extracted, and the work of library preparation is completed.
There are limitations in this study that could be addressed in future work. First, after extraction and library preparation failures, only a modest number of samples had data available from both vendors and with both kits. Second, the vendors may have handled varying library concentrations differently, as seen by the library concentrations in Table 1. Third, we did not have enough recent FFPE material (0-2 year old) for analysis to provide a full spectrum of clinical applicable information as this study was initially designed for selection of sequencing methods for older FFPE samples. Thus, a future study might include relatively new samples as part of a larger study that more fully examines the effects of tumor type and input tissue mass (ie, amount of tissue per slide) on the resulting data.
Conclusions
In summary, the quality and quantity of sequencing data obtained through RNA-Seq were strongly influenced by the choice of library preparation kit. Illumina TruSeq RNA Access library protocol could be a low-cost solution on highly degraded and limited FFPE samples, such as those from clinical studies in which the FFPE quality is severely compromised.
Supplemental Material
Supplemental material, sj-xlsx-1-tct-10.1177_15330338221076304 for Comparison of Two Illumina Whole Transcriptome RNA Sequencing Library Preparation Methods Using Human Cancer FFPE Specimens by Danyi Wang, P. Alexander Rolfe, Dorothee Foernzler, Dennis O’Rourke, Sheng Zhao, Juergen Scheuenpflug and Zheng Feng in Technology in Cancer Research & Treatment
Supplemental material, sj-xlsx-2-tct-10.1177_15330338221076304 for Comparison of Two Illumina Whole Transcriptome RNA Sequencing Library Preparation Methods Using Human Cancer FFPE Specimens by Danyi Wang, P. Alexander Rolfe, Dorothee Foernzler, Dennis O’Rourke, Sheng Zhao, Juergen Scheuenpflug and Zheng Feng in Technology in Cancer Research & Treatment
Supplemental material, sj-xlsx-3-tct-10.1177_15330338221076304 for Comparison of Two Illumina Whole Transcriptome RNA Sequencing Library Preparation Methods Using Human Cancer FFPE Specimens by Danyi Wang, P. Alexander Rolfe, Dorothee Foernzler, Dennis O’Rourke, Sheng Zhao, Juergen Scheuenpflug and Zheng Feng in Technology in Cancer Research & Treatment
{kind=link}
Supplemental material, sj-png-4-tct-10.1177_15330338221076304 for Comparison of Two Illumina Whole Transcriptome RNA Sequencing Library Preparation Methods Using Human Cancer FFPE Specimens by Danyi Wang, P. Alexander Rolfe, Dorothee Foernzler, Dennis O’Rourke, Sheng Zhao, Juergen Scheuenpflug and Zheng Feng in Technology in Cancer Research & Treatment
{kind=link}
Supplemental material, sj-png-5-tct-10.1177_15330338221076304 for Comparison of Two Illumina Whole Transcriptome RNA Sequencing Library Preparation Methods Using Human Cancer FFPE Specimens by Danyi Wang, P. Alexander Rolfe, Dorothee Foernzler, Dennis O’Rourke, Sheng Zhao, Juergen Scheuenpflug and Zheng Feng in Technology in Cancer Research & Treatment
{kind=link}
Supplemental material, sj-png-6-tct-10.1177_15330338221076304 for Comparison of Two Illumina Whole Transcriptome RNA Sequencing Library Preparation Methods Using Human Cancer FFPE Specimens by Danyi Wang, P. Alexander Rolfe, Dorothee Foernzler, Dennis O’Rourke, Sheng Zhao, Juergen Scheuenpflug and Zheng Feng in Technology in Cancer Research & Treatment
{kind=link}
Supplemental material, sj-png-7-tct-10.1177_15330338221076304 for Comparison of Two Illumina Whole Transcriptome RNA Sequencing Library Preparation Methods Using Human Cancer FFPE Specimens by Danyi Wang, P. Alexander Rolfe, Dorothee Foernzler, Dennis O’Rourke, Sheng Zhao, Juergen Scheuenpflug and Zheng Feng in Technology in Cancer Research & Treatment
{kind=link}
Supplemental material, sj-png-8-tct-10.1177_15330338221076304 for Comparison of Two Illumina Whole Transcriptome RNA Sequencing Library Preparation Methods Using Human Cancer FFPE Specimens by Danyi Wang, P. Alexander Rolfe, Dorothee Foernzler, Dennis O’Rourke, Sheng Zhao, Juergen Scheuenpflug and Zheng Feng in Technology in Cancer Research & Treatment
{kind=link}
Supplemental material, sj-png-9-tct-10.1177_15330338221076304 for Comparison of Two Illumina Whole Transcriptome RNA Sequencing Library Preparation Methods Using Human Cancer FFPE Specimens by Danyi Wang, P. Alexander Rolfe, Dorothee Foernzler, Dennis O’Rourke, Sheng Zhao, Juergen Scheuenpflug and Zheng Feng in Technology in Cancer Research & Treatment
{kind=link}
Supplemental material, sj-png-10-tct-10.1177_15330338221076304 for Comparison of Two Illumina Whole Transcriptome RNA Sequencing Library Preparation Methods Using Human Cancer FFPE Specimens by Danyi Wang, P. Alexander Rolfe, Dorothee Foernzler, Dennis O’Rourke, Sheng Zhao, Juergen Scheuenpflug and Zheng Feng in Technology in Cancer Research & Treatment
{kind=link}
Supplemental material, sj-png-11-tct-10.1177_15330338221076304 for Comparison of Two Illumina Whole Transcriptome RNA Sequencing Library Preparation Methods Using Human Cancer FFPE Specimens by Danyi Wang, P. Alexander Rolfe, Dorothee Foernzler, Dennis O’Rourke, Sheng Zhao, Juergen Scheuenpflug and Zheng Feng in Technology in Cancer Research & Treatment
{kind=link}
Supplemental material, sj-png-12-tct-10.1177_15330338221076304 for Comparison of Two Illumina Whole Transcriptome RNA Sequencing Library Preparation Methods Using Human Cancer FFPE Specimens by Danyi Wang, P. Alexander Rolfe, Dorothee Foernzler, Dennis O’Rourke, Sheng Zhao, Juergen Scheuenpflug and Zheng Feng in Technology in Cancer Research & Treatment
{kind=link}
Supplemental material, sj-png-13-tct-10.1177_15330338221076304 for Comparison of Two Illumina Whole Transcriptome RNA Sequencing Library Preparation Methods Using Human Cancer FFPE Specimens by Danyi Wang, P. Alexander Rolfe, Dorothee Foernzler, Dennis O’Rourke, Sheng Zhao, Juergen Scheuenpflug and Zheng Feng in Technology in Cancer Research & Treatment
{kind=link}
Supplemental material, sj-png-14-tct-10.1177_15330338221076304 for Comparison of Two Illumina Whole Transcriptome RNA Sequencing Library Preparation Methods Using Human Cancer FFPE Specimens by Danyi Wang, P. Alexander Rolfe, Dorothee Foernzler, Dennis O’Rourke, Sheng Zhao, Juergen Scheuenpflug and Zheng Feng in Technology in Cancer Research & Treatment
{kind=link}
Supplemental material, sj-png-15-tct-10.1177_15330338221076304 for Comparison of Two Illumina Whole Transcriptome RNA Sequencing Library Preparation Methods Using Human Cancer FFPE Specimens by Danyi Wang, P. Alexander Rolfe, Dorothee Foernzler, Dennis O’Rourke, Sheng Zhao, Juergen Scheuenpflug and Zheng Feng in Technology in Cancer Research & Treatment
{kind=link}
Supplemental material, sj-png-16-tct-10.1177_15330338221076304 for Comparison of Two Illumina Whole Transcriptome RNA Sequencing Library Preparation Methods Using Human Cancer FFPE Specimens by Danyi Wang, P. Alexander Rolfe, Dorothee Foernzler, Dennis O’Rourke, Sheng Zhao, Juergen Scheuenpflug and Zheng Feng in Technology in Cancer Research & Treatment
Acknowledgments
The authors would like to express gratitude to Alice Huang for her great support on the conceptualization and reviewing the manuscript. The authors also would like to express gratitude to Stefan Pinkert for his great support on sequencing data QC.
Glossary
Abbreviations
- FFPE
formalin-fixed, paraffin-embedded
- NGS
next generation sequencing
- RNASeq
RNA sequencing
- RIN
RNA integrity number
- TPMs
transcripts per millions
Footnotes
Author Contributions: DW contributed to the planning and execution of experiments, data analysis, and writing of article. RPA conducted data analysis and writing of article. DF provided scientific expertise. DO provided scientific expertise. SZ provided scientific expertise. JS provided scientific expertise. ZF provided scientific expertise, advised in experimental planning as well as contribution to composing the manuscript.
Declaration of Conflicting Interests: The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This project was supported by the EMD Serono, Merck KGaA, Darmstadt, Germany.
Ethics Approval and Consent to Participate: The written informed consent was received from all subjects used in this study and all samples were collected under the institutional review board approved protocols, as defined by the specimen providers Asterand (AP01338), Cureline (ECR/10/Inst/DC/2013/RR-16) and Conversant Biologics, Inc (5665), respectively. Ethical approval was obtained from Asterand, Cureline and Conversant Biologics, Inc. ethics committee. After collection, all samples were de-identified by the specimen provider so that no identifying information was available to researchers.
ORCID iD: Danyi Wang https://orcid.org/0000-0002-4780-3471
Supplemental Material: Supplemental material for this article is available online.
References
- 1.Cieślik M, Chinnaiyan AM. Cancer transcriptome profiling at the juncture of clinical translation. Nat Rev Genet. 2018;19(2):93–109. [DOI] [PubMed] [Google Scholar]
- 2.Li J, Fu C, Speed TP, Wang W, Symmans WF. Accurate RNA sequencing from formalin-fixed cancer tissue to represent high-quality transcriptome from frozen tissue. JCO Precis Oncol. 2018;2018(2):1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Adiconis X, Borges-Rivera D, Satija R, et al. Comparative analysis of RNA sequencing methods for degraded or low-input samples. Nat Methods. 2013;10(7):623–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cieslik M, Chugh R, Wu YM, et al. The use of exome capture RNA-seq for highly degraded RNA with application to clinical cancer sequencing. Genome Res. 2015;25(9):1372–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Park YS, Kim S, Park DG, et al. Comparison of library construction kits for mRNA sequencing in the Illumina platform. Genes Genomics. 2019;41(10):1233–1240. [DOI] [PubMed] [Google Scholar]
- 6.Schuierer S, Carbone W, Knehr J, et al. A comprehensive assessment of RNA-seq protocols for degraded and low-quantity samples. BMC Genomics. 2017;18(1):442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Consortium. SM-I. A comprehensive assessment of RNA-seq accuracy, reproducibility and information content by the sequencing quality control consortium. Nat Biotechnol. 2014;32(9):903–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dobin A, Davis CA, Schlesinger F, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics (Oxford, England). 2013;29(1):15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Frankish A, Diekhans M, Ferreira AM, et al. GENCODE Reference annotation for the human and mouse genomes. Nucleic Acids Res. 2019;47(D1):D766–d773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011;12:323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.R Core Team (2020). R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. URL https://www.R-project.org. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental material, sj-xlsx-1-tct-10.1177_15330338221076304 for Comparison of Two Illumina Whole Transcriptome RNA Sequencing Library Preparation Methods Using Human Cancer FFPE Specimens by Danyi Wang, P. Alexander Rolfe, Dorothee Foernzler, Dennis O’Rourke, Sheng Zhao, Juergen Scheuenpflug and Zheng Feng in Technology in Cancer Research & Treatment
Supplemental material, sj-xlsx-2-tct-10.1177_15330338221076304 for Comparison of Two Illumina Whole Transcriptome RNA Sequencing Library Preparation Methods Using Human Cancer FFPE Specimens by Danyi Wang, P. Alexander Rolfe, Dorothee Foernzler, Dennis O’Rourke, Sheng Zhao, Juergen Scheuenpflug and Zheng Feng in Technology in Cancer Research & Treatment
Supplemental material, sj-xlsx-3-tct-10.1177_15330338221076304 for Comparison of Two Illumina Whole Transcriptome RNA Sequencing Library Preparation Methods Using Human Cancer FFPE Specimens by Danyi Wang, P. Alexander Rolfe, Dorothee Foernzler, Dennis O’Rourke, Sheng Zhao, Juergen Scheuenpflug and Zheng Feng in Technology in Cancer Research & Treatment
Supplemental material, sj-png-4-tct-10.1177_15330338221076304 for Comparison of Two Illumina Whole Transcriptome RNA Sequencing Library Preparation Methods Using Human Cancer FFPE Specimens by Danyi Wang, P. Alexander Rolfe, Dorothee Foernzler, Dennis O’Rourke, Sheng Zhao, Juergen Scheuenpflug and Zheng Feng in Technology in Cancer Research & Treatment
Supplemental material, sj-png-5-tct-10.1177_15330338221076304 for Comparison of Two Illumina Whole Transcriptome RNA Sequencing Library Preparation Methods Using Human Cancer FFPE Specimens by Danyi Wang, P. Alexander Rolfe, Dorothee Foernzler, Dennis O’Rourke, Sheng Zhao, Juergen Scheuenpflug and Zheng Feng in Technology in Cancer Research & Treatment
Supplemental material, sj-png-6-tct-10.1177_15330338221076304 for Comparison of Two Illumina Whole Transcriptome RNA Sequencing Library Preparation Methods Using Human Cancer FFPE Specimens by Danyi Wang, P. Alexander Rolfe, Dorothee Foernzler, Dennis O’Rourke, Sheng Zhao, Juergen Scheuenpflug and Zheng Feng in Technology in Cancer Research & Treatment
Supplemental material, sj-png-7-tct-10.1177_15330338221076304 for Comparison of Two Illumina Whole Transcriptome RNA Sequencing Library Preparation Methods Using Human Cancer FFPE Specimens by Danyi Wang, P. Alexander Rolfe, Dorothee Foernzler, Dennis O’Rourke, Sheng Zhao, Juergen Scheuenpflug and Zheng Feng in Technology in Cancer Research & Treatment
Supplemental material, sj-png-8-tct-10.1177_15330338221076304 for Comparison of Two Illumina Whole Transcriptome RNA Sequencing Library Preparation Methods Using Human Cancer FFPE Specimens by Danyi Wang, P. Alexander Rolfe, Dorothee Foernzler, Dennis O’Rourke, Sheng Zhao, Juergen Scheuenpflug and Zheng Feng in Technology in Cancer Research & Treatment
Supplemental material, sj-png-9-tct-10.1177_15330338221076304 for Comparison of Two Illumina Whole Transcriptome RNA Sequencing Library Preparation Methods Using Human Cancer FFPE Specimens by Danyi Wang, P. Alexander Rolfe, Dorothee Foernzler, Dennis O’Rourke, Sheng Zhao, Juergen Scheuenpflug and Zheng Feng in Technology in Cancer Research & Treatment
Supplemental material, sj-png-10-tct-10.1177_15330338221076304 for Comparison of Two Illumina Whole Transcriptome RNA Sequencing Library Preparation Methods Using Human Cancer FFPE Specimens by Danyi Wang, P. Alexander Rolfe, Dorothee Foernzler, Dennis O’Rourke, Sheng Zhao, Juergen Scheuenpflug and Zheng Feng in Technology in Cancer Research & Treatment
Supplemental material, sj-png-11-tct-10.1177_15330338221076304 for Comparison of Two Illumina Whole Transcriptome RNA Sequencing Library Preparation Methods Using Human Cancer FFPE Specimens by Danyi Wang, P. Alexander Rolfe, Dorothee Foernzler, Dennis O’Rourke, Sheng Zhao, Juergen Scheuenpflug and Zheng Feng in Technology in Cancer Research & Treatment
Supplemental material, sj-png-12-tct-10.1177_15330338221076304 for Comparison of Two Illumina Whole Transcriptome RNA Sequencing Library Preparation Methods Using Human Cancer FFPE Specimens by Danyi Wang, P. Alexander Rolfe, Dorothee Foernzler, Dennis O’Rourke, Sheng Zhao, Juergen Scheuenpflug and Zheng Feng in Technology in Cancer Research & Treatment
Supplemental material, sj-png-13-tct-10.1177_15330338221076304 for Comparison of Two Illumina Whole Transcriptome RNA Sequencing Library Preparation Methods Using Human Cancer FFPE Specimens by Danyi Wang, P. Alexander Rolfe, Dorothee Foernzler, Dennis O’Rourke, Sheng Zhao, Juergen Scheuenpflug and Zheng Feng in Technology in Cancer Research & Treatment
Supplemental material, sj-png-14-tct-10.1177_15330338221076304 for Comparison of Two Illumina Whole Transcriptome RNA Sequencing Library Preparation Methods Using Human Cancer FFPE Specimens by Danyi Wang, P. Alexander Rolfe, Dorothee Foernzler, Dennis O’Rourke, Sheng Zhao, Juergen Scheuenpflug and Zheng Feng in Technology in Cancer Research & Treatment
Supplemental material, sj-png-15-tct-10.1177_15330338221076304 for Comparison of Two Illumina Whole Transcriptome RNA Sequencing Library Preparation Methods Using Human Cancer FFPE Specimens by Danyi Wang, P. Alexander Rolfe, Dorothee Foernzler, Dennis O’Rourke, Sheng Zhao, Juergen Scheuenpflug and Zheng Feng in Technology in Cancer Research & Treatment
Supplemental material, sj-png-16-tct-10.1177_15330338221076304 for Comparison of Two Illumina Whole Transcriptome RNA Sequencing Library Preparation Methods Using Human Cancer FFPE Specimens by Danyi Wang, P. Alexander Rolfe, Dorothee Foernzler, Dennis O’Rourke, Sheng Zhao, Juergen Scheuenpflug and Zheng Feng in Technology in Cancer Research & Treatment