

Abstract
Simple Summary
Compelling evidence has shown that cancer stem cells (CSCs) are responsible for high resistance to conventional anti-cancer therapies. Here, we demonstrate that the tumor microenvironment protects CR-CSCs from EGFR/HER2, BRAF and PI3K targeting, promoting CD44v6 and Myc expression. Alternatively, as a substitution for HER2 and BRAF, the Myc transcription inhibitor can overcome the protective effects of microenvironmental cytokines, impairing the survival of CR-CSCs. These data highlight the targeting of Myc and PI3K activity as a novel therapeutic strategy against advanced colorectal cancer.
Abstract
Despite advances in the curative approach, the survival rate of advanced colorectal cancer (CRC) patients is still poor, which is likely due to the emergence of cancer cell clones resistant to the available therapeutic options. We have already shown that CD44v6-positive CRC stem cells (CR-CSCs) are refractory toward standard anti-tumor therapeutic agents due to the activation of the PI3K pathway together with high HER2 expression levels. Tumor microenvironmental cytokines confer resistance to CR-CSCs against HER2/PI3K targeting by enhancing activation of the MAPK pathway. Here, we show that the CSC compartment, spared by BRAF inhibitor-based targeted therapy, is associated with increased expression levels of CD44v6 and Myc and retains boosted clonogenic activity along with residual tumorigenic potential. Inhibition of Myc transcription, downstream of the MAPK cascade components, and PI3K pathway activity was able to overcome the protective effects of microenvironmental cytokines, affecting the survival and the clonogenic activity of CR-CSCs, regardless of their mutational background. Likewise, the double targeting induced stabilization of mouse tumor avatars. Altogether, these data outline the rationale for dual kinase targeting of CR-CSCs to prevent their adaptive response, which would lead to disease progression.
Keywords: anti-tumor drug resistance, colorectal cancer, combination therapies, cancer stem cells
1. Introduction
Colorectal cancer (CRC) is the second leading cause of cancer-associated death worldwide [1]. Thanks to the advances in early diagnosis and treatments, the 5-year survival rate for CRC patients with localized disease now reaches about 90%. Nevertheless, the percentage drops to 14% for patients diagnosed with metastatic lesions [2]. Mutations in the Wnt/β-catenin, PI3K/AKT and MAPK pathways lead to their aberrant activation, which drives CRC initiation and progression [3,4]. Compelling evidence has demonstrated that a subset of cancer cells, named cancer stem cells (CSCs), are characterized by a tumor initiation ability and are responsible for recurrence and anticancer drug resistance [5,6]. These CSC peculiarities are sustained by (i) deregulation of key signaling pathways implicated in normal stem cell maintenance, such as WNT, Hedgehog and Notch; (ii) high expression of ABC transporter and anti-apoptotic factors; and (iii) proficient DNA repair machinery [7,8]. The WNT signaling pathway is essential to maintain the homeostasis of colonic crypts, regulating turnover and differentiation of intestinal stem cells (ISCs). Hence, this pathway is aberrantly activated in many tumors and about 90% of CRCs harbor mutations in WNT pathway components [9,10]. We demonstrated that metastatic CR-CSCs express increased levels of CD44v6 and display high Wnt activity [11]. Moreover, cytokines released by the tumor microenvironment (TME) boost CD44v6 expression and WNT signaling activation [11,12]. Although much effort has been directed toward developing targeted therapies that are able to interfere with CRC-related signaling pathways, only a few drugs have successfully advanced from preclinical tests to clinical trials. The reported results of several clinical trials (NCT00154102, NCT00297271, NCT01228734) highlighted that patients bearing BRAFV600E and RAS mutations do not benefit from treatment with anti-EGFR therapies [13,14,15,16]. Recently, we demonstrated that about 50% of BRAF and RAS wt CRC sphere cells (CR-CSphCs) are resistant to cetuximab treatment due to high expression levels of CD44v6 [11,17]. Many other therapeutic strategies, including anti-angiogenic drugs and Met pathway and immune checkpoint inhibitors, are currently under evaluation for managing CRC treatment [2,18,19,20,21]. Another common mechanism of resistance to standard and targeted therapies is gene amplification/overexpression [22,23,24]. The MYC oncogene is deregulated in 70% of cancers, including CRC, and amplified in about 20% of CRC patients [25,26]. Increased Myc expression levels are associated with the onset of tumor growth, constitutive activation of Wnt/β-catenin and an impaired survival rate in CRC patients. Numerous shreds of evidence have demonstrated that a chemoresistant phenotype characterizes tumors expressing high levels of Myc. Likewise, cancer cells surviving the treatment with standard chemotherapeutic compounds displayed higher MYC expression levels [27]. Resistance to Met inhibitors in MET-addicted gastric cancer cell lines is sustained by Myc, and the double blockade of Met and Myc can lessen the tumor growth of MET-amplified patient-derived xenografts tumors [28]. In KRAS mutant CRC cells, the high expression of Myc confers resistance to HDAC inhibitor SAHA [29]. All these data point out that, despite advances in the improvement of targeted therapies for advanced CRC, treatment options for RAS mutated patients remain a challenge.
We previously demonstrated that CD44v6+ CR-CSCs are highly resistant to standard therapies and can be efficiently targeted by PI3K inhibitors only in the absence of a protective TME. Cytokines released by cancer-associated fibroblasts (CAF) counteract the anti-tumoral effect of HER2/PI3K targeting via the upregulation of the MAPK pathway, as indicated by the ability of the MEK inhibitor-based combination to induce tumor shrinkage [17,30]. Here, we demonstrate that the combinatorial treatment of anti-EGFR/HER2, BRAF and PI3K inhibitors induces the selection of a CD44v6+/WNThigh cell population that displays high expression levels of Myc. The dual indirect targeting of CD44v6 and Myc, using PI3K and cyclin-dependent kinase (CDK) inhibitors, reduces, even in the presence of a TME, the survival and clonogenic activity of CR-CSphCs, regardless of the mutational background and/or MYC gene amplification, and stabilizes the growth of tumor xenografts.
2. Materials and Methods
2.1. Isolation and Maintenance of CR-CSphCs and CAFs
Colorectal cancer sphere cells (CR-CSphCs) were obtained from 43 human tumor tissue specimens following the Human Experimentation Policy of Policlinico “Paolo Giaccone”, Palermo (authorization CE9/2015), as previously described. CRC specimens were first minced into small pieces using surgical scissors and then enzymatically digested with collagenase (0.6 mg/mL Sigma-Aldrich, St. Louis, MO, USA) and hyaluronidase (10 µg/mL, Sigma-Aldrich, St. Louis, MO, USA) in DMEM medium with continuous shaking (150 rpm) at 37 °C for 30 min. Thereafter, the obtained suspension was centrifuged at 200× g for 5 min and the cell pellet resuspended in serum-free stem cell medium (SCM) supplemented with recombinant human FGF-basic (100 ng/mL, Peprotech, Cranbury, NJ, USA) and recombinant human EGF (50 ng/mL, Peprotech, Cranbury, NJ, USA) (see also [31]). Cells were seeded in ultra-low attachment culture flasks at a confluence of 1 × 105 cells/mL. Cell propagation was accomplished, dissociating sphere cells with Accutase (ThermoFisher Scientific, Waltham, MA, USA) when they reached a diameter of 50–100 µM. All the experiments were performed with early-passage CR-CSphCs (<20).
CRC spheres were authenticated by comparing them with their patient-related tumor tissues in an analysis of repeated polymorphisms (GlobalFiler™ STR kit, Applied Biosystem, Thermo Fisher Scientific, Waltham, MA, USA) using an ABIPRISM 3130 genetic analyzer (Applied Biosystem, Thermo Fisher Scientific, Waltham, MA, USA). Moreover, every 2 months, CR-CSphC cultures were tested for mycoplasma contamination (MycoAlertTM Plus Mycoplasma Detection Kit, Lonza, Houston, TX, USA). The mutational profiles of the CR-CSphCs were analyzed with a targeted DNA custom panel by sequencing them with the MiSeq platform (Illumina, San Diego, CA, USA). MYC copy number variation (CNV) was evaluated using Digital Droplet PCR (Bio-Rad QX200 reader, Bio-Rad, Hercules, CA, USA) according to the manufacturer’s instructions. The obtained data were normalized with the EIF2C1 gene and a MYC/EIF2C1 ratio ≥4 was used to identify amplified MYC samples [32]. The presence of microsatellite instability (MSI) was assessed with a GENEQUALITY CC-MSI kit according to the manufacturer’s instruction (AB Analitica, Padova, Italy). The presence of four or more unstable markers identified high MSI samples and one to three identified low MSI samples; the absence of unstable markers identified stable samples (Table 1).
Table 1.
CD44v6 expression levels, MYC copy number variation, MSI status and BRAF/KRAS mutational status of colorectal cancer sphere cells (CR-CSphCs).
| CD44v6 expression | ||||||||||||||||||||||||||||||||||||||
| MYC GCN | ||||||||||||||||||||||||||||||||||||||
| MSI status | NA | |||||||||||||||||||||||||||||||||||||
| Site | S | L | R | R | R | R | R | R | S | L | R | R | R | R | L | R | L | S | R | R | M | M | R | NA | L | R | R | R | R | S | S | R | S | R | S | L | L | M |
| BRAF | ||||||||||||||||||||||||||||||||||||||
| KRAS | ||||||||||||||||||||||||||||||||||||||
| CSphC # | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 | 17 | 19 | 20 | 21 | 22 | 23 | 24 | 25 | 27 | 29 | 30 | 33 | 37 | 44 | 49 | 52 | 53 | 55 | 56 | 57 | 58 | 59 |
| CD44v6 expression levels (%) | MYC/EIF2C1 ratio | MSI status | Mutation Type | |||||||||||||||||||||||||||||||||||
| high | >70% | MYC GCN < 4 | stable | wt | ||||||||||||||||||||||||||||||||||
| medium | 30–70% | MYC GCN ≥ 4 | low | Missense | ||||||||||||||||||||||||||||||||||
| low | <30% | high | ||||||||||||||||||||||||||||||||||||
| NA | ||||||||||||||||||||||||||||||||||||||
CAFs were isolated from CRC samples and plated in DMEM medium supplemented with 10% fetal bovine serum (FBS) in an adherent condition. CAFs were detached using Trypsin-EDTA solution when they reached 70–80% confluence and were cultured for at least 10 passages.
2.2. CR-CSphCs In Vitro Treatment
CR-CSphCs were exposed to vemurafenib (1 µM, S1267 Selleckchem, Houston, TX, USA), cetuximab (20 µg/mL) and trastuzumab (10 µg/mL) (provided by Policlinico “Paolo Giaccone”, Palermo, Italy), as well as BKM120 (1 µM, S2247 Selleckchem, Houston, TX, USA), dinaciclib (10 nM, S2768 Selleckchem, Houston, TX, USA) and taselisib (1 µM, GDC-0032 Selleckchem, Houston, TX, USA). All the compounds, with the exception of dinaciclib, were refilled every 48 h. To evaluate the significance of the dinaciclib and taselisib combinatorial treatment, CR-CSphCs were exposed to different concentrations of dinaciclib (5, 10 and 25 nM) and taselisib (0.2, 0.5 and 1 µM) for 72 h and the data were analyzed with Synergy Finder software (version 2.0) [33] (Table 2). To obtain CAF-conditioned medium (CAF CM), CAFs were grown to subconfluence and maintained in SCM for 48 h. Before addition to the CR-CSphCs, the CAF CM was filtered with a 0.22 µm filter to remove any cell debris.
Table 2.
List and diagram illustrating the site of action, type and target of each drug.
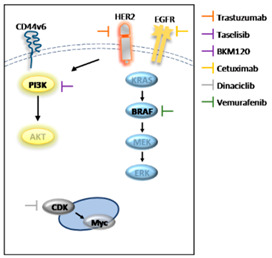
| Name | Type | Target |
|---|---|---|
| Vemurafenib | Small molecule | BRAF V600E |
| Cetuximab | Recombinant human/mouse chimeric IgG(1) monoclonal antibody | EGFR |
| Trastuzumab | Recombinant humanized monoclonal antibody | HER2 |
| BKM120 | dimorpholino pyrimidine derivative | class I PI3K |
| Dinaciclib | small molecule | CDK1/2/5/9 |
| Taselisib | small molecule | PIK3CA |
2.3. Clonogenic, Sphere-Forming and Colony-Formation Assays
The clonogenicity of CR-CSphCs, previously treated as indicated, was assessed by plating them on ultra-low attachment 96-well plates at the single-cell level. SCM was refreshed once a week, and the results were statistically evaluated after 21 days. The sphere-forming assay was performed by culturing xenograft-derived sphere cells at clonal density (1 × 103 cells/mL), avoiding cell–cell contact. Sphere-formation capacity was measured by counting the number of cell clusters that reached the diameter of ≥50 µm after ten days of culture. For the colony-forming assay, 2 × 103 CR-CSphCs were seeded on low-melting agarose (Agarose Sea Plague Agar, Invitrogen, Thermo Fisher Scientific, Waltham, MA, USA) and maintained for 21 days. Crystal violet staining (0.01%) was used to reveal cell colonies and the clonogenic potential of CR sphere cells was assessed using ImageJ software (Bethesda, MD, USA).
2.4. Cell Viability
CR-CSphC viability was evaluated with the CellTiter 96® Aqueous One Solution Cell Proliferation Assay (MTS, Promega, Madison, WI, USA) and, after 2 h, the variation in absorbance was measured with a GDV MPT 370 reader (DV 990 BV6). To validate the obtained data, the percentage of live cells was calculated with the trypan blue exclusion test.
2.5. Lentiviral Production and Transduction
FOP-GFP.mC (Addgene, Watertown, MA, USA, 35492) or TOP-GFP.mC (Addgene, Watertown, MA, USA, 35491) lentiviral supernatants were obtained following the protocol described in [31] and concentrated with the Lenti-X Concentrator reagent (Clontech, Takara Bio, San Jose, CA, USA). A total of 1 × 105 CR-CSphCs were exposed to the obtained lentiviral supernatants with the addition of 8 μg/mL of polybrene (Sigma-Aldrich, St. Louis, MO, USA).
2.6. Flow Cytometry and Cell Sorting
CR-CSphCs were resuspended in PBS and incubated with conjugated antibodies CD44v6-APC (2F10, R&D system, Minneapolis, MN, USA) or the corresponding isotype-matched control (IC002A, mouse IgG1, R&D system, Minneapolis, MN, USA) for 1 h at 4 °C, and 7-AAD staining was used to exclude dead cells (0.25 μg/1 × 106 cells, BD Biosciences, Franklin Lakes, NJ, USA). To enrich TOP-GFP and CD44v6 cell fractions, CR-CSphCs were resuspended in 2% BSA 2 mM EDTA PBS and filtered using a 70 µm mesh, with cell sorting performed with a FACS Melody cell sorter (BD biosciences, Franklin Lakes, NJ, USA).
2.7. Western Blot
Total proteins from CR-CSphCs were isolated as previously described [17]. After performing SDS-PAGE, proteins were blotted on nitrocellulose membranes. Membranes were incubated with blocking solution (5% Blotto, nonfat dry milk, Santa Cruz Biotechnology, Dallas, TX, USA, PBS 0.1% Tween 20) for 45 min at R.T. and then exposed at 4 °C for 16 h with antibodies against phospho-AKT xp (Ser473; D9E, rabbit, IgG, CST, Danvers, MA, USA), AKT (rabbit polyclonal, CST, Danvers, MA, USA), phospho-MEK1/2 (Ser217/221; 41G9, rabbit IgG, CST, Danvers, MA, USA), MEK1/2 (rabbit polyclonal, CST, Danvers, MA, USA), phospho-ERK 1/2 (Thr202/Tyr204; rabbit polyclonal, CST, Danvers, MA, USA), ERK 1/2 (137F5, rabbit IgG, CST, Danvers, MA, USA) or Myc (rabbit polyclonal, CST, Danvers, MA, USA). After incubation with anti-mouse or anti-rabbit HRP-conjugated secondary antibody (goat IgG; Thermo Fisher Scientific, Waltham, MA, USA), chemiluminescence signals were detected with Amersham imager 600 (GE Healthcare, USA). The densitometric analysis of β-actin (8H10D10, mouse, CST, Danvers, MA, USA)-normalized protein levels was undertaken with ImageJ software.
2.8. RNA Extraction and Gene Expression Analysis
Total RNA of xenograft tumors was purified using TRIzol (Thermo Fisher Scientific, Waltham, MA, USA) and, following genomic DNA elimination, 1 µg of RNA was retrotranscribed and analyzed with a PrimePCR custom panel (Bio-Rad, Hercules, CA, USA) (Table 3). GAPDH-normalized data were analyzed using the comparative CT method (2−ΔCt method).
Table 3.
List of cancer stem cell- and metastasis-related genes.
| Gene Name | Assay ID | Gene Name | Assay ID | Gene Name | Assay ID |
|---|---|---|---|---|---|
| AXIN1 | qHsaCID0010131 | ILKAP | qHsaCID0011777 | SERPINE1 | qHsaCID0006432 |
| AXIN2 | qHsaCID0017930 | ITGA5 | qHsaCID0021495 | SFRP1 | qHsaCID0015548 |
| BMP1 | qHsaCID0010875 | ITGAV | qHsaCID0006233 | SFRP4 | qHsaCED0043151 |
| BMP2 | qHsaCID0015400 | ITGB1 | qHsaCED0005248 | SMAD2 | qHsaCID0022031 |
| BMP7 | qHsaCID0011038 | JAG1 | qHsaCID0006831 | SMAD4 | qHsaCID0015670 |
| CCND1 | qHsaCID0013833 | KISS1 | qHsaCED0004976 | SNAI1 | qHsaCED0002998 |
| CD44 | qHsaCID0013679 | KISS1R | qHsaCID0011504 | SNAI2 | qHsaCID0011342 |
| CDH1 | qHsaCID0015365 | KREMEN1 | qHsaCED0047626 | SNAI3 | qHsaCED0047761 |
| CDH2 | qHsaCID0015189 | KRT14 | qHsaCED0047868 | SOX17 | qHsaCED0001884 |
| CDKN2A | qHsaCED0023006 | LRP5 | qHsaCED0045974 | SRC | qHsaCED0004489 |
| COL1A1 | qHsaCED0002181 | LRP6 | qHsaCID0010231 | STAT3 | qHsaCID0010912 |
| CTNNB1 | qHsaCID0010363 | MDM2 | qHsaCID0011000 | TGFB1 | qHsaCID0017026 |
| CXCL12 | qHsaCID0012398 | MET | qHsaCED0002004 | TGFB2 | qHsaCID0018360 |
| CXCR4 | qHsaCED0002020 | MMP2 | qHsaCID0015623 | TGFB3 | qHsaCID0022239 |
| DKK1 | qHsaCED0002060 | MMP3 | qHsaCID0006170 | TIMP1 | qHsaCID0007434 |
| DKK3 | qHsaCED0001115 | MMP7 | qHsaCID0011537 | TIMP2 | qHsaCID0022953 |
| EGFR | qHsaCID0007564 | MMP9 | qHsaCID0011597 | TIMP3 | qHsaCID0015238 |
| EPHB2 | qHsaCID0009881 | MMP10 | qHsaCED0046399 | TIMP4 | qHsaCID0016129 |
| ERBB2 | qHsaCID0012766 | MMP11 | qHsaCID0022136 | TP53 | qHsaCID0013658 |
| ERBB3 | qHsaCID0018397 | MMP13 | qHsaCID0008487 | TSHR | qHsaCID0009606 |
| ERBB4 | qHsaCID0017862 | MYC | qHsaCID0012921 | TWIST1 | qHsaCED0003856 |
| ESR1 | qHsaCED0033920 | NODAL | qHsaCID0006123 | VIM | qHsaCID0012604 |
| FGFBP1 | qHsaCID0015948 | NOTCH1 | qHsaCID0011825 | WNT3A | qHsaCED0045634 |
| FGFR4 | qHsaCED0045915 | OCLN | qHsaCED0038290 | WNT5A | qHsaCID0012240 |
| FN1 | qHsaCID0012349 | PDGFRB | qHsaCID0013272 | WNT5B | qHsaCID0038673 |
| FZD7 | qHsaCED0019290 | PTEN | qHsaCED0036796 | WNT7A | qHsaCID0018523 |
| GAPDH | qHsaCED0038674 | RAC1 | qHsaCED0001330 | WNT7B | qHsaCED0003528 |
| GSK3B | qHsaCID0010097 | RB1 | qHsaCID0007095 | WNT11 | qHsaCID0011927 |
| HGF | qHsaCID0011441 | RGS2 | qHsaCED0001744 | ZEB1 | qHsaCID0009210 |
| HPRT1 | qHsaCID0016375 | RHOA | qHsaCID0008947 |
2.9. Immunofluorescence/Immunohistochemistry
CD44v6/Myc double-staining was performed on 5-µm-thick paraffin-embedded xenograft sections. Antigen retrieval was performed using the PT link system (Dako, Agilent technologies, Santa Clara, CA, USA) with a 10 mM sodium citrate solution (pH 6.0). Before primary antibody incubation, we performed two consecutive blocking steps: one for 5 min with a 3% H2O2 solution to inhibit endogenous peroxidase and the other for 20 min with 10% human serum to reduce unspecific signals. Thereafter, sections were exposed to antibodies specific for CD44v6 (2F10, R&D systems, Minneapolis, MN, USA) and Myc (rabbit polyclonal, CST, Danvers, MA, USA), diluted in antibody diluent solution (Dako, Agilent technologies, Santa Clara, CA, USA). The MACH 2 Double Stain 2 kit (Biocare Medical, Pacheco, CA, USA) was used to reveal primary antibodies, using DAB and Vulcan Fast Red chromogens for detection. Aqueous hematoxylin (Sigma-Aldrich, St. Louis, MO, USA) was used to counterstain cell nuclei.
CR-CSphCs exposed to vemurafenib, trastuzumab and BKM120 were fixed, permeabilized and incubated overnight at 4 °C with CD44v6 and Myc antibodies. Then, cells were stained with Alexa Fluor-488 goat anti-rabbit IgG and Rhodamine Red-x goat anti-mouse IgG1 (Life Technologies, Waltham, MA, USA) secondary antibodies. Toto-3 Iodide (Life Technologies, Waltham, MA, USA) was used to counterstain nuclei.
2.10. In Vivo Experiments
Six- to eight-week-old NOD/SCID mice (Charles River Laboratories, Wilmington, MA, USA) were used for all the in vivo procedures, which were performed following institutional experimental procedure guidelines (Italian Ministry of Health authorization number 154/2017-PR). A total of 2.5 × 105 CR-CSphCs, resuspended in 150 µL of 1:1 SCM/Matrigel (BD) solution, were subcutaneously injected into the flanks of mice. When tumor xenografts reached a volume of 0.03–0.06 cm3, mice were randomly divided into control and treatment groups (six mice/group) and treated for 4 weeks with a vehicle, vemurafenib (20 mg/Kg, twice daily, oral gavage), trastuzumab (5 mg/Kg, weekly, intraperitoneal injection (i.p.)), cetuximab (40 mg/Kg, twice weekly, i.p.), BKM120 (20 mg/Kg, daily, oral gavage), taselisib (5 mg/Kg, daily, oral gavage) and dinaciclib (25 mg/Kg, 3 days/week, i.p.).
To evaluate secondary tumor formation, 1 × 104 sphere cells, freshly isolated from treated primary tumor xenografts, were subcutaneously injected into secondary mice recipients (n = 5). Tumor volumes were calculated as follows: largest diameter × (smallest diameter)2 × π/6. Mice were sacrificed when tumor xenografts reached 2 cm in tumor diameter or when signs of distress were identified, following Directive 2010/63/EU guidelines (D.lgs 26/2016).
2.11. Statistical Analysis
All the results show means ± SD from independent experiments. The means and SD were obtained by analyzing replicates using Prism 5 (GraphPad Software, La Jolla, CA, USA) and applying Student t-test. p-values greater than 0.05 were considered statistically not significant.
3. Results
3.1. CD44v6-Positive CR-CSphCs Are Refractory toward BRAF Inhibition
While treatment with an inhibitor of BRAF (vemurafenib) reduced the clonogenic activity and delayed in vitro growth of BRAF-mutant CR-CSphCs, the clonogenicity of RAS/BRAF-wt (wt) and KRAS-mutant cells showed a trend towards an increase, without altering the proliferation rate (Figure 1A,B and Table 1). Exposure to vemurafenib resulted in minimal or no impact on survival of wt, BRAF- and KRAS-mutant CD44v6+ cells, whereas the CD44v6− fraction of BRAF-mutant cells was significantly sensitive to vemurafenib-induced cell death (Figure 1C and Figure S1A). The exposure to different doses of vemurafenib promoted the activation of RAF signaling in wt and KRAS-mutant CR-CSphCs, leading to the paradoxical activation of ERK (Figure S1B) [34,35]. Alternatively, following exposure to vemurafenib, BRAF-mutant sphere cells attenuated their activation of RAF and strengthened PI3K/AKT signaling (Figure 1D and Figure S1C). Tumorigenic and metastatic CD44v6+ populations express higher HER2 than EGFR or HER3 levels [17]. Given that the refractoriness of BRAF-mutant CD44v6+ cells toward the BRAF inhibitor could be sustained by the activation of RAS and ERK, triggered by EGFR [36], we explored whether EGF deprivation could sensitize this cell compartment to vemurafenib. CD44v6+ cells were found to be inherently more resistant to vemurafenib than the CD44v6− cell fraction, even in the absence of EGF and regardless of their mutational profile (Figure 1E). We also evaluated whether the combinatorial targeting of BRAF and EGFR signaling could affect the cell viability of this cell compartment. We used cetuximab, a chimeric human/mouse monoclonal antibody, and trastuzumab, a humanized monoclonal antibody, to target EGFR and HER2, respectively. BRAF/EGFR andHER2 targeting showed similar effects on the MAPK pathway compared with those observed with the use of vemurafenib monotherapy (Figure 2A and Figure S2). Notably, EGFR-based therapy efficiently killed CD44v6− cells, with a more pronounced effect against the BRAFV600E background. In contrast, CD44v6+ cells were largely spared by this dual targeting, which heightened the CD44v6+/Wnthigh fraction (Figure 2B,C).
Figure 1.

The resistance of CD44v6-positive CR-CSCs to BRAF inhibition is mediated by EGFR-driven MAPK pathway activation. (A) Clonogenic assays for wt (#6, #14, #21, #27, #33), BRAF- (#1, #2, #3, #4, #5) and KRAS-mutant (#9, #10, #11, #13, #16) CSphCs pretreated with a vehicle (Vehicle) or vemurafenib (Vemu, 1 µM) for 120 h. The statistical significance between two groups was determined with a paired two-tailed Student’s t-test. (B) Kinetics of wt (#14, 21), BRAF- (#2, 5) and KRAS-mutant (#11, 16) sphere cell growth following treatment with vemurafenib (Vemu, 1 µM) for up to 120 h. (C) Cell viability percentage of CD44v6+ and CD44v6− wt (#21, #27, #33), BRAF- (#1, #3, #5) and KRAS-mutant (#9, #13, #16) sphere cells treated as in (B). (D) Immunoblot analysis of pAKT, AKT, pMEK, MEK, pERK and ERK on the indicated CR-CSphCs treated with a vehicle (−) or vemurafenib (Vemu, +, 1 µM) for 48 h. (E) Cell viability percentage of CD44v6+ and CD44v6− wt (#21, #27, #33), BRAF- (#1, #3, #5) and KRAS-mutant (#9, #13, #16) sphere cells treated as indicated in the presence or absence (strv) of EGF for up to 120 h. For (B–C,E), data are means ± SD from three independent experiments. ns, not significant; * p ≤ 0.05; ** p ≤ 0.01, **** p ≤ 0.0001. The uncropped blots are shown in supplementary materials.
Figure 2.

PI3K activation protects CD44v6-positive cells from EGFR/HER2 blockade in combination with BRAF inhibition. (A) Immunoblot of pAKT, AKT, pMEK, MEK, pERK and ERK on the indicated CD44v6+ cells exposed to a vehicle (−) or to vemurafenib (Vemu, +, 1 µM) in combination with cetuximab (Cmab, +, 20 µg/mL) or trastuzumab (Tmab, +, 10 µg/mL) for 2 h. (B) Cell viability percentage in CD44v6+ and CD44v6− wt (#21, #27), BRAF- (#1, #5) and KRAS-mutant (#11, #16) CR-CSphCs treated with vemurafenib (Vemu, 1 µM) in combination with cetuximab (Cmab, 20 µg/mL) or trastuzumab (Tmab, 10 µg/mL). Data are shown as means ± SD of three independent experiments for each CR-CSphC line. (C) Fold increase over vehicle (blue dotted line) of CD44v6+/Wnthigh subpopulation in wt (#27), BRAF- (#1) and KRAS-mutant (#9) TOP-GFP transduced sphere cells, treated as indicated for 120 h. Data represent the means ± SD of three independent experiments. ** p ≤ 0.01, **** p ≤ 0.0001.The uncropped blots are shown in supplementary materials.
3.2. CR-CSCs Emerge after Inhibition of HER2, BRAF and PI3K
Given that CD44v6+ cells display enhanced activation of PI3K/AKT signaling that in turn sustains the β-catenin activation [11,37], BKM120, an inhibitor of class I isoforms of PI3K, was supplemented to the anti-EGFR/HER2 and BRAF inhibitor combination. The triple combination transiently decreased the in vitro viability of CR-CSphCs, which restarted growth after treatment suspension (Figure 3A). CR-CSphCs, exposed to this treatment displayed a depletion of CD44v6+ cells together with reduced clonogenic and sphere-forming activity, suggesting that CD44v6+ cells are considerably affected by the addition of BKM120 (Figure 3B–D). We next evaluated the efficacy of this PI3K inhibitor-based therapy in tumor xenografts. This treatment initially, at 5 weeks, produced a stabilizing effect in the growth of BRAF-mutated xenograft tumors and led to a delayed progression of wt and KRAS-mutated xenograft tumors. Eight weeks after treatment suspension, xenograft tumors exhibited consistent regrowth (Figure 3E and Figure S3A). At 5 weeks, the CRC cells surviving this combination treatment displayed tumorigenic activity that was able to generate secondary tumors when extracted and re-injected into immunocompromised mice (Figure 3F). Cells harvested from these secondary tumors showed a comparable percentage of clonogenic cells (Figure S3B), suggesting that resistant tumorigenic cells are spared by PI3K-based combinatorial treatments.
Figure 3.

HER2/BRAF/PI3K combinatorial targeting leads to transient therapeutic response. (A) Cell viability in wt (#21, #27, #33), BRAF- (#1, #3, #5) and KRAS-mutant (# 9, #11, #16) CR-CSphCs treated with Vemu (1 µM) + Tmab (10 µg/mL) + BKM120 (1 µM), Vemu + Cmab (20 µg/mL) + BKM120 or with a vehicle as control. (B) Percentage of CD44v6 positivity evaluated by flow cytometry for wt (#21, #27, #33), BRAF- (#1, #3, #5) and KRAS-mutant (#9, #11, #16) CR-CSphCs treated as indicated for 72 h. (C,D) Clonogenic activity (C) and percentage of sphere-forming cells for wt (#21, #27, #33), BRAF- (#1, #3, #5) and KRAS-mutant (#9, #11, #16) sphere cells pretreated as in (B). For (A–D), data show the means ± SD of three different experiments. (E) Fold change over vehicle of volume of subcutaneous tumor xenograft generated by the injection of wt (#14, 21), BRAF- (#1, 2) or KRAS-mutant (#11, 16) CR-CSphCs and treated with a vehicle (Vehicle), vemurafenib (V, 20 mg/Kg), cetuximab (C, 40 mg/Kg) or trastuzumab (T, 5 mg/Kg) in combination with BKM120 (B, 20 mg/Kg) for 4 weeks. For (A,E), green and red arrows indicate the start and the end of the treatment, respectively. (F) Size of secondary tumors (8 weeks) generated by subcutaneous injection of 1 × 104 freshly purified CR-CSphCs derived from wt (#21), BRAF- (#2) or KRAS-mutant (#11) xenograft tumors. For (D,E), data are expressed as means ± SD (n = 6). ns, not significant; * p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001, **** p ≤ 0.0001.
3.3. Resistance to BRAF-Based Combination Therapy Is Sustained by Myc Expression
To investigate the molecular mechanisms underlying the triple-combination acquired resistance, cancer cells isolated from tumor xenografts were subjected to gene expression analysis of cancer stem cell- and metastasis-related genes. Notwithstanding the fact that the expression levels of genes involved in the Wnt pathway and EMT process were not influenced, cancer cells surviving the in vivo combinatorial treatment showed a markedly increased expression of MYC levels, regardless of the mutational status (Figure 4A). This expression was mainly restricted to CD44v6+ cells surviving the triple combination (Figure 4B and Figure S4A). Although Myc expression levels are altered in many tumors and numerous therapeutic strategies to directly target them have been evaluated, Myc still remains “undruggable” [38]. To study whether the inhibition of CD44v6 and Myc could efficiently target CR-CSCs, we combined dinaciclib, a selective small-molecule inhibitor of CDKs that indirectly downregulates Myc [39], with taselisib, a selective inhibitor of class I PI3K currently used in clinical settings. CR-CSphCs were exposed to taselisib in combination with dinaciclib in a dose–response matrix (Figure S4B). Thus, we sought to investigate whether microenvironmental cytokines, released by cancer-associated fibroblasts (CAFs), would affect the viability of CR-CSphCs treated with dinaciclib-based therapy, which targets downstream of the MEK pathway. In accordance with our previously published data [17], CAF-conditioned medium (CAF CM) played a protective effect in CR-CSphCs exposed to vemurafenib-based therapy, increasing the CD44v6+ cell population expressing Myc (Figure 4C and Figure S4C). This phenomenon was abrogated when using Myc and PI3K inhibitors in CR-CSphCs, the survival of which was significantly reduced regardless of the mutational status (Figure 4C,D). In the presence of CAF CM, dinaciclib in combination with taselisib reduced the cell viability of CR-CSphCs harboring MYC amplification and lowered the activity of the PI3K/AKT pathway and Myc expression (Figure 4E,F and Figure S4D). Notably, this treatment was able to effectively counteract clonogenic and colony-forming activities (Figure 4G,H and Figure S4E). Targeting of Myc transcription and CD44v6 with dinaciclib and taselisib, respectively, significantly affected the growth of tumor xenografts generated by subcutaneous injection of aggressive KRAS-mutant/MYC-amplified CR-CSphCs, without affecting mice weight (Figure 4I and Figure S4F). Altogether, these findings indicate that Myc promotes CSC resistance to BRAF-based combination therapy and its indirect inhibition overwhelms the protective activity of the TME, paving the way for an investigation of dinaciclib in combination with taselisib in clinical regimens.
Figure 4.

Myc and PI3K combinatorial targeting reduces CR-CSphC viability, downregulating Myc and CD44v6 expression levels. (A) Heatmap of cancer stem cell- and metastasis-related genes (2−ΔCt) in freshly purified cells (#21, #2, #10) from xenografts treated with a vehicle or V (20 mg/Kg) + T (5 mg/Kg) + B (20 mg/Kg). (B) Percentage of CD44v6 and Myc expression in xenograft tumors treated for 4 weeks with a vehicle (Vehicle) or V (20 mg/Kg) + T (5 mg/Kg) + B (20 mg/Kg). Data are shown as means ± SD of three different xenografts (#21, #2, #11). (C) Death percentage of CR-CSphCs treated as indicated in the presence of SCM or CAF-conditioned medium (CAF CM) for 72 h. Data represent means ± SD of three independent experiments performed with wt (#21, #27, #33), BRAF- (#1, #3, #5) and KRAS-mutant (#8, #9, #16) CR-CSphCs. (D) Cell viability in wt (#6, #14, #21, #27, #33, #49), BRAF- (#1–#5) and KRAS-mutant (#8, #9, #11, #13, #16, #59) CR-CSphCs treated with vehicle or taselisib (1 µM) and dinaciclib (10 nM) (tas + din) for 72 h. (E) Cell viability in MYC wt (MYC GCN < 4, CSphC#2, #3, #6, #9, #14, #16, #21, #27, #33) and MYC-amplified (MYC GCN ≥ 4, #1, #4, #5, #8, #11, #13, #49, #59) CR-CSphCs treated as in (D). For (C,D), data represent means ± SD of three independent experiments. (F) Analysis of pAKT, AKT and Myc on the indicated CR-CSphCs and treated with a vehicle or tas (1 µM) + din (10 nM) at 48 h. (G,H) Clonogenic activity (G) and colony number (H) of wt (#21, #33), BRAF- (#1, #3) and KRAS-mutant (#9, #16) sphere cells previously treated with a vehicle or tas (1 µM) + din (10 nM) for 72 h, in the presence of CAF CM. For (G,H), data are means ± SD of three independent experiments. For (D–H), CR-CSphCs were exposed to CAF CM. (I) Tumor volume of subcutaneous xenograft generated by the injection of KRAS-mutant (#8) CR-CSphCs and treated with a vehicle (Vehicle) or taselisib (tas, 5 mg/Kg) and dinaciclib (din, 25 mg/Kg) for 4 weeks. Green and red arrows indicate the start and the end of the treatment, respectively. Data are expressed as means ± SD (n = 6). ns, not significant; ** p ≤ 0.01, **** p ≤ 0.0001. The uncropped blots are shown in supplementary materials.
4. Discussion
One of the major challenges in cancer research is preventing the emergence of acquired resistance to targeted therapies. Thus, the discovery of targetable resistance mechanisms may allow the design of new therapeutic strategies to overcome tumor recurrence.
The most frequent deregulated pathways in CRC patients are the Wnt, MAPK and PI3K pathways [40]. Patients harboring RAS/RAF mutations display poor prognosis, highlighting a critical need to improve therapeutic options [41]. CRC patients treated with selective Ras/Raf pathway-targeted agents show limited benefits in contrast to the considerable response observed in BRAF-mutated melanoma patients [2]. Acquired drug resistance to this combination therapy is mediated by feedback reactivation of EGFR via the Ras pathway, triggering cell proliferation and survival [36,42]. Although a clinical advantage was observed in BRAF-mutant CRC patients treated with BRAF, EGFR and MEK inhibitors, the extent of the clinical responses was time-limited, suggesting the urgency of discovering molecular mechanisms driving CRC acquired resistance [43].
As the data show, the vemurafenib treatment produced a short-term inhibition of colony-forming capacity and cell viability in BRAF-mutant CR-CSphCs due to EGFR-mediated activation of ERK signaling. Moreover, a paradoxical activation of the ERK pathway was observed in wt and KRAS-mutant cells, corroborating the finding that the aberrant activity of EGFR signaling may contribute to conferring resistance to vemurafenib in CRC cells.
We recently identified CD44v6 as a functional stem cell-like marker that typifies CR-CSCs characterized by metastatic potential and standard/targeted therapy resistance [11,17,30,44,45]. Independently of their mutational background, CD44v6-negative and -positive fractions displayed different sensitivities to BRAF inhibition in combination with anti-EGFR/HER2 antibodies, limiting their efficacy to the more differentiated cells. This CD44v6+ cell refractoriness is sustained by high expression levels of HER2, associated with an aberrant activation of PI3K-AKT and MAPK signaling [17]. Although PI3K and AKT inhibitors efficiently target disseminating CD44v6+ CR-CSCs, their anti-tumor activity was found to be dramatically reduced in pre-clinical mouse models, where the presence of a protective TME counteracted drug efficacy [11,17]. Likewise, cells spared by BRAF, HER2 and PI3K inhibition maintained the capacity to generate secondary tumors and re-grow following conclusion of treatment, even though reductions of CD44v6 expression and sphere-forming ability were observed in primary tumor xenograft-derived cells [17]. A gene expression analysis of cancer cells exposed to that treatment identified Myc and Rac1, one of the critical factors in the maintenance of the intestinal barrier and upregulated in primary and metastatic CRCs [46], as major candidates driving acquired resistance. This phenomenon could be driven by the adaptive responses to PI3K-AKT signaling pathway inhibition, which leads to Myc induction by activating NOTCH signaling, which is a hallmark of CSCs [47]. Although a good candidate for the development of therapeutic targeted strategies, pharmacological Myc inhibition remains difficult since both its nuclear localization and biological function are critical for tissue homeostasis. JQ1 is a small-molecule inhibitor of BRD4, a member of the bromodomain and extraterminal (BET) family regulating the transcription of MYC. Although JQ1 induces cell death by affecting the growth of several cancers, its bioavailability and short half-life hinder its use in clinical settings [48,49]. Several clinical trials have been designed with analogs of JQ1, such as AZD5153 and GSK525762, to indirectly target Myc, both in hematological and solid neoplasia (i.e., NCT03205176, NCT01587703). Despite their potent anti-tumor activity, BET inhibitors in patients displayed a high toxicity profile [50,51]. Another strategy adopted to interfere with MYC transcription is the use of cyclin-dependent kinase (CDK) inhibitors [50]. A clinical trial involving dinaciclib, a potent multiple CDK inhibitor, in advanced breast cancer patients is currently evaluating the response based on Myc expression (NCT01676753) [51]. Additionally, Myc-dominant negative mutant Omomyc has been recently approved for its first human phase I/III trial to assess its efficacy in cancer patients [52]. Moreover, amplification of MYC has been reported as a key biomarker of response to HER2 inhibitors [53]. The synthetic lethality approach based on dinaciclib in combination with taselisib impairs CR-CSphCs viability and clonogenic capacity, with a concomitant reduction of Myc and PI3K/AKT pathway activity in the presence of TME cytokines, in line with the stabilization of tumor growth in mouse avatars. The possibility of interfering with MYC transcription, a downstream target of MAPK pathway, could provide potential clinical benefits by reducing the side effects of kinase inhibitors and preventing acquired resistance mediated by ERK reactivation. RAS is the most frequently mutated oncogene in cancer and about the 45% of CRCs harbor KRAS mutations. Notably, targeting of Rac1 by depletion of specific Rac-guanine nucleotide exchange factors (Rac-GEFs) fails to hinder the tumor growth in the presence of KRAS mutations, limiting its clinical use for advanced tumors [54].
Despite the efforts that have been made toward the development of efficacious therapeutic regimens, to date no targeted therapies are available for these CRC patients. Recently, several clinical trials have evaluated the anti-tumor activities of small molecules (AMG510 and MRTX84) targeting mutant KRASG12C mutation (NCT03600883, NCT03785249). Adaptive feedback signaling networks that lead to activation of the EGFR pathway after KRASG12C inhibitors were recently identified as the dominant mechanism of resistance in CRC patients [55].
Here, we identified a potential mechanism, shared by CRCs, of acquired resistance to targeted cancer therapies. Under drug treatment pressure, the presence of TME cytokines promotes CD44v6 expression together with high expression levels of Myc, representing an escape mechanism adopted by cancer cells to cope with the action of anti-tumor therapies and favor disease progression. Taken together, these data provide a strong rationale for the use of dinaciclib combined with taselisib for advanced CRCs.
5. Conclusions
Herein, we demonstrated that the combinatorial targeting of MYC transcription and PI3K activity could be a valuable therapeutic option for advanced CRC, regardless of the mutational background and/or MYC gene amplification. We found that the combinatorial targeting of BRAF and EGFR/HER2 spares the CD44v6-positive subpopulation endowed with high PI3K/AKT activity. Although adding a PI3K inhibitor stabilized/delayed the progression of xenograft tumors, following treatment cessation, the tumor microenvironment boosted the expression of CD44v6 and Myc, resulting in tumor xenograft regrowth. The targeting of MYC transcription and PI3K activity impaired CR-CSphCs’ viability and clonogenic capacity, even in the presence of microenvironmental cytokines, and stabilized tumor xenograft growth, supporting the use of this combinatorial treatment for advanced CRC patient management.
Acknowledgments
L.R.M., G.P. (Gaetana Porcelli), C.D. and F.V. are PhD students in the Molecular and Clinical Medicine program. V.D.P. is a PhD student in the Experimental Oncology and Surgery program. A.T. and V.V. are research fellows funded by the European Union—FESR FSE, PON Ricerca e Innovazione 2014–2020 (AIM line 1). We also thank Fancesco Calò for image editing.
Supplementary Materials
The following are available online at https://www.mdpi.com/article/10.3390/cancers14030673/s1, Figure S1: BRAF inhibition leads to paradoxical activation of MAPK pathway in wt and KRAS-mutant CR-CSphCs. Figure S2: The addition of EGFR/HER2 inhibitor to vemurafenib results in no additional effect on the MAPK pathway. Figure S3: Xenograft-derived CR-CSphCs following treatment with HER2/BRAF/PI3K inhibition maintain their sphere-forming capacity. Figure S4: Combinatorial inhibition of Myc and PI3K overcome the protective effect of TME cytokines. The uncropped blots are shown in supplementary materials.
Author Contributions
Conceptualization, M.G., L.R.M. and G.S.; Methodology, C.M., V.D.P., G.P. (Gaetana Porcelli), S.D.F., M.L.I., C.D., F.V., I.P., A.T., V.V., O.R.B. and G.M.; Validation, M.G., L.R.M. and S.D.F.; Formal Analysis, M.G., L.R.M. and C.M.; Writing—Original Draft Preparation, M.G., L.R.M., C.M. and S.D.F.; Writing—Review & Editing, M.T., R.D.M. and G.S.; Supervision, G.P. (Giuseppe Pistone), M.R.B., M.T., R.D.M. and G.S.; Funding Acquisition, M.T., R.D.M. and G.S. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the following grants: AIRC IG (21492) to M.T., RF2018-12367044 to M.T. and R.D.M. and PRIN (Prot. No. 2017WNKSLR) and AIRC IG (21445) to G.S.
Institutional Review Board Statement
The study was conducted according to the guidelines of the Declaration of Helsinki and approved by the Institutional Ethics Committee of the University of Palermo (authorization CE9/2015, Policlinico “Paolo Giaccone”, 14.10.2015, Palermo). The in vivo studies were performed following the animal care committee guidelines of the University of Palermo (Italian Ministry of Health authorization number 154/2017-PR).
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
Data related to the study are included in the article or uploaded as supplementary information. Data are available from the corresponding authors (R.D.M. and G.S.) upon reasonable request.
Conflicts of Interest
The authors declare no conflict of interest.
Footnotes
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Siegel R.L., Miller K.D., Fuchs H.E., Jemal A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021;71:7–33. doi: 10.3322/caac.21654. [DOI] [PubMed] [Google Scholar]
- 2.Xie Y.-H., Chen Y.-X., Fang J.-Y. Comprehensive review of targeted therapy for colorectal cancer. Signal Transduct. Target. Ther. 2020;5:1–30. doi: 10.1038/s41392-020-0116-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Raskov H., Søby J.H., Troelsen J.T., Bojesen R.D., Gögenur I. Driver Gene Mutations and Epigenetics in Colorectal Cancer. Ann. Surg. 2020;271:75–85. doi: 10.1097/SLA.0000000000003393. [DOI] [PubMed] [Google Scholar]
- 4.Matano M., Date S., Shimokawa M., Takano A., Fujii M., Ohta Y., Watanabe T., Kanai T., Sato T. Modeling colorectal cancer using CRISPR-Cas9–mediated engineering of human intestinal organoids. Nat. Med. 2015;21:256–262. doi: 10.1038/nm.3802. [DOI] [PubMed] [Google Scholar]
- 5.Gaggianesi M., Di Franco S., Pantina V.D., Porcelli G., D’Accardo C., Verona F., Veschi V., Colarossi L., Faldetta N., Pistone G., et al. Messing Up the Cancer Stem Cell Chemoresistance Mechanisms Supported by Tumor Microenvironment. Front. Oncol. 2021;11:2847. doi: 10.3389/fonc.2021.702642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Batlle E., Clevers H. Cancer stem cells revisited. Nat. Med. 2017;23:1124–1134. doi: 10.1038/nm.4409. [DOI] [PubMed] [Google Scholar]
- 7.Turdo A., Veschi V., Gaggianesi M., Chinnici A., Bianca P., Todaro M., Stassi G. Meeting the Challenge of Targeting Cancer Stem Cells. Front. Cell Dev. Biol. 2019;7:16. doi: 10.3389/fcell.2019.00016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Di Franco S., Todaro M., Dieli F., Stassi G. Colorectal cancer defeating? Challenge accepted! Mol. Asp. Med. 2014;39:61–81. doi: 10.1016/j.mam.2013.07.001. [DOI] [PubMed] [Google Scholar]
- 9.Cancer Genome Atlas Network Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–337. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.De Sousa e Melo F., Vermeulen L. Wnt Signaling in Cancer Stem Cell Biology. Cancers. 2016;8:60. doi: 10.3390/cancers8070060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Todaro M., Gaggianesi M., Catalano V., Benfante A., Iovino F., Biffoni M., Apuzzo T., Sperduti I., Volpe S., Cocorullo G., et al. CD44v6 Is a Marker of Constitutive and Reprogrammed Cancer Stem Cells Driving Colon Cancer Metastasis. Cell Stem Cell. 2014;14:342–356. doi: 10.1016/j.stem.2014.01.009. [DOI] [PubMed] [Google Scholar]
- 12.Vermeulen L., Felipe de Sousa E.M., van der Heijden M., Cameron K., de Jong J.H., Borovski T., Tuynman J.B., Todaro M., Merz C., Rodermond H., et al. Wnt activity defines colon cancer stem cells and is regulated by the microenvironment. Nat. Cell Bol. 2010;12:468–476. doi: 10.1038/ncb2048. [DOI] [PubMed] [Google Scholar]
- 13.Douillard J.-Y., Oliner K.S., Siena S., Tabernero J., Burkes R., Barugel M., Humblet Y., Bodoky G., Cunningham D., Jassem J., et al. Panitumumab–FOLFOX4 Treatment and RAS Mutations in Colorectal Cancer. N. Engl. J. Med. 2013;369:1023–1034. doi: 10.1056/NEJMoa1305275. [DOI] [PubMed] [Google Scholar]
- 14.Sorich M.J., Wiese M., Rowland A., Kichenadasse G., McKinnon R., Karapetis C. Extended RAS mutations and anti-EGFR monoclonal antibody survival benefit in metastatic colorectal cancer: A meta-analysis of randomized, controlled trials. Ann. Oncol. 2014;26:13–21. doi: 10.1093/annonc/mdu378. [DOI] [PubMed] [Google Scholar]
- 15.Mao C., Liao R.-Y., Qiu L.-X., Wang X.-W., Ding H., Chen Q. BRAF V600E mutation and resistance to anti-EGFR monoclonal antibodies in patients with metastatic colorectal cancer: A meta-analysis. Mol. Biol. Rep. 2010;38:2219–2223. doi: 10.1007/s11033-010-0351-4. [DOI] [PubMed] [Google Scholar]
- 16.Sanz-Garcia E., Argiles G., Elez E., Tabernero J. BRAF mutant colorectal cancer: Prognosis, treatment, and new perspectives. Ann. Oncol. 2017;28:2648–2657. doi: 10.1093/annonc/mdx401. [DOI] [PubMed] [Google Scholar]
- 17.Mangiapane L.R., Nicotra A., Turdo A., Gaggianesi M., Bianca P., Di Franco S., Sardina D.S., Veschi V., Signore M., Beyes S., et al. PI3K-driven HER2 expression is a potential therapeutic target in colorectal cancer stem cells. Gut. 2021;71:119–128. doi: 10.1136/gutjnl-2020-323553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hansen T., Qvortrup C., Pfeiffer P. Angiogenesis Inhibitors for Colorectal Cancer. A Review of the Clinical Data. Cancers. 2021;13:1031. doi: 10.3390/cancers13051031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Overman M.J., Ernstoff M.S., Morse M.A. Where We Stand with Immunotherapy in Colorectal Cancer: Deficient Mismatch Repair, Proficient Mismatch Repair, and Toxicity Management. Am. Soc. Clin. Oncol. Educ. Book. 2018;38:239–247. doi: 10.1200/EDBK_200821. [DOI] [PubMed] [Google Scholar]
- 20.Jácome A.A., Eng C. Role of immune checkpoint inhibitors in the treatment of colorectal cancer: Focus on nivolumab. Expert Opin. Biol. Ther. 2019;19:1247–1263. doi: 10.1080/14712598.2019.1680636. [DOI] [PubMed] [Google Scholar]
- 21.Sartore-Bianchi A., Loupakis F., Argilés G., Prager M.G. Challenging chemoresistant metastatic colorectal cancer: Therapeutic strategies from the clinic and from the laboratory. Ann. Oncol. 2016;27:1456–1466. doi: 10.1093/annonc/mdw191. [DOI] [PubMed] [Google Scholar]
- 22.Sabnis A.J., Bivona T.G. Principles of Resistance to Targeted Cancer Therapy: Lessons from Basic and Translational Cancer Biology. Trends Mol. Med. 2019;25:185–197. doi: 10.1016/j.molmed.2018.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee K.-M., Giltnane J.M., Balko J.M., Schwarz L.J., Guerrero-Zotano A.L., Hutchinson K.E., Nixon M.J., Estrada M.V., Sánchez V., Sanders M.E., et al. MYC and MCL1 Cooperatively Promote Chemotherapy-Resistant Breast Cancer Stem Cells via Regulation of Mitochondrial Oxidative Phosphorylation. Cell Metab. 2017;26:633–647.e7. doi: 10.1016/j.cmet.2017.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Strippoli A., Cocomazzi A., Basso M., Cenci T., Ricci R., Pierconti F., Cassano A., Fiorentino V., Barone C., Bria E., et al. c-MYC Expression Is a Possible Keystone in the Colorectal Cancer Resistance to EGFR Inhibitors. Cancers. 2020;12:638. doi: 10.3390/cancers12030638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shivakumar B.M., Chakrabarty S., Rotti H., Seenappa V., Rao L., Geetha V., Tantry B.V., Kini H., Dharamsi R., Pai C.G., et al. Comparative analysis of copy number variations in ulcerative colitis associated and sporadic colorectal neoplasia. BMC Cancer. 2016;16:1–11. doi: 10.1186/s12885-016-2303-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dong Y., Tu R., Liu H., Qing G. Regulation of cancer cell metabolism: Oncogenic MYC in the driver’s seat. Signal Transduct. Target. Ther. 2020;5:1–11. doi: 10.1038/s41392-020-00235-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Elbadawy M., Usui T., Yamawaki H., Sasaki K. Emerging Roles of C-Myc in Cancer Stem Cell-Related Signaling and Resistance to Cancer Chemotherapy: A Potential Therapeutic Target Against Colorectal Cancer. Int. J. Mol. Sci. 2019;20:2340. doi: 10.3390/ijms20092340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shen A., Wang L., Huang M., Sun J., Chen Y., Shen Y.-Y., Yang X., Wang X., Ding J., Geng M. c-Myc Alterations Confer Therapeutic Response and Acquired Resistance to c-Met Inhibitors in MET-Addicted Cancers. Cancer Res. 2015;75:4548–4559. doi: 10.1158/0008-5472.CAN-14-2743. [DOI] [PubMed] [Google Scholar]
- 29.Wang Q., Tan R., Zhu X., Zhang Y., Tan Z., Su B., Li Y. Oncogenic K-ras confers SAHA resistance by up-regulating HDAC6 and c-myc expression. Oncotarget. 2016;7:10064–10072. doi: 10.18632/oncotarget.7134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Veschi V., Mangiapane L.R., Nicotra A., di Franco S., Scavo E., Apuzzo T., Sardina D.S., Fiori M.E., Benfante A., Colorito M.L., et al. Targeting chemoresistant colorectal cancer via systemic administration of a BMP7 variant. Oncogene. 2019;39:987–1003. doi: 10.1038/s41388-019-1047-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Di Franco S., Zhang L., Gaggianesi M., Iacono M.L., Medema J.P., Stassi G. FACS-based protocol to assess cytotoxicity and clonogenic potential of colorectal cancer stem cells using a Wnt/β-catenin signaling pathway reporter. STAR Protoc. 2021;2:100880. doi: 10.1016/j.xpro.2021.100880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee K.S., Nam S.K., Seo S.H., Park K.U., Oh H.-K., Kim D.-W., Kang S.-B., Kim W.H., Lee H.S. Digital polymerase chain reaction for detecting c-MYC copy number gain in tissue and cell-free plasma samples of colorectal cancer patients. Sci. Rep. 2019;9:1–9. doi: 10.1038/s41598-018-38415-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ianevski A., He L., Aittokallio T., Tang J. SynergyFinder: A web application for analyzing drug combination dose–response matrix data. Bioinformatics. 2017;33:2413–2415. doi: 10.1093/bioinformatics/btx162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Poulikakos P.I., Zhang C., Bollag G., Shokat K.M., Rosen N. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature. 2010;464:427–430. doi: 10.1038/nature08902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Heidorn S.J., Milagre C., Whittaker S., Nourry A., Niculescu-Duvas I., Dhomen N., Hussain J., Reis-Filho J.S., Springer C.J., Pritchard C., et al. Kinase-Dead BRAF and Oncogenic RAS Cooperate to Drive Tumor Progression through CRAF. Cell. 2010;140:209–221. doi: 10.1016/j.cell.2009.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Corcoran R.B., Ebi H., Turke A.B., Coffee E.M., Nishino M., Cogdill A., Brown R.D., Della Pelle P., Dias-Santagata D., Hung K.E., et al. EGFR-Mediated Reactivation of MAPK Signaling Contributes to Insensitivity of BRAF-Mutant Colorectal Cancers to RAF Inhibition with Vemurafenib. Cancer Discov. 2012;2:227–235. doi: 10.1158/2159-8290.CD-11-0341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Di Franco S., Bianca P., Sardina D.S., Turdo A., Gaggianesi M., Veschi V., Nicotra A., Mangiapane L.R., Iacono M.L., Pillitteri I., et al. Adipose stem cell niche reprograms the colorectal cancer stem cell metastatic machinery. Nat. Commun. 2021;12:1–16. doi: 10.1038/s41467-021-25333-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dang C.V., Reddy E.P., Shokat K.M., Soucek L. Drugging the ‘undruggable’ cancer targets. Nat. Cancer. 2017;17:502–508. doi: 10.1038/nrc.2017.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Carey J.P., Karakas C., Bui T., Chen X., Vijayaraghavan S., Zhao Y., Wang J., Mikule K., Litton J.K., Hunt K.K., et al. Synthetic Lethality of PARP Inhibitors in Combination with MYC Blockade Is Independent of BRCA Status in Triple-Negative Breast Cancer. Cancer Res. 2017;78:742–757. doi: 10.1158/0008-5472.CAN-17-1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee C.S., Song I.H., Lee A., Kang J., Lee Y.S., Lee I.K., Song Y.S., Lee S.H. Enhancing the landscape of colorectal cancer using targeted deep sequencing. Sci. Rep. 2021;11:1–26. doi: 10.1038/s41598-021-87486-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tie J., Gibbs P., Lipton L., Christie M., Jorissen R., Burgess A.W., Croxford M., Jones I., Langland R., Kosmider S., et al. Optimizing targeted therapeutic development: Analysis of a colorectal cancer patient population with the BRAFV600E mutation. Int. J. Cancer. 2010;128:2075–2084. doi: 10.1002/ijc.25555. [DOI] [PubMed] [Google Scholar]
- 42.Prahallad A., Sun C., Huang S., Di Nicolantonio F., Salazar R., Zecchin D., Beijersbergen R.L., Bardelli A., Bernards R. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature. 2012;483:100–103. doi: 10.1038/nature10868. [DOI] [PubMed] [Google Scholar]
- 43.Corcoran R.B., André T., Atreya C.E., Schellens J.H., Yoshino T., Bendell J.C., Hollebecque A., McRee A.J., Siena S., Middleton G., et al. Combined BRAF, EGFR, and MEK Inhibition in Patients with BRAFV600E-Mutant Colorectal Cancer. Cancer Discov. 2018;8:428–443. doi: 10.1158/2159-8290.CD-17-1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yang L., Shi P., Zhao G., Xu J., Peng W., Zhang J., Zhang G., Wang X., Dong Z., Chen F., et al. Targeting cancer stem cell pathways for cancer therapy. Signal Transduct. Target. Ther. 2020;5:8. doi: 10.1038/s41392-020-0110-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Turdo A., Glaviano A., Pepe G., Calapà F., Raimondo S., Fiori M., Carbone D., Basilicata M., Di Sarno V., Ostacolo C., et al. Nobiletin and Xanthohumol Sensitize Colorectal Cancer Stem Cells to Standard Chemotherapy. Cancers. 2021;13:3927. doi: 10.3390/cancers13163927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kotelevets L., Chastre E. Rac1 Signaling: From Intestinal Homeostasis to Colorectal Cancer Metastasis. Cancers. 2020;12:665. doi: 10.3390/cancers12030665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Muellner M.K., Uras I.Z., Gapp B.V., Kerzendorfer C., Smida M., Lechtermann H., Craig-Mueller N., Colinge J., Duernberger G., Nijman S.M.B. A chemical-genetic screen reveals a mechanism of resistance to PI3K inhibitors in cancer. Nat. Chem. Biol. 2011;7:787–793. doi: 10.1038/nchembio.695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Delmore J.E., Issa G.C., Lemieux M.E., Rahl P.B., Shi J., Jacobs H.M., Kastritis E., Gilpatrick T., Paranal R.M., Qi J., et al. BET Bromodomain Inhibition as a Therapeutic Strategy to Target c-Myc. Cell. 2011;146:904–917. doi: 10.1016/j.cell.2011.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shorstova T., Foulkes W.D., Witcher M. Achieving clinical success with BET inhibitors as anti-cancer agents. Br. J. Cancer. 2021;124:1478–1490. doi: 10.1038/s41416-021-01321-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chen H., Liu H., Qing G. Targeting oncogenic Myc as a strategy for cancer treatment. Signal Transduct. Target. Ther. 2018;3:5. doi: 10.1038/s41392-018-0008-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gregory G., Hogg S., Kats L., Vidacs E., Baker A.J., Gilan O., Lefebure M., Martin B.P., Dawson M.A., Johnstone R., et al. CDK9 inhibition by dinaciclib potently suppresses Mcl-1 to induce durable apoptotic responses in aggressive MYC-driven B-cell lymphoma in vivo. Leukemia. 2014;29:1437–1441. doi: 10.1038/leu.2015.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Villanueva M.T. Long path to MYC inhibition approaches clinical trials. Nat. Rev. Drug Discov. 2019 doi: 10.1038/d41573-019-00055-2. [DOI] [PubMed] [Google Scholar]
- 53.Risom T., Wang X., Liang J., Zhang X., Pelz C., Campbell L.G., Eng J., Chin K., Farrington C., Narla G., et al. Deregulating MYC in a model of HER2+ breast cancer mimics human intertumoral heterogeneity. J. Clin. Investig. 2019;130:231–246. doi: 10.1172/JCI126390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pickering K.A., Gilroy K., Cassidy J.W., Fey S.K., Najumudeen A.K., Zeiger L.B., Vincent D.F., Gay D.M., Johansson J., Fordham R.P., et al. A RAC-GEF network critical for early intestinal tumourigenesis. Nat. Commun. 2021;12:1–14. doi: 10.1038/s41467-020-20255-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Amodio V., Yaeger R., Arcella P., Cancelliere C., Lamba S., Lorenzato A., Arena S., Montone M., Mussolin B., Bian Y., et al. EGFR Blockade Reverts Resistance to KRASG12C Inhibition in Colorectal Cancer. Cancer Discov. 2020;10:1129–1139. doi: 10.1158/2159-8290.CD-20-0187. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data related to the study are included in the article or uploaded as supplementary information. Data are available from the corresponding authors (R.D.M. and G.S.) upon reasonable request.