

Abstract
Cryo-electron tomography (cryo-ET) is a powerful technique to examine cellular structures as they exist in situ. However, direct imaging by TEM for cryo-ET is limited to specimens up to ~400 nm in thickness, narrowing its applicability to areas such as cellular projections or small bacteria and viruses. Cryo-focused-ion-beam (cryo-FIB) milling has emerged in recent years as a method to generate thin specimens from cellular samples in preparation for cryo-ET. In this technique, specimens are thinned with a beam of gallium ions to gradually ablate cellular material in order to leave a thin, electron-transparent section (a lamella) through the bulk material. The lamella can be used for high-resolution cryo-ET to visualize cells in 3D in a near-native state. This approach has proved to be robust and relatively simple for new users, and exhibits minimal sectioning artifacts. In this chapter, we describe a general approach to cryo-FIB milling for users with prior cryo-EM experience, with extensive notes on operation and troubleshooting.
Keywords: cryo-focused ion beam milling, cryo-electron tomography, sample preparation, lamella, mammalian cells, yeast, bacteria
1. Introduction
A major goal in biological imaging is to visualize macromolecular complexes and the intricate networks they form at high-resolution in situ with minimal artifacts. However, most commonly accessible techniques lack sufficient resolution (such as fluorescence light microscopy) or sufficient cellular context (such as single particle cryo-EM). Cryo-electron tomography is a high-resolution imaging modality that can visualize macromolecular structures in situ in a near-native state in their cellular context [1]. However, specimen thickness is a major limitation with cryo-ET and other electron microscopy techniques. Due to multiple electron scattering, specimen thickness is limited to less than 400 nm at 300 kV [2], which is well below the size of eukaryotic cells and most bacteria.
One approach to overcome this limitation involves sectioning cells under cryogenic conditions using a microtome [3]. However this approach is technically demanding and introduces visual and structural artifacts that arise from the the sectioning process [4]. Recent work has adapted the versatile focused ion beam (FIB) instrument for cryo-electron microscopy applications as a method to generate thin sections through cellular material, yielding unprecedented insight into eukaryotic and prokaryotic cell biology [5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24]. This technique offers significant advantages including relative ease of use, minimal artifacts, and the ability to target specific cells for high-resolution cryo-ET.
Focused ion beam (FIB) microscopy and milling has been used in materials science as a method to thin samples for analysis and to create micro/nanoscale patterns [25]. The operation uses a tightly focused (5–10 nm) beam of ions, typically gallium, that rasters over the sample [26], similar to the beam of electrons in conventional scanning electron microscopy (SEM). Secondary electrons are generated by the ion beam’s interaction with the sample, which can be detected to form images. Furthermore, the relatively large mass of gallium ions can be used as a tool for micro/nano machining by selectively ablating material from a specimen. In this manner, the FIB can be used to precisely remove material above and below a region of interest to leave a thin section—a lamella—that remains supported by the unmilled material around it. FIB milling sample preparation is usually performed in a “Dual-Beam” instrument with both FIB and SEM columns for simultaneous milling and multi-perspective imaging. These instruments are also typically equipped with gas-injection systems that allow targeted metal deposition or modification of milling characteristics [27]. For cryo-FIB applications, these instruments are equipped with a stage cooled to liquid nitrogen temperatures to maintain samples at cryogenic temperatures and an airlock quick-loading system to introduce cryogenic specimens into the chamber under vacuum. This allows cold samples to be transferred into and out of the instrument while avoiding atmospheric ice contamination and to be milled at liquid nitrogen temperatures for several hours.
This chapter will briefly describe practical principles and procedures for cryo-FIB milling including considerations for upstream sample preparation, downstream TEM tomography, and evaluation of sample quality as part of a typical cryo-ET workflow (Fig. 1). A closely related technique using cryo-FIB liftout can be used for much thicker samples such as tissue blocks [28, 29, 30], but will not be discussed in this chapter. At this time of writing, commercial dual-beam instruments can be outfitted with third party cryostages/quickloader systems such as the Quorum PP3006 cryo-stage, the Leica EM VCT500, and the Hummingbird Scientific cryotransfer system. In this chapter, we will describe a typical FIB milling protocol using the Thermo Fisher Scientific (TFS) Aquilos, a dual-beam FIB/SEM platform that includes a cryo-stage/quickloader system. However, many of the general principles should be applicable across instruments.
Figure 1: Typical cryo-FIB-ET workflow.
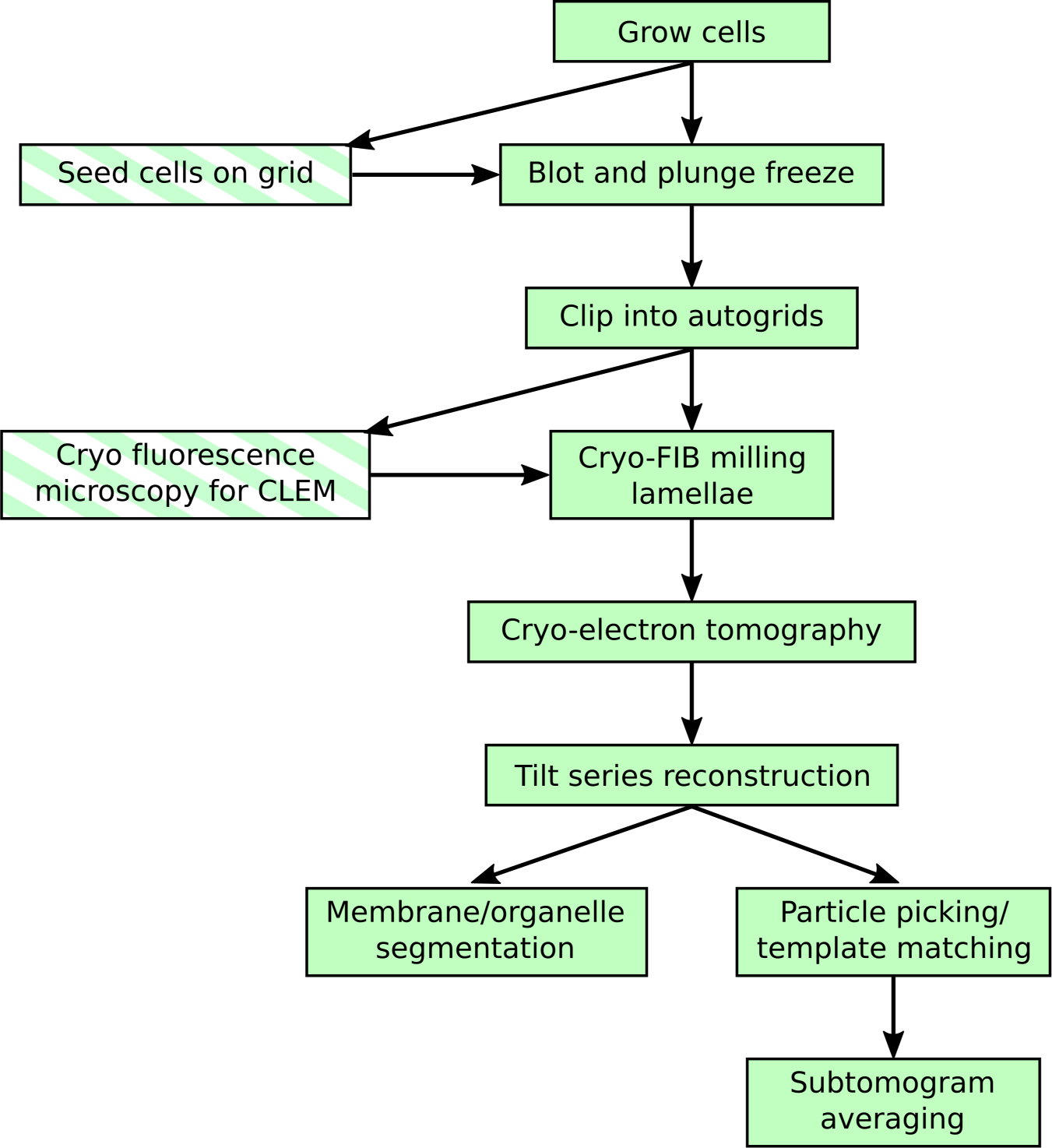
Cryo-FIB-ET is a vertically oriented workflow in which the same sample is processed throughout. This technique is sensitive to the success rate of each step and is limited by low throughput during lamella milling. This chapter seeks to provide practical guidance on increasing success at cryo-FIB milling, thereby increasing throughput for cryo-ET. Typical workflow steps are highlighted with a solid background. Optional steps are highlighted with a striped background.
1.1. Principles of Operation
Cryo-FIB milling is most easily performed using a dual-beam focused-ion-beam/scanning-electron-microscope (FIB/SEM) that allows simultaneous monitoring and processing of the sample. The SEM column is mounted vertically onto the chamber and the FIB column is mounted at a 52° angle relative to the SEM such that the beams intersect. This intersection point, the beam coincidence point, is defined as the working distance of the microscope. In a well aligned system, the beam-coincidence point should be located at the stage eucentric height (Fig. 2).
Figure 2: Schematic of grid and FIB/SEM geometry.
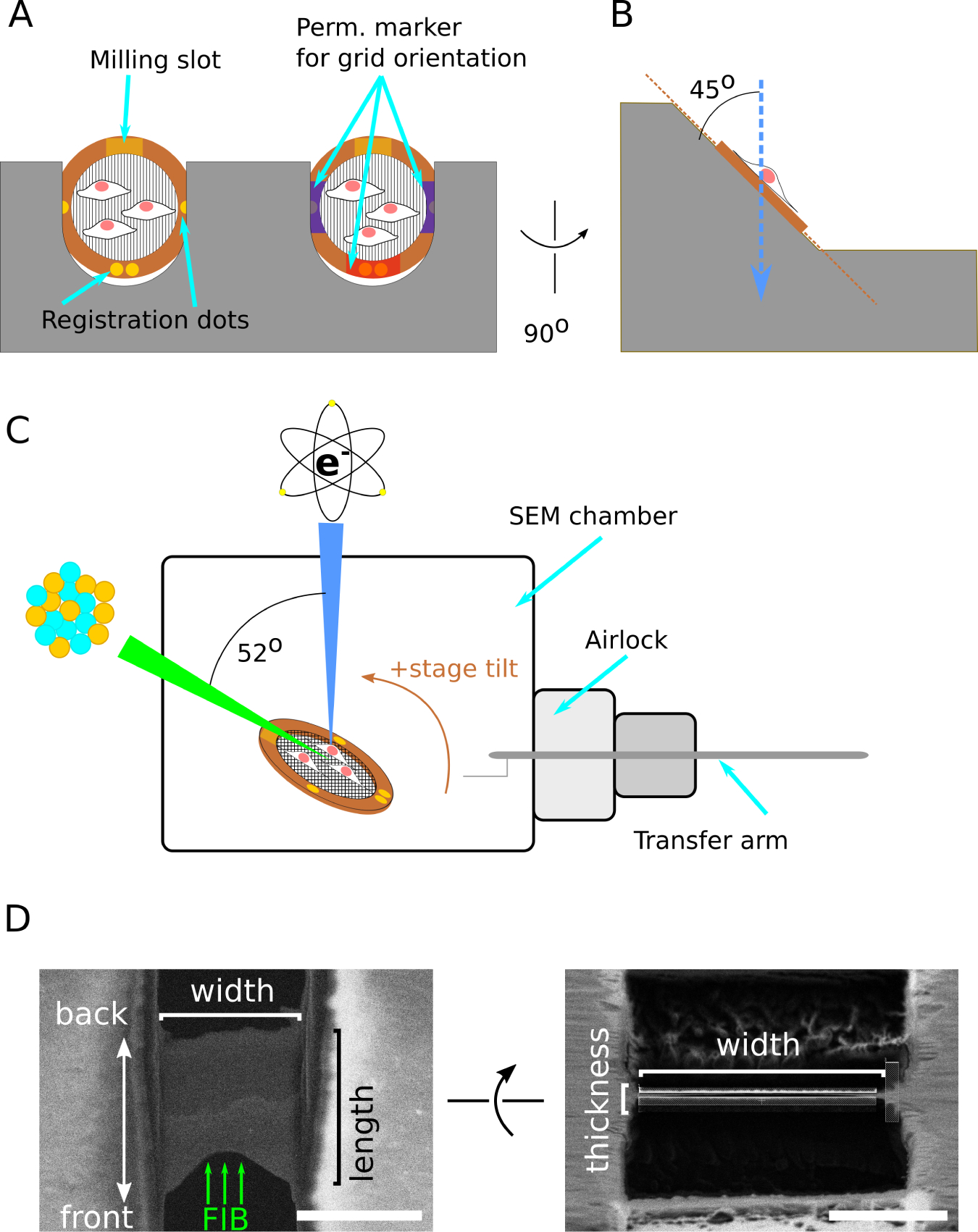
(A) Schematic of two grids clipped into an cryo-FIB-autogrid support and loaded into the shuttle, with cells on the top surface. The cryo-FIB-autogrid has a milling slot that enables lower angle milling and registration dots for aligning the grid when loading at the SEM or TEM. Additional markings with permanent marker can be made to enhance visibility under liquid nitrogen. Compare to Fig. 3E. (B) Side view of grid loaded into shuttle, illustrating the grids are held at a 45° angle relative to vertical. (C) Schematic of relative orientation of the SEM column, FIB column, sample stage, and transfer arm. The grid is positioned at the beam-coincident point, and the milling slot is oriented toward the FIB column. The stage is arranged to have positive tilt towards the FIB column. This results in FIB-sample incident angle 7° smaller than the reported stage angle. SEM beam is indicated in blue, FIB in green, and the specimen in copper. (D) Schematic of terminology used in this chapter to refer to lamella dimensions and orientation. Scale bars: 5 μm
Samples for cryo-FIB milling are typically prepared on TEM grids by plunge-freezing (see Section 3.1). The grid is positioned at the beam-coincidence point and oriented such that its surface forms a low incident angle (typically 5–10 degrees) relative to the FIB column (Fig. 2C). Milling thus results in lamellae that are nearly parallel to the substrate and long enough to contain enough material of interest as detailed in Section 1.2. The FIB is used to mill away material by continually rastering over the sample in a user-specified pattern to gradually ablate material. When starting a lamella, the milling patterns are set ~2 um apart, and are gradually brought closer together to reach the target thickness, <200 nm. Additionally, the ion-beam current is reduced as the lamella becomes thinner in order to afford more control over the milling process and to minimize beam damage. The final target lamella thickness should be informed by the biological question and any downstream analysis (e.g. subtomogram averaging or membrane segmentation) (see Section 3.3). Typical lamellae thicknesses may range from 85 to 250 nm, and typical TEM pixel sizes may range from 0.2 nm to 2 nm (see Section 3.3).
Platinum Sputtering and GIS deposition
Vitrified biological samples are not conductive, and thus often result in beam-induced charging. This charging effect can be detrimental during milling and also during TEM imaging that leads to low quality images. A platinum sputtering system can be used to reduce charging by depositing a thin conductive layer on the sample. The sputtering system may be built into the chamber or immediately outside the chamber as part of a cryo-stage/quickloader installation, both enabling sputtering to be performed on cryogenic samples without excess contamination.
For cryo-FIB milling, an organo-platinum gas injection system (GIS) is used to coat the surface of the cold grid before lamella milling. The gas is released into the chamber close to the sample and immediately condenses on the cold grid. When exposed to the ion beam, the organic component is partially sublimated, leaving a metallic/organic film behind [31]. This residual platinum is essential for protecting the leading edge of the lamella during the milling process to prevent uncontrolled milling that will form “curtain” artifacts [26]. Additionally, the platinum layer acts as structural support that prevents lamella cracking during subsequent handling.
Stage and Shuttle
Up to two grids previously clipped into an autogrid support (Note 3) can be loaded into the specimen shuttle (Fig. 2A,B and Fig. 3E) for insertion into the microscope. The grids are held in place by a metal spring flap that contacts the edge of the autogrid and maintains thermal contact. In order to minimize atmospheric ice contamination, the shuttle is transferred from the loading station to the microscope under vacuum using a sealed transfer arm (Fig. 3A), via a pumped airlock system (the quickloader Fig. 3J). Additionally, the shuttle has a spring-loaded “door” that covers the grids whenever the shuttle is removed from the loading station or the microscope stage to minimize atmospheric ice contamination.
Figure 3: Equipment for FIB/SEM sample preparation and loading.

(A) Loading station with shuttle inserted and in the loading position. Inset shows detailed view of shuttle position. (B) Same as (A) but with the shuttle rotated vertically into the transfer position. Inset shows detailed view of shuttle in vertical transfer position. (C) Transfer arm attached to the transfer lid and positioned over the loading station. (D) Loading station with transfer lid, showing the transfer lid airlock chamber and the registration pins to place the transfer arm. (E) Detail of shuttle showing a loaded grid in the appropriate location and orientation, the locking screw to secure grids, and the anti-contamination door that covers the grids during transfer steps. Users should take care to keep the milling slot unmarked due to potential interactions with FIB imaging. (F) Detail of the transfer arm grabber tip, showing the registration pin and the grabber locking mechanism. (I) Detail of shuttle and grabber together. (G) Detail of transfer arm locking mechanism. The transfer arm lock on the right releases the rod to insert or retract. The smaller grabber release knob on the left opens and closes the grabber. (H) Preparation controller with tools: fine handling tweezers, grid box opening tool, screwdriver. The preparation controller is used to pump or vent the transfer lid and transfer arm assembly when moving specimens to and from the microscope. (J) transfer arm attached to airlock of FIB/SEM quickloader system. The transfer and airlock valves control access to the transfer arm chamber and the microscope chamber, respectively. Airlock pumping and venting actions are controlled by the green ‘P’ and ‘V’ buttons, respectively.
The shuttle holds the grids at a 45° angle relative to the vertically mounted SEM column (Fig 2B and Fig. 3E). During operation, the stage may be tilted anywhere from 0° to 45° relative to the horizontal stage position. The FIB column and platinum GIS needle are mounted 52° relative to the SEM (Fig 2C). The stage is capable of XYZ positioning as well as rotation about the Z-axis (the optical axis of the SEM), and tilt about the X-axis (the direction of sample insertion). Consequently, there is one XYZ position that brings the milling target to the beam-coincidence point. The rotation angle is determined by the mounting position of the FIB column, and tilt is determined by the specific sample and lamella requirements.
Stage Temperature Control
The stage is actively cooled by nitrogen gas flowing through a heat exchanger (Fig. 4A). Nitrogen gas from the source is split into two lines, each passed through a flow regulator, then into a hollow copper coil (Fig. 4B) immersed in a liquid nitrogen dewar (Fig. 4C) to reach liquid nitrogen temperature. The cooled nitrogen gas is then passed to the microscope chamber through a vacuum-isolated tube to minimize thermal loss. Nitrogen gas exits the chamber through return lines through the vacuum tubing, and is vented into the liquid nitrogen dewar to prevent frost formation that may potentially block gas flow and to prevent depositing liquid nitrogen in the lab environment.
Figure 4: Heat exchanger assembly.

(A) Heat exchanger in a stand when not in use. During cryogenic operation, the heat exchanger assembly is immersed into a liquid nitrogen dewar. The heat exchanger assembly is kept under partial vacuum through the clear tubing attached near the top to maintain thermal isolation of cold gas nitrogen. (B) Detail of heat exchanger coils and nitrogen gas lines. Warm nitrogen gas is passed from the blue gas lines into the copper coils and cooled to liquid nitrogen temperatures. Cold gas is routed through the body of the heat exchanger into the microscope stage. (C) Image of heat exchanger assembly inserted into a liquid nitrogen container during cryogenic operation. The thick clear tubing near the top of the heat exchanger unit is part of the vacuum isolation system.
Within the chamber, one of the lines of cooled nitrogen gas is passed through the lower part of the stage. The specimen is located on the upper part of the stage and is cooled by thermal conduction with the lower stage. The second line is passed through the cryo-shield, a separate anti-contamination fixture that has a large surface area to adsorb residual water molecules in the chamber. The stage and shield nitrogen flow rates are regulated separately from the input nitrogen line by the dedicated flow controller. Temperatures are monitored and recorded by computer software through thermocouples embedded in the stage and shield.
1.2. Considerations for Sample Milling Angle and Orientation
The target milling angle depends on the specimen. Typically, a large milling angle relative to the grid surface will result in shorter lamellae (ie measured from the front edge to the back edge) due to reduced cross sectional area (Fig. 2D). High milling angles may be useful to create lamellae in thicker specimens or in specimens where it is difficult to identify a cell at low angles. Conversely, low milling angles result in longer lamellae, but may be more prone to obstruction from surrounding material or may make it difficult to identify a cell due to the nearly parallel viewing angle. Practically, it is best to mill lamellae at as low an angle (i.e., as parallel to the substrate as possible) for two reasons:
The lamella angle limits the tilt range of tomography in the TEM, contributing to resolution anisotropy [32].
Milling at low angles results in long lamellae, maximizing the usable area for tilt-series acquisition.
The lower limit to milling angle is set by sample geometry–at low angles, the edge of the autogrid or the grid bars will block the beam from reaching the grid square surface. This lower limit is about 11° stage tilt, corresponding to a beam incident angle of about 4° with respect to the grid surface. The upper limit to milling angle is set by the TEM stage. Due to the pre-tilt of the lamella, in one tilt direction on the TEM stage, the apparent sample thickness will be greater than compared to the other direction, corresponding to a greater relative tilt between the lamella and the beam. The apparent thickness of the sample increases proportionally to 1/cosine of the stage angle and at high TEM stage tilt angles, the sample will become too thick to image. If the initial lamella angle is too high, it will unnecessarily limit the range of TEM stage angles for tilt series acquisition. This upper limit for lamella milling is typically around 22° on the SEM stage, corresponding to an incident angle of about 15° (see Note 1). In general, ”tall” cells such as yeast and many eukaryotic cells may be milled at a higher angle than unicellular bacteria, which require low milling angles to generate enough usable area.
In order to mill at low angles, special autogrids are available with a cutaway notch that increases accessibility at low stage tilts. The milling notch on the autogrid should be oriented to face towards the FIB source–the clipped grid should be rotated so that the notch is at the top position in the shuttle. To help with orientation, we routinely mark the autogrids with colored permanent marker to create a highly visible reference point (Fig. 2A).
2. Materials
2.1. Equipment
Dual Beam (FIB/SEM) instrument equipped with a temperature controlled stage capable of reaching −175 °C or lower and a vacuum transfer system. This chapter is focused on preparing specimens using the Thermo Fisher Scientific (TFS) Aquilos system with a built-in cryo-stage and quickloader, but general principles should be applicable across instruments.
Sample loading station/equipment and preparation tools compatible with the intended dual-beam instrument (Fig. 3).
Grid boxes.
Grid box opening tools.
Fine manipulation tweezers (e.g., Dumont #5).
Hot plate or hair dryer for warming/drying tools.
Liquid nitrogen transport dewar to hold gridboxes.
4-liter liquid nitrogen storage containers.
Personal protective equipment (PPE) including cryo-gloves and goggles.
2.2. Consumables
Clean liquid nitrogen for sample preparation (NF grade; less than 5ppm moisture).
Pressurized liquid/gas nitrogen (to be regulated to 80 psi with a capacity of 30 L/min, sufficient for 12 hours use). This can be from either an in-house line or from a separate tank, e.g., a 230 L capacity, 230 PSI tank.
EM grids, quantifoil type or with other film substrate (see Note 2).
TFS regular autogrids, or cryo-FIB-autogrids.
TFS c-clips, also called clip-rings or clamprings.
3. Methods
On-grid cryo-FIB milling is suitable for many types of biological specimens including bacteria, yeasts, and mammalian cells. This section describes generalized methods suitable for these three common specimen types with considerations for different approaches required for each specimen. These instructions assume that the reader is familiar with manipulating cryogenic samples to minimize atmospheric ice contamination and risk of sample devitrification. Always use appropriate PPE when working with cryogenic liquids, and always pre-cool tools in liquid nitrogen before handling the specimens.
3.1. Sample Type Considerations
Cryo-FIB milling is adaptable to a wide variety of cell types (Fig 5). Here we offer some considerations on sample preparation and FIB milling for three broad categories of cells. This protocol is not applicable for bulk tissue samples due to major differences in vitrification procedures and FIB milling workflow [28, 29, 30, 20]. The categories are not strictly limited to only those cell types described, and we list some exceptions within the categories. In all cases, grid preparation is critical and users should optimize the following for their particular cell type:
Figure 5: Examples of ideal specimen concentrations.
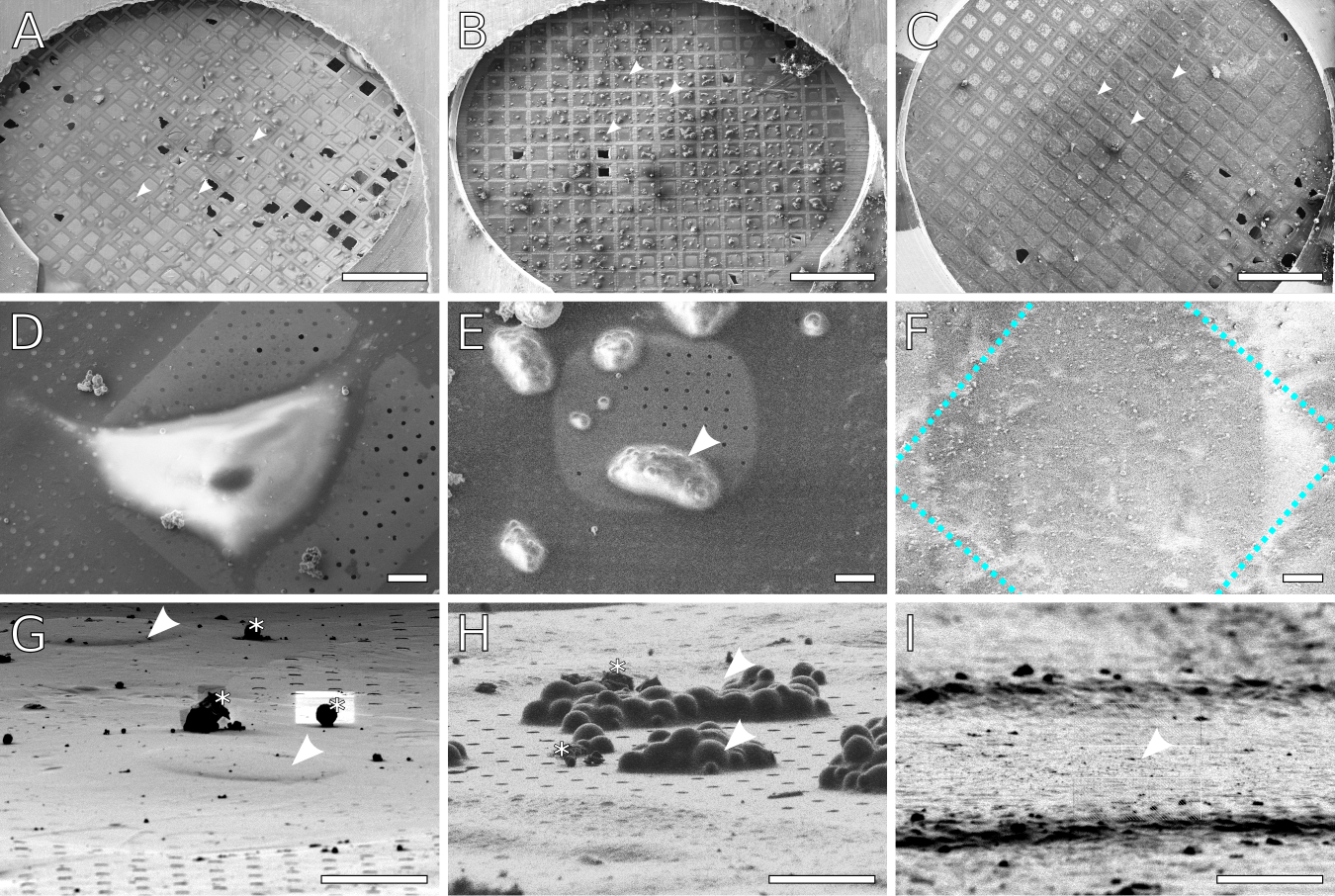
SEM images of (A) Adherent mammalian cells (NIH 3T3) grown on a grid. The cells density is low enough that there are only 1–2 cells per grid square. The surface is sufficiently blotted to reveal the carbon film, while enough liquid remains to cover the cells (some holes remain hydrated). (B) Yeast cells deposited on a grid. The cells form clumps consisting of several cells, with no more than 1–2 large clumps per grid square. As with (A), the surface is blotted to reveal the carbon film, while enough liquid remains to cover each clump. (C) Planktonic bacterial cells deposited on a grid. A monolayer of cells covers each grid square. Enough liquid has been removed to expose the grid bars, but the carbon film is not visible due to coverage by cells and media. Arrow heads indicate examples of cells/grid squares suitable for lamellae milling. (D,E,F) Close up views of individual cells/grid squares for (A,B,C) respectively. In (F), the cells are obscured by the GIS platinum layer. The approximate location of the grid bars is outlined in blue.(G,H,I) Representative FIB views of individual targets for lamella milling, all imaged at 13° stage tilt. Asterisks mark out atmospheric ice contamination. (G) NIH 3T3 cells. Nuclei are indicated by arrowheads. (H) Clumps of yeast, indicated by arrowheads. Notice that yeast clumps have a much taller profile compared to the 3T3 cells or the bacteria. (I) Monolayer of bacteria in a grid square. The darker horizontal ridges are the grid bars. Individual bacteria are not visible as they are embedded in an ice layer and overlaid with platinum from the GIS coating. Scale bars: A-C 500 μm; D-I 10 μm.
Specimens should be thick enough to provide sufficient material to create a lamella. If the sample is too thin, the final generated lamella will be very short and contain almost no cellular features of interest. For these cases, milling wedges can provide a longer electron-transparent window, albeit of varying thickness, on which tomograms can be acquired [21, 33].
Specimens should be thin enough to ensure proper vitrification and minimize crystalline ice domains.
An individual milling target (whether a single cell or clump of cells) should be large enough to support a 5–10 μm wide lamella suitable for cryo-ET.
Appropriate cellular density for the cell type. Note that cell density also affects sample thickness and vitrification.
3.1.1. Mammalian and Flat Eukaryotic Cells
These cells are generally flat and extend several tens of microns in diameter (Fig. 5D,G). Typically, the nucleus appears as a small hill in the center part of the cell. Each lamella made in these samples will be a partial section of a single cell. Examples include fibroblasts and other cells that are cultured on substrates, and some amoebae.
Grid Preparation
Cells may be grown directly on the quantifoil grid by seeding cells and allowing time for them to adhere. The grids should be made of non-cytotoxic material such as gold. Additionally, these grids may be treated with extracellular matrix such as fibronectin or poly-l-lysine to facilitate cell adhesion. If sterility is important, grids may be sterilized under a germicidal UV lamp prior to coating and seeding with cells. The appropriate seeding concentration and recovery periods should be determined empirically.
Some mammalian cells may be deposited on grids immediately before plunging, similarly to protein solutions for single-particle cryo-EM. In this case, adherent cells may be dissociated from the culture surface (using trypsin/EDTA) with the aim to create single cells with minimal clumping. Cell clumps will lead to poor vitrification. Cells should be concentrated or diluted to a pre-determined concentration prior to plunge-freezing.
Evaluating grid quality and milling
In both seeded and deposited cells, the ideal specimen would have on average 1–2 cells per grid square (on a 200 mesh grid, ~6000 μm2 area) with minimal clumps (Fig. 5A). At the SEM, cells should appear well hydrated, and covered in a thin layer of ice (Fig. 5D,G). In some cases, the outline of the nucleus may be visible. Cells that are overblotted (i.e., too dry) may appear to be shrunken, starting to lift off from the substrate (for adherent cells), or have craters appearing on the surface.
Flat eukaryotic cells are typically milled at moderate to high stage tilt angles, from 15° up to 22°. FIB scanning patterns should be positioned to mill through the bulk of the cell in order to have sufficient support material to hold the lamella. Lamellae from cells may extend as far as 15 um in length and may be thicker at the rear due to FIB beam spreading. The lamella thickness can be made uniform through a final milling step with additional stage tilt as described in Section 3.2.
3.1.2. Yeast and Tall Eukaryotic Cells
These cells are smaller than flat cells, typically encompassing less than 10 um in diameter and have a roughly round shape (Fig 5E,H). This is likely due to the presence of a rigid cell wall that holds the cell shape. Each lamella made in these samples will have sections of multiple cells appearing side-by-side. Examples include Saccharomyces cerevisiae, Chlamydomonas. In some cases, large bacteria clusters such as filamentous Anabaena [9] lend themselves to milling in this manner.
Grid Preparation
These cells are typically deposited on the grid before plunging, rather than cultured directly on the grid. Cells should be diluted or concentrated to an empirically determined concentration prior to plunge-freezing. In some cases (such as yeast) the cells are more tolerant of drying due to secreted extracellular polymers or the presence of a cell wall.
Evaluating grid quality and milling
Contrary to the case with flat cells, the ideal yeast specimen should have clumps of 5–10 cells, with 1–2 clumps per grid square (on a 200 mesh grid, Fig. 5B). Due to the relatively small size of yeast, these clumps are necessary to provide enough material to make reasonably sized lamellae. At the SEM, cells should appear relatively dry, but with enough ice to cover the cells and to bridge the gap between neighboring cells (Fig. 5E,H). As a rule of thumb, the clumps should look like steep hills. If there is negative curvature of the clumps where they touch the grid surface, the cells are likely overblotted. If needed, it is possible to mill individual yeast cells, but with a narrow lamella about 3 μm wide instead of 10 μm [34].
Yeast may be milled at moderate to high stage tilt angles, from 15° up to 22°. Milling patterns should be positioned to cut through the center of the clump of cells. As in the case with flat cells, a final milling step with additional stage tilt is recommended to make uniform lamellae.
3.1.3. Bacteria
These cells are generally too small (less than 5 μm in diameter) to mill individually. During blotting, rod-shaped bacteria (e.g E. coli, B. subtilis) will tend to lie down on the grid such that the long axis of the cell is parallel to the grid surface. Examples include most unicellular planktonic bacteria.
Grid Preparation
Bacteria are almost always deposited on the grid immediately prior to plunging. Cells should be diluted or concentrated to an empirically determined concentration prior to plunge-freezing with the aim to make a monolayer of cells embedded in media. The optimal concentration is highly dependent on the specific cell shape and the specific grid hole pattern used because the cells may slip through or become trapped by the holes. Bacteria are generally tolerant of drying because of the large number of closely packed cells and secreted extracellular polymers [35]. However, the relatively large total cell mass can lead to poor vitrification. This issue can be alleviated through the addition of cryoprotectants such as trehalose immediately prior to plunge-freezing.
Evaluating grid quality and milling
The ideal bacterial specimen should have a uniformly flat monolayer of cells over each grid square (Fig. 5C), with the cells packed side-by-side. This arrangement will allow sufficiently long lamella with cells throughout. At the SEM, the ice should be just enough to cover the layer of bacteria, but without excessive hills or valleys (Fig. 5F,I). Generally, if each bacterium is well-defined, the sample is overblotted.
Bacterial grids should be milled at low to moderate stage tilt angles, from 11° to 15°, in order to maximize the number of cells captured in each lamella. This will also result in a long GIS platinum leading edge. If needed, the GIS deposition time can be decreased slightly. A final milling step with additional stage tilt may be useful for some bacterial samples, but is not always necessary for samples milled at moderate stage tilts.
For FIB milling, the lamella should be targeted near the center of the grid square for maximum tilt range at the TEM. Unlike the case with mammalian cells and yeast, it is difficult to target one individual bacterium for milling. Instead, the milling strategy relies on having a near complete grid square coverage to capture as many cells as possible in one single lamella.
3.1.4. Cryo-CLEM
Correlated light and electron microscopy (CLEM) of the same specimen is a powerful technique that allows targeting labeled cellular structures of interest for high-resolution EM [28, 36]. Cryo-CLEM combines the advantage of fluorescence microscopy to specifically locate organelles or proteins of interest and the advantage of TEM to visualize the same object at high-resolution in its native cellular context. A full description of CLEM is outside the scope of this chapter. For FIB milling, correlation can provide great advantages in determining areas to mill, but also provides additional challenges for sample preparation including reduced throughput and increased atmospheric ice contamination due to additional imaging and transfer steps.
For typical mammalian and yeast samples, the cell density described above (about 1–2 cells or clumps per grid square) is typically compatible with cryo-fluorescent microscopy. For bacterial specimens, the cell density should be reduced to somewhat less than full coverage of the grid square in order to be able to resolve individual cells.
During FIB milling, the fluorescent data can be used to guide targeted milling of mammalian and yeast samples. For bacteria, milling is still done in non-targeted manner—while the fluorescent information is useful to screen individual grid squares for suitable cell coverage, ice thickness, and overall quality, the fluorescent data is more critical at the TEM to determine which specific cells should be targeted for tilt series acquisition.
There are commercially available cryo-fluorescence microscopes such as the Corrsight and the Leica Cryo-CLEM system. Additionally, aftermarket cryo-stage additions such as CMS-196 Linkam stage are available in addition to a number of custom made cryo-light microscopes [37, 38]. Recent work has also demonstrated cryo super-resolution microscopy to be compatible with a typical cryo-CLEM workflow [28, 37, 39, 40, 41, 42]. Key points to consider when acquiring cryo-fluorescence microscopes include stage temperature stability, anti-contamination features, and compatibility with existing EM instruments and workflows.
3.2. General FIB milling protocol
3.2.1. Before Starting
These steps should be done before the FIB session.
Culture and plunge-freeze specimens at the appropriate density (see Section 3.1) using your selected grid type (see Note 2). Cells should be frozen on the carbon side of quantifoil grids.
We recommend marking the autogrids with permanent marker before clipping to make sample more visible when under liquid nitrogen (Fig. 2A). Avoid marking the milling notch of the cryo-FIB-autogrid due to potential beam interactions with the marker residue.
While working at liquid nitrogen temperatures, clip specimen grids into a cryo-FIB autogrid support. The grid should be clipped so that the cell side (the carbon side) is facing towards the flat surface of the autogrid so that cells are visible during milling (see Note 3). Clipped grids may be kept for several weeks in a liquid nitrogen storage dewar before subsequent processing.
If fluorescent correlation is required for targeted FIB milling, it should be collected beforehand.
When idle, our microscope is routinely kept in the following manner:
At room temperature, the microscope chamber is pumped down to about 2×10−6 mbar.
Heat exchanger flow controller set to 1 L/min for both stage and shield. This keeps the gas lines free of moisture.
Preferably, the sample transfer arm (Fig. 3C) is attached to the microscope quickloader and pumped. This minimizes dust and moisture contamination inside the transfer arm and the quickloader.
Typically, we keep the FIB source heating is turned off if the system is idle for more than a day, to conserve gallium. Otherwise the source may be left on for the next user. However, practices may vary among facilities depending on instrument usage frequency.
3.2.2. Day of Milling
Microscope Startup and Cooldown
Turn on the FIB and SEM beams, typically done by “waking” the microscope. Start the user interface, and inspect the system status as described in the following steps 2 – 4.
Check that the chamber base pressure is about 2 × 10−6 mbar. (see Note 4)
Check that the stage is empty.
Home the stage to reset stage coordinates and verify range of motion.
Check gas nitrogen source pressure and adjust to 80 psi. If the system is attached to a tank, check that the tank contains sufficient nitrogen for the session. Replace if needed.
Begin purging the heat exchanger gas lines by opening the flow controller to the maximum. Purge for at least 10 minutes.
Begin venting the transfer lid assembly (Fig. 3D). This step is necessary to remove residual moisture inside the lines.
Prepare for loading: turn on the hot plate on the preparation controller (Fig. 3H) and place required tools on it.
Fill a 4-liter dewar with clean liquid nitrogen for sample preparation. Make sure the nitrogen hose attached to the source is dry before filling to minimize ice contamination. Be sure to use appropriate PPE when handling cryogenic liquids.
Fill the heat exchanger tank with liquid nitrogen.
Start the temperature logging software to monitor and record stage and shield temperature.
Slowly insert the heat exchanger into the heat exchanger tank and ensure that it is seated appropriately. Monitor the stage and shield temperature until it stabilizes around −180 °C. Reduce the flow rate to 8.5 L/min to further reduce temperatures by about 3 °C. Allow the system to cool for an additional 30 minutes. (see Note 5) When the stage is at cryogenic temperatures, the chamber vacuum should improve considerably compared to the room temperature base pressure. On our system, the chamber pressure is around 7 × 10−7 mbar when the stage is at cryogenic temperatures.
Sample Preparation and Loading
Retrieve grid boxes containing the frozen, clipped specimens and have them ready in liquid nitrogen.
Insert the shuttle into the loading position in the loading station (Fig. 3A). Fill the loading station with liquid nitrogen, and allow to cool until the liquid is no longer boiling. Refill as needed. Keep the loading station covered with the standard lid to minimize atmospheric contamination while cooling.
Transfer the grid box containing the grids to the loading station and unscrew the lid to access the grids.
Place one grid into each slot of the shuttle, and rotate the grid in position so that the milling slot is at the top (Fig. 2A and Fig. 3E).
Secure the grids in the slots by turning the locking screw (Fig. 3E) at the top of the shuttle. Verify that the grids are secured by turning the shuttle to the vertical position and check that the grids remain in place.
Flip the shuttle into the vertical transfer position (Fig. 3B), and replace the standard lid with the transfer lid (Fig. 3D).
Press ‘V’ to vent the quickloader airlock (Fig. 3J) and retrieve the sample transfer arm. Ensure that the transfer arm valve is closed.
Attach the transfer arm to the transfer lid on the loading station. If present, secure the transfer arm with the locking clamps (Fig. 3C). Otherwise, securely hold the base of the transfer arm. Check that the shuttle grabber is in the ‘open’ position (Fig. 3G).
Pump the transfer lid airlock for 35–40 seconds, then open the transfer arm valve.
Vent the transfer lid airlock for 3 seconds, then close the venting line.
Open the sliding valve on the transfer lid to allow access to the shuttle.
While observing the shuttle, insert the transfer arm and close the grabber to hold the shuttle. Verify the hold by pulling the shuttle back slightly by a few millimeters. The next two steps should be done quickly in succession to minimize contamination.
Quickly and smoothly withdraw the shuttle completely to the back position, lock the transfer arm, and close the sliding valve on the transfer lid.
Pump the transfer lid airlock for 35–40 seconds to evacuate both the transfer lid airlock and the transfer arm, then close the transfer arm valve. Check that the transfer arm is in the ‘locked’ position.
Vent the transfer lid airlock, then release and lift the transfer arm from the transfer lid.
Attach the transfer arm to the microscope airlock and pump the airlock by pressing the ‘P’ button. This will pump the airlock and move the stage to the loading position.
Wait for the airlock vacuum to reach an acceptable level–the ‘OK’ button will turn on and the quickloader valve will unlock (see Note 6). Listen for a click.
Open the microscope airlock gate valve, then open the transfer arm valve.
Unlock the transfer arm and insert it all the way to dock the shuttle into the stage. Release the shuttle grabber and retract the arm. Check that the shuttle is no longer attached to the arm. Lock the transfer arm.
Close the microscope gate valve to isolate the microscope chamber. Leave the transfer arm attached to the quickloader. The transfer arm valve may be left open.
Verify that the stage temperature is around −180 °C and that the chamber pressure is similar to the value before the transfer (within 2–3 × 10−7 mbar).
Sample Inspection and Milling Preparation
While milling, the user should periodically check the stage temperature and chamber pressure for appropriate values. If there is any large deviation, the user may need to respond rapidly to salvage the sample. The heat exchanger dewar will last about 8 hours after initial cooldown, depending on gas flow rate. If the instrument needs to be operated for longer, the dewar can be refilled by slightly propping the heat exchanger aside and using a funnel to pour liquid nitrogen into the tank.
See Tables 1 and 2 for FIB/SEM imaging and scanning conditions. The preset positions for mapping, sputtering, and GIS deposition should be determined beforehand (e.g. during initial system installation) and kept for all users.
Table 1:
Typical imaging conditions for SEM and FIB for frozen hydrated biological specimens.
| Voltage (kV) | Current (pA) | Detector | Mode | |
|---|---|---|---|---|
| SEM | 2-5 | 25 | ETD | Secondary Electrons |
| FIB | 30 | 1.5*; 10–500* | ETD | Secondary Electrons |
1.5 pA for live imaging. 10–500 pA for active milling.
Table 2:
Typical scan settings for SEM and FIB views for frozen hydrated specimens. ‘Live scan’ and ‘snapshot’ are used to monitor lamella milling. The ‘photo’ preset is typically used to generate a low magnification grid overview due to the high electron dose. Snapshots and photos are not used in FIB view due to the potential for beam damage.
| Live (FIB/SEM) | Snapshot (SEM) | Photo (SEM) | |
|---|---|---|---|
| Dwell Time | 200 ns | 200 ns | 2 μs |
| Line Integration | 1 | 1 | 1 |
| Resolution | 1536 × 1024 | 3072 × 2048 | 6144 × 4096 |
| Bit Depth | 8 | 8 | 8–16 |
Switch on the SEM and FIB beams. If the FIB was previously turned off, it may require 10–15 minutes to start up.
Set scan rotation to 180° for both SEM and FIB. (see Note 7).
On the SEM view, change to the lowest magnification and find the grids to ensure that they are present. Check that the grids are seated appropriately in the shuttle.
Set the working distance: Start live SEM imaging and roughly focus the sample. Increase magnification to 5000x and refine focus and astigmatism. While actively imaging at focus, click ‘Link Stage to Z’ to set the working distance in the microscope coordinate frame.
Move the stage to the ‘Mapping Position’ for one of the grids—at 45° tilt at about 7.5 mm working distance. This will place the sample perpendicular with respect to the SEM column.
Acquire and save an SEM ‘Photo’ preset image at a low enough magnification (80x) to see the majority of the grid. Evaluate the grid quality and pick out appropriate target cells or squares for lamella milling (see Section 3.1, Fig. 5, and Note 8).
If the samples are good, discard the nitrogen from the loading station and let the station dry. Otherwise, you may unload the shuttle and load new specimens.
Move the stage to the sputtering position and activate the platinum sputtering feature. (see Note 9). When finished, click ‘Recover from Sputtering’ to return to the mapping position.
Move the stage to the platinum deposition position (GIS position), insert the GIS needle, and deposit the organo-platinum compound for a pre-determined amount of time (typically 5–10 seconds, see Note 10). When finished, ensure the GIS needle is retracted and return to the mapping position.
Lamella Milling
Lamella milling can broadly be divided into three phases: initial rough milling, thinning to about 400 nm thickness, and final milling to target thickness (Fig. 6).
Figure 6: Overview of typical FIB milling progression.

SEM (left column) and FIB (middle column) views of one lamella milled from beginning to end. As material is removed, the lamella becomes more uniform and electron transparent seen from the SEM view. Additionally, the leading edge of the lamella becomes thinner seen from the FIB view. Third column: Schematic of milling patterns overlaid onto lamella FIB views (Table 4). Bottom row: schematic of final milling step with a +0.5° stage tilt to make lamellae of uniform thickness using only the top milling pattern and an additional +0.5° stage tilt. Green arrow represents the direction of the ion-beam milling. For the final step, only the top pattern is activated to selectively mill the thicker portion in the back. The milled area is indicated in grey. Scale bars: SEM images 10 μm; FIB images 5 μm.
In the rough milling phase, the goal is to develop a lamella that has been completely cleared of material throughout its length by the bottom and top milling patterns, respectively. We commonly refer to this as the lamella ”breaking through” and is characterized by the appearance of empty space bordering the front and back of the lamella. This step is crucial because the lamellae may otherwise be obscured at high tilts in the TEM. Additionally, if the lamella has not broken through, the sample is likely too thick. A recent report has demonstrated that milling “expansion joints” adjacent to the lamella improves milling stability and quality [43]. In our hands, the use of expansion joints has increased overall lamella quality and is now a routine part of our cryo-FIB-ET workflow.
In the thinning phase, the goal is to gradually mill the lamella without introducing curtaining artifacts (see Section 3.4.3). Before reducing the milling pattern gap, each step should be allowed to continue until no biological material appears in the FIB milling pattern with increased software contrast. Additionally, the lamella surface should be monitored at regular intervals using the SEM to check for even texture.
In the final milling phase, the FIB current should be reduced to 30 pA or 10 pA. The milling patterns should be gradually moved closer to reach the target thickness. The lamella should be monitored more frequently by SEM to check that milling is proceeding evenly and that the GIS platinum layer is still sufficient to protect the leading edge of the lamella. The SEM can also be used to estimate lamella thickness by comparing the relative transparency of the lamella at 5 kV and 3 kV [13]. During this phase, the user should monitor the FIB milling pattern for any image drift and compensate accordingly with the X/Y shift knobs. Continue milling until the lamella is judged to be thin enough (see Note 11). Sometimes, the lamella will not be able to survive continued milling at which point milling should be stopped (e.g. lamella bending/bulging, loss of GIS platinum layer, or holes in the lamella from uneven milling).
Bring a selected milling target to the beam-coincidence point (see Note 12). Make sure sample is at desired tilt (see Section 1.2 and Section 3.1) to determine appropriate milling angle).
Import the saved milling pattern into the FIB view (see Table 3 for typical parameters). Adjust the milling position to create a lamella through the desired target. If needed, change the dimensions and gap between the milling patterns.
Change the FIB current to the desired beam current (Table 4).
After changing to a new FIB current, you may need to run the auto contrast routine for the FIB view. Additionally, take several brief FIB images to allow any image/beam drift to settle and to focus at the lamella position (See Note 13).
Start rastering the patterns. Monitor the milling progress regularly with the FIB and SEM. If desired, you may save SEM snapshots of intermediate milling steps to judge progress.
When milling at this current is finished, switch to a lower current, reduce the milling pattern separation, and adjust the pattern dimensions. (see Table 4 and Fig. 6)
Continue milling while reducing pattern separation and FIB current until you reach a pattern separation of ~200nm. (see Note 14 and Table 4)
On some specimens, it may be necessary to perform a final milling step with a stage tilt of ±0.5° from the desired angle to compensate for the slightly diverging FIB beam (Fig. 6 ‘Final Mill’). In this case, only enable milling on the side corresponding to the tilt direction (i.e. only enable the top pattern if milling with a +0.5° tilt. See Note 15) [13].
Continue on to other lamella locations as desired.
Table 3:
Milling pattern parameters.
| Milling Pattern Basic Properties | |
|---|---|
| Application | Si |
| X Size | 12–10 μm |
| Y Size | * |
| Z Size | ∼10 μm |
| Scan Direction | **Outside to inside |
| Dwell Time | 1 μs |
| Beam | Ion |
The Y size for both patterns should be adjusted throughout milling to become smaller, just enough to encompass the remaining unmilled material.
The scan direction for each pattern (ie. top-to-bottom or bottom-to-top) should be set so that the milling direction is towards the lamella. For very sensitive samples, the dwell time may be reduced.
Table 4:
A typical scheme for performing step-wise milling starting from rough to final polish. FIB current and pattern X size are gradually reduced as the lamella becomes thinner.
| Rough I | Rough II | Middle | Fine I | Fine II | Final Polish | |
|---|---|---|---|---|---|---|
| FIB current | 0.5 nA | 0.3 nA | 0.1 nA | 30 pA | 10 pA | 10 pA |
| Pattern Separation | 3 μm | 1.5 μm | 750 nm | 400 nm | 200 nm | *n/a |
| Pattern X size | 12 μm | 11.5 μm | 11 μm | 10.5 μm | 10 μm | 10 μm |
| Typical time | 10 min | 10 min | 15 min | 20 min | 20 min | 10 min |
Note that the pattern separation does not correspond exactly to lamella thickness due to FIB beam spread. At low currents, milling time increases but there is greater control over lamella quality.
For final polishing, the stage is tilted by an additional 0.5 degrees and only one pattern is used to mill.
Sample Retrieval
About 1 hour before retrieval, begin purging the loading station with dry nitrogen gas. If the loading station is at liquid nitrogen temperatures, increase the purge time to 2 hours. The sequence of events for sample retrieval is essentially the reverse of sample loading, but it is reproduced here for the sake of completeness.
After completing all lamellae, perform a final cleaning step to remove amorphous ice buildup (see Note 16). When finished, close the SEM and FIB column valves.
Make sure the shuttle holder in the loading station is in the vertical position.
Stop purging the loading station and fill with liquid nitrogen. Allow to cool until the liquid is no longer boiling. Refill as needed. Place the transfer lid on the loading station.
Open the transfer arm valve, then press the ‘P’ button to pump the airlock and move the stage to the loading position.
Wait for the airlock gate valve to unlock. Open the airlock gate valve.
Insert the transfer arm fully and close the grabber. Test the hold by pulling back slightly.
Fully retract the transfer arm and lock the arm in place. Close the transfer arm valve and the airlock gate valve.
Press ‘V’ to vent the quickloader airlock. Retrieve the transfer arm and place it onto the transfer lid.
Pump the transfer lid airlock for 35–40 seconds (make sure transfer lid sliding valve is closed), then open the transfer arm valve. The next three steps should be done quickly in succession to minimize contamination.
Open the transfer arm valve, then vent the transfer lid airlock for 3 seconds. Stop venting.
Unlock the transfer arm.
Open the transfer lid slider and quickly and gently insert the transfer arm to dock the shuttle into the loading station.
Release the shuttle grabber and retract the arm. If desired, pump down the transfer arm as described above.
Remove the transfer lid from the loading station and replace with the standard lid.
Using pre-cooled tools, flip the shuttle into the horizontal loading position, turn the locking screw to release the grids, then transfer the grids to a pre-cooled, labeled grid box.
Store the grid boxes with your specimens in liquid nitrogen.
End of Session Tasks
Remove heat exchanger from dewar and allow the stage and shield to warm to room temperature. (30–60 minutes)
Home the stage for the next user.
If the microscope will not be used in the next day, sleep the system to conserve gallium. Otherwise, keep it on but ensure the SEM and FIB column valves are closed.
Turn off hot plates and store tools.
When the stage and shield are near room temperature, turn the flow rates to 1 L/min to prevent moisture accumulation inside the heat exchanger gas lines.
3.3. Considerations for TEM
The specimen should be loaded such that the FIB milling axis is perpendicular to the TEM tilt axis (Fig. 7). This will allow maximum tilt range for tomography without the unmilled material blocking the field of view at high tilts. Due to FIB milling geometry, the lamella will have a measurable pre-tilt at the TEM ranging from 5° to 15° difference compared to reported stage goniometer angle. This pre-tilt will influence the tilt series acquisition scheme because in one tilt direction the specimen will appear to be much thicker at high tilt angles compared to the other direction. (See Section 1.2) Additionally, sample orientation is important for accurate focus and tracking when acquiring data with low-dose techniques. TEM data acquisition is mostly handled through automated routines from software packages such as SeriaEM [44, 45].
Figure 7: Representative lamella and reconstructed tomogram.
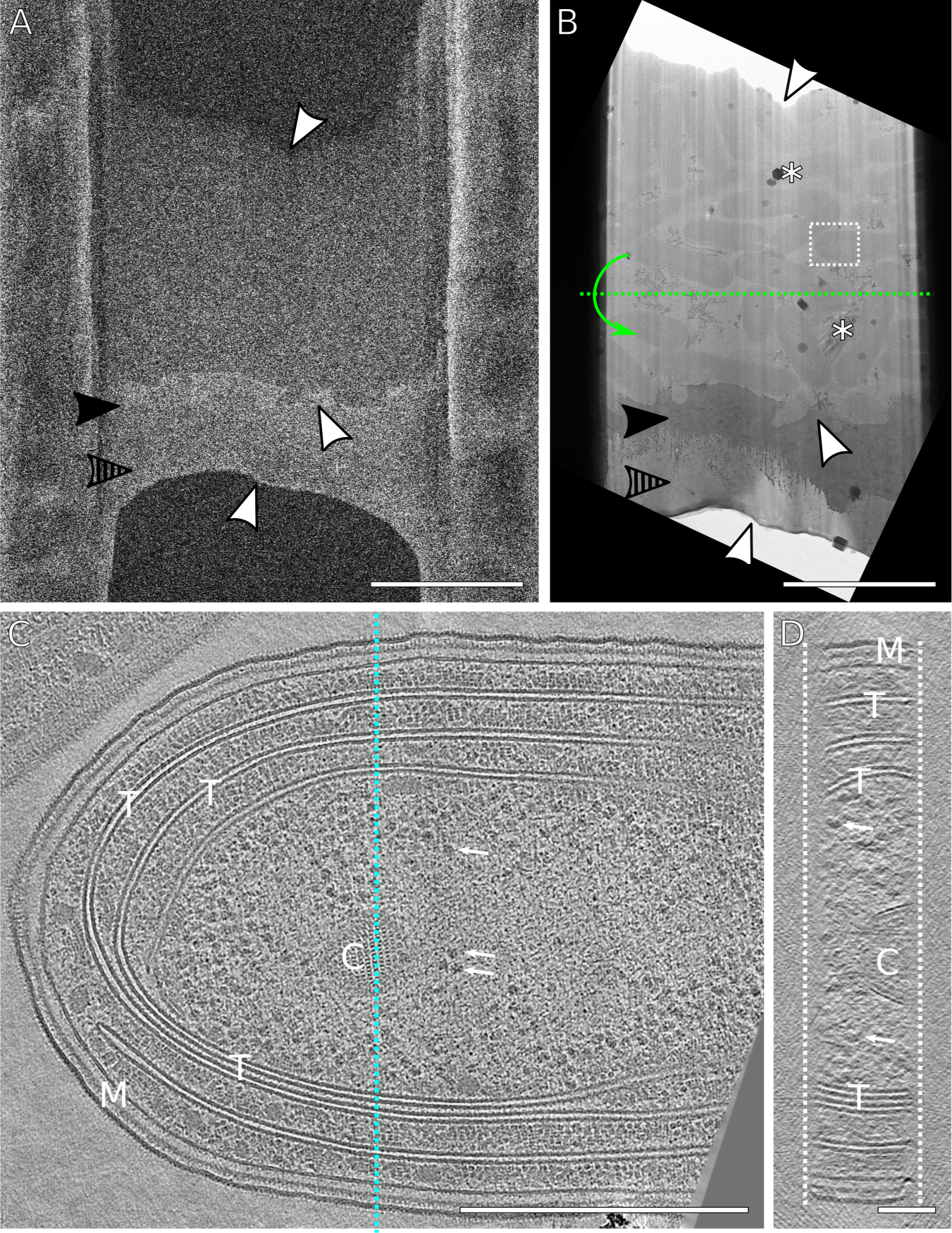
(A) SEM image of a finished lamella through a lawn of cyanobacteria. The milling direction was from bottom to top. The lamella was milled with expansion joints to reduce buckling (not visible here) [43]. (B) Corresponding TEM image of (A) showing a field of bacteria. White arrowheads mark areas of correlation between the SEM and TEM image. The solid and striped arrowheads point out the platinum sputtering and GIS platinum layers, respectively. Asterisks mark areas of atmospheric ice contamination and poor vitrification. The white box indicates the field of view for tilt-series acquisition. Green dotted line represents the TEM tilt axis. (C) XY view of a reconstructed tomogram of a single cyanobacterium from the lamella. M: inner/outer membrane. T: thylakoid membranes. C: carboxysomes. Arrows: ribosomes. (D) YZ view of reconstructed tomogram corresponding to the slice indicated by the blue line in (C) with structures labeled identically. The lamella thickness is approximately 150 nm. The surfaces of lamella are indicated by the dashed white lines. Scale bars: A,B 5 μm; C, 500 nm; D, 100 nm
When beginning a new project, it will be necessary to iteratively optimize samples between the TEM and FIB. For example, the final lamella thickness should be guided by the intended TEM data acquisition pixel size. This is important in order to limit the total dose applied to a specimen during tilt series acquisition. Doubling the TEM magnification while maintaining a similar beam intensity on the camera will result in four times the applied dose (in electrons per square angstrom) to the sample. A higher desired TEM magnification will therefore require thinner lamellae in order reduce the dose applied to the specimen while maintaining an appropriate beam intensity on the camera. Conversely, thick specimens will require additional radiation, resulting in lower signal-to-noise ratio and lower resolution, but allow imaging of a larger volume for increased cellular context.
For reasons described above, it is important to track and orient grids during FIB milling and through to TEM data acquisition. This is most easily accomplished by using marked autogrid supports that allow the user to orient the sample at the aquilos loading station and at the TEM loading station. Examples of marked autogrids include the commercially available cryo-FIB-autogrids that have laser etched dots in the surface of the ring to indicate sample orientation. These marks may be difficult to see under liquid nitrogen, so we routinely mark the autogrids with a colored permanent marker.
While cryo-FIB milling exhibits fewer artifacts compared to sections from microtomy, the ion beam will damage the surface of the lamella as it ablates material [46]. Additionally, repeated use of the SEM for monitoring milling progress also contributes electron damage. In particular, sensitive structures such as bacterial polyphosphate bodies, accumulated damage from the FIB/SEM can be seen as localized bubbling confined to the surface of the specimen to several nanometers deep. However, previous work has demonstrated that FIB milled samples do not exhibit heat-induced devitrification beyond the immediate interacting surface [46].
3.4. Common Troubleshooting Items
3.4.1. System Maintenance
In addition to manufacturer suggested regular instrument upkeep, we recommend following a regular maintenance schedule for cryo-FIB/SEM systems focused on checking stage temperature, cleaning the stage of platinum buildup, and cleaning seals/o-rings of dust. These tasks should be done every 3–4 months. Additionally, the stage components should be inspected for wear and defects regularly due to the large temperature cycles that these parts experience.
System consumables include the gallium liquid-metal ion-source and the FIB aperture strip, both of which may need replacing every six months if the instrument is used routinely.
3.4.2. Equipment Troubleshooting
Poor chamber vacuum.
The system should reach better than 2 × 10−6 mbar at room temperature and better than 7 × 10−7 mbar at cryogenic temperatures. If these are not satisfied, the chamber seals especially those on the front door and the quickloader should be inspected and cleaned. Poor vacuum may lead to an increased rate of water vapor accumulation on the sample resulting in an increase in the lamella thickness that reduces imaging quality.
Frost on heat exchanger tubing.
Frost buildup on the heat exchanger tubing is due to loss of vacuum insulation. The typical causes are pump failure, a leak in the outer tubing, or a leak in the internal nitrogen lines. Continued use of the system with frost buildup may lead to poor stage cooling
Amorphous ice contamination.
From our experience, poor stage cooling will lead to accumulation of amorphous ice or even “leopard skin” ice when observed at the TEM. The temperature of the top of the stage (where the shuttle is inserted) should be measured using a thermocouple. Starting from room temperature, the stage top should be able to reach below −170 °C within 30 minutes. If this is not the case, the stage should be disassembled and thoroughly cleaned. We have found that buildup from the sputter coater and GIS can be responsible for stage temperature issues.
Crystalline/Atmospheric ice contamination.
Check that the o-rings on the transfer arm and transfer lid are clean and in good condition. Ensure that all tools and loading equipment are dry before handling samples. Ensure that the venting line on the transfer lid has been purged for at least 10 minutes, and keep purging continuously for the duration of the session. Ensure that any liquid nitrogen having direct contact with samples is free of frost.
Inaccurate ion beam currents.
When the beam is blanked, the FIB is deflected into a faraday cup to measure the beam current. If the reported value is much higher (10–15%) than the nominal value, the ion beam aperture strip is likely worn out (Fig. 8). Typical FIB milling may use one particular aperture (eg 10 or 30 pA) much more than the others, which may cause them to wear out earlier and become much larger than the initial size. This leads to drastically increased ion beam currents and poor control over milling. On certain microscopes, it may be necessary to switch to an unused “parking position” FIB aperture when the microscope is idle but the ion source is left on, due to the continual exposure of the aperture strip to the ion beam.
Figure 8: FIB aperture wear.

Basic light microscope image of a FIB aperture strip retrieved after about 6 months of 4 sessions/week use. The smallest apertures used for milling (10 and 30 pA) have suffered extensive damage to the edge of the aperture hole leading to high beam currents, compared to an infrequently used aperture (50 pA).
3.4.3. Sample Troubleshooting
FIB image instability.
The image may appear to drift or distort in the FIB view, especially at high beam currents or magnifications. Check that there are no ice or autogrid obstructions near the field of view that may be deflecting the ion beam. Check that the FIB accelerating voltage is at 30 kV. Increasing platinum sputtering time may help with drift.
Lamella appears skewed relative to milling axis.
The grid surface in the FIB view is not perfectly flat. Use additional scan rotation to make the FIB image appear horizontal. (see Note 7 and Fig. 9)
Figure 9: Correcting skewed FIB milling with scan rotation.
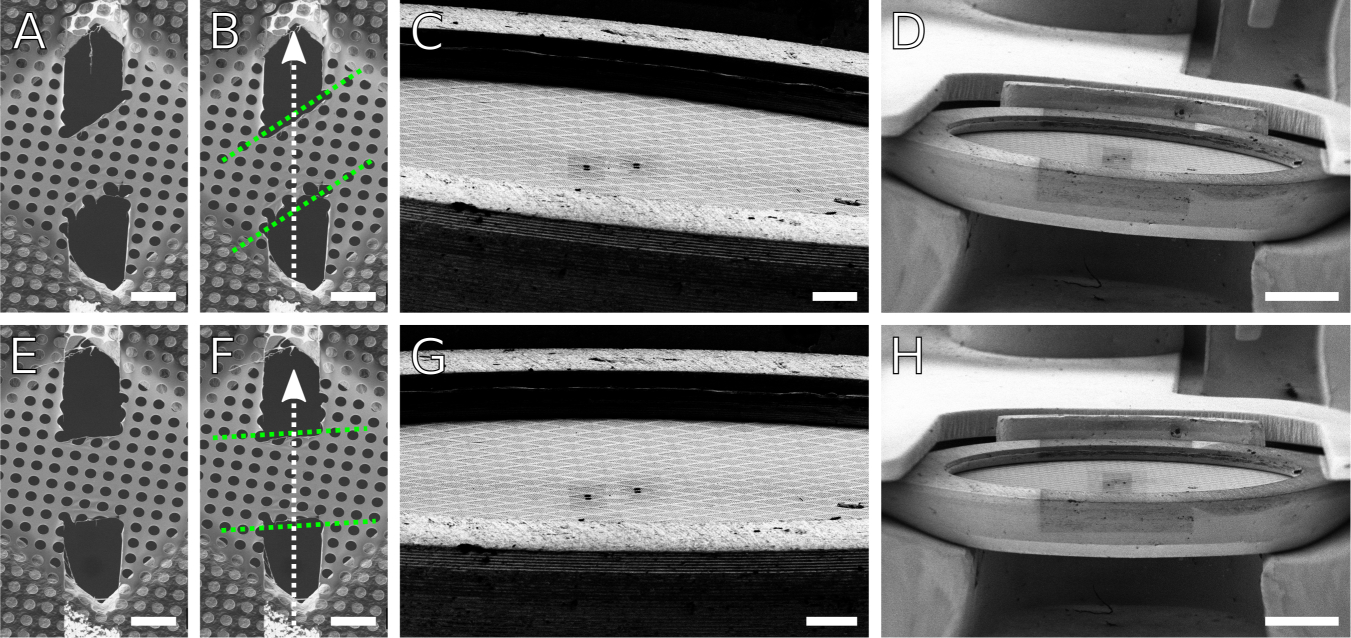
(A,B) SEM images of a skewed lamella made in carbon foil. Dotted green lines mark the approximate geometry of the lamella relative to the milling axis as indicated by the white arrow. (C,D) FIB views of the clipped grid at low magnifications, showing an apparent tilt of the grid relative to the horizontal. (G,H) By adjusting the apparent tilt using additional scan rotation applied to the FIB view, the lamella skew angle can be corrected (E,F) to become nearly perpendicular to the milling axis. These demonstrative lamellae were milled on a room temperature carbon film grid. Results are directly applicable for cryogenic samples. Scale bars: A,B,E,F 10 μm; C,G 100 μm; D,H 500 μm.
Curtaining artifacts.
These artifacts manifest as ridges and grooves on the surface of the lamella parallel to the milling axis. The FIB current is too high for the lamella thickness or milling has not completed before narrowing the patterns. Reduce the current and extend the milling time to ensure a flat lamella surface. Monitor the quality of the lamella at regular intervals with the SEM. If the GIS platinum layer is wearing away too quickly, the initial deposition time may be increased.
For tall cells, the profile is presented such that the cell surface is nearly perpendicular to the beam. This is not the case for flat cells or bacteria, where the surface is much more oblique (compare Fig 5H and 5I). The tall profile can lead to issues with insufficient GIS platinum deposition that would otherwise be acceptable. These cells may require a longer GIS platinum deposition time and rotating the stage to optimize deposition thickness.
Broken lamellae at the TEM.
The finished lamellae are relatively fragile and are susceptible to damage from rough handling. Slightly increasing the GIS platinum deposition time may proved extra support and make handling easier. It is important to only handle lamellae grids by the very edge of the autogrid support. Another option is to reduce the width of the lamellae to make them less susceptible to breakage.
Lamella appears to bend during milling.
Recent reports have suggested that lamella bending can be attributed to tension in the bulk material arising from the vitrification process. The proposed solution is to mill additional “micro-expansion joints” near the lamella to reduce the impact of surrounding material motion [43]. In our hands, the micro-expansion joints also reduce bending and subsequent breaking at the TEM. Silicon dioxide grids may also offer a stiffer support that may reduce lamella bending or breaking [47].
5. Acknowledgments
We would like to acknowledge the following people: Reika Watanabe and Miles Paszek for providing images of NIH3T3 and yeast grids, respectively. Reika Watanabe, Kanika Khanna, and Sergey Suslov for fruitful discussion of FIB/SEM. Mario Aguilera for photography of microscope equipment.
This work was supported by an NIH Director’s New Innovator Award 1DP2GM123494-01 (to E.V.) and NIH 5T32GM7240-40 (to V.L.). Images of cyanobacteria are from projects supported by NIH R35GM118290 awarded to Susan S. Golden. This work was performed in part at the San Diego Nanotechnology Infrastructure (SDNI) of UCSD, a member of the National Nanotechnology Coordinated Infrastructure, which is supported by the National Science Foundation (Grant ECCS-1542148).
Footnotes
Because the grid is held at 45° and the FIB column angle relative to the SEM column is 52°, there is a 7° difference between the reported stage angle and the incident milling angle. To estimate the incident milling angle, subtract 7° from the reported stage angle.
The EM grid should be chosen such that each grid square is large enough to contain the widest desired lamella, but small enough to have sufficient sample rigidity so that the lamella will not bend and break during subsequent handling. We routinely use 200 mesh quantifoil grids for FIB milling. The specific pattern of holes will determine blotting speed and sample dryness. We are routinely using R1/4 grids for mammalian cells and R2/1 grids for bacteria. Grid material should be: gold for cells cultured directly on the grid, copper for cells deposited immediately before plunge freezing. See Section 3.1 for sample type considerations.
The autogrid is a rigid ring support in which TEM grids are placed and secured with a spring clip. This arrangement allows improved handling and stability of samples. Grids may be clipped either using the provided loading station, or in any other suitable device that will minimize atmospheric contamination. Grids should be clipped so that the cell side is facing towards the flat side of the autogrid. We have found clipping grids under a very low level of nitrogen to be most efficient, with minimal grid square breakage. Once clipped, the grid is permanently mounted in the autogrid and is essentially impossible to remove at cryogenic temperatures without damaging the grid.
On our system, the chamber base pressure at room temperature is typically 2 × 10−6 mbar or better. If the base pressure is too high, the chamber seals, airlock seals, and nitrogen lines should be cleaned and inspected (See Section 3.4.2).
The reported stage temperature is measured from the lower stage, which is quickly cooled by cold nitrogen gas. However, the upper stage (where the sample sits) usually lags behind due to its thermal mass and reduced contact area with the lower stage. Waiting at least 30 minutes after full cooldown is important to ensure that the upper stage is well below devitrification temperature.
The microscope quickloader gate valve is kept locked to prevent accidental opening and venting of the chamber. The valve will only unlock if the user has issued the ‘pump’ command through the button interface AND if the pressure in the quickloader airlock is low enough. Additionally, the gate valve is only unlocked for about 90 seconds before the action times out and relocks. The valve must be opened within this timeframe. When closing the valve, wait for it to lock before reopening or re-issuing the pump command. Otherwise, we have found that this may lead to the gate valve being stuck in the open state.
Setting the scan rotation to 180° is optional, but it orients the SEM and FIB views to appear more natural such that the FIB view shows cells ”sitting” on the grid, rather than ”hanging” from the grid. In some cases, the scan rotation of the FIB column is not perfectly parallel to the grid surface. In this case, the resulting lamellae will be tilted relative to the milling axis (Fig. 9A–D). Such tilted lamellae become more difficult for tomography due to limited focus and tracking area (see 3.3). This issue can be corrected by rotating the scan direction of the FIB column until the grid surface appears horizontal (Fig. 9E–H).
In addition to sample specific considerations for milling, users should consider the accessible area of the grid at the TEM for tilt series acquisition. This depends on the specific TEM instrument, but is typically limited to the grid squares that are in the center 1 mm of the grid. For a 200 mesh grid, this is about 10-by-10 grid squares centered over the grid. Lamellae should be milled within this region. To minimize occlusion from the grid bars at high tilt at the TEM, it is generally best to make lamellae that are centered in the grid square.
Sputter coating is used to improve the image quality of non-conductive samples by depositing a layer of metal to allow charge to quickly dissipate. The Aquilos has a built-in sputter coater. Set the number of purge cycles to 0 (which would otherwise lead to sample contamination) and click ‘Prepare for Sputtering’. The microscope will enter a low vacuum state (about 0.1 mbar) by partially venting with dry argon, and the stage temperature will increase slightly to about −170 °C. The stage will move to the sputtering position. When the preparation actions have finished, select the sputtering parameters or the preset values and run the process. Typical sputter conditions for initial rough sputtering are 1 kV, 30 mA, for 15 seconds at 0.10 mbar chamber pressure. You can see the plasma glow using the chamber camera. When sputtering is complete, click ‘Recover from Sputtering’ to return the stage to the mapping position and return to high vacuum
The GIS deposition time depends on the sample type, the intended deposition thickness, and the preset deposition stage coordinate. If the stage is closer to the tip of the GIS needle, a shorter deposition time will yield the same thickness of GIS platinum as a longer deposition time at a further distance. The deposition stage position/coordinates can be adjusted to allow long/short deposition times, or can be adjusted to be centered between the two grids such that both the left and right grids are coated at the same time.
We have found that judging appropriate lamella thickness and when to stop milling requires some experience and iterative comparison between the SEM and TEM to ‘calibrate’ the user. One helpful exercise is to make a lamella, estimate its thickness at the SEM, then measure the actual thickness from a tomogram.
To manually find the beam coincidence point, tilt to the desired milling angle and go to a low mag view for both SEM and FIB. Find your target in both SEM and FIB, and center the target in the FIB view using XY moves. Switch to SEM view and move the stage in Z so that the target is about halfway closer to the center of the SEM field of view. (e.g. if the target was 100 um away from the center of the SEM, move the stage in Z so that the target is now about 50 um away.) Switch to the FIB view and recenter the target using XY moves. Switch to SEM and check positioning. Reiterate between XY moves in FIB view and Z moves in SEM view until the target is centered in both SEM and FIB. Alternatively, the proprietary MAPS software package from TFS allows correlation between fluorescent, SEM, and TEM images. Additionally, MAPS may be used as a microscope control program. This software package has the ability to calculate eucentric height for a given point on a sample, which should be very close to the beam-coincidence point. To do so, add a lamella object, then go to its mapping position. Select ‘Calculate Eucentric Height’ which will ask you to keep the object centered at several tilts. The specific internal calibration for accurate eucentricity should be determined during software installation and kept for all users.
This apparent drift is not due to physical specimen movement, but due to slight movement in the beam due to sample interaction. Especially at high FIB currents, the sample may acquire charging that influences the FIB [48].
Step milling improves lamella stability by forming a gradual transition from the unmilled material to the lamella. Without step milling, the lamellae are more prone to developing cracks at the edge where they connect to the bulk material.
The FIB view has a shallow depth of field, which can be observed when the the front of the lamella is in sharp focus, but the back of the lamella is out of focus. This leads to heterogeneity in milling efficiency over the surface of the lamella. At the TEM, this can be observed where the back of the lamella is thicker than the front. To thin only the back portion of the lamella, the stage should be tilted approximately +0.5° which will expose the back for milling without disturbing the front edge of the lamella [13].
Given the large chamber volume, poor vacuum compared to the TEM, and cryogenic temperatures, amorphous ice buildup is inevitable inside the cryo-FIB/SEM. This can be minimized with proper instrument maintenance, but in our experience, long operating times of more than 6 to 8 hours lead to ice buildup from the chamber and from milling redeposition. Before sample retrieval, it is recommended to perform a short five minute lamella cleaning step with a low FIB current for all lamellae immediately prior to sample retrieval (Fig. 6). This step is essentially identical to the fine milling step, but with the pattern set to skim over the surface of the lamella without removing too much material.
References
- [1].Koning RI, Koster AJ, and Sharp TH (2018) Advances in cryo-electron tomography for biology and medicine. Annals of Anatomy - Anatomischer Anzeiger 217:82–96 [DOI] [PubMed] [Google Scholar]
- [2].Russo CJ and Passmore LA (2016) Progress towards an optimal specimen support for electron cryomicroscopy. Current Opinion in Structural Biology 37:81–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Al-Amoudi A, Chang JJ, Leforestier A, McDowall AW, Michel Salamin L, Norlén L, Richter K, Sartori Blanc N, Studer D, and Dubochet J (2004) Cryo-electron microscopy of vitreous sections. The EMBO Journal 23:3583–3588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Al-Amoudi A, Studer D, and Dubochet J (2005) Cutting artefacts and cutting process in vitreous sections for cryo-electron microscopy. Journal of Structural Biology 150(1):109–121 [DOI] [PubMed] [Google Scholar]
- [5].Mahamid J, Tegunov D, Maiser A, Arnold J, Leonhardt H, Plitzko JM, and Baumeister W (2019) Liquid-crystalline phase transitions in lipid droplets are related to cellular states and specific organelle association. Proceedings of the National Academy of Sciences 116(34):16866–16871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Chaikeeratisak V, Khanna K, Nguyen KT, Sugie J, Egan ME, Erb ML, Vavilina A, Nonejuie P, Nieweglowska E, Pogliano K, Agard DA, Villa E, and Pogliano J (2019) Viral capsid trafficking along treadmilling tubulin filaments in bacteria. Cell 177(7):1771–1780.e12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Khanna K, Lopez-Garrido J, Zhao Z, Watanabe R, Yuan Y, Sugie J, Pogliano K, and Villa E (2019) The molecular architecture of engulfment during Bacillus subtilis sporulation. eLife 8:e45257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Rast A, Schaffer M, Albert S, Wan W, Pfeffer S, Beck F, Plitzko JM, Nickelsen J, and Engel BD (2019) Biogenic regions of cyanobacterial thylakoids form contact sites with the plasma membrane. Nature Plants 5(4):436–446 [DOI] [PubMed] [Google Scholar]
- [9].Weiss GL, Kieninger AK, Maldener I, Forchhammer K, and Pilhofer M (2019) Structure and function of a bacterial gap junction analog. Cell 178(2):374–384.e15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Lopez-Garrido J, Ojkic N, Khanna K, Wagner FR, Villa E, Endres RG, and Pogliano K (2018) Chromosome translocation inflates bacillus forespores and impacts cellular morphology. Cell 172(4):758–770.e14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Noble JM, Lubieniecki J, Savitzky BH, Plitzko J, Engelhardt H, Baumeister W, and Kourkoutis LF (2018) Connectivity of centermost chromatophores in rhodobacter sphaeroides bacteria. Molecular Microbiology 109(6):812–825 [DOI] [PubMed] [Google Scholar]
- [12].Mosalaganti S, Kosinski J, Albert S, Schaffer M, Strenkert D, Salomé PA, Merchant SS, Plitzko JM, Baumeister W, Engel BD, and Beck M (2018) In situ architecture of the algal nuclear pore complex. Nature Communications 9(1):2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Schaffer M, Mahamid J, Engel BD, Laugks T, Baumeister W, and Plitzko JM (2017) Optimized cryo-focused ion beam sample preparation aimed at in situ structural studies of membrane proteins. Journal of Structural Biology 197(2):73–82 [DOI] [PubMed] [Google Scholar]
- [14].Chaikeeratisak V, Nguyen K, Khanna K, Brilot AF, Erb ML, Coker JKC, Vavilina A, Newton GL, Buschauer R, Pogliano K, Villa E, Agard DA, and Pogliano J (2017) Assembly of a nucleus-like structure during viral replication in bacteria. Science 355(6321):194–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Rosenzweig ESF, Xu B, Cuellar LK, Martinez-Sanchez A, Schaffer M, Strauss M, Cartwright HN, Ronceray P, Plitzko JM, Förster F, Wingreen NS, Engel BD, Mackinder LC, and Jonikas MC (2017) The eukaryotic co2-concentrating organelle is liquid-like and exhibits dynamic reorganization. Cell 171(1):148–162.e19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Bäuerlein FJ, Saha I, Mishra A, Kalemanov M, Martínez-Sánchez A, Klein R, Dudanova I, Hipp MS, Hartl FU, Baumeister W, and Fernández-Busnadiego R (2017) In situ architecture and cellular interactions of polyq inclusions. Cell 171(1):179–187.e10 [DOI] [PubMed] [Google Scholar]
- [17].Mahamid J, Pfeffer S, Schaffer M, Villa E, Danev R, Kuhn Cuellar L, Förster F, Hyman AA, Plitzko JM, and Baumeister W (2016) Visualizing the molecular sociology at the hela cell nuclear periphery. Science 351(6276):969–972 [DOI] [PubMed] [Google Scholar]
- [18].Zhang J, Ji G, Huang X, Xu W, and Sun F (2016) An improved cryo-fib method for fabrication of frozen hydrated lamella. Journal of Structural Biology 194(2):218–223 [DOI] [PubMed] [Google Scholar]
- [19].Engel BD, Schaffer M, Kuhn Cuellar L, Villa E, Plitzko JM, and Baumeister W (2015) Native architecture of the Chlamydomonas chloroplast revealed by in situ cryo-electron tomography. eLife 4:e04889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Harapin J, Börmel M, Sapra KT, Brunner D, Kaech A, and Medalia O (2015) Structural analysis of multicellular organisms with cryo-electron tomography. Nature Methods 12:634–636 [DOI] [PubMed] [Google Scholar]
- [21].Villa E, Schaffer M, Plitzko JM, and Baumeister W (2013) Opening windows into the cell: focused-ion-beam milling for cryo-electron tomography. Current Opinion in Structural Biology 23(5):771–777. Protein-carbohydrate interactions / Biophysical methods [DOI] [PubMed] [Google Scholar]
- [22].Wang K, Strunk K, Zhao G, Gray JL, and Zhang P (2012) 3d structure determination of native mammalian cells using cryo-fib and cryo-electron tomography. Journal of Structural Biology 180(2):318–326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Rigort A, Bäuerlein FJB, Villa E, Eibauer M, Laugks T, Baumeister W, and Plitzko JM (2012) Focused ion beam micromachining of eukaryotic cells for cryoelectron tomography. Proceedings of the National Academy of Sciences 109(12):4449–4454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Marko M, Hsieh C, Schalek R, Frank J, and Mannella C (2007) Focused-ion-beam thinning of frozen-hydrated biological specimens for cryo-electron microscopy. Nature Methods 4:215. [DOI] [PubMed] [Google Scholar]
- [25].Rajput NS and Luo X (2015) Chapter 3 - FIB Micro-/Nano-fabrication. In Micromanufacturing Engineering and Technology, (ed.) Qin Y, Micro and Nano Technologies, pp. 61–80. William Andrew Publishing, Boston, second edition [Google Scholar]
- [26].Volkert CA and Minor AM (2007) Focused ion beam microscopy and micromachining. MRS Bulletin 32(5):389–399 [Google Scholar]
- [27].Chyr I and Steckl AJ (2001) Gan focused ion beam micromachining with gas-assisted etching. Journal of Vacuum Science & Technology B: Microelectronics and Nanometer Structures Processing, Measurement, and Phenomena 19(6):2547–2550 [Google Scholar]
- [28].Schaffer M, Pfeffer S, Mahamid J, Kleindiek S, Laugks T, Albert S, Engel BD, Rummel A, Smith AJ, Baumeister W, and Plitzko JM (2019) A cryo-fib lift-out technique enables molecular-resolution cryo-et within native caenorhabditis elegans tissue. Nature Methods 16(8):757–762 [DOI] [PubMed] [Google Scholar]
- [29].Zhang J, Zhang D, Sun L, Ji G, Huang X, Niu T, and Sun F (2019) VHUT-cryo-FIB, a method to fabricate frozen-hydrated lamella of tissue specimen for in situ cryo-electron tomography. bioRxiv [DOI] [PubMed] [Google Scholar]
- [30].Mahamid J, Schampers R, Persoon H, Hyman AA, Baumeister W, and Plitzko JM (2015) A focused ion beam milling and lift-out approach for site-specific preparation of frozen-hydrated lamellas from multicellular organisms. Journal of Structural Biology 192(2):262–269 [DOI] [PubMed] [Google Scholar]
- [31].Stevie FA, Griffis DP, and Russell PE (2005) Focused Ion Beam Gases for Deposition and Enhanced Etch, pp. 53–72. Springer US, Boston, MA [Google Scholar]
- [32].Diebolder C, Faas F, Koster A, and Koning R (2015) Conical fourier shell correlation applied to electron tomograms. Journal of Structural Biology 190(2):215–223 [DOI] [PubMed] [Google Scholar]
- [33].Strunk K, Wang K, Ke D, Gray J, and Zhang P (2012) Thinning of large mammalian cells for cryo-TEM characterization by cryo-FIB milling. Journal of Microscopy 247(3):220–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Medeiros JM, Böck D, Weiss GL, Kooger R, Wepf RA, and Pilhofer M (2018) Robust workflow and instrumentation for cryo-focused ion beam milling of samples for electron cryotomography. Ultramicroscopy 190:1–11 [DOI] [PubMed] [Google Scholar]
- [35].Guo YS, Furrer JM, Kadilak AL, Hinestroza HF, Gage DJ, Cho YK, and Shor LM (2018) Bacterial extracellular polymeric substances amplify water content variability at the pore scale. Frontiers in Environmental Science 6:93 [Google Scholar]
- [36].Arnold J, Mahamid J, Lucic V, de Marco A, Fernandez JJ, Laugks T, Mayer T, Hyman AA, Baumeister W, and Plitzko JM (2016) Site-specific cryo-focused ion beam sample preparation guided by 3D correlative microscopy. Biophysical Journal 110(4):860–869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Nahmani M, Lanahan C, DeRosier D, and Turrigiano GG (2017) High-numerical-aperture cryogenic light microscopy for increased precision of superresolution reconstructions. Proceedings of the National Academy of Sciences 114(15):3832–3836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Carlson DB and Evans JE (2011) Low-cost cryo-light microscopy stage fabrication for correlated light/electron microscopy. JoVE (52):e2909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Tuijtel MW, Koster AJ, Jakobs S, Faas FGA, and Sharp TH (2019) Correlative cryo super-resolution light and electron microscopy on mammalian cells using fluorescent proteins. Scientific Reports 9(1):1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Schellenberger P, Kaufmann R, Siebert CA, Hagen C, Wodrich H, and Grünewald K (2014) High-precision correlative fluorescence and electron cryo microscopy using two independent alignment markers. Ultramicroscopy 143:41–51. SI: Correlative Microscopy [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Chang YW, Chen S, Tocheva EI, Treuner-Lange A, Löbach S, Søgaard-Andersen L, and Jensen GJ (2014) Correlated cryogenic photoactivated localization microscopy and cryo-electron tomography. Nature Methods 11(7):737–739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Kaufmann R, Schellenberger P, Seiradake E, Dobbie IM, Jones EY, Davis I, Hagen C, and Grünewald K (2014) Super-resolution microscopy using standard fluorescent proteins in intact cells under cryo-conditions. Nano Letters 14(7):4171–4175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Wolff G, Limpens RWAL, Zheng S, Snijder EJ, Agard DA, Koster AJ, and Bárcena M (2019) Mind the gap: micro-expansion joints drastically decrease the bending of FIB-milled cryo-lamellae. bioRxiv [DOI] [PubMed] [Google Scholar]
- [44].Mastronarde DN (2005) Automated electron microscope tomography using robust prediction of specimen movements. Journal of Structural Biology 152(1):36–51 [DOI] [PubMed] [Google Scholar]
- [45].Schorb M, Haberbosch I, Hagen WJH, Schwab Y, and Mastronarde DN (2019) Software tools for automated transmission electron microscopy. Nature Methods 16(6):471–477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Marko M, Hsieh C, Moberlychan W, Mannella CA, and Frank J (2006) Focused ion beam milling of vitreous water: prospects for an alternative to cryo-ultramicrotomy of frozen-hydrated biological samples. Journal of Microscopy 222(1):42–47 [DOI] [PubMed] [Google Scholar]
- [47].Toro-Nahuelpan M, Zagoriy I, Senger F, Blanchoin L, Théry M, and Mahamid J (2019) Tailoring cryo-electron microscopy grids by photo-micropatterning for in-cell structural studies. bioRxiv [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Stokes DJ, Vystavel T, and Morrissey F (2007) Focused ion beam (FIB) milling of electrically insulating specimens using simultaneous primary electron and ion beam irradiation. Journal of Physics D: Applied Physics 40(3):874–877 [Google Scholar]