

Keywords: acid-base; ammonia; Na+-bicarbonate cotransporter, electrogenic, isoform 1 (SLC4A4); proximal tubule
Abstract
The molecular mechanisms regulating ammonia metabolism are fundamental to acid-base homeostasis. Deletion of the A splice variant of Na+-bicarbonate cotransporter, electrogenic, isoform 1 (NBCe1-A) partially blocks the effect of acidosis to increase urinary ammonia excretion, and this appears to involve the dysregulated expression of ammoniagenic enzymes in the proximal tubule (PT) in the cortex but not in the outer medulla (OM). A second NBCe1 splice variant, NBCe1-B, is present throughout the PT, including the OM, where NBCe1-A is not present. The purpose of the present study was to determine the effect of combined renal deletion of NBCe1-A and NBCe1-B on systemic and PT ammonia metabolism. We generated NBCe1-A/B deletion using Cre-loxP techniques and used Cre-negative mice as controls. As renal NBCe1-A and NBCe1-B expression is limited to the PT, Cre-positive mice had PT NBCe1-A/B deletion [PT-NBCe1-A/B knockout (KO)]. Although on a basal diet, PT-NBCe1-A/B KO mice had severe metabolic acidosis, yet urinary ammonia excretion was not changed significantly. PT-NBCe1-A/B KO decreased the expression of phosphate-dependent glutaminase and phosphoenolpyruvate carboxykinase and increased the expression of glutamine synthetase, an ammonia-recycling enzyme, in PTs in both the cortex and OM. Exogenous acid loading increased ammonia excretion in control mice, but PT-NBCe1-A/B KO prevented any increase. PT-NBCe1-A/B KO significantly blunted acid loading-induced changes in phosphate-dependent glutaminase, phosphoenolpyruvate carboxykinase, and glutamine synthetase expression in PTs in both the cortex and OM. We conclude that NBCe1-B, at least in the presence of NBCe1-A deletion, contributes to PT ammonia metabolism in the OM and thereby to systemic acid-base regulation.
NEW & NOTEWORTHY The results of the present study show that combined deletion of both A and B splice variants of electrogenic Na+-bicarbonate cotransporter 1 from the proximal tubule impairs acid-base homeostasis and completely blocks changes in ammonia excretion in response to acidosis, indicating that both proteins are critical to acid-base homeostasis.
INTRODUCTION
Ammonia metabolism is central to acid-base homeostasis, which is critical to optimal health. Renal ammonia1 generation and excretion results in equimolar bicarbonate generation and is the primary component of net acid excretion both under basal conditions and in response to acid-base disturbances (1–3). In patients with chronic kidney disease, which affects as many as 14% of adults in Western countries, acid-base disturbances are frequently present, and they are associated with numerous adverse consequences, including mortality, progressive nephropathy, need for renal replacement therapy, skeletal muscle atrophy and weakness, insulin resistance, and thyroid hormone resistance (4–8). Inadequate ammonia excretion correlates with adverse outcomes (9, 10), suggesting that impaired renal ammonia metabolism directly contributes to these outcomes.
Recent studies have expanded our understanding of the molecular mechanisms that regulate proximal tubule (PT) and systemic ammonia metabolism. Bicarbonate transporter, Na+-bicarbonate cotransporter, electrogenic, isoform 1 (NBCe1, SLC4A4) is present exclusively in the PT, where it is a primary mechanism of the basolateral bicarbonate exit necessary for filtered bicarbonate reabsorption (11–14). In addition, NBCe1 also mediates a critical role in regulating PT ammonia generation and renal ammonia excretion. Its deletion results in impaired ammonia excretion and abnormal expression of essential proteins involved in PT ammonia generation (15). In mice, but not humans, NBCe1 deletion results in 100% mortality shortly after weaning (15–17), indicating critical roles of NBCe1 in addition to acid-base homeostasis.
NBCe1 has multiple splice variants. The A splice variant, NBCe1-A, is the primary splice variant present in the kidney and is found exclusively in the PT in the cortex (13, 18–23). Deletion of just NBCe1-A is not a lethal mutation, but it does lead to spontaneous metabolic acidosis, impaired ammonia excretion responses to acidosis, and blunted acid loading-induced changes in cortical PT ammoniagenic enzyme responses (19). However, in the proximal straight tubule in the outer medulla (PST-OM), not only did NBCe1-A deletion not inhibit ammoniagenic enzyme responses, it actually led to accentuated responses to metabolic acidosis (19). The accentuated PST-OM response indicates that mechanisms other than NBCe1-A appear to regulate PT ammonia metabolism.
A second NBCe1 splice variant is present in the kidney. The B splice variant, NBCe1-B, is expressed and exhibits exclusive basolateral expression in the PT (24). Importantly, NBCe1-B is found in the PST-OM, where NBCe1-A is not present (19, 24, 25). Thus, NBCe1-B is a candidate for the NBCe1-A-independent regulation of PT ammonia metabolism.
The purpose of the present study was to determine the effect of combined NBCe1-A/B deletion on renal acid-base homeostasis, ammonia excretion, and PT ammoniagenic enzyme expression. To do so, we used mice with loxP sites flanking critical exons in the gene structure for NBCe1 (26) and mice expressing Cre-recombinase under control of a constitutively active kidney-specific Pax8 promoter (27). Since the only NBCe1 splice variants in the kidney are NBCe1-A and NBCe1-B, and they are expressed only in the PT (24), this approach is designed to yield PT-specific NBCe1-A/B deletion [PT-NBCe1-A/B knockout (KO)]. We then used these mice to determine the role of NBCe1-A/B in basal and exogenous acidosis-stimulated acid-base homeostasis, ammonia excretion, and PT ammoniagenic enzyme expression.
METHODS
Animals
We generated PT-NBCe1-A/B KO mice using standard Cre-loxP techniques. Mice with a floxed NBCe1 allele have been previously described (26). Using Cre-recombinase under control of a constitutively active Pax8 promoter (27), we generated mice with kidney-specific NBCe1 deletion. Since the only NBCe1 splice variants present in the kidney are NBCe1-A and NBCe1-B, and they are found solely in the PT, this leads to PT-NBCe1-A/B KO. Trained personnel performed all animal breeding in the University of Florida Cancer and Genetics Transgenic Animal Facility, and all mice were genotyped using tail-clip DNA samples. The Institutional Animal Care and Use Committee of the Gainesville Veterans Affairs Medical Center, and the University of Florida College of Medicine approved all experiments. Mice used in these experiments averaged 5.0 ± 0.5 mo of age, and the age did not differ significantly between genotypes. Both male and female mice were studied in all protocols. All mice used were on a C57/Bl6 background.
Antibodies
Table 1 shows all antibodies used in these experiments.
Table 1.
Antibodies used
| Target | Supplier | Location | Catalog Number | Dilution |
|---|---|---|---|---|
| GS | Abcam | Cambridge, MA | ab73593 | W: 1:1,500I: 1:20,000 |
| NBCe1-A | Michael Romero, PhD | Mayo Clinic, Rochester, MN | Α-333 | W: 1:1,000I: 1:20,000 |
| Pan-NBCe1 | ProteinTech | Rosemont, IL | 11885-1-AP | W: 1:2,000I: 1:20,000 |
| PDG | Norman Curthoys, PhD | Colorado State University | N/A | W: 1:3,000I: 1:10,000 |
| PEPCK | Cayman Chemical | Ann Arbor, MI | 10004943 | W: 1:2,000I: 1:10,000 |
GS, glutamine synthetase; I, dilution used for immunohistochemistry; NBCe1, Na+-bicarbonate cotransporter, electrogenic, isoform 1; PDG, phosphate-dependent glutaminase; PEPCK, phosphoenolpyruvate carboxykinase; W, dilution used for Western blot analysis.
Metabolic Cage Experiments
Mice were placed in metabolic cages for 2–3 days and fed a powdered normal diet (18% protein, Harlan Teklad) mixed 1:1 with water. Daily food consumption was measured, and daily 24-h urine samples were collected. Urine samples were collected in tubes containing water-equilibrated mineral oil. After collection, daily urine volume and pH were recorded, and urine samples were then stored at −20°C until analyzed further.
Blood Analysis
Blood was obtained by cannulation of the abdominal aorta in mice anesthetized with isoflurane, drawn into a heparinized syringe, and immediately analyzed for Na+, K+, and bicarbonate concentrations using a Siemens Microanalytic Blood Gas Analyzer (RAPIDLab 348 Analyzer, Siemens, Munich, Germany).
Urinary Analysis
Urine ammonia was measured using a modification of a commercially available kit (A7553, Pointe Scientific, Canton, MI), as previously described (28, 29). Urine pH was measured using a micro-pH electrode (ROSS semi-micro pH, Orion 8103BN, Thermo Scientific, Waltham, MA). Titratable acid excretion was measured as previously described (28, 29).
Acid Loading
Animals were acid loaded using our standard techniques (19, 30). Briefly, HCl (0.4 M) was added to powdered rodent chow, which was provided ad libitum to the mice.
Protein Preparation
Animals were anesthetized with inhalant isoflurane, and kidneys were rinsed by in vivo cardiac perfusion with PBS (pH 7.4) containing 6,000 U/L of Na-heparin and 120 mg/L of lidocaine. The right kidney was removed rapidly, and the cortex and outer stripe of the OM (OSOM) were isolated on a cold stage under a dissecting microscope. Samples were snap frozen in liquid nitrogen and stored frozen at −80°C until use. Tissues were homogenized in T-PER tissue protein extraction reagent (Pierce Biotechnology, Rockford, IL) using microtube pestles (USA Scientific, Ocala, FL), and protein was extracted according to the manufacturer’s recommendations. An aliquot was used for total protein quantification using a bicinchoninic acid (BCA) assay, and the remainder was stored at −80°C until use.
Immunoblot Procedures
Immunoblot analysis was performed as previously detailed (31–33). Briefly, 5 μg of renal protein was electrophoresed on 10% PAGE ReadyGel (Bio-Rad, Hercules, CA). Gels were then transferred electrophoretically to nitrocellulose membranes, blocked with 5 g/dL nonfat dry milk in Blotto buffer (50 mM Tris, 150 mM NaCl, 5 mM Na2EDTA, and 0.05% Tween 20, pH 7.6), and incubated at 4°C overnight with primary antibody diluted in nonfat dry milk. Loading and transfer equivalence were assessed with Ponceau S staining. After being washed, membranes were exposed to goat anti-rabbit IgG secondary antibody (Cell Signaling Technology, Beverly, MA) conjugated to horseradish peroxidase at a dilution of 1:5,000. Results were visualized using enhanced chemiluminescence (SuperSignal West Pico Substrate, Pierce) and a Kodak Image Station 440CF digital imaging system. In selected experiments, blots were stripped. Band density was normalized such that the mean density in a region (cortex or OSOM) in wild-type (WT) male mice on the basal diet was 100. The absence of saturation was confirmed by examining pixel intensity distribution in all immunoblots. In all experiments, we assessed loading and transfer equivalence using Ponceau S staining, as previously described (17, 19, 24, 25, 28, 30, 32–40). Gels with evident differences between lanes were discarded and not used for analysis. If the same sample was analyzed on different blots, the mean expression from the different blots was calculated and used as a single measurement for statistical analysis. The calculated molecular weight of the identified protein is shown in each figure.
Tissue Preparation for Immunohistochemistry
We obtained tissues for immunohistochemistry using previously detailed techniques (31–33). Briefly, under isoflurane anesthesia, kidneys were preserved by in vivo cardiac perfusion with PBS (pH 7.4) containing Na-heparin (6,000 U/L) and lidocaine (120 mg/L) followed by periodate-lysine-2% paraformaldehyde (PLP), cut transversely into 2- to 3-mm-thick slices, and immersed for 24–30 h at 4°C in the same fixative. Samples were embedded in polyester wax made with polyethylene glycol 400 distearate (Polysciences, Warrington, PA) with 10% 1-hexadecanol, and 2-µm thick sections were cut and mounted on gelatin-coated slides.
Immunohistochemistry
We performed immunohistochemistry using our standard procedures (28, 36, 39–42). Briefly, sections were dewaxed in ethanol, rehydrated, and then rinsed in PBS. Antigen retrieval was accomplished by immersing slides in Trilogy (Cell Marque, Rocklin, CA) at 96°C for 1 h. Endogenous peroxidase activity was blocked by incubating the sections in 3% H2O2 for 45 min. After being washed in PBS, sections were treated with 0.5% Triton X-100 in PBS for 15 min. They then underwent several washes in PBS containing 1% BSA, 0.05% saponin, and 0.2% gelatin followed by incubation with Serum-Free Protein Block (Dako Cytomation) for 15 min and then incubated overnight at 4°C with primary antibody diluted in Dako antibody diluent. After several washes in PBS containing 0.1% BSA, 0.05% saponin, and 0.2% gelatin, sections were washed in PBS and incubated for 1 h with undiluted polymer-linked, peroxidase-conjugated goat anti-rabbit IgG (MACH2, Biocare Medical, Concord, CA), as per the manufacturer’s instructions, washed again with PBS, and then exposed to diaminobenzidine for 5 min. Sections were washed with distilled water, dehydrated with xylene, mounted, and observed by light microscopy. When comparing immunohistochemistry results, we used tissues from the same immunohistochemistry experiment. Each experiment included a section exposed to the immunolabeling procedure without primary antibody to ensure that the label was due to the primary antibody.
Statistical Analysis
Results are presented as means ± SE; n refers to the number of animals studied. All experiments used a similar number of male and female mice in each protocol. Statistical analyses were performed using ANOVA. If ANOVA indicated significant differences, individual comparisons were performed using unpaired t tests. P < 0.05 was considered statistically significant. All statistical analyses included both male and female mice, and sex was included as an independent variable in the ANOVA. The number of male and female mice in each analysis is reported as xM/yF. If sex altered the effect of genotype, this is reported specifically. Quantitative measures over time, such as urine ammonia and pH, were analyzed using general linear modeling with repeated measures to determine genotype effects. The statistical significance of specific day measures was determined using a Student’s t test. We performed statistical analysis using SPSS software and Microsoft Excel.
RESULTS
Validation of NBCe1-A/B Deletion
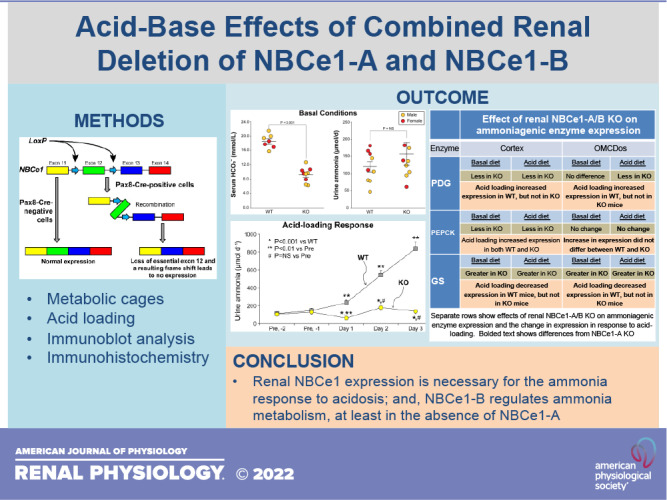
We first determined the extent of deletion of NBCe1-A and NBCe1-B in PT-NBCe1-A/B KO mice. To assess both NBCe1-A and NBCe1-B expression, we used an antibody directed against all NBCe1 splice variants (pan-NBCe1), and we used an antibody specific to NBCe1-A to evaluate the expression of just NBCe1-A (Table 1). Detection of pan-NBCe1 but not NBCe1-A immunolabel indicates NBCe1-B expression. Immunohistochemistry in control, Cre-negative mice showed that basolateral pan-NBCe1 immunolabel was present throughout the entire PT, which parallels that which we have previously observed in WT mice (24). In Cre-positive mice, basolateral pan-NBCe1 immunolabel was present only in rare, scattered cells in the PT (Fig. 1A), indicating that gene deletion was highly efficient, with very few cells retaining a functional allele. Immunoblot analysis showed nearly complete loss of pan-NBCe1 protein expression (Fig. 1B). For NBCe1-A, Cre-negative mice exhibited basolateral immunolabel throughout the PT in the cortex but not in the OSOM. This pattern parallels our previous report (19). In Cre-positive mice, only rare, scattered NBCe1-A-expressing PT cells were present. Moreover, serial sections showed complete concordance between pan-NBCe1 and NBCe1-A-specific immunolabel responses in Cre-positive mice (Fig. 1C). Immunoblot analysis showed nearly complete loss of NBCe1-A protein expression (Fig. 1D). These results demonstrate the successful development of mice with combined renal deletion of NBCe1-A and NBCe1-B. The lack of 100% efficacy of the Cre-LoxP system is consistent with our previous findings (37).
Figure 1.
Demonstration of Na+-bicarbonate cotransporter, electrogenic, isoform 1 (NBCe1) deletion. A: micrographs of pan-NBCe1 immunolabel in Cre-negative and Cre-positive NBCe1fl/fl kidneys. Low-power micrograph (a) and high-power micrographs from the proximal convoluted tubule (PCT; b), proximal straight tubule (PST) in the medullary ray (PST-MR; c), and PST in the outer stripe of the outer medulla (OSOM) (PST-OSOM; d). Basolateral pan-NBCe1 immunolabel was present throughout the proximal tubule. e–h show parallel findings in Cre-positive mice, with the low-power micrograph (e) demonstrating the near-complete absence of pan-NBCe1 immunolabel and high-power micrographs of the PCT (f), PST-MR (g), and PST-OSOM (h) showing the lack of detectable pan-NBCe1 immunolabel in these segments. B: pan-NBCe1 immunoblot demonstrating the near-complete absence of NBCe1 protein in Cre-positive [knockout (KO)] kidneys. C: micrographs demonstrating NBCe1-A immunolabel. i−l: NBCe1-A immunolabel in a control Cre-negative NBCe1fl/fl kidney. Low-power micrograph (i) and high-power micrographs from the PCT (j), PST-MR (k), and PST-OSOM (l). Basolateral NBCe1-A immunolabel was present in cortical proximal tubule segments, PCT (j) and PST-MR (k). NBCe1-A immunolabel was not detectable in the PST-OSOM (l). m–p: findings in Cre-positive NBCe1fl/fl mice. m: low-power micrograph demonstrating the near-complete absence of NBCe1-A immunolabel. High-power micrographs of the PCT (n), PST-MR (o), and PST-OSOM (p), showing the lack of detectable NBCe1-A immunolabel in these segments. Micrographs for Cre-negative mice (a–d and i–l) were taken from serial tissue sections, as were the micrographs for Cre-positive mice (e–h and m–p). D: NBCe1-A immunoblot. Immunohistochemistry micrographs are representative of findings in n = 4 male (M)/4 female (F) mice of each genotype. Immunoblot analysis results are representative of findings in n = 4M/3F mice for each genotype on the basal diet and 6M/3F wild-type (WT) mice and 4M/3F KO mice on the acid diet. Statistical analysis used ANOVA with genotype, diet, and sex as independent variables. Sex did not significantly alter the effects of genotype or diet. P values show results of specific unpaired t test comparisons. NS, not significant. *Proximal tubule segments.
Effect on Basal Electrolytes
Our subsequent experiments determined the effect of PT-NBCe1-A/B KO on baseline serum and urine electrolytes. Figure 2 shows these results. Serum K+ was not significantly altered [WT: 3.90 ± 0.12 mmol/L and KO: 3.66 ± 0.16 mmol/L, n = 4M/3F and 5M/4F, respectively, P = not significant (NS)], while bicarbonate was dramatically decreased (WT: 18.6 ± 0.7 mmol/L and KO: 9.2 ± 0.7 mmol/L, n = 4M/3F and 5M/4F, respectively, P < 0.001) (Fig. 1, A and B). Serum Na+ did not differ significantly (WT: 155 ± 1 mmol/L and KO, 152 ± 1 mmol/L, n = 4M/3F and 5M/4F, respectively, P = NS). Body weight was significantly less in KO mice (WT: 22.3 ± 0.8 g and KO: 18.7 ± 1.1, n = 5M/4F in each group, P < 0.05). Although body weight was less in females than in males, sex did not alter the effect of genotype on weight (n = 5M/4F in each group, P = NS by ANOVA).
Figure 2.

Effect of proximal tubule (PT)-specific Na+-bicarbonate cotransporter, electrogenic, isoform 1 (NBCe1) deletion [PT-NBCe1-A/B knockout (KO)] on serum and urine chemistries. A: serum K+ measurement. PT-NBCe1-A/B KO did not alter serum K+ significantly. B: serum measurement, demonstrating that serum was significantly lower in KO mice than in wild-type (WT) mice. C: urine ammonia excretion, calculated as the average of two consecutive 24-h urine collections. PT-NBCe1-A/B KO did not alter urine ammonia excretion significantly. D: urine pH calculated as the average pH of two consecutive 24-h urine collections. PT-NBCe1-A/B KO significantly decreased urine pH. n = 4 male (M)/3 female (F) WT mice and 5M/4F KO mice for serum K+ and serum . n = 5M/4F mice of each genotype for urine ammonia excretion and urine pH. Statistical analysis used ANOVA with genotype and sex as independent variables. Sex did not significantly alter the effects of genotype on any of the reported analyses. NS, not significant.
Despite this severe metabolic acidosis, urine ammonia excretion did not differ significantly between WT and KO mice (WT: 119 ± 15 µmol/day and KO: 158 ± 33 µmol/day, n = 5M/4F in each group, P = NS; Fig. 2C). Because PT-NBCe1-A/B KO mice had decreased body weight, we also evaluated urine ammonia excretion adjusted for body weight, which did not differ significantly between WT and PT-NBCe1-A/B KO mice (WT: 5.53 ± 0.82 µmol/g and KO: 8.69 ± 2.10 µmol/g, n = 5M/4F in each group, P = NS).
Urine pH is an essential determinant of ammonia excretion. Urine pH was significantly more acidic (lower) in PT-NBCe1-A/B KO mice (WT: 6.18 ± 0.06 and KO: 5.53 ± 0.10, n = 5M/4F in each group, P < 0.001; Fig. 2D). Thus, PT-NBCe1-A/B KO results in normokalemic metabolic acidosis associated with an absence of the expected increase in urine ammonia excretion despite an intact ability to acidify urine.
A second method used by the kidneys to maintain acid-base homeostasis involves titratable acid excretion. In contrast to ammonia, titratable acid excretion was significantly greater in KO than in WT mice (WT: 64 ± 7 µmol/day and KO: 132 ± 25 µmol/day, n = 3M/3F in each group, P < 0.05).
Ammoniagenic Enzyme Expression on the Basal Diet
Phosphate-dependent glutaminase.
The fact that PT-NBCe1-A/B KO results in severe metabolic acidosis without altering ammonia excretion despite an intact ability to acidify the urine suggests that there is abnormal intrarenal ammonia generation. To evaluate this, we determined the effect of PT-NBCe1-A/B KO on PT ammoniagenic enzyme expression. We evaluated critical enzymes in ammoniagenesis, mitochondrial phosphate-dependent glutaminase (PDG), and cytosolic phosphoenolpyruvate carboxykinase (PEPCK), which are involved in ammonia generation, and glutamine synthetase (GS), which catalyzes the reaction of ammonium with glutamate to regenerate glutamine and thus serves as an ammonia recycling enzyme.
The first enzyme we evaluated was PDG, which catalyzes the initial enzymatic step in ammonia generation. The expected response to the spontaneous metabolic acidosis present in PT-NBCe1-A/B KO mice is increased PDG expression. In the cortex, however, PDG expression was significantly less in spontaneously acidotic KO mice than in WT mice (n = 4M/3F in each genotype, P < 0.05 by ANOVA). Inclusion of sex as an independent variable did not alter this analysis. In the OSOM, PT-NBCe1-A/B KO also did not alter PDG expression significantly (n = 4M/3F in each genotype, P = NS by ANOVA). Figure 3 shows these results. Inclusion of sex as an independent variable did not alter this analysis.
Figure 3.

Effect of proximal tubule (PT)-specific Na+-bicarbonate cotransporter, electrogenic, isoform 1 (NBCe1) deletion [PT-NBCe1-A/B knockout (KO)] on phosphate-dependent glutaminase (PDG) expression under basal conditions and after acid loading. A: immunoblot analysis of the renal cortex and outer stripe of the outer medulla (OSOM) on the basal diet and after the acid-loading diet. B: quantification of results from the cortex. NBCe1-A/B deletion decreased PDG expression significantly in the cortex while on the basal diet. Acid loading increased PDG expression significantly in wild-type (WT) mice but not in KO mice, and expression was significantly less in acid-loaded KO mice than in acid-loaded WT mice. C: quantification of results from the OSOM. PDG expression did not differ between WT and KO mice on the basal diet. Acid loading increased PDG expression in WT mice but not in KO mice, and expression was significantly less in acid-loaded KO mice than in acid-loaded WT mice. Immunoblot analysis results are representative of findings in n = 4 male (M)/3 female (F) mice of each genotype on the basal diet and 6M/3F WT mice and 4M/3F KO mice on the acid-loading diet. Statistical analysis used ANOVA with genotype, diet, and sex as independent variables. Sex did not significantly alter the effects of genotype on any of the reported analyses. P values show the results of specific unpaired t test comparisons. NS, not significant.
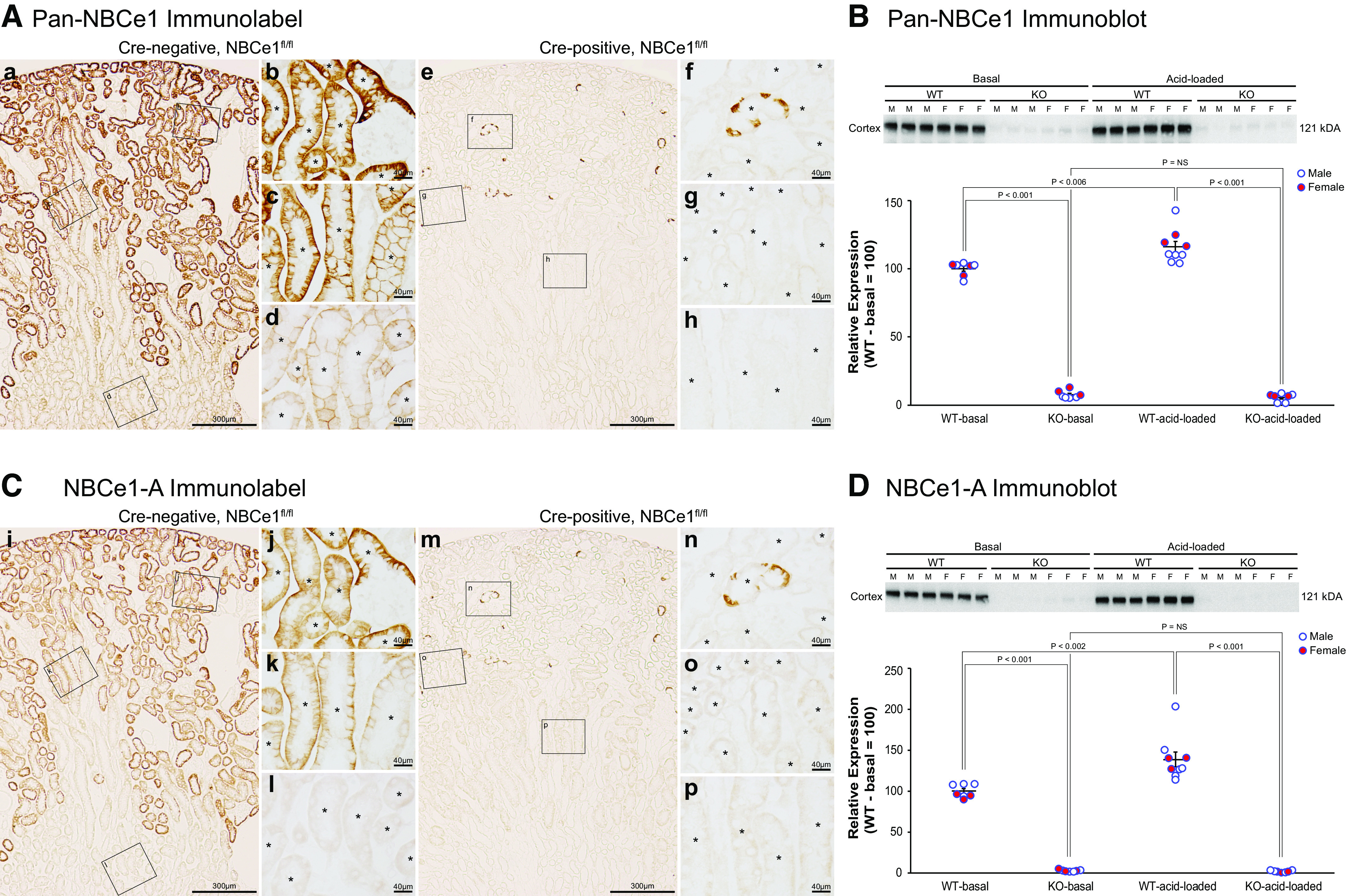
We then used immunohistochemistry to examine the effect of PT-NBCe1-A/B KO specifically on PT PDG expression. In WT mice, there was moderate-intensity PDG immunolabel in the proximal convoluted tubule (PCT), in the proximal straight tubule (PST) in the medullary ray (PST-MR), and the PST in the OSOM (PST-OSOM) (Fig. 4, column 1). Strong PDG immunolabel intensity was evident in the thick ascending limb of the loop of Henle (TAL), as we have previously reported in abstract form (43).
Figure 4.
Phosphate-dependent glutaminase (PDG) immunolabel in wild-type (WT) and proximal tubule (PT)-specific Na+-bicarbonate cotransporter, electrogenic, isoform 1 (NBCe1) deletion [PT-NBCe1-A/B knockout (KO)] kidneys on basal and acid-loading diets. Column 1 shows micrographs of a WT kidney on the basal diet. Separate rows show the proximal convoluted tubule (PCT) in the cortical labyrinth (top row), proximal straight tubule (PST) in the medullary ray (PST-MR; middle row) and PST in the outer stripe of the outer medulla (PST-OSOM; bottom row). PT segments were identified by the presence of an apical brush border on high-power observation and are denoted with * in the tubule lumen. Intermediate-intensity PDG immunolabel was present throughout the entire PT. Column 2 shows representative micrographs from PT-NBCe1-A/B KO mice on the basal diet. PDG immunolabel intensity was less intense in the PCT, PST-MR, and PST-OSOM of KO mice than in WT mice. Column 3 shows PDG immunolabel in acid-loaded WT mice. A dramatic increase in PT PDG immunolabel intensity compared with findings in WT mice on the basal diet (column 1) was evident in the PCT, PST-MR, and PST-OSOM. Column 4 shows findings from acid-loaded KO mice. PDG immunolabel intensity was low in all PT segments and not detectably different than that observed in KO mice on the basal diet. Immunolabel micrographs are representative of findings in n = 4 male (M)/4 female (F) for each genotype on the basal diet and 6M/6F WT mice and 4M/5F KO mice on the acid-loading diet. *PT segments; #thick ascending limb.
The expected PT PDG response to acidosis is a dramatic increase in PT expression. In contrast, PDG immunolabel intensity was less intense in PT-NBCe1-A/B KO mice throughout the PCT, PST-MR, and PST-OSOM than in corresponding portions of the PT in WT mice (Fig. 4, WT: column 1 and KO: column 2). The impact of PT-NBCe1-A/B KO on PDG expression was specific to the PT; there was no detectable change in TAL PDG expression.
Phosphoenolpyruvate carboxykinase.
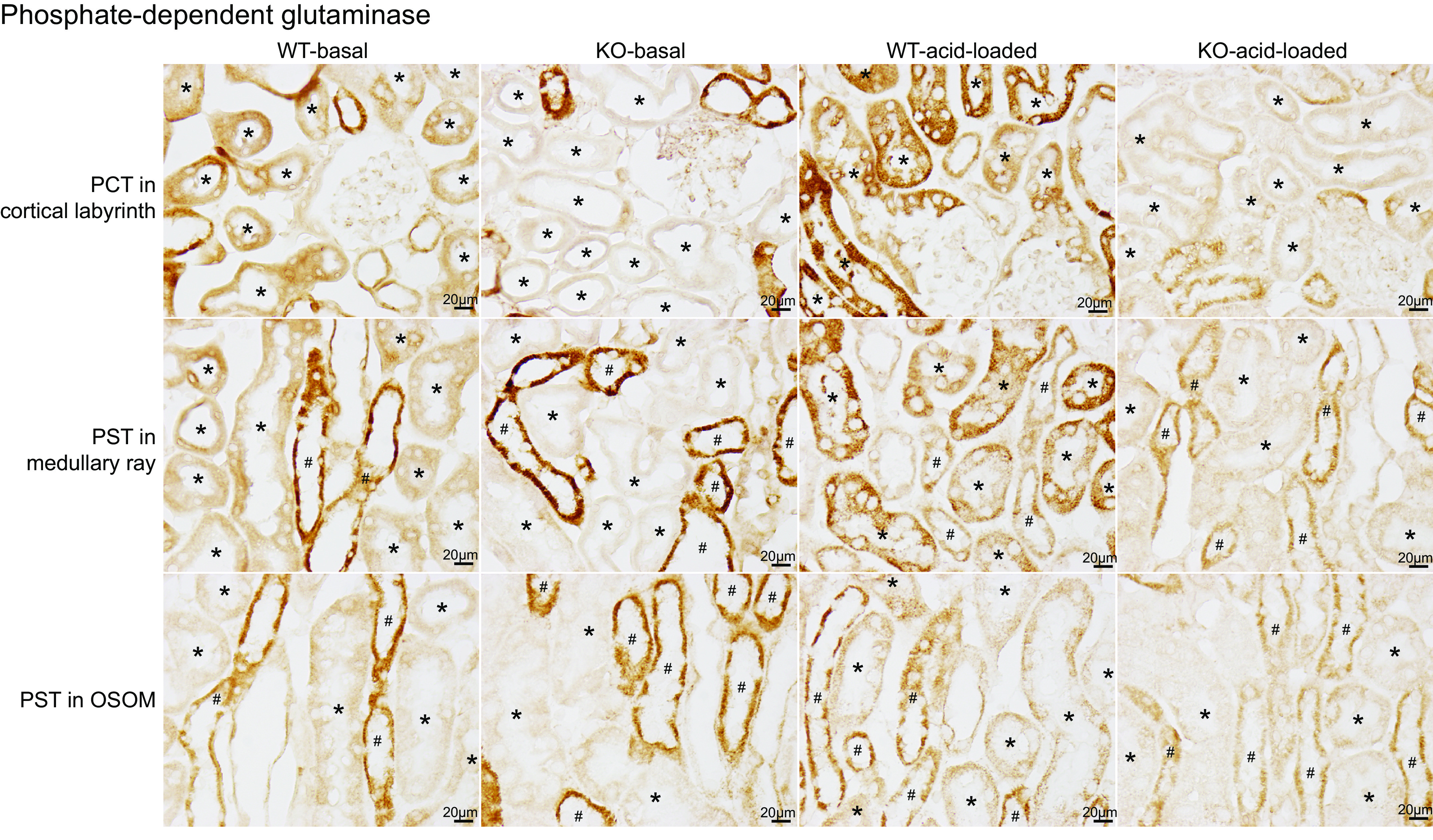
Next, we evaluated PEPCK. PEPCK is a cytosolic enzyme critical in ammoniagenesis and is expressed solely in the PT. While on a basal diet, WT mice exhibited moderate to strong PEPCK immunolabel intensity throughout the entire PT, i.e., PCT, PST-MR, and PST-OSOM (Fig. 5, column 1). There was no detectable immunolabel in non-PT epithelial cells. In KO mice, only low-intensity immunolabel was present in the PCT, PST-MR, and PST-OSOM (Fig. 5, column 2), and it was less intense than in WT mice in the PCT, PST-MR, and PST-OSOM (Fig. 5, WT: column 1 vs. KO: column 2). Since the expected response of PEPCK to acidosis is to increase expression, these results indicate an abnormal response in spontaneously acidotic KO mice.
Figure 5.
Phosphoenolpyruvate carboxykinase (PEPCK) immunolabel in wild-type (WT) and proximal tubule (PT)-specific Na+-bicarbonate cotransporter, electrogenic, isoform 1 (NBCe1) deletion [PT-NBCe1-A/B knockout (KO)] kidneys on basal and acid-loading diets. Column 1 shows micrographs of a WT kidney on the basal diet. Separate rows show the proximal convoluted tubule (PCT) in the cortical labyrinth (top row), proximal straight tubule (PST) in the medullary ray (PST-MR; middle row), and PST in the outer stripe of the outer medulla (PST-OSOM; bottom row). PT segments were identified by the presence of an apical brush border on high-power observation and are identified with an * in the tubule lumen. Intermediate-intensity PEPCK immunolabel was present throughout the entire PT in WT mice on the basal diet. Column 2 shows representative micrographs from PT-NBCe1-A/B KO mice on the basal diet. PEPCK immunolabel intensity was less intense in the PCT, PST-MR, and PST-OSOM than in WT mice (column 1). Column 3 shows PEPCK immunolabel in acid-loaded WT mice. PEPCK immunolabel was more intense than in WT mice on the basal diet in the PCT, PST-MR, and PST-OSOM. Column 4 shows findings from acid-loaded KO mice. PEPCK immunolabel intensity was more intense than in nonacid-loaded KO mice on the basal diet in each region but was less intense than in acid-loaded WT mice in the PCT and PST-MR. Immunolabel micrographs are representative of findings in n = 4 male (M)/4 female (F) mice for each genotype on the basal diet and 6M/6F WT mice and 4M/5F KO mice on the acid-loading diet. *PT segments.
Immunoblot analysis for PEPCK protein expression quantified the effects observed with immunohistochemistry. In mice on the basal diet, PT-NBCe1-A/B KO decreased PEPCK protein expression significantly in the cortex (Fig. 6). In the OSOM, in contrast, PT-NBCe1-A/B KO did not alter PEPCK expression significantly (Fig. 6, bottom). In both the cortex and OSOM, the expected increase in PEPCK expression due to the spontaneous metabolic acidosis induced by combined PT-NBCe1-A/B KO was not present.
Figure 6.

Effect of proximal tubule (PT)-specific Na+-bicarbonate cotransporter, electrogenic, isoform 1 (NBCe1) deletion [PT-NBCe1-A/B knockout (KO)] on phosphoenolpyruvate carboxykinase (PEPCK) expression under basal conditions and after acid loading. A: immunoblot analysis of the renal cortex and outer stripe of the outer medulla (OSOM) on both the basal diet and after the acid-loading diet. B: quantification of results from the cortex. NBCe1-A/B deletion decreased PEPCK expression significantly in the cortex while on the basal diet. Acid loading increased PEPCK expression significantly in both genotypes, but expression was less in acid-loaded KO mice than in acid-loaded wild-type (WT) mice. C: quantification of results from the OSOM. PEPCK expression did not differ between WT and KO mice on the basal diet. Acid loading increased PEPCK expression in both WT and KO mice, but expression did not differ in acid-loaded KO mice compared with acid-loaded WT mice. Immunoblot analysis results are representative of findings in n = 4 male (M)/3 female (F) mice for each genotype (OSOM, n = 4M/4F for KO mice) on the basal diet and 6M/3F WT mice and 4M/3F KO mice on the acid diet. Statistical analysis used ANOVA with genotype, diet, and sex as independent variables. Sex did not significantly alter the effects of genotype on any of the reported analyses. P values show results of specific unpaired t test comparisons. NS, not significant.
GS Expression on the Basal Diet
GS is a cytosolic enzyme that catalyzes the regeneration of glutamine from glutamate and . Its expression typically changes in the opposite direction of that observed with PDG and PEPCK, thereby enabling assessment of whether changes in PDG and PEPCK expression reflect coordinated regulation of ammoniagenesis or whether they reflect a generalized decrease in PT enzyme expression. Immunoblot analysis showed that PT-NBCe1-A/B KO increased GS significantly in the cortex (n = 4M/3F in each genotype, P < 0.001; Fig. 7). Given the metabolic acidosis present in PT-NBCe1-A/B KO mice, this is an inappropriate response. In the OSOM, immunoblot analysis similarly showed that PT-NBCe1-A/B KO increased GS expression significantly (n = 4M/3F in each group, P < 0.05; Fig. 7). The increase in GS expression occurred in each sex (P < 0.05 for each sex) but was significantly greater in female mice than in male mice (n = 4M/3F, P < 0.02 by ANOVA).
Figure 7.

Effect of proximal tubule (PT)-specific Na+-bicarbonate cotransporter, electrogenic, isoform 1 (NBCe1) deletion [PT-NBCe1-A/B knockout (KO)] on glutamine synthetase (GS) expression under basal conditions and after acid loading. A: immunoblot analysis of the renal cortex and outer stripe of the outer medulla (OSOM) on both the basal diet and after the acid-loading diet. B: quantification of results from the cortex. NBCe1-A/B deletion increased GS expression significantly in the cortex while on the basal diet. Acid loading decreased GS expression significantly in wild-type (WT) mice but not in KO mice, and expression was significantly greater in acid-loaded KO mice than in acid-loaded WT mice. C: quantification of results from the OSOM. Total GS expression was significantly greater in KO mice than in WT mice on the basal diet. Acid loading decreased GS expression in WT mice but not in KO mice, and expression was significantly greater in acid-loaded KO mice than in acid-loaded WT mice. Immunoblot analysis results are representative of findings in n = 4 male (M)/3 female (F) mice for each genotype on the basal diet and 6M/3F WT mice and 4M/3F KO mice on the acid-loading diet. Statistical analysis used ANOVA with genotype, diet, and sex as independent variables. Sex did not significantly alter the effects of genotype in the cortex. Effects of sex in the OSOM are detailed in the text. P values show results of specific unpaired t test comparisons. NS, not significant.
We then examined axial differences in GS expression using immunohistochemistry. There was moderate-level immunolabel intensity in WT mice throughout the PCT, PST-MR, and PST-OSOM (Fig. 8, column 1). In PT-NBCe1-A/B KO mice, immunolabel intensity was greater than in WT mice in each of these portions of the PT (Fig. 8, WT: column 1 and KO: column 2). Since PT-NBCe1-A/B KO mice are acidotic, and acidosis decreases GS expression (42, 44), increased expression in PT-NBCe1-A/B KO mice indicates abnormal regulation of GS expression.
Figure 8.
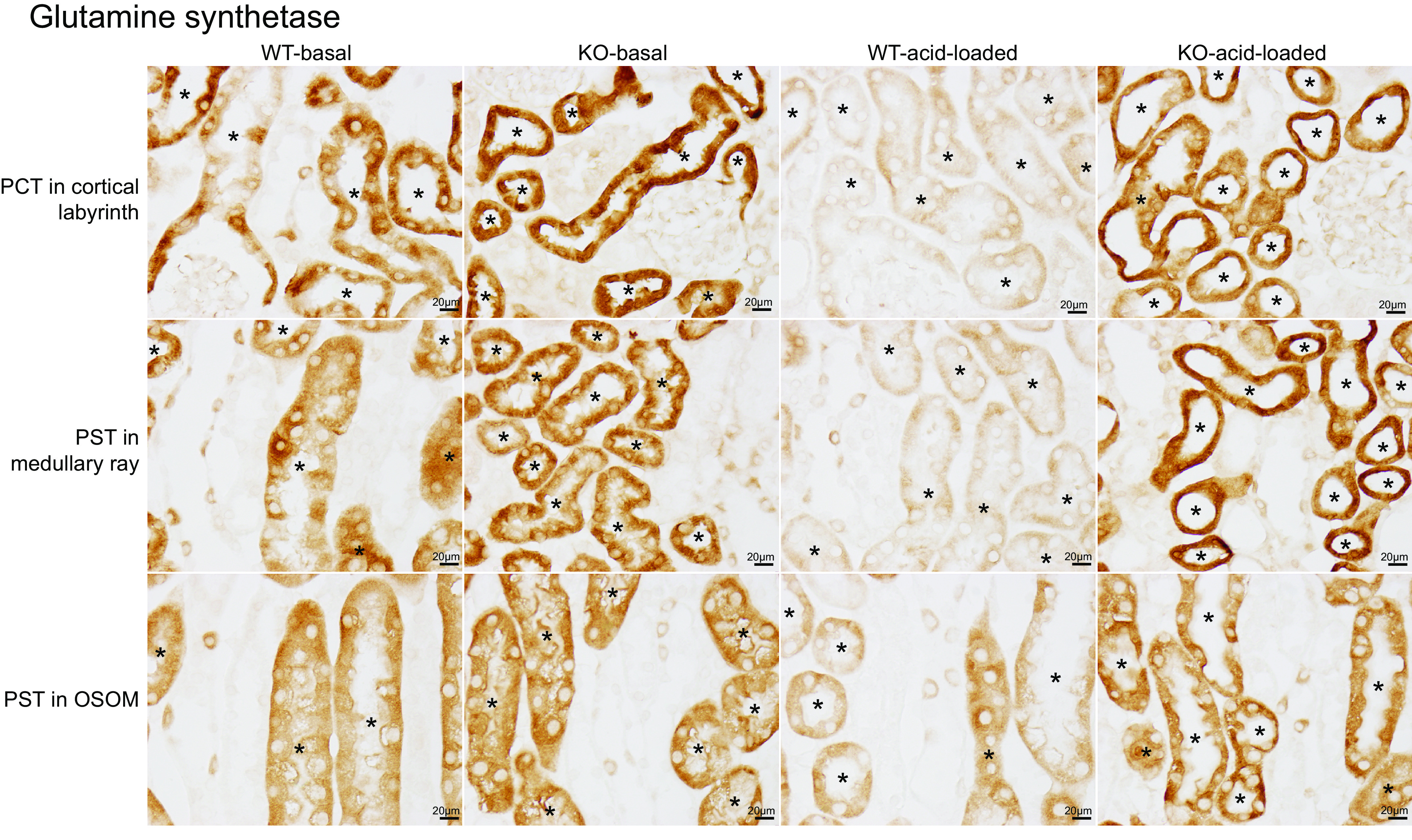
Glutamine synthetase (GS) immunolabel in wild-type (WT) and proximal tubule (PT)-specific Na+-bicarbonate cotransporter, electrogenic, isoform 1 (NBCe1) deletion [PT-NBCe1-A/B knockout (KO)] kidneys on basal and acid-loading diets. Column 1 shows micrographs of the proximal convoluted tubule (PCT; top row), proximal straight tubule (PST) in the medullary ray (PST-MR; middle row), and PST in the outer stripe of the outer medulla (PST-OSOM; bottom row). PT segments were identified by the presence of an apical brush border on high-power observation. In WT mice on the basal diet, there was intermediate-intensity GS immunolabel throughout the PT. Column 2 shows representative micrographs from a PT-NBCe1-A/B KO kidney on the basal diet. GS immunolabel intensity was substantially greater in the PCT, PST-MR, and PST-OSOM of KO mice than in WT mice. Column 3 shows GS immunolabel in acid-loaded WT mice. GS immunolabel intensity was very low throughout the PT, including the PCT, PST-MR, and PST-OSOM. Column 4 shows findings in acid-loaded PT-NBCe1-A/B KO kidneys. GS immunolabel intensity was substantially greater than in acid-loaded WT mice in the PCT, PST-MR, and PST-OSOM, and it was not detectably different from that observed in KO mice on the basal diet. Immunolabel micrographs are representative of findings in n = 4 male (M)/4 female (F) mice for each genotype on the basal diet and 6M/6F WT mice and 4M/5F KO mice on the acid-loading diet. *PT segments.
Response to Acid Loading
The kidneys respond to an exogenous acid load by increasing ammonia excretion, and NBCe1-A deletion partially, but not wholly, inhibits this response (19). The subsequent experiments examined whether NBCe1-B contributes to this residual response by determining the effect of combined deletion of both NBCe1-A and NBCe1-B. For baseline measurements, WT and KO mice were placed on a basal diet for 2 days and then were changed to an acid-loading diet. After 3 days, PT-NBCe1-A/B KO mice exhibited progressive weight loss, and we terminated acid loading. Serum K+ in acid-loaded WT mice (4.10 ± 0.08 mmol/L, n = 6M/6F) did not differ significantly (P = NS by an unpaired t test) from the serum K+ observed in basal diet-fed WT mice (see Effect on Basal Electrolytes). Serum K+ in acid-loaded KO mice (4.56 ± 0.24 mmol/L, n = 2M/4F) was slightly greater (P < 0.05 by an unpaired t test) than observed in basal diet-fed KO mice (see Effect on Basal Electrolytes). Serum K+ did not differ significantly between WT and KO mice (P = NS).
Serum in acid-loaded WT mice (13.1 ± 0.7 mmol/L, n = 6M/6F) was significantly less than in WT mice on the basal diet (see Effect on Basal Electrolytes for WT data, P < 0.001 by an unpaired t test). In acid-loaded KO mice, serum averaged 5.6 ± 0.9 mmol/L (n = 2M/4F), which was significantly less than in KO mice on the basal diet (P < 0.01 vs. the basal diet). Serum was significantly lower in acid-loaded KO mice than in WT mice (n = 6M/6F and 2M/4F, respectively; P < 0.001). Figure 9 shows these results.
Figure 9.

Effect of proximal tubule (PT)-specific Na+-bicarbonate cotransporter, electrogenic, isoform 1 (NBCe1) deletion [PT-NBCe1-A/B knockout (KO)] on serum and urine electrolyte responses to acid loading. A: serum K+, which did not differ significantly between wild-type (WT) and KO mice [WT: 4.10 ± 0.08 mmol/L and KO: 4.56 ± 0.24 mmol/L, n = 6 male (M)/6 female (F) and 2M/4F mice, respectively, P = not significant (NS)]. B: serum , which was significantly lower in KO mice than in WT mice (WT: 13.1 ± 0.7 mmol/L and KO: 5.6 ± 0.9 mmol/L, n = 6M/6F and 2 M/4F mice, respectively, P < 0.001). C: urine ammonia excretion measured in 24-h urine samples. Baseline ammonia excretion did not differ between WT and KO mice. WT mice responded to the acid-loading diet with a rapid increase in ammonia excretion. In KO mice, ammonia excretion was significantly less than in WT mice on each day of acid loading. Furthermore, in KO mice, ammonia excretion decreased slightly from baseline on day 1 of acid loading and then did not increase significantly from baseline rates during days 2 or 3 of acid loading. n = 4−6M/5−6F mice for each genotype at each time point. D: urine pH. Urine pH was significantly more acidic in KO mice than in WT mice on each day prior to acid loading and on each day of acid loading. n = 4−6M/5−6F mice for each genotype at each day. Statistical analysis used ANOVA with genotype, diet, and sex as independent variables. Sex did not significantly alter the effects of genotype. Quantitative measures over time, such as urine ammonia and pH, were analyzed using general linear modeling with repeated measures to determine genotype effects. The statistical significance of specific day measures was determined using Student’s t test.
We then examined urinary ammonia excretion. Baseline ammonia excretion did not differ significantly between WT and KO mice. WT mice responded to an acid-loading diet with a rapid increase in ammonia excretion (Fig. 9C). The response in KO mice differed significantly (n = 6M/6F WT mice and 4M/5F KO mice, P < 0.001 by ANOVA with repeated measures). In KO mice, ammonia excretion decreased slightly from baseline on day 1 of acid loading and then did not increase significantly above baseline rates during days 2 or 3 of acid loading (n = 4M/5F–6M/6F per day, P = NS for each). On each day of acid loading, ammonia excretion was significantly less in KO mice than in WT mice.
The difference in ammonia excretion was not due to failure to acidify the urine. Urine pH was significantly more acidic (lower) in KO mice than in WT mice before acid loading and on each day of acid loading (Fig. 9D). Thus, the inability of PT-NBCe1-A/B KO mice to increase urine ammonia excretion is not due to defective urine acidification.
Titratable acid excretion, which was greater in KO mice than in WT mice on a basal diet, remained greater after acid loading. On the last day of acid loading, titratable acid excretion differed significantly between WT mice (23 ± 4 µmol/day) and KO mice (56 ± 5 µmol/day) (n = 3M/3F in each group, P < 0.001). Sex did not significantly alter titratable acid excretion after acid loading (P = NS by ANOVA).
Effect of PT-NBCe1-A/B KO on the Ammoniagenic Enzyme Response to Acid Loading
Phosphate-dependent glutaminase.
The lack of a significant increase in ammonia excretion in PT-NBCe1-A/B KO mice in response to acid loading suggests a defect in the regulation of PT ammoniagenesis. To test this, we determined the effect of PT-NBCe1-A/B KO on the reaction of the critical ammoniagenic enzyme PDG to exogenous acid loading. The expected PDG response to acid loading is increased total protein expression. PT-NBCe1-A/B KO significantly altered this response (P < 0.005 by ANOVA; Fig. 3). In the cortex, acid loading increased PDG expression significantly in WT mice (n = 6M/3F WT, P < 0.001) but not in KO mice (n = 4M/3F, P = NS). Consistent with these findings, total cortical PDG expression was significantly less in the cortex of acid-loaded KO mice than in acid-loaded WT mice (P < 0.001).
Similar findings were observed when we examined the OSOM. Acid loading increased OSOM PDG expression in WT mice (n = 6M/3F, P < 0.05) but not in KO mice (n = 4M/3F, P = NS) (Fig. 3). This difference led to total PDG expression being significantly less in acid-loaded KO mice than in acid-loaded WT mice (P < 0.001). Inclusion of sex as an independent variable in the ANOVA did not alter these conclusions.
We then used immunohistochemistry to evaluate the effects of PT-NBCe1-A/B KO on PT PDG expression. In WT mice, acid loading increased PT PDG immunolabel intensity in the PCT, PST-MR, and PST-OSOM (Fig. 4, WT-basal diet: column 1 vs. WT-acid loading: column 3). In KO mice, in contrast, acid loading did not result in a detectable difference in PT PDG immunolabel intensity (Fig. 4, KO-basal diet: column 2 vs. KO-acid loading: column 4). When we compared acid-loaded WT and PT-NBCe1-A/B KO mice, PDG immunolabel intensity was substantially less intense in KO mice than in WT mice in the PCT, PST-MR, and PST-OSOM (Fig. 4, WT-acid loading: column 3 vs. KO-acid loading: column 4). Thus, combined expression of NBCe1-A and NBCe1-B is necessary for the normal PT PDG response to exogenous acid loading.
Phosphoenolpyruvate carboxykinase.
We then determined the effect on PEPCK expression. Immunoblot analysis of proteins from WT mice showed that acid loading increased expression significantly in both the cortex and OSOM (Fig. 6). In KO mice, acid loading increased PEPCK expression in the cortex and OSOM, but expression remained significantly less than in the WT mice in the cortex (Fig. 6).
We used immunohistochemistry to evaluate the PT segment-specific PEPCK response. In WT mice, acid loading increased PEPCK immunolabel intensity in the PCT, PST-MR, and PST-OSOM (Fig. 5, WT-basal diet: column 1 vs. WT-acid loading: column 3). In PT-NBCe1-A/B KO mice, acid loading increased PEPCK immunolabel intensity slightly in the PCT, PST-MR, and PST-OSOM (Fig. 5, KO-basal diet: column 2 vs. KO-acid loading: column 4), but it remained less intense than in WT mice in the PCT and PST-MR of the PT (Fig. 5, WT-acid loading: column 3 vs. KO-acid loading: column 4). Since PT-NBCe1-A/B KO mice have significantly more severe metabolic acidosis than WT mice, PT-NBCe1-A/B KO results in an impaired PT PEPCK expression response to systemic acid-base status.
Effect of PT-NBCe1-A/B KO on the GS Response to Acid Loading
As noted earlier, GS catalyzes ammonia recycling and decreases net PT ammonia generation. In WT mice, acid loading decreased GS expression in both the cortex (n = 4M/3F with the basal diet and 6M/3F with the acid diet, P < 0.001) and OSOM (n = 4M/3F with the basal diet and 6M/3F with the acid diet, P < 0.05) (Fig. 7). In KO mice, in contrast, acid loading did not alter GS expression significantly in either the cortex (n = 4M/3F in each genotype, P = NS) or OSOM (n = 4M/3F in each genotype, P = NS) (Fig. 7).
ANOVA showed the effects of sex on these findings in the OSOM but not in the cortex. In female mice, acid loading did not result in a statistically significant difference in GS expression in either WT or KO mice (WT-basal: 128.6 ± 7.6, WT-HCl: 118.9 ± 11.7, KO-basal: 271.1 ± 41.5, and KO-HCl: 173.8, n = 3 in each group). ANOVA showed significant effects of both genotype (P < 0.005) and diet (P < 0.03), and the interaction of genotype with diet exhibited a statistical significance value of P < 0.053. In male mice, acid loading decreased GS expression in WT mice (WT-basal: 101.0 ± 4.7 and WT-HCl: 51.0 ± 6.5, n = 4 and 6, respectively, P < 0.001). In KO mice, in contrast, GS expression did not change (KO-basal: 134.9 ± 4.7 and KO-HCl: 151.6 ± 11.7, n = 4 in each group, P = NS).
Immunohistochemistry showed that acid loading decreased GS immunolabel intensity significantly in the WT kidney throughout the PT, including the PCT, PST-MR, and PST-OSOM (Fig. 8, WT-basal diet: column 1 vs. WT-acid loading: column 3), which is the expected response (42). In acid-loaded PT-NBCe1-A/B KO mice, in contrast, GS immunolabel intensity was substantially greater than in acid-loaded WT mice in the PCT, PST-MR, and PST-OSOM (Fig. 8, WT-acid loading: column 3 vs. KO-acid loading: column 4). Moreover, in KO mice, acid loading did not detectably alter PT GS immunolabel intensity in any region of the PT (Fig. 8, KO-basal diet: column 2 vs. KO-acid loading: column 4). Thus, the expression of NBCe1-A and NBCe1-B is necessary for the PT GS response required for the maximal acid-base in ammonia metabolism response to acid loading.
DISCUSSION
This study provides important new information regarding the cellular mechanisms that regulate renal ammonia excretion and the expression of critical enzymes involved in PT ammonia generation. Combined deletion of both NBCe1-A and NBCe1-B from the kidney results in severe spontaneous metabolic acidosis without an adaptive change in ammonia excretion, and it completely blocks the ammonia excretion response to an exogenous acid load. On a basal diet, combined NBCe1-A/B deletion decreased the expression of the critical proteins PDG and PEPCK, required for PT ammoniagenesis, and it increased the expression of the ammonia recycling enzyme GS. Combined NBCe1-A/B deletion completely blocked the PDG and GS response to acid loading and substantially blocked the PEPCK response. Importantly, the effects of combined NBCe1-A/B deletion differ significantly in several aspects from the effects of NBCe1-A deletion alone. Tables 2 and 3 show these findings in the cortex and OSOM, respectively. Thus, these results demonstrate that NBCe1-B can mediate an essential role in the regulation of ammonia metabolism under basal conditions and in response to acid loading.
Table 2.
Differences between NBCe1-A KO and PT-NBCe1-A/B KO in the cortex
| Protein | NBCe1-A KO (19) | PT-NBCe1-A/B KO (Present Study) | ||
|---|---|---|---|---|
| PDG | Basal Diet | Acid Diet | Basal Diet | Acid Diet |
| Less in KO mice | Less in KO mice | Less in KO mice | Less in KO mice | |
| Acid loading increased expression in both WT and KO mice | Acid loading increased expression in WT mice but not in KO mice* | |||
| PEPCK | Basal Diet | Acid Diet | Basal Diet | Acid Diet |
| Less in KO mice | Less in KO mice | Less in KO mice | Less in KO mice | |
| Acid loading increased expression in both WT and KO mice but less in KO mice | Acid loading increased expression in both WT and KO mice | |||
| GS | Basal Diet | Acid Diet | Basal Diet | Acid Diet |
| No effect of KO | Greater expression in KO mice | Greater expression in KO mice* | Greater expression in KO mice | |
| Acid loading decreased expression in both WT and KO mice, but the decrease was less in KO mice than in WT mice | Acid loading decreased expression in WT mice but not in KO mice* | |||
GS, glutamine synthetase; KO, knockout; NBCe1, Na+-bicarbonate cotransporter, electrogenic, isoform 1; PDG, phosphate-dependent glutaminase; PEPCK, phosphoenolpyruvate carboxykinase; WT, wild type.
*Difference between NBCe1-A deletion [NBCe1-A knockout (KO)] and proximal tubule (PT)-specific NBCe1 deletion (PT-NBCe1-A/B KO).
Table 3.
Differences between NBCe1-A and PT-NBCe1-A/B KO in the OSOM
| Protein | NBCe1-A KO (19) | PT-NBCe1-A/B KO (Current Study) | ||
|---|---|---|---|---|
| PDG | Basal Diet | Acid Diet | Basal Diet | Acid Diet |
| WT and KO mice did not differ | WT and KO mice did not differ | WT and KO mice did not differ | Less expression in KO mice* | |
| Acid loading increased expression in both WT and KO mice | Acid loading increased expression in WT mice but not in KO mice* | |||
| PEPCK | Basal Diet | Acid Diet | Basal Diet | Acid Diet |
| WT and KO mice did not differ | Greater in KO mice than in WT mice | WT and KO mice did not differ | WT and KO mice did not differ* | |
| Acid loading increased expression in both WT and KO mice; the increase was greater in KO mice than in WT mice | Increase in expression did not differ between WT and KO mice* | |||
| GS | Basal Diet | Acid Diet | Basal Diet | Acid Diet |
| WT and KO mice did not differ | Decreased by KO | Increased by KO* | Increased by KO* | |
| Acid loading decreased expression in both WT and KO mice; the difference was greater in KO mice | Acid loading decreased expression in WT mice but not in KO mice* | |||
GS, glutamine synthetase; KO, knockout; NBCe1, Na+-bicarbonate cotransporter, electrogenic, isoform 1; OSOM, outer stripe of the outer medulla; PDG, phosphate-dependent glutaminase; PEPCK, phosphoenolpyruvate carboxykinase; PT, proximal tubule; WT, wild type.
*Difference between NBCe1-A deletion [NBCe1-A knockout (KO)] and proximal tubule (PT)-specific NBCe1 deletion (PT-NBCe1-A/B KO).
The first finding in this study is that combined deletion of NBCe1-A/B does not cause early mortality. There was no spontaneous mortality in our observation of PT-NBCe1-A/B KO mice through as much as 10 mo of age. In contrast, deletion of all splice variants leads to 100% mortality by an age of ∼30 days (15–17). This early mortality is not the result of NBCe1-A deletion, which does not cause premature mortality (19, 25). Combined renal deletion of NBCe1-A and NBCe1-B, the only NBCe1 splice variants expressed in the kidney (24), does not cause early mortality (present study), indicating the premature mortality induced by global NBCe1 deletion does not result from its roles in the kidney. In addition, global deletion of NBCe1-B and NBCe1-C results in early mortality (45). Thus, the mortality associated with global NBCe1 or with global NBCe1-B/C deletion likely result from critical extrarenal functions of either NBCe1-B or NBCe1-C. These roles may be related to its expression in the pancreas, central nervous system, colonic epithelial cells, heart, or thyroid (26, 46–51).
The second finding in this study is that the combined renal deletion of NBCe1-A and NBCe1-B induces severe metabolic acidosis. This likely reflects the critical roles of NBCe1-A and NBCe1-B in multiple aspects of renal acid-base homeostasis. Basolateral Na+-coupled, electrogenic bicarbonate transport is a significant component of basolateral bicarbonate transport necessary for filtered bicarbonate reabsorption in cortical PT segments and the PST in the OSOM (52–55). Since detectable NBCe1-A is not present in the PST-OSOM (19), whereas NBCe1-B is expressed (24), NBCe1-B likely contributes to filtered bicarbonate reabsorption in the PST-OSOM. Combined NBCe1-A/B deletion also results in impaired regulation of urinary ammonia excretion and PT ammoniagenic enzyme expression, which likely also contributes to this severe metabolic acidosis.
A third, and critical component, of the present study is that it advances our understanding of the role of NBCe1-B in ammonia metabolism. NBCe1-B differs from NBCe1-A in that it results from alternative transcription initiation, resulting in a differing 5′-end of the transcribed mRNA, which results in the differing initial amino acid sequence (56, 57). This different amino acid sequence results in several differences, including basal transport activity, the presence of an autostimulatory domain in NBCe1-A and an autoinhibitory domain in NBCe1-B (58), and responsiveness to specific signaling regulation, most prominently IRBIT-1 (59). In addition, NBCe1-A and NBCe1-B have different expression patterns; NBCe1-A expression is limited to the PCT and PST-MR (13, 18–23), whereas NBCe1-B expressed is found throughout the entire PT (24, 48). Thus, differences between the effects of NBCe1-A deletion (19) and combined PT-NBCe1-A/B KO (present study) in the PST-OSOM can be attributed to NBCe1-B.
Under basal conditions, combined NBCe1-A/B deletion resulted in severe metabolic acidosis, yet no increase in ammonia excretion. The lack of change in ammonia excretion was associated with, and likely resulted from, decreases in the expression of the ammoniagenic enzymes PDG and PEPCK and increases in GS throughout the entire PT. Similar effects were seen in cortical portions of the PT with NBCe1-A deletion (19), precluding specific conclusions about the role of NBCe1-B in these segments. However, in the PST in the OSOM, combined NBCe1-A/B KO decreased PDG and PEPCK expression while increasing GS expression (present study), whereas NBCe1-A deletion alone did not alter the expression of either of these (19). We conclude that NBCe1-B mediates a critical role in regulating ammoniagenic enzyme expression in the PST-OSOM. Moreover, the observation that NBCe1-A deletion increased ammonia excretion on a per gram weight basis (19) but that combined NBCe1-A/B KO did not suggests that these effects of NBCe1-B on PST-OSOM ammoniagenic and ammonia recycling enzyme expression contributes to basal ammonia excretion.
NBCe1-B also likely contributes to the ammonia excretion response to acid loading. In the present study, combined NBCe1-A/B KO completely blocked the ability to increase ammonia excretion significantly. In contrast, mice with NBCe1-A deletion retained a partial (∼30% of normal) ability to increase ammonia excretion (19). Thus, NBCe1-B appears to mediate a critical role in the ability to increase ammonia excretion in response to acid loading, at least in the absence of NBCe1-A.
The role of NBCe1-B in acid loading-induced increases in ammonia excretion likely results from its role regulating ammoniagenic enzymes PDG and PEPCK and ammonia recycling enzyme GS expression in the PST-OSOM. As shown in the present study, in PT-NBCe1-A/B KO mice, acid loading did not increase PT PDG or decrease GS in either the PCT, PST in the cortex, or PST in the OSOM. These effects differ from those observed in mice with deletion of just NBCe1-A, where acid loading-induced changes in ammoniagenic enzyme expression were blocked in cortical segments, but in the PST-OSOM, they were accentuated (19). Because NBCe1-B is present in the PST-OSOM, whereas NBCe1-A is not, we suggest that NBCe1-B contributes to an essential component of the effects of acid loading on ammoniagenic and ammonia recycling enzyme expression, at least in the PST-OSOM.
The most likely mechanism through which NBCe1-A/B deletion alters ammoniagenic enzyme expression involves intracellular pH. Na+-dependent, Cl−-independent basolateral bicarbonate transport activity, i.e., NBCe1 activity, is the primary mechanism of basolateral bicarbonate transport and is the major determinant of intracellular pH (52). Inhibition of this NBCe1 activity by deletion of NBCe1-A/B likely impairs base exit and alkalinizes PT cell pH. Indeed, pharmacological NBCe1 inhibition, with disulfonic stilbenes, alkalinizes PT cells (52). Intracellular alkalinization is likely to decrease PDG and PEPCK expression through pH response elements regulating PDG expression (60) and effects on p38 stress-activated protein kinase and subsequent phosphorylation of transcription factor ATF-2 that regulate PEPCK expression (61). However, we also note that the effect of PT-NBCe1-A/B KO on PDG and GS expression differed from its effects on PEPCK expression. For PDG and GS, PT-NBCe1-A KO completely blocked responsiveness to acid loading throughout the PT, whereas for PEPCK, there was a persistent ability to increase expression. This suggests that mechanisms regulating PEPCK expression differ, at least in some respects, from those that regulate PDG and GS expression.
Urine pH was more acidic in NBCe1-A/B KO mice than in their Cre-negative controls. This indicates that a lack of luminal acidification does not explain the failure of appropriate ammonia response to spontaneous acidosis. Instead, the increased urine acidification likely results from inadequate availability of NH3 for collecting duct secretion. Moreover, the lower urine pH may enable unchanged ammonia excretion despite inhibition of ammoniagenic enzyme expression. Studies in humans have shown that only ∼50% of ammonia generated in the kidneys enters the urine (62, 63). More acidic urine, which shifts the NH3 + H+ ↔ equilibrium toward , decreases luminal NH3 concentration, thereby increasing the transepithelial gradient in the collecting duct for NH3 secretion. Through these mechanisms, the more acidic urine pH in PT-NBCe1-A/B KO mice might increase the relative secretion of ammonia into the urine, enabling unchanged urinary ammonia despite inhibition of ammoniagenic enzyme expression.
The comparison of the effect of NBCe1-A/B KO with those of NBCe1-A KO demonstrates the contribution of NBCe1-B in the absence of NBCe1-A. However, NBCe1-B may contribute to acid-base and ammonia metabolism even in the presence of NBCe1-A. First, acid loading increases NBCe1-B expression (24), suggesting a role of NBCe1-B in acid-base homeostasis. Second, the greatest difference between NBCe1-A and combined NBCe1-A/B KO mice is in the OSOM, a site in which NBCe1-A is not present. Importantly, acid loading induces changes in OSOM PDG, PEPCK, and GS expression that are either unaffected or accentuated by NBCe1-A deletion (19) but are blocked by combined NBCe1-A/B deletion (present study). Thus, it is likely that NBCe1-B contributes to ammonia regulation even in the presence of NBCe1-A. Definitive data that NBCe1-B contributes to the regulation of ammonia metabolism will require study of mice with NBCe1-B deletion; however, the likelihood of adaptive response of NBCe1-A expression and/or regulation to the absence of NBCe1-B might make a negative finding in such a study difficult to interpret definitively.
The change in serum bicarbonate in response to acid loading did not differ between WT mice and PT-NBCe1-A/B KO mice, even though urine ammonia excretion increased in WT mice but did not in KO mice. In humans, it has been observed that the volume of distribution for bicarbonate and acid loads increases as the severity of metabolic acidosis increases (64). This might occur in response to greater activation of tissue buffering mechanisms, such as increased bone buffering involving increased osteoclast activity (65).
We have shown that there are differences in ammonia handling between male and female mice. This includes differences in basal ammonia excretion, with female mice excreting more than male mice (28), differences in the expression of several proteins involved in ammonia generation and transport under baseline conditions (24), and differences in the response to acid loading (30). These differences are at least in part due to testosterone-mediated effects (36) and involve signaling through the canonical testosterone receptor, the androgen receptor (34). In the present study, there were statistically significant differences between males and females in the regulation of GS in the OSOM, particularly with respect to the effects of acid loading in WT mice, where the expected decrease in expression was not observed in female mice. Sex did not statistically alter the effect of genotype on diet in any of the other analyses examined. Understanding the mechanism of this sex difference in the regulation of OSOM GS expression will be an important issue in future studies.
In summary, the present study provides critical new insights into the molecular regulation of ammonia metabolism. Combined NBCe1-A/B deletion results in severe metabolic acidosis under basal conditions, blocks the ammonia excretion response to this spontaneous acidosis, and blocks the increase in response to exogenous acid loading. It also blocks the normal PDG, PEPCK, and GS expression responses to spontaneous acidosis and exogenous acid loading in PT segments both in the cortex and in the OSOM. Because these effects differ from the effects of NBCe1-A deletion, which did not inhibit PST-OSOM responses, they indicate that NBCe1-B has a critical role regulating ammonia metabolism, particularly in the PST in the OSOM, where NBCe1-A is not present.
GRANTS
This work was supported by National Institute of Diabetes and Digestive and Kidney Diseases Grants R01DK107798 (to I.D.W. and J.W.V.) and K08DK120873 (to A.N.H.).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
H-W.L., J.W.V., A.N.H., and I.D.W. conceived and designed research; H-W.L. and J.W.V. performed experiments; H-W.L., J.W.V., and I.D.W. analyzed data; H-W.L., J.W.V., A.N.H., and I.D.W. interpreted results of experiments; H-W.L. prepared figures; H-W.L., J.W.V., G.E.S., A.N.H., and I.D.W. edited and revised manuscript; H-W.L., J.W.V., G.E.S., A.N.H., and I.D.W. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Dr. Sharon W. Matthews and Chao Chen of the University of Florida College of Medicine Electron Microscopy Core Laboratory for excellent tissue processing for our immunohistochemical experiments.
Footnotes
Ammonia exists in two molecular forms, NH3 and , which are in equilibrium with each other. We use the term “ammonia” to refer to both molecular species. If referring to a specific molecular form, we explicitly state either “NH3” or “.”
REFERENCES
- 1.Weiner ID, Mitch WE, Sands JM. Urea and ammonia metabolism and the control of renal nitrogen excretion. Clin J Am Soc Nephrol 10: 1444–1458, 2015. doi: 10.2215/CJN.10311013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Weiner ID, Verlander JW. Emerging features of ammonia metabolism and transport in acid-base balance. Semin Nephrol 39: 394–405, 2019. doi: 10.1016/j.semnephrol.2019.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Weiner ID, Verlander JW. Recent advances in understanding renal ammonia metabolism and transport. Curr Opin Nephrol Hypertens 25: 436–443, 2016. doi: 10.1097/MNH.0000000000000255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gojowy D, Skiba K, Bartmanska M, Kolonko A, Wiecek A, Adamczak M. Is metabolic acidosis a novel risk factor for a long-term graft survival in patients after kidney transplantation? Kidney Blood Press Res 45: 702–712, 2020. doi: 10.1159/000508476. [DOI] [PubMed] [Google Scholar]
- 5.Mannon EC, O’connor PM. Alkali supplementation as a therapeutic in chronic kidney disease: what mediates protection? Am J Physiol Renal Physiol 319: F1090–F1104, 2020. doi: 10.1152/ajprenal.00343.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Navaneethan SD, Shao J, Buysse J, Bushinsky DA. Effects of treatment of metabolic acidosis in CKD: a systematic review and meta-analysis. Clin J Am Soc Nephrol 14: 1011–1020, 2019. doi: 10.2215/CJN.13091118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Raphael KL. Metabolic acidosis in CKD: core curriculum 2019. Am J Kidney Dis 74: 263–275, 2019. doi: 10.1053/j.ajkd.2019.01.036. [DOI] [PubMed] [Google Scholar]
- 8.Tangri N, Reaven NL, Funk SE, Ferguson TW, Collister D, Mathur V. Metabolic acidosis is associated with increased risk of adverse kidney outcomes and mortality in patients with non-dialysis dependent chronic kidney disease: an observational cohort study. BMC Nephrol 22: 185, 2021. doi: 10.1186/s12882-021-02385-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Raphael KL, Carroll DJ, Murray J, Greene T, Beddhu S. Urine ammonium predicts clinical outcomes in hypertensive kidney disease. J Am Soc Nephrol 28: 2483–2490, 2017. doi: 10.1681/ASN.2016101151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Scialla JJ, Asplin J, Dobre M, Chang AR, Lash J, Hsu C-Y, Kallem RR, Hamm LL, Feldman HI, Chen J, Appel LJ, Anderson CA, Wolf M; Chronic Renal Insufficiency Cohort Study Investigators, et al. Higher net acid excretion is associated with a lower risk of kidney disease progression in patients with diabetes. Kidney Int 91: 204–215, 2017. doi: 10.1016/j.kint.2016.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Aalkjaer C, Frische S, Leipziger J, Nielsen S, Praetorius J. Sodium coupled bicarbonate transporters in the kidney, an update. Acta Physiol Scand 181: 505–512, 2004. doi: 10.1111/j.1365-201X.2004.01324.x. [DOI] [PubMed] [Google Scholar]
- 12.Alper SL. Familial renal tubular acidosis. J Nephrol 23 Suppl 16: S57–S76, 2010. [PubMed] [Google Scholar]
- 13.Endo Y, Yamazaki S, Moriyama N, Li Y, Ariizumi T, Kudo A, Kawakami H, Tanaka Y, Horita S, Yamada H, Seki G, Fujita T. Localization of NBC1 variants in rat kidney. Nephron Physiol 104: p87–p94, 2006. doi: 10.1159/000094003. [DOI] [PubMed] [Google Scholar]
- 14.Kurtz I, Zhu Q. Structure, function, and regulation of the SLC4 NBCe1 transporter and its role in causing proximal renal tubular acidosis. Curr Opin Nephrol Hypertens 22: 572–583, 2013. doi: 10.1097/MNH.0b013e328363ff43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Handlogten ME, Osis G, Lee H-W, Romero MF, Verlander JW, Weiner ID. NBCe1 expression is required for normal renal ammonia metabolism. Am J Physiol Renal Physiol 309: F658–F666, 2015. doi: 10.1152/ajprenal.00219.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gawenis LR, Bradford EM, Prasad V, Lorenz JN, Simpson JE, Clarke LL, Woo AL, Grisham C, Sanford LP, Doetschman T, Miller ML, Shull GE. Colonic anion secretory defects and metabolic acidosis in mice lacking the NBC1 Na+/HCO3− cotransporter. J Biol Chem 282: 9042–9052, 2007. doi: 10.1074/jbc.M607041200. [DOI] [PubMed] [Google Scholar]
- 17.Osis G, Handlogten ME, Lee H-W, Hering-Smith KS, Huang W, Romero MF, Verlander JW, Weiner ID. Effect of NBCe1 deletion on renal citrate and 2-oxoglutarate handling. Physiol Rep 4: e12778, 2016. doi: 10.14814/phy2.12778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Abuladze N, Lee I, Newman D, Hwang J, Pushkin A, Kurtz I. Axial heterogeneity of sodium-bicarbonate cotransporter expression in the rabbit proximal tubule. Am J Physiol Renal Physiol 274: F628–F633, 1998. doi: 10.1152/ajprenal.1998.274.3.F628. [DOI] [PubMed] [Google Scholar]
- 19.Lee H-W, Osis G, Harris AN, Fang L, Romero MF, Handlogten ME, Verlander JW, Weiner ID. NBCe1-A regulates proximal tubule ammonia metabolism under basal conditions and in response to metabolic acidosis. J Am Soc Nephrol 29: 1182–1197, 2018. doi: 10.1681/ASN.2017080935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Maunsbach AB, Vorum H, Kwon T-H, Nielsen S, Simonsen B, Choi I, Schmitt BM, Boron WF, Aalkjær C. Immunoelectron microscopic localization of the electrogenic Na/HCO3 cotransporter in rat and ambystoma kidney. J Am Soc Nephrol 11: 2179–2189, 2000. doi: 10.1681/ASN.V11122179. [DOI] [PubMed] [Google Scholar]
- 21.Schmitt BM, Biemesderfer D, Romero MF, Boulpaep EL, Boron WF. Immunolocalization of the electrogenic Na+-HCO3− cotransporter in mammalian and amphibian kidney. Am J Physiol Renal Physiol 276: F27–F38, 1999. doi: 10.1152/ajprenal.1999.276.1.F27. [DOI] [PubMed] [Google Scholar]
- 22.Wang G, Li C, Kim SW, Ring T, Wen J, Djurhuus JC, Wang W, Nielsen S, Frøkiaer J. Ureter obstruction alters expression of renal acid-base transport proteins in rat kidney. Am J Physiol Renal Physiol 295: F497–F506, 2008. doi: 10.1152/ajprenal.00425.2007. [DOI] [PubMed] [Google Scholar]
- 23.Yamada H, Yamazaki S, Moriyama N, Hara C, Horita S, Enomoto Y, Kudo A, Kawakami H, Tanaka Y, Fujita T, Seki G. Localization of NBC-1 variants in human kidney and renal cell carcinoma. Biochem Biophys Res Commun 310: 1213–1218, 2003. doi: 10.1016/j.bbrc.2003.09.147. [DOI] [PubMed] [Google Scholar]
- 24.Fang L, Lee H-W, Chen C, Harris AN, Romero MF, Verlander JW, Weiner ID. Expression of the B splice variant of NBCe1 (SLC4A4) in the mouse kidney. Am J Physiol Renal Physiol 315: F417–F428, 2018. doi: 10.1152/ajprenal.00515.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee H-W, Harris AN, Romero MF, Welling PA, Wingo CS, Verlander JW, Weiner ID. NBCe1-A is required for the renal ammonia and K+ response to hypokalemia. Am J Physiol Renal Physiol 318: F402–F421, 2020. doi: 10.1152/ajprenal.00481.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vairamani K, Prasad V, Wang Y, Huang W, Chen Y, Medvedovic M, Lorenz JN, Shull GE. NBCe1 Na+-HCO3− cotransporter ablation causes reduced apoptosis following cardiac ischemia-reperfusion injury in vivo. World J Cardiol 10: 97–109, 2018. doi: 10.4330/wjc.v10.i9.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bouchard M, Souabni A, Busslinger M. Tissue-specific expression of cre recombinase from the Pax8 locus. Genesis 38: 105–109, 2004. doi: 10.1002/gene.20008. [DOI] [PubMed] [Google Scholar]
- 28.Harris AN, Lee H-W, Osis G, Fang L, Webster KL, Verlander JW, Weiner ID. Differences in renal ammonia metabolism in male and female kidney. Am J Physiol Renal Physiol 315: F211–F222, 2018. doi: 10.1152/ajprenal.00084.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim H-Y, Baylis C, Verlander JW, Han K-H, Reungjui S, Handlogten ME, Weiner ID. Effect of reduced renal mass on renal ammonia transporter family, Rh C glycoprotein and Rh B glycoprotein, expression. Am J Physiol Renal Physiol 293: F1238–F1247, 2007. doi: 10.1152/ajprenal.00151.2007. [DOI] [PubMed] [Google Scholar]
- 30.Harris AN, Lee HW, Fang L, Verlander JW, Weiner ID. Differences in acidosis-stimulated renal ammonia metabolism in the male and female kidney. Am J Physiol Renal Physiol 317: F890–F905, 2019. doi: 10.1152/ajprenal.00244.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bishop JM, Verlander JW, Lee H-W, Nelson RD, Weiner AJ, Handlogten ME, Weiner ID. Role of the Rhesus glycoprotein, Rh B glycoprotein, in renal ammonia excretion. Am J Physiol Renal Physiol 299: F1065–F1077, 2010. doi: 10.1152/ajprenal.00277.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee H-W, Verlander JW, Bishop JM, Igarashi P, Handlogten ME, Weiner ID. Collecting duct-specific Rh C glycoprotein deletion alters basal and acidosis-stimulated renal ammonia excretion. Am J Physiol Renal Physiol 296: F1364–F1375, 2009. doi: 10.1152/ajprenal.90667.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee H-W, Verlander JW, Bishop JM, Nelson RD, Handlogten ME, Weiner ID. Effect of intercalated cell-specific Rh C glycoprotein deletion on basal and metabolic acidosis-stimulated renal ammonia excretion. Am J Physiol Renal Physiol 299: F369–F379, 2010. doi: 10.1152/ajprenal.00120.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Harris AN, Castro RA, Lee HW, Verlander JW, Weiner ID. Role of the renal androgen receptor in sex differences in ammonia metabolism. Am J Physiol Renal Physiol 321: F629–F644, 2021. doi: 10.1152/ajprenal.00260.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Harris AN, Grimm PR, Lee H-W, Delpire E, Fang L, Verlander JW, Welling PA, Weiner ID. Mechanism of hyperkalemia-induced metabolic acidosis. J Am Soc Nephrol 29: 1411–1425, 2018. doi: 10.1681/ASN.2017111163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Harris AN, Lee HW, Verlander JW, Weiner ID. Testosterone modulates renal ammonia metabolism. Am J Physiol Renal Physiol 318: F922–F935, 2020. doi: 10.1152/ajprenal.00560.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lee H-W, Osis G, Handlogten ME, Lamers WH, Chaudhry FA, Verlander JW, Weiner ID. Proximal tubule-specific glutamine synthetase deletion alters basal and acidosis-stimulated ammonia metabolism. Am J Physiol Renal Physiol 310: F1229–F1242, 2016. doi: 10.1152/ajprenal.00547.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lee HW, Osis G, Handlogten ME, Verlander JW, Weiner ID. Proximal tubule glutamine synthetase expression is necessary for the normal response to dietary protein restriction. Am J Physiol Renal Physiol 313: F116–F125, 2017. doi: 10.1152/ajprenal.00048.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee H-W, Verlander JW, Bishop JM, Handlogten ME, Han K-H, Weiner ID. Renal ammonia excretion in response to hypokalemia: effect of collecting duct-specific Rh C glycoprotein deletion. Am J Physiol Renal Physiol 304: F410–F421, 2013. doi: 10.1152/ajprenal.00300.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee HW, Verlander JW, Handlogten ME, Han KH, Weiner ID. Effect of collecting duct-specific deletion of both Rh B glycoprotein (Rhbg) and Rh C glycoprotein (Rhcg) on renal response to metabolic acidosis. Am J Physiol Renal Physiol 306: F389–F400, 2014. doi: 10.1152/ajprenal.00176.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bishop JM, Lee H-W, Handlogten ME, Han K-H, Verlander JW, Weiner ID. Intercalated cell-specific Rh B glycoprotein deletion diminishes renal ammonia excretion response to hypokalemia. Am J Physiol Renal Physiol 304: F422–F431, 2013. doi: 10.1152/ajprenal.00301.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Verlander JW, Chu D, Lee HW, Handlogten ME, Weiner ID. Expression of glutamine synthetase in the mouse kidney: localization in multiple epithelial cell types and differential regulation by hypokalemia. Am J Physiol Renal Physiol 305: F701–F713, 2013. doi: 10.1152/ajprenal.00030.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lee HW, Osis G, Harris AN, Fang L, et al. Axial heterogeneity of phosphate-dependent glutaminase expression and response to metabolic acidosis (Abstract). J Am Soc Nephrol 28: 367, 2017. [Google Scholar]
- 44.Conjard A, Komaty O, Delage H, Boghossian M, Martin M, Ferrier B, Baverel G. Inhibition of glutamine synthetase in the mouse kidney: a novel mechanism of adaptation to metabolic acidosis. J Biol Chem 278: 38159–38166, 2003. doi: 10.1074/jbc.M302885200. [DOI] [PubMed] [Google Scholar]
- 45.Salerno EE, Patel SP, Marshall A, Marshall J, Alsufayan T, Mballo CSA, Quade BN, Parker MD. Extrarenal signs of proximal renal tubular acidosis persist in nonacidemic Nbce1b/c-null mice. J Am Soc Nephrol 30: 979–989, 2019. doi: 10.1681/ASN.2018050545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Abuladze N, Lee I, Newman D, Hwang J, Boorer K, Pushkin A, Kurtz I. Molecular cloning, chromosomal localization, tissue distribution, and functional expression of the human pancreatic sodium bicarbonate cotransporter. J Biol Chem 273: 17689–17695, 1998. doi: 10.1074/jbc.273.28.17689. [DOI] [PubMed] [Google Scholar]
- 47.Barmeyer C, Ye JH, Soroka C, Geibel P, Hingsammer LM, Weitgasser L, Atway D, Geibel JP, Binder HJ, Rajendran VM. Identification of functionally distinct Na-HCO3 co-transporters in colon. PLoS One 8: e62864, 2013. doi: 10.1371/journal.pone.0062864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Brandes A, Oehlke O, Schümann A, Heidrich S, Thévenod F, Roussa E. Adaptive redistribution of NBCe1-A and NBCe1-B in rat kidney proximal tubule and striated ducts of salivary glands during acid-base disturbances. Am J Physiol Regul Integr Comp Physiol 293: R2400–R2411, 2007. doi: 10.1152/ajpregu.00208.2007. [DOI] [PubMed] [Google Scholar]
- 49.Rickmann M, Orlowski B, Heupel K, Roussa E. Distinct expression and subcellular localization patterns of Na+/HCO3− cotransporter (SLC 4A4) variants NBCe1-A and NBCe1-B in mouse brain. Neuroscience 146: 1220–1231, 2007. doi: 10.1016/j.neuroscience.2007.02.061. [DOI] [PubMed] [Google Scholar]
- 50.Roussa E, Nastainczyk W, Thevenod F. Differential expression of electrogenic NBC1 (SLC4A4) variants in rat kidney and pancreas. Biochem Biophys Res Commun 314: 382–389, 2004. doi: 10.1016/j.bbrc.2003.12.099. [DOI] [PubMed] [Google Scholar]
- 51.Virreira M, Jin L, Djerbib S, De Deken X, Miot F, Massart C, Svoboda M, Van Sande J, Beauwens R, Dumont JE, Boom A. Expression, localization, and regulation of the sodium bicarbonate cotransporter NBCe1 in the thyroid. Thyroid 29: 290–301, 2019. doi: 10.1089/thy.2017.0576. [DOI] [PubMed] [Google Scholar]
- 52.Alpern RJ, Chambers M. Cell pH in the rat proximal convoluted tubule. Regulation by luminal and peritubular pH and sodium concentration. J Clin Invest 78: 502–510, 1986. doi: 10.1172/JCI112602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Biagi BA, Sohtell M. Electrophysiology of basolateral bicarbonate transport in the rabbit proximal tubule. Am J Physiol Renal Physiol 250: F267–F272, 1986. doi: 10.1152/ajprenal.1986.250.2.F267. [DOI] [PubMed] [Google Scholar]
- 54.Preisig PA, Alpern RJ. Basolateral membrane H-OH-HCO3 transport in the proximal tubule. Am J Physiol Renal Physiol 256: F751–F765, 1989. doi: 10.1152/ajprenal.1989.256.5.F751. [DOI] [PubMed] [Google Scholar]
- 55.Sasaki S, Shiigai T, Yoshiyama N, Takeuchi J. Mechanism of bicarbonate exit across basolateral membrane of rabbit proximal straight tubule. Am J Physiol Renal Physiol 252: F11–F18, 1987. doi: 10.1152/ajprenal.1987.252.1.F11. [DOI] [PubMed] [Google Scholar]
- 56.Boron WF, Chen L, Parker MD. Modular structure of sodium-coupled bicarbonate transporters. J Exp Biol 212: 1697–1706, 2009. doi: 10.1242/jeb.028563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Romero MF, Chen AP, Parker MD, Boron WF. The SLC4 family of bicarbonate (HCO3−) transporters. Mol Aspects Med 34: 159–182, 2013. doi: 10.1016/j.mam.2012.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mcalear SD, Liu X, Williams JB, Mcnicholas-Bevensee CM, Bevensee MO. Electrogenic Na/HCO3 cotransporter (NBCe1) variants expressed in Xenopus oocytes: functional comparison and roles of the amino and carboxy termini. J Gen Physiol 127: 639–658, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shirakabe K, Priori G, Yamada H, Ando H, Horita S, Fujita T, Fujimoto I, Mizutani A, Seki G, Mikoshiba K. IRBIT, an inositol 1,4,5-trisphosphate receptor-binding protein, specifically binds to and activates pancreas-type Na+/HCO3− cotransporter 1 (pNBC1). Proc Natl Acad Sci USA 103: 9542–9547, 2006. doi: 10.1073/pnas.0602250103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ibrahim H, Lee YJ, Curthoys NP. Renal response to metabolic acidosis: role of mRNA stabilization. Kidney Int 73: 11–18, 2008. doi: 10.1038/sj.ki.5002581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Curthoys NP, Gstraunthaler G. Mechanism of increased renal gene expression during metabolic acidosis. Am J Physiol Renal Physiol 281: F381–F390, 2001. doi: 10.1152/ajprenal.2001.281.3.F381. [DOI] [PubMed] [Google Scholar]
- 62.Deferrari G, Robaudo C, Garibotto G, Saffioti S, Sala MR, Tizianello A. Determinants of the partition of renal ammonia production between urine and venous blood in man with metabolic acid-base disturbances. Contrib Nephrol 92: 109–113, 1991. [DOI] [PubMed] [Google Scholar]
- 63.Weiner ID, Verlander JW. Ammonia transporters and their role in acid-base balance. Physiol Rev 97: 465–494, 2017. doi: 10.1152/physrev.00011.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Garella S, Dana CL, Chazan JA. Severity of metabolic acidosis as a determinant of bicarbonate requirements. N Engl J Med 289: 121–126, 1973. doi: 10.1056/NEJM197307192890303. [DOI] [PubMed] [Google Scholar]
- 65.Arnett TR, Dempster DW. Effect of pH on bone resorption by rat osteoclasts in vitro. Endocrinology 119: 119–124, 1986. doi: 10.1210/endo-119-1-119. [DOI] [PubMed] [Google Scholar]