

Abstract
All the articles in this issue, with the exception of this one, focus on roles for OGT, the O-GlcNAc-transferase that transfers GlcNAc to serine (Ser) or threonine (Thr) of cytoplasmic and nuclear proteins. Here we describe a much more recently discovered O-GlcNAc-transferase termed EOGT for EGF-domain specific O-GlcNAc-transferase. EOGT transfers GlcNAc to Ser or Thr in secreted and membrane proteins that contain one or more epidermal growth factor-like (EGF) repeats with a specific consensus sequence. Thus, OGT and EOGT are in separate cellular compartments and have mostly distinct substrates. This review will describe known substrates of EOGT, and biological roles for EOGT in Drosophila and humans. Mutations in EOGT that give rise to Adams-Oliver Syndrome in humans will also be discussed.
Introduction
Proteins with a covalently attached O-GlcNAc residue were first reported in 1984 [1]. Subsequently, it was shown that the relevant O-GlcNAc-transferase OGT, resides in the cytoplasm and nucleus and acts on cytosolic and nuclear proteins [2]. This includes the cytosolic domains of transmembrane proteins such as the inositol 1,4,5-triphosphate receptor type 1 (InsP3) [3]. Many functions have now been ascribed to the O-GlcNAc modification of nuclear and cytosolic proteins [4, 5]. Given the extensive investigations of intracellular O-GlcNAc since 1984, it was a surprise to discover a “new” O-GlcNAc residue on the EGF repeat of a secreted protein. This secretory pathway O-GlcNAc modification was first identified attached to a Drosophila Notch extracellular domain fragment containing EGF20 that was secreted from S2 cells [6]. Subsequently, several other glycoproteins including Dumpy (Dp), Notch, and the Notch ligands Delta and Serrate were shown to carry O-GlcNAc in Drosophila [7, 8]. In mammals, NOTCH1, NOTCH2, heparan sulfate proteoglycan 2 (HSPG2), neural epidermal growth factor-like I (NELL1), laminin subunit alpha-5 (LAMA5), peptidase domain containing associated with muscle regeneration1 (PAMR1), and aminoacyl-tRNA synthase complex-interacting multifunctional protein 1 (AIMP1) were shown to carry the O-GlcNAc modification [9–11]. This review focuses on the nature of the EGF-domain specific O-GlcNAc-transferase EOGT, and biological roles for EOGT and O-GlcNAc in Drosophila and in humans with a genetic disorder associated with inherited mutations in EOGT.
Identification of a novel O-GlcNAc-transferase
Following the discovery of O-GlcNAc by mass spectrometry on Drosophila Notch EGF20 [6], an O-GlcNAc-transferase was sought by in silico screening of databases, including the CAZy database for established and putative glycosyltransferases [12, 13]. Candidate genes encoding proteins with characteristic features of a glycosyltransferase were knocked down in Drosophila S2 cells, and the loss of O-GlcNAc from secreted Notch EGF20 was monitored by mass spectrometry or western analysis [7, 8]. The Drosophila gene identified was CG9867, which generates a single transcript. The predicted Eogt amino acid sequence is shown in Fig. 1 and compared to three mammalian EOGT sequences. EOGT is highly conserved across species, and shares only ~12% similarity with OGT. EOGT open reading frames encode a protein of 524–527 amino acids, with a signal peptide at the amino terminus, and a KDEL sequence for endoplasmic reticulum (ER) retention at the carboxyl terminus (Fig. 2A). EOGT also contains a nucleotide sugar binding “DXD” motif, mutations in which result in decreased EOGT activity [8]. For optimal activity, EOGT requires the divalent cation Mn2+ and a neutral pH of 7.0–7.5. Additionally, EOGT utilizes only uridine diphosphate (UDP)-GlcNAc as sugar donor, with a Km of ~25 μm. It has a low affinity for UDP-GlcNAc compared to OGT, the enzyme responsible for the O-GlcNAc modification of intracellular proteins [7, 10].
Figure 1.

The EOGT sequence is highly conserved among different species. Identical amino acids are highlighted in black and similar amino acids are shown in red.
Figure 2.
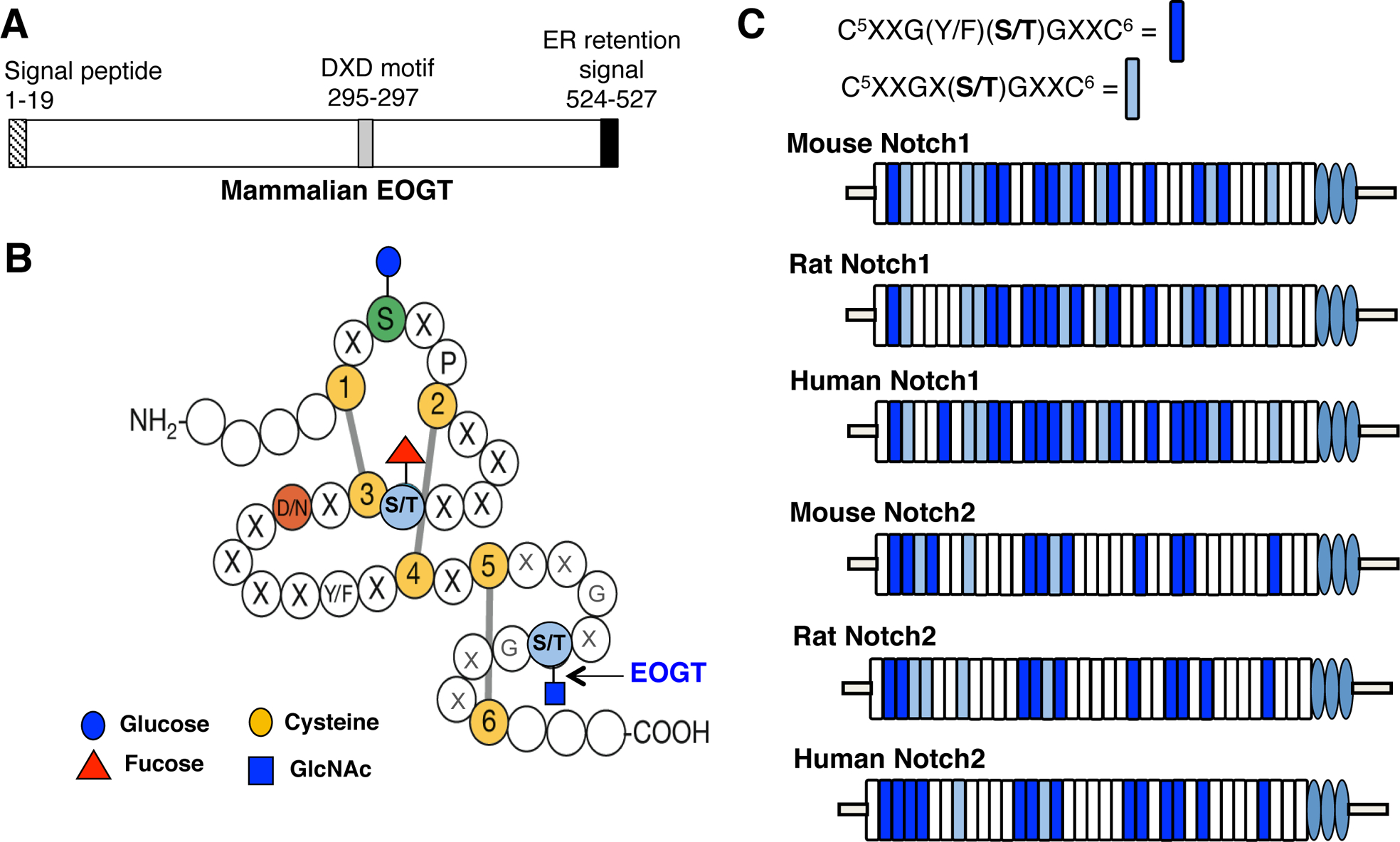
EOGT conserved motifs and consensus sites for O-GlcNAc in Notch receptors. A) Schematic diagram of EOGT structural motifs. B) An EGF domain depicting the consensus site for the addition of O-GlcNAc by EOGT. EGF repeat modified with permission from Cambell and Baron [14] C) Representation of the conserved EGF domains with an O-GlcNAc consensus site in the extracellular domain of NOTCH1 and NOTCH2 in mammals.
EOGT is an ER resident enzyme, as predicted by the HDEL retention signal at the C-terminus [7]. EOGT transfers GlcNAc to a Ser or Thr that occurs between the fifth and sixth cysteines of an EGF repeat in the broad consensus sequence CXXG(Y/F)(T/S)GX2–3C (Fig. 2B [14]) [6, 7, 9]. Although, OGT and EOGT O-GlcNAc transferases act in different cellular compartments and target mostly distinct groups of proteins, they are both regulated by the hexosamine biosynthetic pathway (HBP) [15]. The HBP synthesizes UDP-GlcNAc in the cytosol, where it is a substrate of OGT. Cytoplasmic UDP-GlcNAc is transported into the Golgi complex and endoplasmic reticulum (ER) via the nucleotide sugar transporters SLC35A3 [16] and SLC35B4 [17], respectively. The levels of glucose and glutamine in the cell regulate the amount of UDP-GlcNAc synthesized by the HBP [18], which in turn influences the amount of UDP-GlcNAc transported into the secretory pathway. Thus, the cytosolic concentration of these metabolites are expected to affect the levels of O-GlcNAc on proteins of the secretory pathway that contain appropriate EGF domains.
Substrates of EOGT
The first O-GlcNAc on a secreted protein was found on Thr38 located between the fifth and the sixth cysteine of Drosophila Notch EGF20 [6], In vitro assays in Drosophila S2 cells and Chinese hamster ovary (CHO) cells identified other members of the Notch pathway to be O-GlcNAcylated, including Drosophila Notch ligands Delta and Serrate [8] and mammalian NOTCH1 [10, 11]. In Drosophila, the large membrane-anchored cell surface protein termed Dumpy (Dp) was shown to carry O-GlcNAc and to contain 86 EGF repeats with an O-GlcNAc consensus site amongst the 308 EGF repeats of its extracellular domain [7]. Dp plays an important role in maintaining the integrity of the apical extracellular matrix. In mouse cerebellum, mass spectrometric analysis identified five additional proteins with O-GlcNAc on EGF repeats, including HSPG2, NELL1, LAMA5, PAMR1, AIMP1 and NOTCH2 [9]. Additionally, in platelets an EGF repeat in thrombospondin-1 (TSP-1) was shown to be O-GlcNAcylated [19]. Studies in human embryonic kidney HEK293T cells identified O-GlcNAc that was further extended by a galactose (Gal) residue [10].
Notch receptors represent a common target for O-GlcNAc modification across species. Of the 36 mouse NOTCH1 EGF repeats, 17 have the O-GlcNAc consensus site CXXG(Y/F)(T/S)GX2–3C. Rat and human NOTCH1 have 19 and 21 O-GlcNAc consensus sites respectively. Altogether, 17 of the EGF repeat consensus sites are conserved across the three species. Similarly, mouse, rat and human NOTCH2 have 14, 14 and 15 EGF repeats with an O-GlcNAc consensus site respectively, and 14 of these EGF repeats are conserved between species (Fig. 2C). Interestingly, mass spectrometry of Drosophila Notch purified from larval lysate identified O-GlcNAc on fewer sites than predicted by the consensus sequence [20]. This may reflect the tissue or time of development at which Notch was obtained. Alternatively, O-GlcNAc may have been lost during purification by the action of glycosidases in the lysate, or potentially during mass spectrometry.
Extracellular domain EGF repeats of Notch receptors also carry O-glycans other than O-GlcNAc (Fig. 2B) [21, 22]. O-glucose glycans are initiated by the protein O-glucosyltransferase Rumi (in flies) or POGLUT1 (in mammals). The O-glucose is usually extended by one or two xylose residues. O-fucose glycans are initiated by the protein O-fucosyltransferase Ofut1 (in flies) or POFUT1 (in mammals). The O-fucose is extended by Fringe N-acetylglucosaminyltransferases - Fng (in flies) or Lunatic, Manic and Radical Fringe in mammals (LFNG, MFNG, RFNG).
Roles of EOGT in Drosophila development
The expression of the Eogt gene is developmentally and spatially regulated in Drosophila. Eogt expression is highest at the embryonic preblastoderm-stage and declines with age [7]. Disruption of the Eogt gene in Drosophila is pupal lethal, while knockdown of eogt in the wing causes wing blisters, and knockdown in the thorax causes vortices and commata [7, 8]. Genetic interaction studies revealed that the wing blistering and other phenotypes in eogt knockdown tissues are exacerbated in flies that have also lost one allele of Dumpy [7, 8]. In addition, removal of one copy of wingblister that encodes laminin α chain with an EGF repeat that might receive an O-GlcNAc, enhances blister formation in eogt knockdown wings [8].
Most eogt null flies die during the second/third instar interface, with a few mutant larvae surviving until early third-instar [7, 8]. The surviving eogt mutants exhibit defects in the wings, cuticle, notum, vortex and larval trachea, similar to mutants lacking proteins required to maintain the integrity of epithelial cell-extracellular matrix interactions like Piopio, Zona pellucida and Dumpy. Notably, although Drosophila Notch EGF repeats are O-GlcNAcylated [20], the phenotype of eogt null flies does not include deficiencies typical of flies with reduced Notch signaling. Thus, larvae lacking eogt do not exhibit significant disruptions in Notch mediated processes such as neurogenesis, wing margin formation and wing vein specification [23]. Nevertheless, genetic interaction studies showed that inactivation of one allele of Notch or Notch pathway members including the ligands Serrate and Delta, as well as transcription regulators including suppressor of hairless (Su(H)) and mastermind (mam), suppress the wing blistering phenotype observed in eogt knockdown wings [8]. This suggests that Notch with low O-GlcNAc promotes blister formation in eogt knockdown wings, and that loss of one copy of Notch suppresses wing blistering. This study also identified a link between Eogt and pyrimidine metabolism [8]. Inactivation of one copy of genes encoding enzymes responsible for the production of UTP and UDP-GlcNAc suppress the wing blistering phenotype, consistent with reduced levels of UDP-GlcNAc further reducing O-GlcNAc on Notch in eogt knockdown wings. Interestingly, the genetic loss of one allele of genes encoding enzymes involved in uracil catabolism enhanced the blistering phenotype, suggesting that uracil levels increase as a result of reduced eogt and an increase in cytosolic UDP-GlcNAc, and promote blister formation [8].
Genetic interaction approaches are helpful in implicating gene products that interact to promote or suppress a phenotype, but interpretation of mechanism is necessarily hypothetical. The best insight to date of a function for O-GlcNAc in Drosophila comes from investigations of Dp in eogt null flies [7]. Focusing on larval tracheae, Sakaidani et al. [7] showed that Dp is modified by O-GlcNAc in wild type but not in eogt null tracheae lysates. Dp is localized apically and adjacent to the cuticle in trachea, and this localization is disrupted in eogt null tracheae. In addition, some tracheal tubes were twisted or bent and dorsal trunks broken in the absence of Eogt. Therefore, certain defective epithelial cell-extracellular matrix interactions observed in eogt mutants appear to arise from the lack of O-GlcNAc on Dp.
Roles of EOGT in mammals
In situ hybridization of Eogt transcripts was first performed in mouse embryos where it was shown that Eogt expression is enriched in the caudal presomitic mesoderm at E9.5 [24]. In embryos at E9.5 and E10.5, Eogt is robustly expressed in the apico-ectodermal ridge of developing limbs [25]. At E12.5, Eogt is expressed in the digits of developing limbs [25]. In adult mouse, Eogt is ubiquitously present in most tissues, with highest expression in lungs [10],
Functional roles of EOGT in mammals were first identified in humans [25, 26]. Exome sequencing of patients diagnosed with Adams-Oliver Syndrome (AOS; MIM 100300) revealed homozygous autosomal recessive mutations in EOGT - two missense mutations (W207S and R377Q), and a frame shift mutation (G359 Dfs*28) that generates a premature stop codon [25, 26]. The premature stop codon results in a truncated form of EOGT that lacks the putative catalytic domain, and hence abolishes enzyme activity. Co-expression of EOGT missense variants (EOGTW207S, EOGTR337Q) and the frame shift mutation (EOGTG359*) and a soluble NOTCH1 extracellular domain fragment (NOTCH1-EGF1–36) in HEK293T cells, demonstrated that all three mutations lead to a functionally inactive enzyme [27]. The W207S and G359* mutations result in EOGT protein degradation via the ubiquitin-proteasome pathway, whereas the R377Q mutation does not affect the stability or the localization of the enzyme, but adversely affects binding of UDP-GlcNAc substrate [27].
AOS is a rare congenital disorder with highly variable clinical features. Most common defects in AOS include terminal limb malformations, congenital cutis aplasia characterized by scalp defects, brain anomalies, cardiac malformations and vasculopathy, suggesting that vascular defects might underlie many of the other pathologies. AOS also arises from mutations in a number of additional genes, including heterozygous mutations in Rho GTPase activating protein 31 (ARHGAP31), and biallelic mutations in dedicator of cytokinesis 6 (DOCK6), and heterozygous mutations in NOTCH1, the Notch ligand delta-like 4 (DLL4), and recombination signal binding protein for immunoglobulin kappa J region (RBPJ) [28–32]. Several different autosomal dominant mutations have been identified in the NOTCH1, DLL4 and RBPJ genes (Table1), and they give rise to a spectrum of AOS clinical pathologies.
Table 1:
EOGT and Notch pathway mutations in AOS patients.
| Gene | Type of mutation | Mutation | References |
|---|---|---|---|
| EOGT | Missense | W207S, R377Q | [25] |
| Frameshift | G359 Dfs*28 | [25, 26] | |
| NOTCH1 | Deletion | 85kb deletion spanning the promoter region and exon 1 | [29] |
| Missense | P407R, C429R, R448Q, C449R, C456Y, C1374R, C1496Y, A1740S, D1989N | [29, 30] | |
| Frameshift | Y550*,M1580lfs*30, S2017Tfs*9 | [30] | |
| Nonsense | E1555* | [30] | |
| DLL4 | Missense | A121P, R186C, F195L, P267T, C390R/Y, C455W | [31] |
| Nonsense | Q554, R558 | [31] | |
| RBPJ | Missense | Z63G, K169Z | [32] |
Studies in mice have demonstrated an indispensable role for Notch signaling in somitogenesis, bone morphogenesis, vascular and cardiac development, retinal angiogenesis and arterio-venous differentiation. These phenotypes include a subset of pathologies observed in AOS patients, which might, at least in part, be the result of sub-optimal Notch signaling. Interestingly, autosomal dominant mutations in the genes POFUT1 [33] and POGLUT1 [34] responsible for the addition of O-fucose or O-glucose to Notch receptor EGF repeats (Fig. 2B), have to date not been associated with AOS, but instead cause Dowling Degos Disease (DDD; MIM 179850). Null mutants of Pofut1 [35] or Poglut1 [36] are embryonic lethal in mouse. The loss of O-fucose glycans markedly reduces Notch ligand binding whereas the loss of O-glucose glycans reduces Notch activation due to reduced stability of Notch at the cell surface. Mice null for Eogt have been generated and are viable and fertile with no obvious deficiencies [37]. Mouse models of DDD or AOS have not yet been reported. A mouse model for AOS may need to be generated by knocking a human mutation into the mouse Eogt gene. It is possible that the respective mutant EOGT protein would give rise to clinical symptoms typical of AOS.
Conclusion
This review focuses on the recently identified ER-resident enzyme, EOGT that is responsible for the addition of O-GlcNAc to secreted and membrane proteins. This modification and EOGT are conserved from C. elegans to humans. In Drosophila larvae the major O-GlcNAcylated protein is Dp, and O-GlcNAcylation of Dp is required for interactions between apical epithelial cells and cuticle in trachea. Drosophila Notch is also modified by O-GlcNAc, and genetic interaction studies revealed that reduced Notch signaling suppresses wing blisters induced by eogt knockdown. In humans, mutations in EOGT lead to autosomal recessive AOS. Other mutations that give rise to AOS are autosomal dominant mutations in NOTCH1, DLL4 and RBPJ, again suggesting that loss of EOGT and reduced Notch signaling might be functionally related. Further studies in mice will help to elucidate the role of EOGT in Notch receptor trafficking, processing, ligand binding and signaling. Additionally, since the EOGT gene and O-GlcNAc consensus sites on secreted and membrane proteins are evolutionarily conserved, they are likely to contribute to signaling pathways yet to be identified.
Funding
This work was supported by funding from National Institutes of Health grant RO1 GM106417 to PS.
Abbreviations
- O-GlcNAc
O-linked N-acetylglucosamine
- OGT
O-GlcNAc-transferase
- GlcNAc
N-acetylglucosamine
- EGF
epidermal growth factor-like
- EOGT
EGF domain-specific O-GlcNAc-transferase
- InsP3
inositol 1,4,5-triphosphate receptor type 1
- AIMP1
aminoacyl-tRNA synthase complex-interacting multifunctional protein1
- HSPG2
heparan sulfate proteoglycan 2
- NELL1
neural epidermal growth factor-like 1
- LAMA5
laminin α5
- PAMR1
peptidase domain containing associated with muscle regeneration 1
- ER
endoplasmic reticulum
- HBP
hexosamine biosynthesis pathway
- CHO
Chinese hamster ovary
- TSP-1
thrombospondin-1
- Fng
Fringe N-acetylglucosaminyltransferase
- LFNG
Lunatic fringe
- MFNG
Manic FNG
- RFNG
Radical FNG
- Su(H)
suppressor of hairless
- Mam
mastermind
- POFUT1
protein fucosyltransferase 1
- POGLUT1
protein glucosyltransferase 1
- ARHGAP31
Rho GTPase activating protein 31
- Dll4
Delta-like 4
- RBPJ
recombination signal binding protein for immunoglobulin kappa J region
- AOS
Adams-Oliver syndrome
- DDD
Dowling Degos Disease
Footnotes
Competing Interests
The authors declare that there are no competing interests associated with the manuscript.
References
- 1.Torres CR and Hart GW (1984) Topography and polypeptide distribution of terminal N-acetylglucosamine residues on the surfaces of intact lymphocytes. Evidence for O-linked GlcNAc. J Biol Chem. 259, 3308–3317 [PubMed] [Google Scholar]
- 2.Kreppel LK, Blomberg MA and Hart GW (1997) Dynamic glycosylation of nuclear and cytosolic proteins. Cloning and characterization of a unique O-GlcNAc transferase with multiple tetratricopeptide repeats. J Biol Chem. 272, 9308–9315 [DOI] [PubMed] [Google Scholar]
- 3.Rengifo J, Gibson CJ, Winkler E, Collin T and Ehrlich BE (2007) Regulation of the inositol 1,4,5-trisphosphate receptor type I by O-GlcNAc glycosylation. J Neurosci. 27, 13813–13821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Butkinaree C, Park K and Hart GW (2010) O-linked beta-N-acetylglucosamine (O-GlcNAc): Extensive crosstalk with phosphorylation to regulate signaling and transcription in response to nutrients and stress. Biochim Biophys Acta. 1800, 96–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rexach JE, Clark PM, Mason DE, Neve RL, Peters EC and Hsieh-Wilson LC (2012) Dynamic O-GlcNAc modification regulates CREB-mediated gene expression and memory formation. Nat Chem Biol. 8, 253–261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Matsuura A, Ito M, Sakaidani Y, Kondo T, Murakami K, Furukawa K, Nadano D, Matsuda T and Okajima T (2008) O-linked N-acetylglucosamine is present on the extracellular domain of notch receptors. J Biol Chem. 283, 35486–35495 [DOI] [PubMed] [Google Scholar]
- 7.Sakaidani Y, Nomura T, Matsuura A, Ito M, Suzuki E, Murakami K, Nadano D, Matsuda T, Furukawa K and Okajima T (2011) O-linked-N-acetylglucosamine on extracellular protein domains mediates epithelial cell-matrix interactions. Nat Commun. 2, 583. [DOI] [PubMed] [Google Scholar]
- 8.Muller R, Jenny A and Stanley P (2013) The EGF repeat-specific O-GlcNAc-transferase Eogt interacts with notch signaling and pyrimidine metabolism pathways in Drosophila. PLoS One. 8, e62835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Alfaro JF, Gong CX, Monroe ME, Aldrich JT, Clauss TR, Purvine SO, Wang Z, Camp DG, 2nd, Shabanowitz J, Stanley P, Hart GW, Hunt DF, Yang F and Smith RD (2012) Tandem mass spectrometry identifies many mouse brain O-GlcNAcylated proteins including EGF domain-specific O-GlcNAc transferase targets. Proc Natl Acad Sci U S A. 109, 7280–7285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sakaidani Y, Ichiyanagi N, Saito C, Nomura T, Ito M, Nishio Y, Nadano D, Matsuda T, Furukawa K and Okajima T (2012) O-linked-N-acetylglucosamine modification of mammalian Notch receptors by an atypical O-GlcNAc transferase Eogt1. Biochem Biophys Res Commun. 419, 14–19 [DOI] [PubMed] [Google Scholar]
- 11.Tashima Y and Stanley P (2014) Antibodies that detect O-linked beta-D-N-acetylglucosamine on the extracellular domain of cell surface glycoproteins. J Biol Chem. 289, 11132–11142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cantarel BL, Coutinho PM, Rancurel C, Bernard T, Lombard V and Henrissat B (2009) The Carbohydrate-Active EnZymes database (CAZy): an expert resource for Glycogenomics. Nucleic Acids Res. 37, D233–238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lombard V, Golaconda Ramulu H, Drula E, Coutinho PM and Henrissat B (2014) The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 42, D490–495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Campbell ID and Baron M (1991) The structure and function of protein modules. Philos Trans R Soc Lond B Biol Sci. 332, 165–170 [DOI] [PubMed] [Google Scholar]
- 15.Ogawa M, Furukawa K and Okajima T (2014) Extracellular O-linked beta-N-acetylglucosamine: Its biology and relationship to human disease. World J Biol Chem. 5, 224–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Maszczak-Seneczko D, Sosicka P, Olczak T, Jakimowicz P, Majkowski M and Olczak M (2013) UDP-N-acetylglucosamine transporter (SLC35A3) regulates biosynthesis of highly branched N-glycans and keratan sulfate. J Biol Chem. 288, 21850–21860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Maszczak-Seneczko D, Olczak T and Olczak M (2011) Subcellular localization of UDP-GlcNAc, UDP-Gal and SLC35B4 transporters. Acta Biochim Pol. 58, 413–419 [PubMed] [Google Scholar]
- 18.Swamy M, Pathak S, Grzes KM, Damerow S, Sinclair LV, van Aalten DM and Cantrell DA (2016) Glucose and glutamine fuel protein O-GlcNAcylation to control T cell self-renewal and malignancy. Nat Immunol. 17, 712–720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hoffmann BR, Liu Y and Mosher DF (2012) Modification of EGF-like module 1 of thrombospondin-1, an animal extracellular protein, by O-linked N-acetylglucosamine. PLoS One. 7, e32762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Harvey BM, Rana NA, Moss H, Leonardi J, Jafar-Nejad H and Haltiwanger RS (2016) Mapping Sites of O-Glycosylation and Fringe Elongation on Drosophila Notch. J Biol Chem. 291, 16348–16360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stanley P and Okajima T (2010) Roles of glycosylation in Notch signaling. Curr Top Dev Biol. 92, 131–164 [DOI] [PubMed] [Google Scholar]
- 22.Takeuchi H and Haltiwanger RS (2014) Significance of glycosylation in Notch signaling. Biochem Biophys Res Commun. 453, 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Artavanis-Tsakonas S, Matsuno K and Fortini ME (1995) Notch signaling. Science. 268, 225–232 [DOI] [PubMed] [Google Scholar]
- 24.Sewell W, Sparrow DB, Smith AJ, Gonzalez DM, Rappaport EF, Dunwoodie SL and Kusumi K (2009) Cyclical expression of the Notch/Wnt regulator Nrarp requires modulation by Dll3 in somitogenesis. Dev Biol. 329, 400–409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shaheen R, Aglan M, Keppler-Noreuil K, Faqeih E, Ansari S, Horton K, Ashour A, Zaki MS, Al-Zahrani F, Cueto-Gonzalez AM, Abdel-Salam G, Temtamy S and Alkuraya FS (2013) Mutations in EOGT confirm the genetic heterogeneity of autosomal-recessive Adams-Oliver syndrome. Am J Hum Genet. 92, 598–604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cohen I, Silberstein E, Perez Y, Landau D, Elbedour K, Langer Y, Kadir R, Volodarsky M, Sivan S, Narkis G and Birk OS (2014) Autosomal recessive Adams-Oliver syndrome caused by homozygous mutation in EOGT, encoding an EGF domain-specific O-GlcNAc transferase. Eur J Hum Genet. 22, 374–378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ogawa M, Sawaguchi S, Kawai T, Nadano D, Matsuda T, Yagi H, Kato K, Furukawa K and Okajima T (2015) Impaired O-linked N-acetylglucosaminylation in the endoplasmic reticulum by mutated epidermal growth factor (EGF) domain-specific O-linked N-acetylglucosamine transferase found in Adams-Oliver syndrome. J Biol Chem. 290, 2137–2149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Southgate L, Machado RD, Snape KM, Primeau M, Dafou D, Ruddy DM, Branney PA, Fisher M, Lee GJ, Simpson MA, He Y, Bradshaw TY, Blaumeiser B, Winship WS, Reardon W, Maher ER, FitzPatrick DR, Wuyts W, Zenker M, Lamarche-Vane N and Trembath RC (2011) Gain-of-function mutations of ARHGAP31, a Cdc42/Rac1 GTPase regulator, cause syndromic cutis aplasia and limb anomalies. Am J Hum Genet. 88, 574–585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stittrich AB, Lehman A, Bodian DL, Ashworth J, Zong Z, Li H, Lam P, Khromykh A, Iyer RK, Vockley JG, Baveja R, Silva ES, Dixon J, Leon EL, Solomon BD, Glusman G, Niederhuber JE, Roach JC and Patel MS (2014) Mutations in NOTCH1 cause Adams-Oliver syndrome. Am J Hum Genet. 95, 275–284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Southgate L, Sukalo M, Karountzos AS, Taylor EJ, Collinson CS, Ruddy D, Snape KM, Dallapiccola B, Tolmie JL, Joss S, Brancati F, Digilio MC, Graul-Neumann LM, Salviati L, Coerdt W, Jacquemin E, Wuyts W, Zenker M, Machado RD and Trembath RC (2015) Haploinsufficiency of the NOTCH1 Receptor as a Cause of Adams-Oliver Syndrome With Variable Cardiac Anomalies. Circ Cardiovasc Genet. 8, 572–581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Meester JA, Southgate L, Stittrich AB, Venselaar H, Beekmans SJ, den Hollander N, Bijlsma EK, Helderman-van den Enden A, Verheij JB, Glusman G, Roach JC, Lehman A, Patel MS, de Vries BB, Ruivenkamp C, Itin P, Prescott K, Clarke S, Trembath R, Zenker M, Sukalo M, Van Laer L, Loeys B and Wuyts W (2015) Heterozygous Loss-of-Function Mutations in DLL4 Cause Adams-Oliver Syndrome. Am J Hum Genet. 97, 475–482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hassed SJ, Wiley GB, Wang S, Lee JY, Li S, Xu W, Zhao ZJ, Mulvihill JJ, Robertson J, Warner J and Gaffney PM (2012) RBPJ mutations identified in two families affected by Adams-Oliver syndrome. Am J Hum Genet. 91, 391–395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen M, Li Y, Liu H, Fu X, Yu Y, Yu G, Wang C, Bao F, Liany H, Wang Z, Shi Z, Zhang D, Zhou G, Liu J and Zhang F (2014) Analysis of POFUT1 gene mutation in a Chinese family with Dowling-Degos disease. PLoS One. 9, e104496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Basmanav FB, Oprisoreanu AM, Pasternack SM, Thiele H, Fritz G, Wenzel J, Grosser L, Wehner M, Wolf S, Fagerberg C, Bygum A, Altmuller J, Rutten A, Parmentier L, El Shabrawi-Caelen L, Hafner C, Nurnberg P, Kruse R, Schoch S, Hanneken S and Betz RC (2014) Mutations in POGLUT1, encoding protein O-glucosyltransferase 1, cause autosomal-dominant Dowling-Degos disease. Am J Hum Genet. 94, 135–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shi S and Stanley P (2003) Protein O-fucosyltransferase 1 is an essential component of Notch signaling pathways. Proc Natl Acad Sci U S A. 100, 5234–5239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fernandez-Valdivia R, Takeuchi H, Samarghandi A, Lopez M, Leonardi J, Haltiwanger RS and Jafar-Nejad H (2011) Regulation of mammalian Notch signaling and embryonic development by the protein O-glucosyltransferase Rumi. Development. 138, 1925–1934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ogawa M, Varsney S, Sawaguchi S, Sakaidani YY,H, Takeshita K, Murohara T, Kato K, Stanley P and Okajima T (2016) Extracellular O-GlcNAc is required for retinal vascular development and Dll4-Notch signaling. Glycobiology, in press. [DOI] [PMC free article] [PubMed] [Google Scholar]