

ABSTRACT
Antibody-mediated cancer immunotherapy targets inhibitory surface molecules, such as PD1, PD-L1, and CTLA-4, aiming to re-invigorate dysfunctional T cells. We purified and characterized tumor-infiltrating lymphocytes (TILs) and their patient-matched non-tumor counterparts from treatment-naïve NSCLC patient biopsies to evaluate the effect of PD1 expression on the functional and molecular profiles of tumor-resident T cells. We show that PD1+ CD8+ TILs have elevated expression of the transcriptional regulator ID3 and that the cytotoxic potential of CD8 T cells can be improved by knocking down ID3, defining it as a potential regulator of T cell effector function. PD1+ CD4+ memory TILs display transcriptional patterns consistent with both helper and regulator function, but can robustly facilitate B cell activation and expansion. Furthermore, we show that expanding ex vivo-prepared TILs in vitro broadly preserves their functionality with respect to tumor cell killing, B cell help, and TCR repertoire. Although purified PD1+ CD8+ TILs generally maintain an exhausted phenotype upon expansion in vitro, transcriptional analysis reveals a downregulation of markers of T-cell dysfunction, including the co-inhibitory molecules PD1 and CTLA-4 and transcription factors ID3, TOX and TOX2, while genes involved in cell cycle and DNA repair are upregulated. We find reduced expression of WNT signaling components to be a hallmark of PD1+ CD8+ exhausted T cells in vivo and in vitro and demonstrate that restoring WNT signaling, by pharmacological blockade of GSK3β, can improve effector function. These data unveil novel targets for tumor immunotherapy and have promising implications for the development of a personalized TIL-based cell therapy for lung cancer.
KEYWORDS: PD1, memory, TILs, ID3, GSK3β
Background
The immune checkpoint receptor programmed cell death 1 (PD1) marks clonally expanding, antigen-specific T cells1. In cancer, T cells are exposed to chronic antigen stimulation, causing CD8+ tumor-infiltrating lymphocytes (TILs) to enter a state of dysfunction. This “exhausted” state is characterized by loss of cytotoxic effector functions and sustained surface expression of PD1 and other co-inhibitory receptors, such as CTLA-4, LAG3, and TIM3, as well as overexpression of transcription factors TOX and TOX2.2 Immune checkpoint blockade (ICB) therapies employ antibodies that prevent engagement of PD1 with its cognate ligand programmed cell death ligand 1 (PDL1), thereby triggering the expansion and reactivation of exhausted tumor-reactive PD1hi CD8+ TILs.3 Despite their lack of cytotoxicity, the presence of PD1hi CD8+ TILs predicts response to ICB and correlates with reduced tumor burden and increased overall survival in resectable non-small cell lung cancer (NSCLC).4,5 However, the majority of patients with lung cancer treated with antibodies targeting PD1/PDL1 axis do not derive lasting clinical benefit from ICB.6 Whether the lack of response to ICB is due to an inability to reinvigorate tumor-resident CD8+ T cells,7 a failure to recruit such naïve cells from the periphery, or both remains unclear.8 A deeper understanding of the functional and transcriptional consequences of PD1 expression in tumor-derived CD8+ T cells is required to address this unresolved question.
Although rarely mentioned in the context of immunotherapy, CD4+ memory T cells play an essential yet complex part in the host’s defense against cancer,9 as they have been assigned both immune-activating and immune-suppressive roles. Compared to their CD8+ counterparts, we know much less about the gene expression programs, molecular features and function of CD4+ T cells in lung cancer patients. The multifaceted role of CD4+ T cells has important clinical implications when it comes to developing an adoptive TIL-based therapy using naturally occurring autologous TILs,10 as it is unclear whether the functional identity of CD4+ TILs, such as surface receptors, transcriptional program, and TCR repertoire are maintained following in vitro expansion.11
Here, we examine the effect of PD1 expression on the functional and transcriptional profiles of CD45RO+ memory CD8+ and CD4+ T cells obtained ex vivo from the treatment of naive NSCLC resected tissue and compare them with T cells derived from matched uninvolved normal lung tissue. While PD1+CD8+ memory TILs show hallmark features of exhaustion, PD1+CD4+ memory TILs are both highly proliferative and cytotoxic. Moreover, we demonstrate that PD1+CD4+ memory TILs are located in close proximity to CD19+ B cells and provide B cell help despite sharing expression of several co-inhibitory receptors and a core set of transcription factors that are central regulators of exhaustion with PD1+CD8+ memory TILs. Transcriptional analysis revealed that the transcriptional regulator ID3 is specifically upregulated in PD1+CD8+ TILs and knockdown of ID3 improves tumor cell killing in wild-type CD8+ T cells. We demonstrate that PD1+CD8+ and PD1+CD4+ memory TILs generated from in vitro expanded cultures exhibit generally similar functional patterns compared with their matched ex vivo counterparts but display some key transcriptional differences, such as downregulation of components of the WNT signaling pathway. Compensating the lack of WNT signaling through pharmacological inhibition of GSK3β improves the effector function of in vitro expanded populations.
Methods
Collection of lung tumor samples
Fresh tumor samples were collected from patients with NSCLC undergoing primary surgical resection with a curative intent at Bern University Hospital, Division of General Thoracic Surgery. Immediately following surgical resection of tumor, specimens were sent to the Institute of Pathology, University of Bern, where a pathologist (SB) dissected tumor and matched uninvolved lung tissue for further analysis. Uninvolved lung tissue (tumor-free) was collected from an area at least 5 cm from the tumor margin. For all specimens, a piece of tumor sample was fixed in 10% formalin for histopathological analysis. The remaining tissue (tumor and matched uninvolved) was collected and used to generate single-cell suspensions to perform flow cytometric, functional and transcriptomic profiling, as described below. All patients provided informed written consent for use of their material for research purposes, which was approved by Ethics Commission of the Canton of Bern (KEK-BE:2018–01801). See supplement for detailed Materials and methods and additional references.
Tissue processing, flow cytometric analysis and cell sorting
Single-cell suspensions were generated from surgically resected tumor and matched uninvolved lung tissue, as previously described.12 Single cells were stained in buffer containing Fc block (eBioscience, San Diego, CA, USA) and incubated with the following fluorescently conjugated human monoclonal antibodies: CD45, CD3, CD4, CD8, CD45RO, CD56, CD16, PD1, PDL1 (see Table S2). For fluorescence-activated cell sorting (FACS), cells were separated into PD1high or PD1low subsets following gating on CD45RO+ T cells for both CD8 and CD4 T cell compartment using either a MoFLo Astrios EQ 6way sorter (Beckman Coulter, Indianapolis, IN, USA)) or BD FACS Aria III (BD Biosciences, San Jose, CA, USA). Cells from tumor and matched uninvolved lung tissue were sorted directly into collection tubes containing 500 µl lysis buffer (4 M guanidinium thiocyanate, 30 mM Tris pH 8.0, 1% Triton-X-100) and processed for RNA isolation, as previously described.13 Total RNA was treated with DNase I (Qiagen). RNA quantity was assessed on a ND-1000 spectrophotometer (NanoDrop Technologies, Thermo Scientific). Integrity of RNA (RIN > 8) was assessed with an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA). For in vitro expansion of intratumoral CD4 and CD8 T cells, stained single cells from tumor and matched uninvolved lung were separated into PD1high or PD1low subsets following gating on CD45RO+ T cells for both CD8 and CD4 T cell compartment. Sorted T cell subsets were plated in ultra-low adherent Nunclon sphere 24-well plates (Thermo Scientific) in 500 µl of ImmunoCult™-XF T-cell media supplemented with rhIL-2 (10 ng/ml), rhIL7 (10 ng/ml) and rhIL15 (15 ng/ml) with 3% human AB serum. CD3/CD28/CD2 activator beads (StemCell Technologies) were included. Fresh media changes including CD3/CD28/CD2 activator beads were made every 5 days. From day 20, expanded T cell subsets were used for downstream experiments or cryopreserved for later use.
Results
PD1 expression is associated with exhaustion in CD8 but not CD4 memory TILs
To functionally characterize T cell populations from NSCLC, we dissociated surgically resected tumor and FACS-sorted tumor-infiltrating lymphocytes (TILs) and their matching counterparts from adjacent histologically uninvolved lung tissue (NILs) from 18 NSCLC specimens (8 adenocarcinoma, 10 squamous cell carcinoma). Table 1 presents the clinical and pathological features for this particular dataset. Following isolation of CD45RO+ memory CD4+ and CD45RO+ memory CD8+ memory T cells, we further stratified both cell types by expression level of PD1, resulting in four subpopulations (PD1+CD4+, PD1−CD4+, PD1+CD8+ and PD1−CD8+) for each patients tumor and matched normal sample (Figure 1(a,b) and Figure S1A). We found that memory PD1+ T cells were more abundant in tumors than in uninvolved tissues (50% versus 31% in the CD4 and 45% versus 27% in the CD8 fraction), suggesting an activated and potentially exhausted phenotype of TILs (Figure 1(c)). In matched normal controls, PD1+CD4+ cells were less abundant than their PD1−CD4+ counterparts (31% versus 42%), while PD1+CD8+ and PD1−CD8+ cells were found with about equal frequency (27% versus 29%) (Figure 1(c)).
Table 1.
Patient sets and baseline characteristics of all patients included in this study
| Variable | Total (n = 35) |
Flow cytometric T cell analysis set (n = 18) |
RNAseq set (n = 8) |
|---|---|---|---|
| Gender Male Female |
26 9 |
15 3 |
6 2 |
| Histology Adenocarcinoma Squamous |
17 18 |
8 10 |
5 3 |
| Smoking history Current Former Never Unknown |
18 29 2 5 |
12 15 1 2 |
6 8 0 |
| Pack years ≥50 <50 Unknown |
15 15 5 |
9 7 2 |
5 3 |
| Tumor Grade G1 G2 G3 Unknown |
1 18 11 5 |
1 8 6 3 |
4 3 1 |
| Tumor stage IIA IIIIA IB IIB IIIB IV Unknown |
3 14 6 8 1 2 1 |
3 8 1 4 0 1 1 |
3 1 3 1 |
| Tumor size (cm) <4 cm ≥4 cm |
12 23 |
6 12 |
3 5 |
| COPD | 4 | 3 | 1 |
| Prior Tx Chemotherapy |
11 | 7 | 1 |
| Survival Alive Dead |
24 11 |
10 8 |
4 4 |
Figure 1.
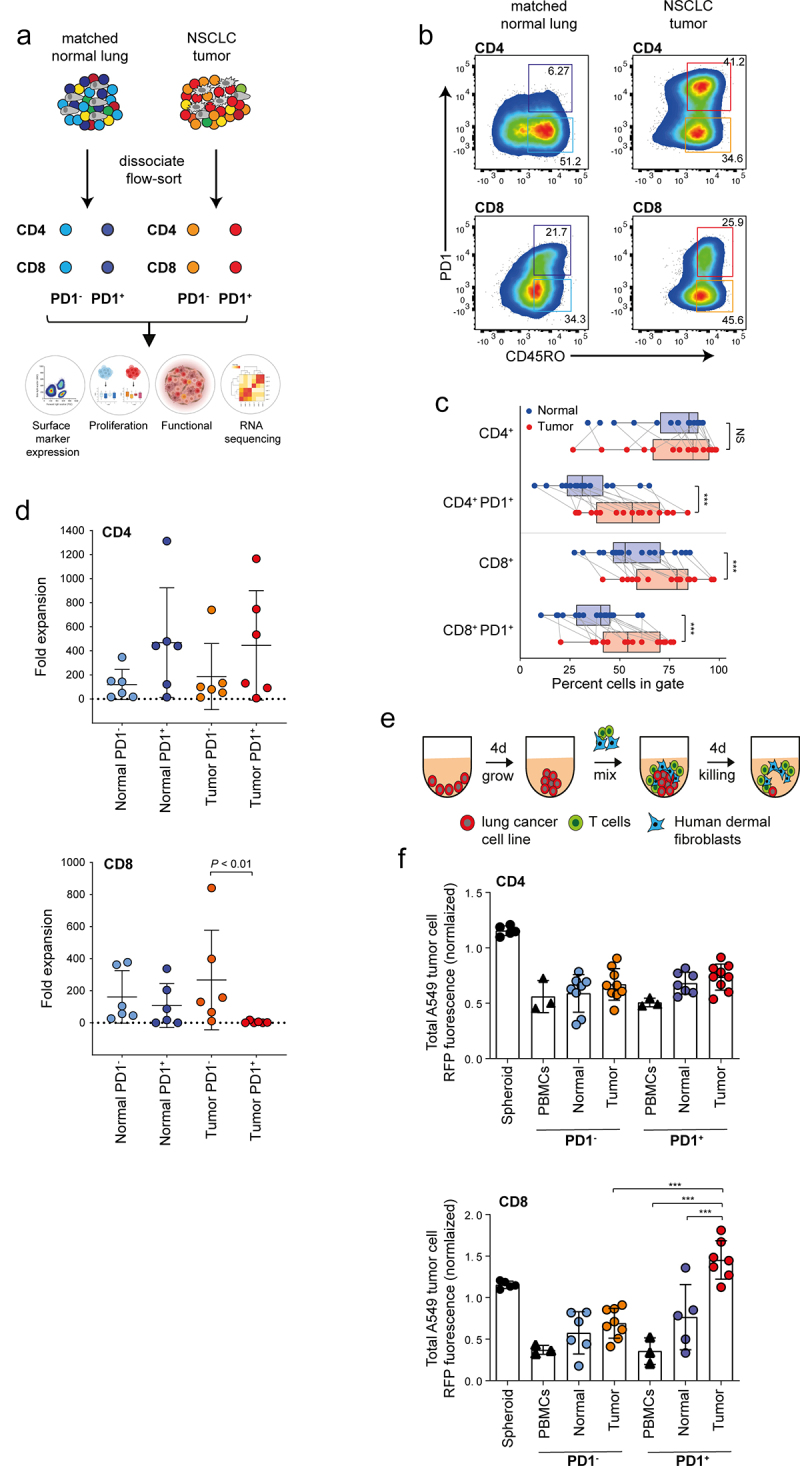
Functional characterization of CD4 and CD8 memory T cells based on PD1 expression in resectable NSCLC. (a) Graphical overview of the four populations of T cells (TC) isolated for characterization from tumor and matched uninvolved normal lung tissue and analysis strategy. (b) Representative panels showing gating strategy to isolate CD45RO+ (memory) CD4 and CD8 T cells based on PD1 status from tumor and matched uninvolved lung tissue. (c) Box and whisker plots showing breakdown of immune cell populations in tumor compared with matched uninvolved lung tissue. Line represents the median (n = 18, biological replicates). (d) Panels showing cumulative growth curves of eight TC subsets from patient donors in CD4 (upper) and CD8 compartment (lower) (n = 6, biological replicates). (e) Schematic illustration showing method to assess T cell reactivity. (f) Histograms showing cell death of tumor spheroids generated from red fluorescent protein (RFP)-nuclear labeled A549 human lung adenocarcinoma cells co-cultured without or with the addition of various T cell subsets at a 1:1 effector-to-target ratio. Tumor cell killing was monitored in real time and analyzed as a loss of RFP intensity (n = 6 biological replicates). Data are normalized to spheroids with no T cells. Data are displayed as mean ± SD. Data in C by paired Students t-test, two-tailed. Data in d, f, by one-way ANOVA followed by post-hoc Newman-Keuls. **P < .01, ***P < .001; ns, not significant.
Next, we determined the effector potential of the T cell subpopulations by measuring their ability to expand in vitro and cytotoxic activity against lung cancer cell lines in a three-dimensional (3D) tumor spheroid assay14 (Figure 1(e)). All four CD4+ memory T cell subpopulations expanded well in vitro (Figure 1(d)) and effectively eliminated A549 lung adenocarcinoma cells, irrespective of their PD1 status and whether they originated from tumor or uninvolved tissue, indicating that their effector potential was intact (Figure 1(f) and Figure S1B,D). Conversely, amongst the CD8 subpopulations, only tumor-derived PD1+CD8+ memory TILs showed diminished growth capacity in vitro (Figure 1(d)) and failed to kill A549 lung adenocarcinoma cells (Figure 1(f) and Figure S1C). Similar patterns of tumor cell killing capacity were also confirmed against H157 lung squamous cell carcinoma cells (Figure S1E-F). In contrast, tumor killing capacity of CD8+ T cells from the peripheral blood of healthy donors was intact irrespective of PD1 status (Figure 1(f) and Figure S1D). We conclude that high PD1 expression correlates with an exhausted phenotype in CD8+TILs, but not CD4+ TILs or PD1+ CD8+ or PD1+ CD4+ T cells derived from adjacent normal tissue.
Transcriptional characterization of NSCLC memory T cell populations
Gene expression patterns of T cell populations derived from tumor tissue and adjacent matched uninvolved lung from 8 NSCLC specimens was then assessed by RNA Seq. Table 2 presents the clinical and pathological characteristics of patients included for the sequencing of CD4+ and CD8+ memory T cells based on PD1 expression. Two-dimensional projection of the transcriptomics data using T-distributed Stochastic Neighbor Embedding (t-SNE) demonstrated clear separation of CD4 and CD8 subpopulations into two distinct clusters (Figure 2(a)). Within those clusters, the projection distinguished samples from tumor and matched uninvolved tissue, indicating a strong effect of the microenvironment on both CD4+ and CD8+ memory T cells. The expression level of PD1 has a small, but visible influence on global gene expression, particularly in sorted TILs.
Table 2.
Clinical characteristics of patients included for the sequencing of TILs and NTILs
| ID | Tumor type | Sex | Age | Former smoker |
Pack years | Tumor grade |
Tumor Stage |
Tumor status |
Tumor size (cm) | COPD | Prior tx |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | LUAD | M | 67 | y | 110 | G3 | IIA | unk | 6.4 | n | none |
| 2 | LUSC | F | 72 | y | 100 | G2 | IIA | unk | 1.6 | n | none |
| 3 | LUSC | M | 62 | y | 60 | G2 | IIB | TP53 | 6.8 | n | chemo* |
| 4 | LUAD | F | 64 | y | 30 | G3 | IIA | unk | 2.6 | n | none |
| 5 | LUSC | M | 67 | y | 35 | G3 | IIIA | unk | 7.5 | n | none |
| 6 | LUSC | M | 85 | y | 30 | G2 | N/A | unk | 3.4 | Gold II | none |
| 7 | LUAD | M | 69 | y | 80 | N/A | IIB | unk | 5.2 | n | none |
| 8 | LUSC | M | 58 | y | 60 | G2 | IIB | unk | 9.8 | n | none |
LUAD, lung adenocarcinoma; LUSC, lung squamous cell carcinoma; N/A, not available; unk, unkown; n, no; y, yes.
*Platinol/Navelbine.
Figure 2.
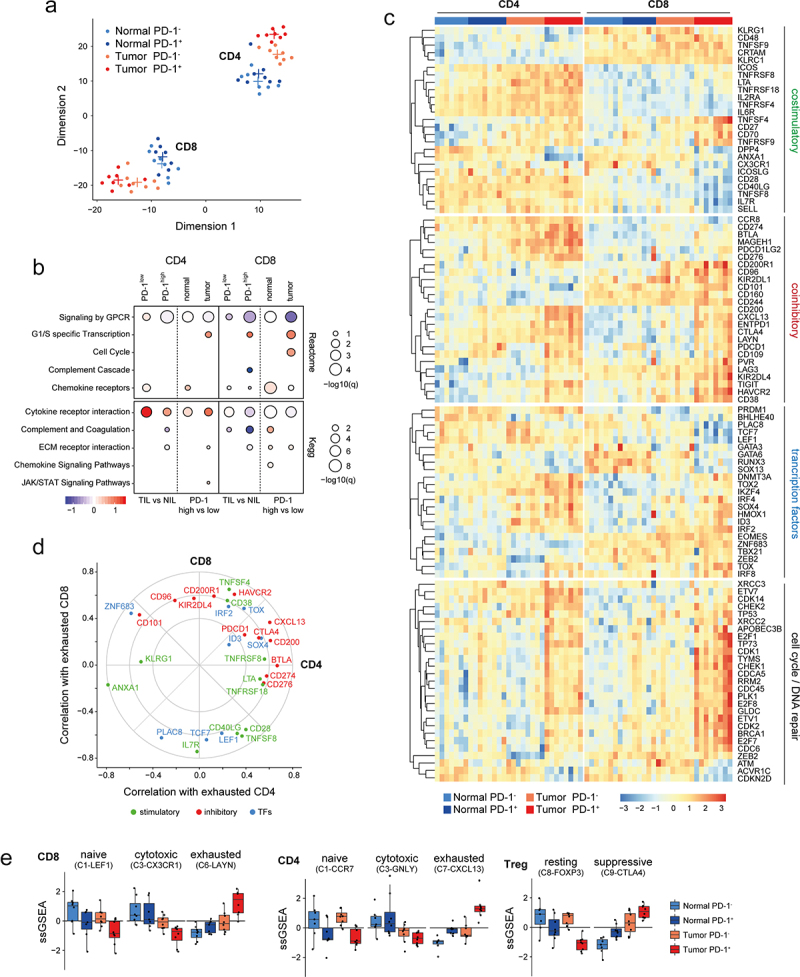
Transcriptional profile of sorted CD8 and CD4 memory T cells based on PD1 expression from matched patient samples in NSCLC. (a) tSNE plot showing the separation of 8 cell subsets based on biological genotypes, cell type and PD1 expression. Each dot represents a patient (n = 8, biological replicates). (b) Functional annotation of the differentially expressed genes via GSEA dot plot showing KEGG and reactome pathway enrichment comparing biological genotypes (tumor versus normal), cell type (CD4 and CD8) and PD1 status (high vs low). (c) Functional heatmap of differentially expressed candidate genes in memory T cells for the following modules (coinhibitory, stimulatory, transcription factors and cell cycle/DNA repair) separated based on biological genotypes, cell type and PD1 expression. Each bar represents one patient. (d) Radar plot showing deregulated (up and down) gene programs in CD8 and CD4 TILs based on PD1 status that correlate with exhaustion based on single cell TIL data in NSCLC from Guo et al.15 (e) Cluster barplot showing the correlation of prospectively sorted cell fractions to single cell TIL clusters in NSCLC based on Guo et al.15 Each dot represents one patient.
The difference between CD4+ and CD8+ memory T cells is also reflected in the number of differentially expressed genes. Although we determined 664 genes significantly up- or downregulated (FDR ≤ 0.05) in both CD4 and CD8 cells sorted from tumor tissue compared to matched normal tissue, we found a roughly equal number of CD4- and CD8-specific genes (727 and 821, respectively) (Figure S2B). Interestingly, high PD1 expression had a much more pronounced effect on the number of significantly changed genes in TILs compared to NILs from matched uninvolved tissue, in particular in CD8+ T cells (820 tumor-specific genes, 74 shared, 33 normal-specific) (Figure S2C). We also noted a significant overlap of differentially expressed genes in PD1hi TILs between CD8+ and CD4+ T cells, compared to PD1lo (259 and 33 genes shared, respectively). PD1 expression is part of the T cell activation transcriptional program, thus it is conceivable that this observation relates to T cells encountering more antigen in the tumor but less so in matched uninvolved lung tissue.16
Gene set enrichment confirmed that downregulation of GPCR signaling is associated with tumor residence and high PD1 expression in CD4+ and CD8+ memory T cells, suggesting reduced T cell activation in chronically activated TILs (Figure 2(b)). CD4 and CD8 cells differed with respect to genes involved in cytokine receptor interaction, with CD4 TILs generally showing upregulation of such genes, while CD8+ TILs, particularly PD1+ cells, showing downregulation (Figure 2(b) and Figure S2A). A possible explanation is an increased number of helper or regulatory T cells within the CD4+ TIL population.
To provide a concise overview of the gene expression patterns found in the different T cell populations, we curated lists of T cell co-stimulatory and co-inhibitory genes, transcription factors playing a part in T cell function, as well as genes involved in cell cycle, mitosis and DNA repair, and plotted their expression as a heatmap (Figure 2(c)). PD1+CD4+ and PD1+CD8+ memory TILs displayed high expression of known exhaustion-associated genes, CTLA4, ENTPD1 (CD39), CD200, LAG3, TIGIT, HAVCR2 (TIM3),17 as well as CD38, and LAYN.15 In addition, PD1+CD4+ memory TILs showed elevated expression of PDCD1LG1 (CD274/PDL1) and PDCD1LG2 (CD273/PDL2), which negatively regulate T cell effector function by engaging with PD11. Moreover, we observed upregulation of MAGEH1 and CCR8, two genes associated with CD4 regulatory T cell function in breast18 and lung cancer patients.19 Unlike PD1+CD4+ TILs, PD1+CD8+ memory TILs are characterized by downregulation of co-stimulatory molecules, including CD28, CD40LG, TNFSF8 (CD30LG) and memory-associated genes IL7R (CD127), SELL (CD62L) and LEF1 (Figure 2(c)). These changes coincided with downregulation of the memory precursor gene TCF7 (TCF-1). Furthermore, key transcription factors associated with T cell exhaustion, including TOX,20–22 TOX2,23 and ID3,24 were upregulated in PD1+CD8+ memory TILs, whereas RUNX3, which controls expression of cytotoxicity-related genes, was downregulated. Expression of cell cycle and DNA repair genes was generally elevated in both PD1+ TILs, particularly in CD8 TILs (Figure 2(c) and Figure S2A). This finding is interesting considering the reduced ability of PD1+CD8 TILs to expand in vitro (Figure 1(d)). Furthermore, WNT signaling was downregulated specifically in tumor-derived PD1+ CD8 cells (Figure S2D). Overexpression of several canonical exhaustion-associated genes (TOX, TOX2, IRF4, CD200, CD38 and CTLA4) were shared between PD1+CD8+ and PD1+CD4+ memory TILs (Figure 2(d)). Additionally, we confirmed co-expression of multiple inhibitory receptors associated with chronic T cell activation and exhaustion at the protein level by flow cytometric analysis of PD1+CD8+ and PD1+CD4+ memory TILs compared with PD1−CD8+ and PD1−CD4+ memory TILs (Figure S3A-B). We confirmed that a subset of PD1+CD8+ memory TILs that co-expressed CD38 and CD101 (CD38+CD101+PD1+CD8+ memory TILs) (Figure S3C), which were previously shown to be associated with a dysfunctional chromatin state in NSCLC,7 exhibited diminished proliferation compared with CD38−CD101+PD1+CD8+ memory TILs (Figure S3C).
We obtained gene signatures corresponding to functional T cell clusters from a recent single-cell sequencing analysis in NSCLC15 and used them to determine the relative abundance of those clusters in our FACS sorted T cell populations with single sample gene set enrichment (ssGSEA). We found that in both CD4+ and CD8+ memory TILs, tumor residence and PD1 expression correlated with an exhausted state (C7-CXCL13 and C6-LAYN clusters, respectively) (Figure 2(e) and Figure S2E). Conversely, effector cell clusters (C3-CX3CR1 and C3-GNLY) showed a higher correlation in matched uninvolved lung tissue, compared with tumor, indicating an immunosuppressive tumor microenvironment. Naïve clusters tracked with both PD1− TILs in both CD4 and CD8 compartments. Notably, naïve regulatory T cells were most enriched in matched normal CD4 populations, while the suppressive regulatory T cell signature (C9-CTLA4) strongly correlated with tumor PD1+ status in CD4+ memory TILs (Figure 2(e) and Figure S2E).
Spatial immune profiling shows PD1+ TILs localized to CD19+ B cells and tumor stroma
The presence and degree of infiltration of CD8+ T cells within tumors has been proposed to be associated with prognosis,25 as well as response to treatment.26 However, less is known about CD4 TILs in NSCLC. Solid tumors are complex ecosystems whereby TILs interact with tumor cells and cells within the tumor stroma including tertiary lymphoid structures (TLS) composed of CD19+ B cells.27 Based on the molecular and functional data generated for PD1+ TILs (Figures 1 and 2), we next performed multiplexed quantitative immunofluorescence and IHC-based spatial analysis to detect and quantify the density (number/mm2) of TILs and their spatial arrangement (location) in tumor core and stromal areas, as well as TLS using whole slide images following staining with CD19, PD1, CD4 and CD8 (Figure 3(a) and Figure S4-S6). We observed a greater density of PD1+CD4+ TILs associated CD19+ B cell clusters compared with PD1+CD8+ TILs, which may indicate formation of TLS in both LUAD and LUSC (Figure 3(b,e) and Figure S4-S6). The density of both CD4 and CD8 TILs were found to be increased in the stromal compartment compared with the tumor core in both LUAD and LUSC (Figure 3(c,f)). This was also the case for the localization of CD19+ B cells (Figure 3(c,f)). When we combine both CD4 and CD8 TILs into one metric, we also find a greater density of TILs within the stroma compared with tumor core (Figure 3(d,f)). Interestingly, both absolute number of PD1+ TILs (both CD4 and CD8) and percentage of PD1+ TILs also showed greater density in stromal areas compared with tumor core (Figure 3(d,g)).
Figure 3.
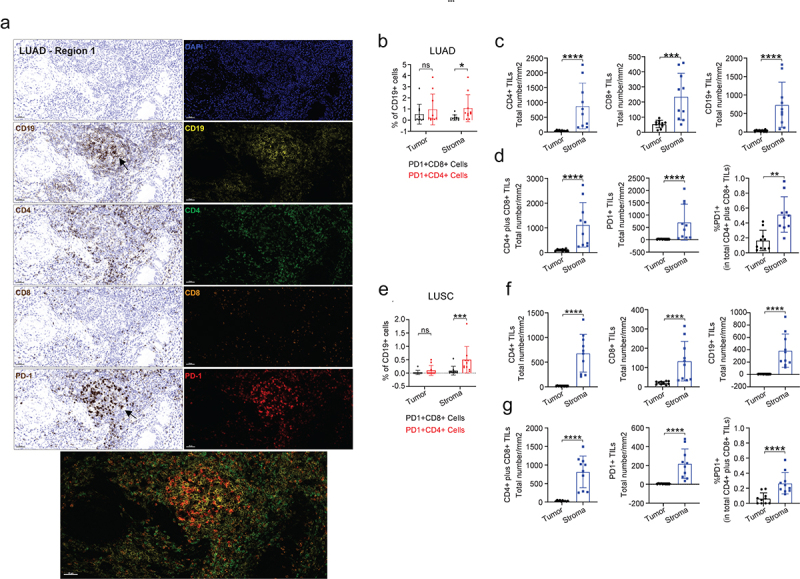
Multiplexed IHC shows the spatial profiles of immune cell infiltrates in LUAD. (a) Representative regions (200x) showing the spatial expression of the indicated immune markers (CD19, CD4, CD8 and PD-1) in LUAD (pT2bN0M0). (b–d) Individual or selected combinations of the immune markers in A were quantified (10 randomly selected regions, refer to Figure S3A) and shown. **p < .01; ***p < .001 ****p < .0001 by two-sided student’s t-test. ns, not significant. (e–g) Individual or selected combinations of the immune markers in LUSC (pT2bN0M0) sample were quantified (10 randomly selected regions, refer to Figure S3B) and shown. ***p < .001 ****p < .0001 by two-sided student’s t-test. ns, not significant.
Knock-down of ID3 in CD8 T cells increases their tumor killing capacity
Re-invigoration of exhausted CD8 T cells is a key goal of cancer immunotherapy, so we searched our gene expression data for candidates that could potentially regulate T cell exhaustion. Recent work has shown that the transcriptional regulator ID3 is enriched in dysfunctional TILs in human melanoma and NSCLC.24 We found increased expression of ID3 in CD4+ and CD8+ TIL populations, compared to their matched controls (Figure 2(c)). Furthermore, ID3 expressing cells were most prominently represented in the exhausted CD8 cluster (C6-LAYN) at the single-cell level (Figure 4(a,b)). Notably, the expression of ID3 in some cells in the naïve cluster (C1-LEF1) (Figure 4(b)), may reflect the described role of ID3 in T cell differentiation.28 Conversely, the effector cluster (C3-CX3CR1) did not show any expression of ID3 (Figure 4(b)). To test if ID3 had a functional role in T cell exhaustion, we knocked-down ID3 in peripheral blood-derived CD8 T cells using siRNA and evaluated their capacity to kill A549 lung adenocarcinoma cells (Figure 1(e)). We found that reduction of ID3 mRNA levels led to significantly increased killing compared to scrambled siRNAs and no siRNA controls (Figure 4(c,d)). Similar results were obtained in Jurkat cells (a leukemic T cell line) following the knockdown of ID3 (Figure S7A and B). We conclude that ID3 suppresses the effector potential of CD8 cells. Therefore, strategies aimed at decreasing ID3 expression in CD8 TILs could help to re-invigorate them from their exhausted state.
Figure 4.
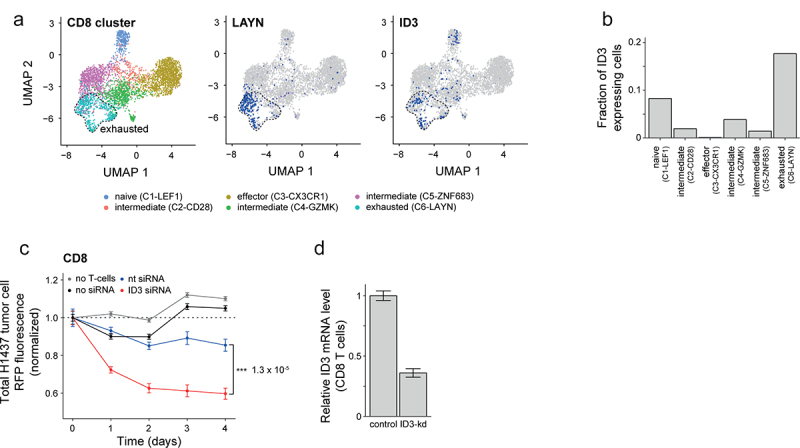
Downregulation of ID3 enhances effector function of CD8 T cells. (a) Uniform Manifold Approximation and Projection (UMAP) panels showing projection of clusters of CD8 TILs marked by color-code based on single cell data from NSCLC tumor lesions by Guo et al.15 Single cells are shown as dots. (b) Bar graphs showing fraction of ID3 expressing cells enriched within the different single cell CD8 TIL clusters as shown in a. (c) Line graphs showing the kinetics of tumor cell death quantified as a loss of red fluorescence protein (RFP) intensity following coculture with activated CD8+ T cells from blood of healthy donors. Total RFP were normalized to H1437 lung tumor cells cultured with no TILs at time = 0. (d) Bar graphs showing knockdown of ID3 expression in CD8+ T cells derived from PBMCs of healthy donors. Data are displayed as mean ± SD. Data in c, Data in d.
PD1+CD4+ memory TILs facilitate B cell activation and expansion in vitro
Our transcriptional analysis suggested that PD1+CD4 memory TILs displayed features of both helper and regulatory T cells (Figure 2(c)). Zappasodi et al.29 recently identified a population of CD4 PD1+ cells (4PD-1Hi) that were transcriptionally similar to T follicular helper (TFH)-like cells, but were functionally immunosuppressive and correlated with poor patient survival in NSCLC. Visual inspection of the expression of the signature genes from this study showed enrichment in CD4+ memory TILs (Figure 5(a)) and ssGSEA analysis demonstrated a relative enrichment of the T follicular regulatory (TFR) signature in the PD1+CD4+ memory TIL population (Figure 5(b)). Many of the genes most enriched in PD1+CD4+ memory TILs, such as CXCL13, CD200, TIGIT and SH2D1A, are also part of a TFH cell gene signature31 (Figure 5(a)). Moreover, PD1+CD4+ memory TILs were enriched in other CD4 helper-related genes, TNFRSF18 (GITR), TNFRSF4 (OX40) and TOX2.32 Using ssGSEA, we found equally strong enrichment of the TFH and the TFR signatures in PD1+CD4 TILs (Figure 5(b)).
Figure 5.
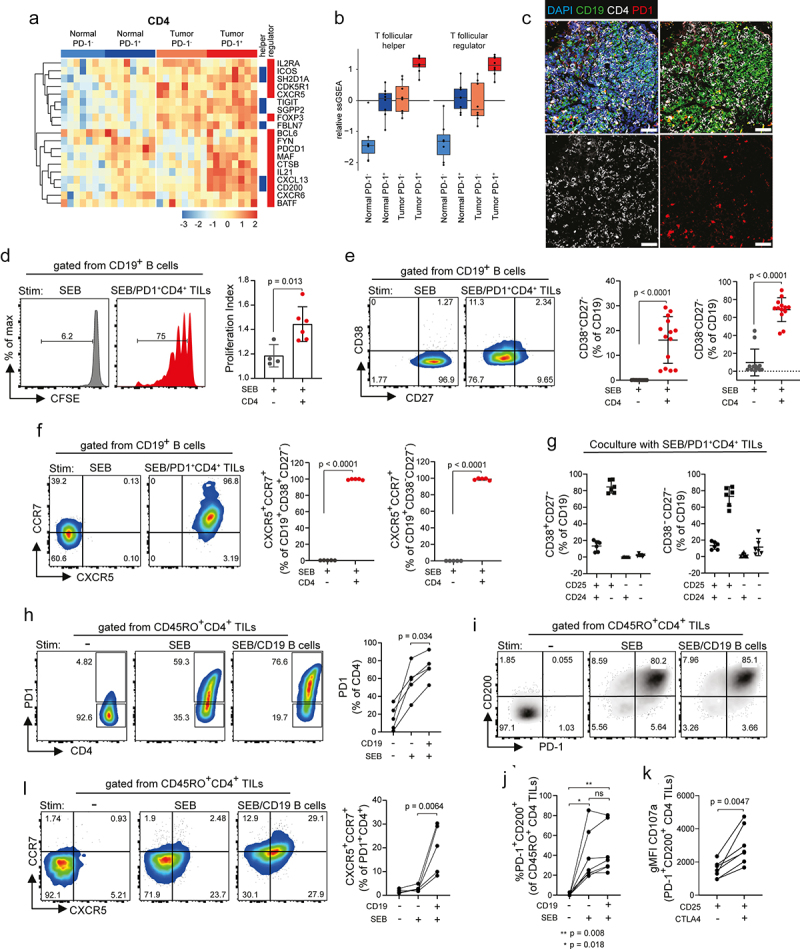
PD1+CD4+ memory TILs show features of TFH-like cells and are poised to help B cells. Heatmap with unsupervised clustering in (a), and ssGSEA scores in (b), showing enrichment of a 4PD-1hi TIL immunosuppressive signature based on a published dataset by Zappasodi et al.30 and TFH gene signature by Gu-Trantien et al.31 in CD4 TILs separated based on genotype (tumor versus normal) and PD1 status (PD1+ and PD1−). Each column in a represents a patient. (c) Representative panels illustrating localization of CD4/PD-1 in relation to CD19+ B cells (green) in tumor via confocal imaging. (d) Histograms and proliferation index of carboxyfluorescein succinimidyl ester (CFSE)-labeled CD19+ B cells stimulated (Stim) with staphylococcal enterotoxin B (SEB, 500 ng/ml) alone or SEB cocultured with PD1+CD4 TILs (n = 4–6, biological replicates). (e) Histograms and frequency of CD38/CD27 subsets in CD19+ B cells after stimulation with SEB (500 ng/ml) alone or SEB in presence of PD1+ CD4 TILs (n = 13, biological replicates). (f) Histograms and frequency of CXCR5+CCR7+ expression in stimulated CD19+ B cells. (g) Frequencies of CD38/CD27 subsets for expression of CD24/CD25 stimulated with SEB in the presence of PD1+CD4 TILs. (h) Histograms and change in PD1 expression in CD4 TILs following stimulation with SEB alone or SEB in presence of CD19+ B cells. (i and j) Histograms (i) and frequencies (j) of PD1+CD200+ cells as a % of CD4 TILs stimulated with SEB alone or SEB in the presence of CD19+ B cells (n = 6, biological replicates). (k) gMFI of degranulation marker CD107a in PD1+CD200+ CD4 TILs co-expressing CD25 and CTLA4 (n = 6, biological replicates). (l) Histogram and frequencies of CXCR5+CCR7+ CD4 TILs stimulated with SEB alone or SEB in the presence of CD19 B cells. Data are displayed as mean ± SD. Data in d, e, f, and k by paired Students t-test, two-tailed. Data in h, j and l by one-way ANOVA followed by post-hoc Newman-Keuls. ns, not significant.
Our spatial profiling demonstrating that CD19+ B cell clusters were surrounded by PD1+CD4+ TILs is suggestive of functional interaction providing support for the development of tertiary lymphoid structures (TLS) (Figure 3). This was also confirmed following in situ staining in additional donors (Figure 5(c) and Figure S8A). Due to the challenges in differentiating TFR from TFH cells solely based on gene expression data, we utilized a more functional approach. We FACS-purified two subsets of PD1+CD4 memory TILs, co-expressing the tumor-specific antigen receptor CD39,33 based on CD25 (IL2RA) and CD200 expression and demonstrated both subsets exhibited robust expansion in culture (Figure S8B-C). In vitro expanded PD1+CD4+ memory TILs were co-cultured with carboxyfluorescein succinimidyl ester (CFSE)-labeled CD19+ B cells, derived from dissected mediastinal lymph nodes of lung cancer patients and stimulated with the TCR activator staphylococcal enterotoxin B (SEB) (Figure S8D). As compared to B cells stimulated with SEB alone, culture with SEB-activated PD1+CD4+ memory TILs induced B cell proliferation (38 ± 13 versus 2.6 ± 0.6%, respectively) (Figure 5(d)). Proliferating CD19+ B cells were observed to downregulate expression of CD27 and upregulate CD38 (Figure 5(e) and Figure S8F-H). This coincided with an upregulation of homing molecules CXCR5 and CCR7 (Figure 5(f)). Importantly, the majority of CD19+ B cells also upregulate CD25 but not CD24 (Figure 5(g) and Figure S8F-H) or IL10 (Figure S8E), markers of B regulatory cells. FACS-sorted PD1+CD45RO+CD4+ TILs decrease PD1, CD200 and CD25 expression in culture, as shown in Figure 5(h) (left panel). However, exposure to SEB in the presence of CD19+ B cells reactivates PD1 expression along with CD200 (Figure 5(i,j)) and CD25 but not CTLA4 (Figure S8I,J) and showed an intact ability to degranulate, based on increased expression of CD107a (Figure 5(k)). In contrast, activation of PD1+CD4+ memory TILs with SEB alone did not result in any change in expression of homing receptors CXCR5 and CCR7 (Figure 5(l)). However, similar to PD1, exposure to SEB in the presence of CD19+ B cells increased co-expression of CXCR5 and CCR7, which is required to migrate to B cell zones (Figure 5(l)). Despite the fact that the sorted PD1+CD4+ memory TIL population likely represents a mixture of both TFH and TFR cells, we were able to show these cells are competent to engage B cells and thus functionally behave more like TFH rather than TFR cells.
Patient T cells expanded in vitro retain their cytotoxic and helper functions
Adoptive cell therapy (ACT) using autologous TILs expanded from patient tumors is a promising approach to treat solid tumors, including metastatic melanoma34,35 and NSCLC.36 Presently, it is unclear if and to what extent T cells change their phenotype under persistent TCR stimulation. To address this, we generated primary T cell cultures from resected tissue of NSCLC specimens and expanded T cells under feeder-free conditions via repetitive rounds of T cell receptor (TCR) ligation, using anti CD3/CD28/CD2 beads in the presence of the gamma chain (γc) cytokines IL-2, IL-7, and IL-15 (Figure 6(a)). T cells from single-cell suspensions of uninvolved tissues proliferated more readily than their matched tumor counterparts (38 × 106 ± 12.8 versus 26 × 106 ± 7 T cells, respectively) (Figure S9A). Bulk primary T cell cultures were then FACS sorted into populations, as previously described (Figure 1(a)). Compared with the native tumor, in vitro expansion altered the percentage of CD4 and CD8 subsets, as well as CD4/CD8 ratio with the extent and direction depending on the donor (Figure 6(b)). Despite this, there was a consistent decrease in PD1+ expression in both CD8 and CD4 memory TILs following expansion (Figure 6(b) and Figure S9B). To quantify shifts in the composition within the CD4 and CD8 subpopulations, we performed transcriptomics analysis on each of the in vitro expanded subpopulation within the CD45RO memory compartment, calculated the enrichment of the T cell cluster signatures used previously (Figure 2(e)) and compared the in vitro patterns with the ex vivo prepared samples for which there were matching donors. Patterns of cell-type composition were preserved well upon expansion in vitro, although the sharp distinctions between populations observed in vivo were less pronounced. Importantly, the dysfunctional clusters in both CD4 (C7-CXCL13) and CD8 (C6-LAYN) remain prominent in the PD1+ TIL population (Figure 6(d)). The PD1+CD4+ memory TIL subset re-isolated from expanded bulk cultures retained their ability to help B cells (Figure S9F-H). To test the composition of the TCR clonotype between matched ex vivo prepared and in vitro expanded populations, we in silico re-constructed the TCR repertoire from samples with overlapping donors (four for CD4 and three for CD8 populations). Quantification of the overlap of TCR alpha (Figure S9E) and beta chains (Figure 6(e)) demonstrated that the donor-specific repertoire was stable in vitro.
Figure 6.
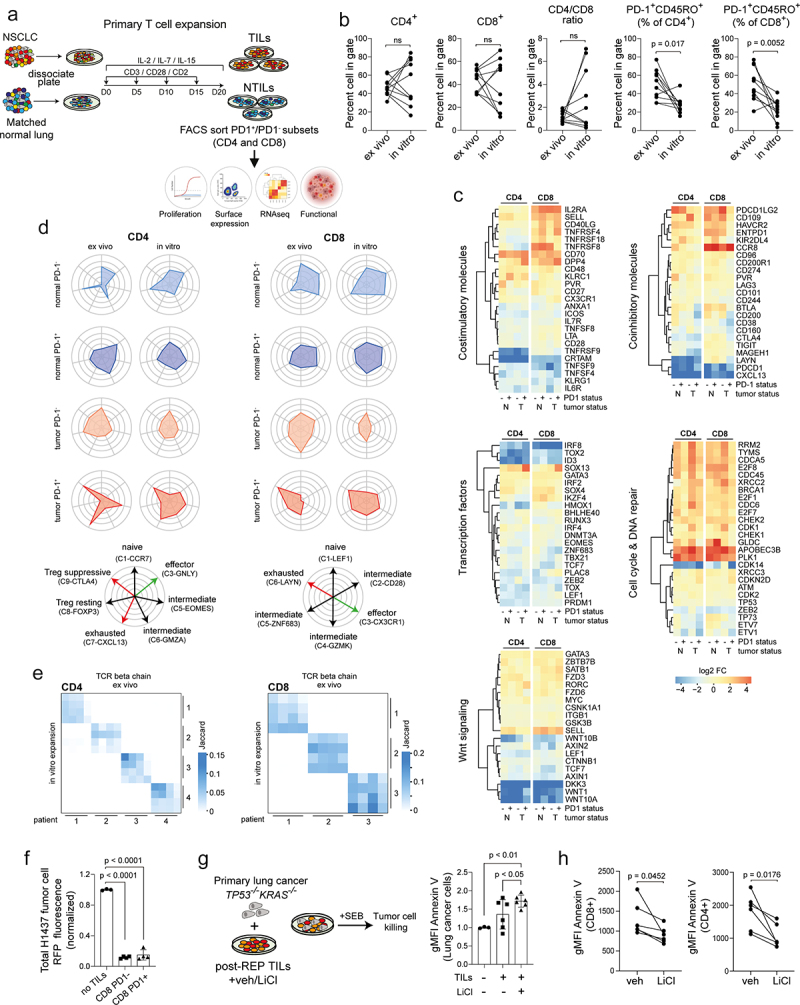
Transcriptional and functional analysis of memory T cells obtained after in vitro expansion from bulk tumor and matched normal specimens in NSCLC. (a) Graphical overview showing method of the experimental design used to expand T cells from single cell suspension, culture and analysis strategy. (b) Scatter plots showing % change in CD4 and CD8 subsets, CD4/CD8 ratio and memory TILs based on PD1 expression for ex vivo (primary) compared with in vitro (culture expanded) TILs from matched patients (n = 10, biological replicates). (c) Functional heatmaps showing relative gene expression levels of selected candidate genes in memory T cells for the following modules (stimulatory molecules, inhibitory molecules, transcription factors, cell cycle/DNA repair, Wnt signaling) comparing ex vivo and in vitro memory T cell subsets. Each column represents a matched patient. (d) Cell type radar plots correlating expression profile of ex vivo and in vitro memory T cell subsets to single cell TC clusters described by Guo et al.15 (e) TCRβ repertoire analysis showing the degree to which clonal cells in both the CD4 and CD8 memory compartments between groups are maintained. Comparison between ex vivo and in vitro samples based on PD1 status. (f) Histograms comparing tumor killing capacity of in vitro expanded CD8 TILs. Killing capacity measured as loss of RFP intensity. Total RFP normalized to H1437 lung tumor cells cultured with no TILs (n = 4, biological replicates). (g) Schematic representation of experimental setup and histograms comparing tumor killing capacity of in vitro expanded TILs without or with LiCl (2 mM) treatment quantified as the frequency of Annexin V expression in EpCAM+ primary lung cancer cells. Data were normalized to lung tumor cells cultured without TILs. (n = 6, biological replicates). (h) Scatter plots showing change in Annexin V expression in CD8 and CD4 TILs in coculture with primary lung tumor cells without or with LiCl (2 mM) (n = 6 biological replicates). Data in b, g, by paired students t-test, two-tailed. Data in f, g, by one-way ANOVA followed by post-hoc Newman-Keuls. ns, not significant.
Surprisingly we observed a general decrease of PD1 transcript levels in all in vitro expanded subpopulations following persistent TCR stimulation (Figure 6(c)), a finding that was confirmed at the protein level (Figure 6(b)). This trend toward a less “exhausted” gene expression profile was further exemplified by downregulation of the co-inhibitory molecules CD200, CD38, CD160, LAYN, and CTLA4 and concomitant upregulation of co-stimulatory molecules, such as IL2RA (CD25), TNFRSF4 (CD134) and CD40LG, in in vitro expanded PD1+CD8+ memory TILs (Figure 6(c)). Extensive phenotyping of expanded TILs with a panel of antibodies targeting several suppressive immune checkpoint molecules using flow cytometry confirmed these changes (Figure S9C,D). Furthermore, all T cell populations downregulated the transcriptional regulators IRF8, TOX, TOX2, and ID3, which are associated with an exhausted phenotype. PD1+CD8+ TILs upregulate memory/quiescence-associated genes (SELL, LEF1, CCR8) as well as cell cycle and DNA repair genes (PLK1, BRCA1) indicating a more proliferative phenotype. Both in vitro expanded PD1− and PD1+ populations displayed robust killing ability (Figure 6(f)), indicating that in vitro expansion of CD8+ memory TILs produces functionally intact cells.
Downregulation of the components of the WNT pathway had already been observed in PD1+ CD8+ memory TILs in vivo (Figure S2D) and we observed a further downregulation of WNT1, WNT10A, and DKK3 upon in vitro expansion in all T cell populations (Figure 6(c)). As WNT signaling is known to arrest the development of effector CD8+ T cells,37 we examined whether restoring WNT signaling via inhibition of GSK3β could further improve T cell function. We found that treating bulk cultures during the expansion period with the GSK3 β inhibitor lithium chloride (LiCl) resulted in a reduction in the CD8 compartment (Figure S10A). However, when bulk TIL cultures were challenged with a pool of MHC Class I human peptides including type A influenza, Epstein-Barr virus, and cytomegalovirus, both CD4 and CD8 TILs increased IFNγ secretion (Figure S10B) and upregulated expression of PD1 and CD80 (Figure S10C). We also found that treating TILs during expansion with LiCl significantly increased the number of patient-derived primary lung tumor cells undergoing apoptosis, as judged by Annexin V staining, suggestive of enhanced TIL killing capacity (Figure 6(g)). Moreover, we observed an upregulation of PD-L1 expression in primary lung cancer cells when combined with TILs treated with vehicle but not LiCl (Figure S10D-E). Subpopulations of CD8 and CD4 TILs based on PD1 expression also were observed, irrespective of treatment (Figure S10F-G). Interestingly, TILs with intermediate and high levels of PD1 were shown to upregulate PD-L1, irrespective of treatment group (Figure S10F). Despite observing no difference in PD-L1 expression on TILs between vehicle and LiCl treatment, only TILs pretreated with LiCl displayed downregulation of Annexin V (Figure 6(h)) suggestive of cell protection. We conclude that expansion of T cells in vitro could increase the number of autologous tumor-specific T cells for adoptive transfer and potentially also relieve some of the tumor-induced dysfunction, partially via restoring WNT signaling.
Discussion
Adoptive cell transfer of naturally occurring antigen-specific TILs has great potential as an antitumor therapy. Despite recent reports of clinical benefit in a number of solid tumors,38–40 important open questions remain, that include the extent of in vivo T cell persistence,41 the contribution of other cell types, such as CD4+ cells,42 and whether the antitumor activity after PD1 checkpoint blockade originates from re-activation of exhausted TILs or migration of distinct clones from the periphery. In this report, we compare the phenotypic and transcriptomic profiles of resident memory CD8 and CD4 T cell populations prepared from NSCLC patient resections with their in vitro expanded counterparts. T cell expansion using chronic TCR stimulation resulted in highly heterogeneous expansion rates for different T cell subtypes, yet the expanded culture largely maintained their phenotypic and transcriptomic features, compared to the tissue-resident cells. Importantly, they retained their ability to efficiently kill tumor targets and promote B cell proliferation. Some important differences include downregulation of PD1 expression, both at the protein and mRNA level, in re-expanded CD8+ and CD4+ memory TILs compared with native tumor. In contrast, co-inhibitory receptors, TIM3 (HAVCR2) and LAG3, increased at the mRNA level, indicating at least a partial maintenance of an exhausted phenotype upon in vitro expansion, in line with previous data on ovarian cancer.38 Furthermore, the stem-like gene TCF7 (TCF1), a T-cell-specific HMG-box containing transcription factor, remained downregulated in the in vitro expanded PD1+CD8+ memory compartment, even in the presence of homeostatic γc cytokines IL-7/IL-15,43 coinciding with an overall decrease in WNT signaling,7,37 an indication of T cell dysfunction.44 We show that restoring WNT signaling via a GSK3β inhibitor LiCl45 may provide an important means to improve the functional capacity of in vitro expanded TILs for ACT. Lastly, the pool of re-expanded CD8+ memory TILs downregulated TOX, TOX2 and KLRG1, while simultaneously upregulating CD27, CD28 and CD62L (SELL), arguing against formation of a terminal or short-lived effector state due to chronic TCR stimulation. The broad conservation of phenotype and functionality of expanded TILs is promising. In support of this, Creelan and colleagues46 provided evidence of safety and efficacy of patient-derived ex-vivo expanded TILs in phase 1 trial in a small cohort of patients with advanced stage NSCLC with progression on nivolumab. This study demonstrates that adoptive TIL therapy is clinically beneficial in patients with ICB-resistant NSCLC, and lends further support to our approach used in this study of characterizing the ex-vivo expanded TIL product. Moving forward, these results suggest that the success of ACT will be critically dependent on an understanding of the functional composition of the expanded culture and the careful selection of the appropriate T cell subpopulation.
Although CD4+ memory T cell contribution is essential to coordinating host immunity against viral pathogens and chronic infection, the function of CD4 memory TILs in cancer control is less well studied, compared to their CD8 counterparts. The ability of CD4+ T cells to substantially modulate anti-tumor activity is exemplified in a recent finding that high PD1 expression in CD4+ TILs negatively correlates with patient survival in NSCLC.30 Here, we show that the gene expression signature in PD1+CD4+ memory TILs highly correlate with a suppressor Treg signature,15 although when purified, at least a subset of PD1+CD4+ memory TILs are able to proliferate and effectively kill tumor cells. This argues that the PD1+CD4+ memory T cell compartment is a mixture of cell types, with potentially antagonistic functionality. Indeed, we show that PD1+CD4+ memory TILs are enriched in genes related to both TFH31 and TFR cells.19 One of the most upregulated genes in PD1+CD4+ memory TILs is the B lymphocyte chemoattractant CXCL13,47 which, in conjunction with IL-21, triggers the enrichment of TFH in germinal centers to coordinate B cell help. We demonstrate that activation of PD1+CD4+ memory TILs with the enterotoxin SEB induces mediastinal lymph node-derived B cells to proliferate and upregulate CXCR5, the receptor of CXCL13 ligand that is highly upregulated in PD1+CD45RO+ CD4 TILs (Figure 5(a)), and the CC-chemokine receptor CCR7 while they themselves simultaneously re-express PD1, CD200, CD25 and CXCR5, known markers of TFH cells. Importantly, proliferating B cells do not express CD24 or IL10, two common markers of regulatory B cells. Interestingly, re-activated PD1+CD4+ TILs also upregulate CCR7. Spatial profiling supports a role for PD1+CD4+ TILs in B cell help, CD19+ B cells clusters were surrounded by PD1+CD4+ TILs compared with PD1+CD8+ TILs. There is emerging evidence in melanoma that CD8+CD20+ TLS improve the response to immunotherapy resulting in better overall survival.48,49 Recently, Thommen and colleagues demonstrated that TLS and their components, including PD1+CD4+ TILs and CXCL13 expression, were predictive for response to PD1 blockade in NSCLC.50 Therefore, further studies are necessary to decipher the critical involvement of the CD19+ B cell-PD1+CD4+ axis in NSCLC prognosis and response to ICB, which to date is lacking.51
PD1+CD8+ TILs prepared from NSCLC patients display the major hallmarks of T cell dysfunction, a reduced proliferative potential and low tumor-killing activity. A key finding of our study, is that knockdown of ID3 in CD8+ T cells or Jurkat cells results in enhanced effector function. At the mRNA level, ID3 is enriched in dysfunctional CD8 TILs in both melanoma and NSCLC.24 In chronic lymphocytic choriomeningitis virus (LCMV) infection in mice, ID3 overexpression was linked with chronic TCR stimulation and correlated with CD8+ T cell exhaustion.20 ID3 is a member of the helix-loop-helix family of proteins that act as transcriptional regulators, preventing E proteins, e.g. E2A and HEB, from binding to DNA and repressing transcription. ID3 has been shown to be critical for the developmental progression of T cell differentiation52 as well as formation of CD8+ memory subsets.53,54 The transcription factor TOX has recently emerged as a regulator of T cell exhaustion and as a potential target for immunotherapy.20–22 TOX is thought to antagonize the function of E proteins through the upregulation of proteins of the ID family, including ID3.55 Notably, scRNA-sequencing data of TILs in lung cancer patients also demonstrates that TOX is enriched in the exhausted CD8 T cell cluster.15 Further, ablation of TOX in CD8+ T cells partially restores function.21,22 Taken together, ID3 and TOX may have functionally redundant roles in promoting a dysfunctional CD8 T cell phenotype resulting from chronic TCR stimulation, making them potential therapeutic targets for immune oncology. It should be noted that we found that many transcription factors acting as central regulators of T cell exhaustion (e.g. ID3, IRF4, TOX and TOX2) and other exhaustion-associated-genes, such as CD38, ENTPD1 or TIGIT overexpressed in PD1+CD8+ memory TILs, are also upregulated in PD1+CD4+ memory TILs. Recently it has been shown that ablation of TOX2 and TOX impairs TFH cell differentiation32 acting as a reminder that drug development efforts against such factors must consider that positive effects in one cell type may be mitigated by adverse effects in other cell types, as TFH cells are central for a robust host anti-tumor immune response. The transcriptional regulator ID3 was also increased in CD4 TILs. ID3 appears to be necessary for maintenance of FoxP3 expression and CD4 Treg suppressive function.56 Loss of ID3 leads to an increase in CD4 TH9 phenotype that exhibit potent anti-tumor immunity57 and adoptive TIL therapy using tumor-specific CD4 TH9 cells demonstrates potent anti-tumor activity.58 Therefore, ID3 may be a more appropriate target for anti-tumor immunotherapy in lung cancer especially in the adoptive cell therapy setting. Despite our efforts to address the molecular and functional aspects of TIL dysfunction based on PD1 expression, our study has several limitations. First, our study was descriptive in nature and based on a small sample size, which requires careful interpretation. Second, we did not correlate molecular signatures in memory PD1+ TILs with advanced disease in NSCLC and whether memory PD1+ TIL phenotypes were associated with favorable outcomes in patients undergoing ICB. Bernatchez and colleagues59 recently demonstrated that the molecular state of both infiltrating CD8 and CD4 TILs together with infiltrating B cells not overall number of infiltrating T cells dictate clinical outcome in NSCLC. Finally, despite our in vitro functional assays demonstrating an interplay between PD1+CD4+ memory TILs and CD19+ B cells leading to generation of distinct T cell and B cell phenotypes, we still do not know whether PD1+CD4+ TILs direct CD19+ B cells to form new TLS and whether therapeutic strategies to induce the formation of PD1+CD4+CD19+ tumors would improve responses to ICB in NSCLC, as recently described in melanoma.48
Conclusions
Here, we show shared and distinct transcriptional profiles between CD4 and CD8 PD1+ memory TILs in NSCLC. Importantly, reducing ID3 expression improved CD8 T cell effector function. Therefore, depending upon the degree to which the original expression profiles observed in tissue-resident TILs are represented in the in vitro group following prolonged expansion in culture, future protocols may need to be modified to direct or redirect cellular differentiation to ensure enrichment of memory-precursor/stem-like T cells, which may correlate with persistence and improved clinical outcomes. Moreover, defining the optimal conditions for targeting WNT pathway to improve expanded autologous TIL product for ACT in NSCLC warrants further investigation.
Supplementary Material
Acknowledgments
Tissue were provided by the Tissue Bank Bern. The lung cancer cell line PF139 was established in collaboration with the West-German Biobank Essen (WBE).
Funding Statement
This work has been supported by the Innovative Medicines Initiative Joint undertaking under grant agreement n° 115188, resources of which are composed of financial contribution from the European Union’s Seventh Framework Programme (FP7/2007-2013) and EFPIA companies in kind contribution.
Abbreviations
PD1, programmed cell death 1; TILs, Tumor-infiltrating lymphocytes; ICB; immune checkpoint blockade; PDL1; programmed cell death ligand 1; FACS, fluorescence-activated cell sorting; NILs, normal-infiltrating lymphocytes; 3D, three-dimensional; t-SNE; T distributed Stochastic Neighbor Embedding; ssGSEA, single sample gene set enrichment; TFH; T follicular helper; TFR, T follicular regulatory; CFSE; carboxyfluorescein succinimidyl ester; SEB, Staphylococcal enterotoxin B; ACT, Adoptive cell therapy; TCR, T cell Receptor; γc, gamma chain.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Availability of data and material
The data that support the findings of this study are available within the paper and its Supplementary Information files. The RNA sequencing data have been deposited in the Gene Expression Omnibus (GEO) with the accession code GSExxxxxx. Any other material including the R code used for data analysis is available upon reasonable request to the authors (JL and SRRH).
Authors’ contributions
Conception & design – JL, WS, SRRH; tissue and data acquisition – JL, LW, HY, FY, NH, SM, SB, PD, TMM, GJK, BH, AS, WS, RAS, SRRH; Data interpretation & analysis – JL, LW, NH, SB, BH, AS, WS, SRRH; Drafting of Manuscript – JL, SRRH; Editing of manuscript – JL, LW, HY, FY, NH, SM, SB, PD, TMM, GJK, BH, AS, SC, MAP, WS, RAS, SRRH; Final Approval of manuscript – JL, WS, SRRH.
Ethics approval and consent to participate
The protocol used for this study was approved by the Ethics Commission of the Canton of Bern (KEK-BE:2018-01801). All patients provided informed written consent for use of their material for research purposes. The cell-line generation was performed in collaboration with the Westgerman Biobank Essen (WBE) and approved by the Ethics Committee of the University Duisburg-Essen (#18–8208-BO). All patients provided informed consent.
Consent for publication
All authors agreed on the manuscript.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website
References
- 1.Sharpe AH, Pauken KE.. The diverse functions of the PD1 inhibitory pathway. Nat Rev Immunol. 2018. Mar;18(3):153–15. doi: 10.1038/nri.2017.108. [DOI] [PubMed] [Google Scholar]
- 2.McLane LM, Abdel-Hakeem MS, Wherry EJ. CD8 t cell exhaustion during chronic viral infection and cancer. Annu Rev Immunol. 2019. Apr 26;37(1):457–495. doi: 10.1146/annurev-immunol-041015-055318. [DOI] [PubMed] [Google Scholar]
- 3.Kamphorst AO, Pillai RN, Yang S, Nasti TH, Akondy RS, Wieland A, Sica GL, Yu K, Koenig L, Patel NT, et al. Proliferation of PD-1+ CD8 T cells in peripheral blood after PD-1-targeted therapy in lung cancer patients. Proc Natl Acad Sci U S A. 2017. May 9;114(19):4993–4998. doi: 10.1073/pnas.1705327114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, Lee W, Yuan J, Wong P, Ho TS, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015. Apr 3;348(6230):124–128. doi: 10.1126/science.aaa1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thommen DS, Koelzer VH, Herzig P, et al. A transcriptionally and functionally distinct PD-1(+) CD8(+) T cell pool with predictive potential in non-small-cell lung cancer treated with PD-1 blockade. Nat Med. 2018. Jul;24(7):994–1004. doi: 10.1038/s41591-018-0057-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Garon EB, Hellmann MD, Rizvi NA, et al. Five-year overall survival for patients with advanced nonsmall-cell lung cancer treated with pembrolizumab: results from the phase I KEYNOTE-001 study. J Clin Oncol. 2019. Oct 1;37(28):2518–2527. doi: 10.1200/JCO.19.00934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Philip M, Fairchild L, Sun L, Horste EL, Camara S, Shakiba M, Scott AC, Viale A, Lauer P, Merghoub T, et al. Chromatin states define tumour-specific T cell dysfunction and reprogramming. Nature. 2017. May 25;545(7655):452–456. doi: 10.1038/nature22367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wu TD, Madireddi S, de Almeida PE, Banchereau R, Chen YJJ, Chitre AS, Chiang EY, Iftikhar H, O’Gorman WE, Au-Yeung A, et al. Peripheral T cell expansion predicts tumour infiltration and clinical response. Nature. 2020. Feb 26;579(7798):274–278. doi: 10.1038/s41586-020-2056-8. [DOI] [PubMed] [Google Scholar]
- 9.Borst J, Ahrends T, Babala N, Melief CJM, Kastenmuller W. CD4(+) T cell help in cancer immunology and immunotherapy. Nat Rev Immunol. 2018. Oct;18(10):635–647. doi: 10.1038/s41577-018-0044-0. [DOI] [PubMed] [Google Scholar]
- 10.Creelan B, Teer J, Toloza E, Mullinax J, Landin A, Gray J, Tanvetyanon T, Taddeo M, Noyes D, Kelley L, et al. OA05.03 safety and clinical activity of adoptive cell transfer using tumor infiltrating lymphocytes (TIL) combined with nivolumab in NSCLC. Journal of Thoracic Oncology. 2018;13(10):S330–S331. doi: 10.1016/j.jtho.2018.08.259. [DOI] [Google Scholar]
- 11.Utzschneider DT, Charmoy M, Chennupati V, Pousse L, Ferreira DP, Calderon-Copete S, Danilo M, Alfei F, Hofmann M, Wieland D, et al. T Cell Factor 1-expressing memory-like CD8(+) T Cells sustain the immune response to chronic viral infections. Immunity. 2016. Aug 16;45(2):415–427. doi: 10.1016/j.immuni.2016.07.021. [DOI] [PubMed] [Google Scholar]
- 12.Wang L, Dorn P, Zeinali S, Froment L, Berezowska S, Kocher GJ, Alves MP, Brügger M, Esteves BIO, Blank F, et al. CD90+CD146+ identifies a pulmonary mesenchymal cell subtype with both immune modulatory and perivascular-like function in postnatal human lung. Am J Physiol Lung Cell Mol Physiol. 2020. Apr 1;318(4):L813–L830. doi: 10.1152/ajplung.00146.2019. [DOI] [PubMed] [Google Scholar]
- 13.Oberli A, Popovici V, Delorenzi M, Baltzer A, Antonov J, Matthey S, Aebi S, Altermatt HJ, Jaggi R. Expression profiling with RNA from formalin-fixed, paraffin-embedded material. BMC Med Genomics. 2008. Apr 19;1:9. doi: 10.1186/1755-8794-1-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Osswald A, Hedrich V, Sommergruber W. 3D-3 tumor models in drug discovery for analysis of immune cell infiltration. Methods Mol Biol. 2019;1953:151–162. doi: 10.1007/978-1-4939-9145-7_10. [DOI] [PubMed] [Google Scholar]
- 15.Guo X, Zhang Y, Zheng L, Zheng C, Song J, Zhang Q, Kang B, Liu Z, Jin L, Xing R, et al. Global characterization of T cells in non-small-cell lung cancer by single-cell sequencing. Nat Med. 2018. Jul;24(7):978–985. doi: 10.1038/s41591-018-0045-3. [DOI] [PubMed] [Google Scholar]
- 16.Reuben A, Zhang J, Chiou SH, Gittelman RM, Li J, Lee W-C, Fujimoto J, Behrens C, Liu X, Wang F, et al. Comprehensive T cell repertoire characterization of non-small cell lung cancer. Nat Commun. 2020. Jan 30;11(1):603. doi: 10.1038/s41467-019-14273-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Clarke J, Panwar B, Madrigal A, Singh D, Gujar R, Wood O, Chee SJ, Eschweiler S, King EV, Awad AS, et al. Single-cell transcriptomic analysis of tissue-resident memory T cells in human lung cancer. J Exp Med. 2019. Sep 2;216(9):2128–2149. doi: 10.1084/jem.20190249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang L, Simons DL, Lu X, Tu TY, Solomon S, Wang R, Rosario A, Avalos C, Schmolze D, Yim J, et al. Connecting blood and intratumoral Treg cell activity in predicting future relapse in breast cancer. Nat Immunol. 2019. Sep;20(9):1220–1230. doi: 10.1038/s41590-019-0429-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.De Simone M, Arrigoni A, Rossetti G, Gruarin P, Ranzani V, Politano C, Bonnal RP, Provasi E, Sarnicola M, Panzeri I, et al. Transcriptional landscape of human tissue lymphocytes unveils uniqueness of tumor-infiltrating T regulatory cells. Immunity. 2016. Nov 15;45(5):1135–1147. doi: 10.1016/j.immuni.2016.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Alfei F, Kanev K, Hofmann M, Wu M, Ghoneim HE, Roelli P, Utzschneider DT, Von Hoesslin M, Cullen JG, Fan Y, et al. TOX reinforces the phenotype and longevity of exhausted T cells in chronic viral infection. Nature. 2019. Jul;571(7764):265–269. doi: 10.1038/s41586-019-1326-9. [DOI] [PubMed] [Google Scholar]
- 21.Khan O, Giles JR, McDonald S, Manne S, Ngiow SF, Patel KP, Werner MT, Huang AC, Alexander KA, Wu JE, et al. TOX transcriptionally and epigenetically programs CD8(+) T cell exhaustion. Nature. 2019. Jul;571(7764):211–218. doi: 10.1038/s41586-019-1325-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Scott AC, Dundar F, Zumbo P, Chandran SS, Klebanoff CA, Shakiba M, Trivedi P, Menocal L, Appleby H, Camara S, et al. TOX is a critical regulator of tumour-specific T cell differentiation. Nature. 2019. Jul;571(7764):270–274. doi: 10.1038/s41586-019-1324-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Seo H, Chen J, Gonzalez-Avalos E, Samaniego-Castruita D, Das A, Wang YH, López-Moyado IF, Georges RO, Zhang W, Onodera A, et al. TOX and TOX2 transcription factors cooperate with NR4A transcription factors to impose CD8 + T cell exhaustion. Proc Natl Acad Sci U S A. 2019. Jun 18;116(25):12410–12415. doi: 10.1073/pnas.1905675116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li H, van der Leun AM, Yofe I, Lubling Y, Gelbard-Solodkin D, van Akkooi ACJ, van Den Braber M, Rozeman EA, Haanen JBAG, Blank CU, et al. Dysfunctional CD8 t cells form a proliferative, dynamically regulated compartment within human melanoma. Cell. 2019. Feb 7;176(4):775–789 e18. doi: 10.1016/j.cell.2018.11.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pages C, Tosolini M, Camus M, Berger A, Wind P, et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science. 2006. Sep 29;313(5795):1960–1964. doi: 10.1126/science.1129139. [DOI] [PubMed] [Google Scholar]
- 26.Liu H, Zhang T, Ye J, Li H, Huang J, Li X, Wu B, Huang X, Hou J. Tumor-infiltrating lymphocytes predict response to chemotherapy in patients with advance non-small cell lung cancer. Cancer Immunol Immunother. 2012. Oct;61(10):1849–1856. doi: 10.1007/s00262-012-1231-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sautes-Fridman C, Petitprez F, Calderaro J, Fridman WH. Tertiary lymphoid structures in the era of cancer immunotherapy. Nat Rev Cancer. 2019. Jun;19(6):307–325. doi: 10.1038/s41568-019-0144-6. [DOI] [PubMed] [Google Scholar]
- 28.Omilusik KD, Shaw LA, Goldrath AW. Remembering one’s ID/E-ntity: e/ID protein regulation of T cell memory. Curr Opin Immunol. 2013. Oct;25(5):660–666. doi: 10.1016/j.coi.2013.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zappasodi R, Budhu S, Hellmann MD, Postow MA, Senbabaoglu Y, Manne S, Gasmi B, Liu C, Zhong H, Li Y, et al. Non-conventional inhibitory CD4(+)Foxp3(-)PD-1(hi) T Cells as a biomarker of immune checkpoint blockade activity. Cancer Cell. 2018. Jun 11;33(6):1017–1032 e7. doi: 10.1016/j.ccell.2018.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zappasodi R, Budhu S, Hellmann MD, Postow MA, Senbabaoglu Y, Manne S, Gasmi B, Liu C, Zhong H, Li Y, et al. Non-conventional inhibitory CD4(+)Foxp3(-)PD-1(hi) T Cells as a biomarker of immune checkpoint blockade activity. Cancer Cell. 2018. Oct 8;34(4):691. doi: 10.1016/j.ccell.2018.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gu-Trantien C, Loi S, Garaud S, Equeter C, Libin M, de Wind A, Ravoet M, Le Buanec H, Sibille C, Manfouo-Foutsop G, et al. CD4(+) follicular helper T cell infiltration predicts breast cancer survival. J Clin Invest. 2013. Jul;123(7):2873–2892. doi: 10.1172/JCI67428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xu W, Zhao X, Wang X, Feng H, Gou M, Jin W, Wang X, Liu X, Dong C. The transcription factor tox2 drives t follicular helper cell development via regulating chromatin accessibility. Immunity. 2019. Nov 19;51(5):826–839 e5. doi: 10.1016/j.immuni.2019.10.006. [DOI] [PubMed] [Google Scholar]
- 33.Simoni Y, Becht E, Fehlings M, Loh CY, Koo S-L, Teng KWW, Yeong JPS, Nahar R, Zhang T, Kared H, et al. Bystander CD8(+) T cells are abundant and phenotypically distinct in human tumour infiltrates. Nature. 2018. May;557(7706):575–579. doi: 10.1038/s41586-018-0130-2. [DOI] [PubMed] [Google Scholar]
- 34.Dafni U, Michielin O, Lluesma SM, Tsourti Z, Polydoropoulou V, Karlis D, Besser MJ, Haanen J, Svane IM, Ohashi PS, et al. Efficacy of adoptive therapy with tumor-infiltrating lymphocytes and recombinant interleukin-2 in advanced cutaneous melanoma: a systematic review and meta-analysis. Ann Oncol. 2019. Dec 1;30(12):1902–1913. doi: 10.1093/annonc/mdz398. [DOI] [PubMed] [Google Scholar]
- 35.Nguyen LT, Saibil SD, Sotov V, Le MX, Khoja L, Ghazarian D, Bonilla L, Majeed H, Hogg D, Joshua AM, et al. Phase II clinical trial of adoptive cell therapy for patients with metastatic melanoma with autologous tumor-infiltrating lymphocytes and low-dose interleukin-2. Cancer Immunol Immunother. 2019. May;68(5):773–785. doi: 10.1007/s00262-019-02307-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Federico L, Haymaker CL, Forget M-A, Ravelli A, Bhatta A, Karpinets T, Zhang R, Weissferdt A, Fang B, Zhang J, et al. A preclinical study of tumor-infiltrating lymphocytes in NSCLC. Journal of Clinical Oncology. 2018;36(5_suppl):161. doi: 10.1200/JCO.2018.36.5_suppl.161. [DOI] [Google Scholar]
- 37.Gattinoni L, Zhong XS, Palmer DC, Ji Y, Hinrichs CS, Yu Z, Wrzesinski C, Boni A, Cassard L, Garvin LM, et al. Wnt signaling arrests effector T cell differentiation and generates CD8+ memory stem cells. Nat Med. 2009. Jul;15(7):808–813. doi: 10.1038/nm.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Owens GL, Price MJ, Cheadle EJ, Hawkins RE, Gilham DE, Edmondson RJ. Ex vivo expanded tumour-infiltrating lymphocytes from ovarian cancer patients release anti-tumour cytokines in response to autologous primary ovarian cancer cells. Cancer Immunol Immunother. 2018. Oct;67(10):1519–1531. doi: 10.1007/s00262-018-2211-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rosenberg SA, Yang JC, Sherry RM, Kammula US, Hughes MS, Phan GQ, Citrin DE, Restifo NP, Robbins PF, Wunderlich JR, et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin Cancer Res. 2011. Jul 1;17(13):4550–4557. doi: 10.1158/1078-0432.CCR-11-0116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stevanovic S, Helman SR, Wunderlich JR, Langhan MM, Doran SL, Kwong MLM, Somerville RPT, Klebanoff CA, Kammula US, Sherry RM, et al. A Phase ii study of tumor-infiltrating lymphocyte therapy for human papillomavirus-associated epithelial cancers. Clin Cancer Res. 2019. Mar 1;25(5):1486–1493. doi: 10.1158/1078-0432.CCR-18-2722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lu YC, Jia L, Zheng Z, Tran E, Robbins PF, Rosenberg SA. Single-Cell transcriptome analysis reveals gene signatures associated with T-cell persistence following adoptive cell therapy. Cancer Immunol Res. 2019. Nov;7(11):1824–1836. doi: 10.1158/2326-6066.CIR-19-0299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Inderberg EM, Walchli S. Long-term surviving cancer patients as a source of therapeutic TCR. Cancer Immunol Immunother. 2020. Jan 8;69(5):859–865. doi: 10.1007/s00262-019-02468-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cieri N, Camisa B, Cocchiarella F, Forcato M, Oliveira G, Provasi E, Bondanza A, Bordignon C, Peccatori J, Ciceri F, et al. IL-7 and IL-15 instruct the generation of human memory stem T cells from naive precursors. Blood. 2013. Jan 24;121(4):573–584. doi: 10.1182/blood-2012-05-431718. [DOI] [PubMed] [Google Scholar]
- 44.van Loosdregt J, Coffer PJ. The role of WNT signaling in mature T Cells: T Cell factor is coming home. J Immunol. 2018. Oct 15;201(8):2193–2200. doi: 10.4049/jimmunol.1800633. [DOI] [PubMed] [Google Scholar]
- 45.Ohteki T, Parsons M, Zakarian A, Jones RG, Nguyen LT, Woodgett JR, Ohashi PS. Negative regulation of T cell proliferation and interleukin 2 production by the serine threonine kinase GSK-3. J Exp Med. 2000. Jul 3;192(1):99–104. doi: 10.1084/jem.192.1.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Creelan BC, Wang C, Teer JK, et al. Tumor-infiltrating lymphocyte treatment for anti-PD-1-resistant metastatic lung cancer: a phase 1 trial. Nat Med. 2021. Aug;27(8):1410–1418. doi: 10.1038/s41591-021-01462-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shi J, Hou S, Fang Q, Liu X, Liu X, Qi H. PD-1 controls follicular T helper cell positioning and function. Immunity. 2018. Aug 21;49(2):264–274 e4. doi: 10.1016/j.immuni.2018.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cabrita R, Lauss M, Sanna A, Donia M, Skaarup Larsen M, Mitra S, Johansson I, Phung B, Harbst K, Vallon-Christersson J, et al. Tertiary lymphoid structures improve immunotherapy and survival in melanoma. Nature. 2020. Jan;577(7791):561–565. doi: 10.1038/s41586-019-1914-8. [DOI] [PubMed] [Google Scholar]
- 49.Helmink BA, Reddy SM, Gao J, Zhang S, Basar R, Thakur R, Yizhak K, Sade-Feldman M, Blando J, Han G, et al. B cells and tertiary lymphoid structures promote immunotherapy response. Nature. 2020. Jan;577(7791):549–555. doi: 10.1038/s41586-019-1922-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Voabil P, de Bruijn M, Roelofsen LM, et al. An ex vivo tumor fragment platform to dissect response to PD-1 blockade in cancer. Nat Med. 2021. Jul;27(7):1250–1261. doi: 10.1038/s41591-021-01398-3. [DOI] [PubMed] [Google Scholar]
- 51.Miggelbrink AM, Jackson JD, Lorrey SJ, Srinivasan ES, Waibl-Polania J, Wilkinson DS, Fecci PE. CD4 T-cell exhaustion: does it exist and what are its roles in cancer? Clin Cancer Res. 2021. Jun 14;27(21):5742–5752. doi: 10.1158/1078-0432.CCR-21-0206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kee BL. E and ID proteins branch out. Nat Rev Immunol. 2009. Mar;9(3):175–184. doi: 10.1038/nri2507. [DOI] [PubMed] [Google Scholar]
- 53.Ji Y, Pos Z, Rao M, et al. Repression of the DNA-binding inhibitor Id3 by Blimp-1 limits the formation of memory CD8+ T cells. Nat Immunol. 2011. Nov 6;12(12):1230–1237. doi: 10.1038/ni.2153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yang CY, Best JA, Knell J, Yang E, Sheridan AD, Jesionek AK, Li HS, Rivera RR, Lind KC, D’Cruz LM, et al. The transcriptional regulators Id2 and Id3 control the formation of distinct memory CD8+ T cell subsets. Nat Immunol. 2011. Nov 6;12(12):1221–1229. doi: 10.1038/ni.2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Xiong Y, Bosselut R. The enigma of CD4-lineage specification. Eur J Immunol. 2011. Mar;41(3):568–574. doi: 10.1002/eji.201041098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rauch KS, Hils M, Lupar E, Minguet S, Sigvardsson M, Rottenberg ME, Izcue A, Schachtrup C, Schachtrup K. Id3 maintains Foxp3 expression in regulatory T Cells by controlling a transcriptional network of E47, Spi-B, and SOCS3. Cell Rep. 2016. Dec 13;17(11):2827–2836. doi: 10.1016/j.celrep.2016.11.045. [DOI] [PubMed] [Google Scholar]
- 57.Nakatsukasa H, Zhang D, Maruyama T, et al. The DNA-binding inhibitor Id3 regulates IL-9 production in CD4(+) T cells. Nat Immunol. 2015. Oct;16(10):1077–1084. doi: 10.1038/ni.3252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Xue G, Zheng N, Fang J, Jin G, Li X, Dotti G, Yi Q, Lu Y. Adoptive cell therapy with tumor-specific Th9 cells induces viral mimicry to eliminate antigen-loss-variant tumor cells. Cancer Cell. 2021. Oct 5;39(12):1610–1622.e9. doi: 10.1016/j.ccell.2021.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Federico L, McGrail DJ, Bentebibel SE, et al. Distinct tumor-infiltrating lymphocyte landscapes are associated with clinical outcomes in localized non-small cell lung cancer. Ann Oncol. 2021. Oct 12. doi: 10.1016/j.annonc.2021.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available within the paper and its Supplementary Information files. The RNA sequencing data have been deposited in the Gene Expression Omnibus (GEO) with the accession code GSExxxxxx. Any other material including the R code used for data analysis is available upon reasonable request to the authors (JL and SRRH).