

ABSTRACT
This study explored the anti-tumor effect of ginkgetin, an extract from ginkgo biloba, on human hepatocellular carcinoma cell lines and the underlying mechanisms. Cell viability was measured by MTT assay. Apoptotic cell morphology was observed under an inverted microscope after Hoechst 33,258 staining, and the ratio of apoptotic and necrotic cells was examined by flow cytometry after FITC/PI staining. Cell cycle changes were analyzed using flow cytometry. Cytochrome c release and caspase 3 and 8 activities were monitored using the relevant reagent kits. The levels of cell cycle-related proteins were detected by Western blot. MTT results indicated that ginkgetin significantly reduced HepG2 cell viability in a dose-dependent manner. Cellular morphology observation revealed that ginkgetin induced typical apoptotic morphological features of HepG2 cells, such as increased apoptotic bodies and cell shrinkage. Cell cycle analysis showed that ginkgetin increased the proportion of cells in the S phase. S-phase cell accumulation could be attributed to the decreased expression of cell cycle regulatory factors. Similarly, ginkgetin also induced the apoptosis and S phase cell accumulation of another human HCC cell line SK-HEP-1. Furthermore, ginkgetin treatment increased caspase-3 activity and cytochrome c release but not caspase-8 activity, implying that ginkgetin might mediate cell apoptosis through the mitochondrial pathway. In addition, the tumor formation experiment in nude mice showed that ginkgetin administration inhibited tumor growth. These results suggest that ginkgetin could be a cell apoptosis stimulator by affecting the balance between cell proliferation and apoptosis, suggesting that ginkgetin might be suitable for human HCC treatment.
KEYWORDS: Ginkgetin, cell cycle, apoptosis, hepatocellular carcinoma
Introduction
Ginkgo bilobal, an abundant plant in China, is the oldest species in the existing seed plants. Due to its long history, it has the reputation of “living fossils” [1,2]. In recent years, with the deepening of research in traditional Chinese medicine, the medicinal value of ginkgo has attracted much attention [3]. Studies have confirmed that ginkgetin, a ginkgo flavonoids-rich extract from the leaves of Ginkgo biloba, possesses complex and diverse pharmacological activities, such as anti-oxidation and anti-aging effects, promoting blood circulation, and preventing cardiovascular diseases [4–6]. Moreover, studies have also found that ginkgetin could enhance the body’s immunity and has an anti-tumor function [7,8].
Signal transducer and activator of transcription 3 (STAT3) is structurally activated in human tumors and a therapeutic target of tumor drug discovery. It is reported that ginkgo flavonoids exert their anti-tumor activity by inhibiting STAT3 in prostate tumor cells [7]. Ginkgetin enhances the therapeutic effect of cisplatin via ferroptosis-mediated disruption of the Nrf2/HO-1 axis in EGFR wild-type non-small-cell lung cancer [9]. Because of its biological activities, it has been widely recognized worldwide. At present, research on the potential pharmacological mechanism of flavonoids has become an important part of the world’s medical field. Scientists believed that flavonoids should have a unique pharmacological effect and will eventually become the most promising natural drug [10,11].
It is well known that primary carcinoma of the liver (PCL) refers to a highly malignant tumor developed from hepatocytes in the liver or epithelial cells of the inner bile duct system [12,13]. At present, most studies believe that many factors are involved in the process of hepatocellular carcinoma (HCC) formation, such as cirrhosis (chronic liver damage caused by fibrosis), hepatitis B virus (HBV) infection, hepatitis C virus (HCV) infection, alcohol abuse, and metabolic syndrome [3]. Other co-factors, such as tobacco inhalation and aflatoxin B1 (a fungal carcinogen present in food supplies associated with mutations in the tumor suppressor gene TP53) intake are well-characterized contributors to HCC. The outcome of HCC is the compiled action of many factors, which eventually lead to gene changes of cells, imbalance of regulatory functions, and induction tumors through their respective pathways [14,15]. Over the past decades, our understanding of the molecular pathogenesis of the disease has been significantly improved [16]. Genomic analysis has provided a clear picture of the main drivers responsible for tumor initiation and progression. Each HCC has an average of 40 genomic aberrations, among which few are considered drivers. Common mutations affect telomere maintenance (mutations in telomere reverse transcriptase (TERT)), Wnt pathway activation (mutations in catenin beta 1 (CTNNB1)), inactivation of cellular tumor antigen p53 (p53, encoded by TP53), chromatin remodeling (mutations in AT-rich interaction domain 1A (ARID1A)), Ras signaling, mammalian target of rapamycin (mTOR) signaling, and oxidative stress pathway activation. Only a handful of these drivers are currently druggable targets, such as amplification of fibroblast growth factor 19 (FGF19) [17]. It has been found that abnormalities in cell proliferation, apoptosis, and cell cycle are the key steps in tumorigenesis. Therefore, how to effectively induce or promote tumor cell apoptosis in vivo is a hot research spot and also the pharmacological mechanism and action pathway of chemotherapeutic drugs in the treatment of tumors [18,19].
At present, there is a great breakthrough in the laboratory and clinical research of Chinese medicine treatment of tumors. Traditional Chinese medicine can inhibit abnormal liver cell growth and proliferation by regulating the expression of abnormal genes and changing the cell cycle and cytokine levels [20,21]. Recent studies have shown that ginkgo biloba induces apoptosis of oral cavity cancer cells and activates caspase-3 [22,23]. Other studies have also shown that ginkgo biloba may inhibit tumor cell apoptosis by affecting the expression of c-myc, bcl-2, and c-fos genes [24]. Bcl-2 families are divided into two categories, one is anti-apoptosis, mainly including Bcl-2, Bcl-XL, Bcl-W, Mcl-1, and CED9, and the other is pro-apoptosis, including Bax, Bak, Bcl-XS, Bad, Bik, and Bid. A number of studies have shown that the anticancer effects of Ginkgo biloba extracts are probably associated with the arrestment of cell cycle transition from G2-M phase to G1 phase, interference of DNA synthesis in S phase, and induction of cancer cell apoptosis [25]. At present, Western medicines are not effective in reversing or blocking precancerous lesions. However, in recent years, many studies at home and abroad have shown that Ginkgo biloba extracts can intervene the development of precancerous lesions at various stages and canceration [26,27]. Laboratory studies on Ginkgo biloba extracts indicated that a variety of Ginkgo biloba extracts play important roles in inhibiting proliferation and inducing apoptosis of liver cancer cells. This may also provide a direction for the treatment of liver cancer.
In this study, the effects of Ginkgo biloba flavonoids on human HCC cell lines were observed, and their effects on cell apoptosis and cell cycle were explored. On this basis, the expression of cyclin A, cyclin B, CDK1, CDK2, and retinoblastoma (Rb) proteins (Rb) in human HCC cells induced by ginkgetin was further studied. Rb gene is susceptible to retinoblastoma. It is the first tumor suppressor gene cloned and sequenced in the world. Rb protein is distributed in the nucleus and a DNA-binding protein. In the G1 phase of the cell cycle, Rb protein is phosphorylated by cyclin-CDK (usually cyclin D/CDK4 or cyclin E/CDK2) to become pRb, releasing the transcription factor E2F1 from its binding complex to promote the transcription of cyclin and CDK proteins necessary for S phase, and eventually resulting in transition of cells from G1 phase to S phase. However, E2F1 protein preferentially binds to the retinoblastoma protein pRb in a cell cycle-dependent manner, which could mediate cell proliferation and p53-dependent/independent apoptosis at the same time. The ultimate goal of the study was to explore the molecular mechanism of ginkgetin inducing apoptosis of human HCC cells and to provide a scientific theoretical basis for the development and utilization of ginkgo.
Materials and methods
Culture and reagents
Human HCC cell lines, HepG2 and SK-HEP-1, were obtained from the China Center for Type Culture Collection, The First Affiliated Hospital of University of Southern China. Cells were cultured in high glucose Dulbecco’s Modified Eagle Medium (DMEM, GIBCO Corporation, USA) containing 10% fetal bovine serum (FBS, Hyclone, Logan, UT, USA) and 1% solution containing 100 units/ml penicillin and 100 mg/ml streptomycin at 37℃ in an incubator containing 5% CO2. Ginkgetin (purity ≥ 98%, Sigma-Aldrich, St. Louis, MO) was dissolved in dimethyl sulfoxide (DMSO, Sigma-Aldrich, St. Louis, MO) to obtain a 1 mM stock solution and added to the medium with specified concentrations. The final concentration of DMSO was less than 0.1%. Figure 1(a) shows the chemical structure of ginkgetin.
Figure 1.
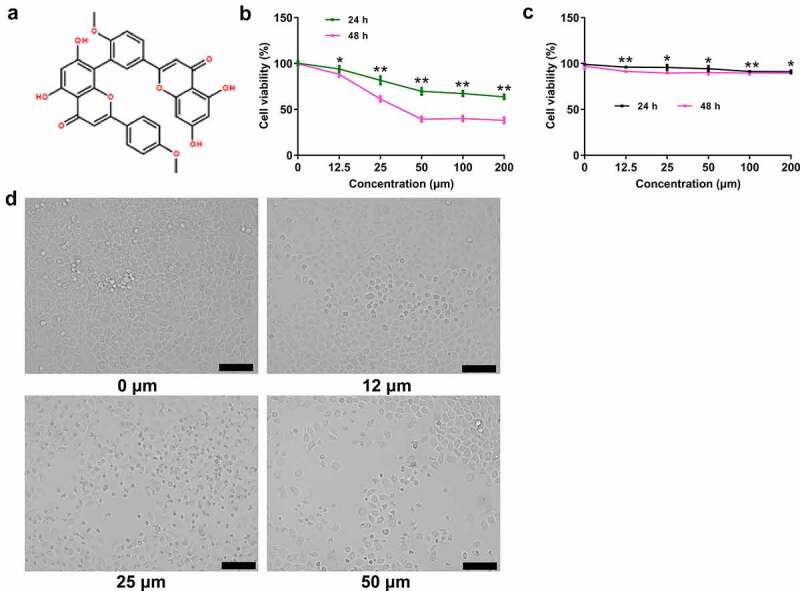
The effect of ginkgetin on HepG2 cell viability. (a) Chemical structure of ginkgetin, which has a molecular weight of 566.51. (b-c) The effects of ginkgetin on the viability of HepG2 and normal cells. Cells were treated with ginkgetin at the concentration of 0, 5, 10, 20, 30, 40, 50, and 60 μM for 24 and 48 h. The data are expressed as the mean ± SD (n = 3). *P < 0.05, **P < 0.01 compared with the control. (d) The effect of ginkgetin on morphological changes of HepG2 cells.
Cell viability assay
The treated cells were incubated with 3-(4,5-dimethylthiazole-2-yl)-2,5-diphenyltetrazole bromide (MTT) solution (5 mg/ml in PBS, ThermoFisher, USA) to induce the formation of methyl crystals. Then, the methyl crystals were dissolved in 100 μl DMSO, and the optical density (OD) at 570 nm was measured using an iMarkTM Microplate Reader (Bio-Rad, Richmond, CA). Cell viability was calculated as ODexperiment/ODcontrol × 100%.
Xenograft nude mice model
The establishment of xenograft nude mouse model and ginkgetin preparation were conducted as previously described [28]. Briefly, when tumors on the transplanted nude mice reached around 100 mm3, the mice were randomized divided into the control and ginkgetin groups. Both physiological saline (30 mg/kg) and ginkgetin (30 mg/kg) were administered by intraperitoneal injection once per 2–3 d, respectively. After 31 d, the nude mice were sacrificed, and tumors were removed and weighed. Animal experiments were performed in accordance with the National Institutes of Health Guide for Care and Use of Laboratory Animals.
Observation of morphological changes
Cells were prepared as 1×105/mL suspension and treated with or without ginkgetin. After 48 h of culture, cell morphology was observed under an inverted microscope (Jiangnan New Optics Co., Ltd. China).
Hoechst 33,258 staining
After further culture, apoptotic cells were stained with 10 g/mL Hochest 332 solution at 37°C for 10–15 min. After washout of the residual Hochest staining solution by PBS, the morphological changes of apoptotic cells were observed under a laser confocal microscope, and the cell images were collected.
Determination of the ratio of apoptotic and necrotic cells after staining with annexin V-fluorescein isothiocyanate (FITC)/propidium iodide (PI)
The treated cells were washed with PBS, resuspended, stained with 10 μL of Annexin V-FITC and 5 μL of PI in 300 μL of binding buffer at room temperature for 15 min, and immediately subjected to flow cytometric analysis.
Cell cycle analysis
The treated cells were digested, centrifuged, washed with pre-cooled PBS solution, and pelleted using centrifugation. The cells were fixed with 70% glacial ethanol at 4°C for 18 h. After centrifugation to discard the fixative solution, cells were treated with ribonuclease solution (1.0 g/L) at 37°C in a water bath for 30 min and stained with 250 µL of 75 µM propidium iodide (PI) for 30 min at 4 C in the dark. Cell cycles were analyzed by flow cytometry based on colored cells with excitation wavelength at 488 nm and the emission wavelength at 630 nm.
Release of cytochrome c
Cytoplasmic proteins were collected using nuclear and cytoplasmic extraction reagents (Pierce, Rockford, IL). Proteins were normalized using the BCA Protein Assay Kit (Pierce), and cytochrome c in the cytoplasm was determined using the Quantikine Cytochrome c Immunoassay Kit (R&D Systems).
Measurement of caspase activity
Caspase 3 and 8 activities were assessed by Caspase 3 and 8 Colorimetric Assay Kits (Keygen, Nanjing, China). After treatment with ginkgetin at different doses (0, 12.5, 25, and 50 μM) for different times (0 h, 12 h, 24 h, and 48 h), cells were collected, resuspended, and prepared as a suspension of 5 × 106 cells in 50 μl lysates. After normalized using the BCA protein kit (Abcam, UK), cells were incubated with 50 μl of reaction buffer and 5 μl of substrate solution for 4 h at 37°C in the dark. The optical density (OD) at 405 nm was measured using an iMark Microplate Reader (Bio-Rad, USA).
Western blot analysis
Cells were decomposed with protein extraction for 30 min and centrifuged at 12,000 r/min for 5 min at 4℃. After measuring protein concentration using BCA protein quantitative kits, 50 μg proteins were mixed with SDS Loading Buffer and fully denatured at 100℃ for 5 min, separated by SDS-PAGE electrophoresis and transferred onto PVDF membranes (ThermoFisher, USA). The membranes were blocked with 1% BSA (Amresco, USA) and incubated with polyclonal antibodies against GAPDH, Bax, Bcl-2, Bcl-XL, Rb, phosphorylated-Rb, E2F1 cyclin A, cyclin B, CDK1, and CDK2 (1:1000, Cell Signaling Technology) overnight at 4°C. After washed with 0.05% TBST (Sigma, USA) for 3 times, membranes were further incubated with 1:5000 HRP-labeled anti-rabbit secondary antibody for 1 h at room temperature and washed 3 times with TBS. After that, the gray values of the target bands and the internal reference band were recorded by ECL chemiluminescence (ThermoFisher, USA). The gray ratio of the target bands to the internal reference bands was used to analyze the relative expression of the target proteins.
Statistical methods
The monitoring data were analyzed by SPSS19.0 statistical software. All data were expressed as mean ± standard deviation (mean ± SD). Differences between two groups were analyzed using t test and among multiple groups were based using one-way ANOVA followed by the least significant difference analysis. P < 0.05 indicated a statistical significance.
Results
Ginkgetin decreased cell viability of human hepatocellular carcinoma cell
To study the effects of ginkgetin on the growth and survival of HCC cells, HepG2 cells were treated with ginkgetin at 0, 12.5, 25, 50, 100, and 200 μM for 24 and 48 h, respectively. The results shown in Figure 1(b) indicated that ginkgetin significantly reduced HepG2 cell viability in a dose-dependent manner (P < 0.05, P < 0.01). However, ginkgetin did not reduce the viability of normal cells (P > 0.05) (Figure 1(c)). Moreover, there were no significant differences in body weight and heart, liver, and renal indexes between mice in the control and ginkgetin treatment groups (P > 0.05) (Table 1).
Table 1.
Body mass growth rate, heart, liver, and kidney index (mean ± SD)
| Groups | n | Body mass growth rate | Heart index | Liver index | Kidney index |
|---|---|---|---|---|---|
| Control | 10 | 29.40 ± 22.35 | 0.41 ± 0.07 | 3.89 ± 0.60 | 0.80 ± 0.23 |
| Ginkgetin | 10 | 28.50 ± 12.78 | 0.38 ± 0.06 | 3.65 ± 0.72 | 0.79 ± 0.08 |
Figure 1(d) shows the morphological features of HCC cells. The cultured human HCC HepG-2 cells adhered well to the culture cells, stretched into a fusiform or polygonal shape, and tightly fused. After 12.5 μM ginkgetin treatment, cell adherence ability decreased, intercellular contour became more clear and loosened, proliferative ability weakened, more cells were round and floating, and the number of viable cells decreased. When the concentration of ginkgetin was 50.0 μM, the number of fragmented cells increased, and the number of living cells was further decreased.
Ginkgetin induced apoptosis of human HCC cells
To investigate the effect of ginkgetin on apoptosis of HCC cells, the apoptosis was detected by Hoechst staining and Annexin V-PI double staining. Figure 2(a) shows the results of Hochest33342 staining of HepG2 cells treated with ginkgetin at different concentrations. The cells in the control group were structurally intact and evenly colored, showing a pale blue fluorescence. When treated with 12.5 μM ginkgetin, the nuclear chromatin was concentrated, and some dark, blue fluorescent particles were visible in the cells. When treated with 25.0 μM ginkgetin, deep, bright blue fluorescent particles were seen in the cells, and the nuclei began to aggregate. When treated with 50.0 μM ginkgetin, many dense and densely stained bright blue granules appeared in the cells, and the nuclei were further concentrated and fragmented. When treated with ginkgetin for 48 h, the apoptotic rate of cells increased in a dose-dependent manner (Figure 2(b)). Moreover, when treated with 50 μM ginkgetin, the apoptotic rate increased significantly with treatment time increasing. These results indicated that ginkgetin affected HepG2 cell viability by inducing apoptosis.
Figure 2.
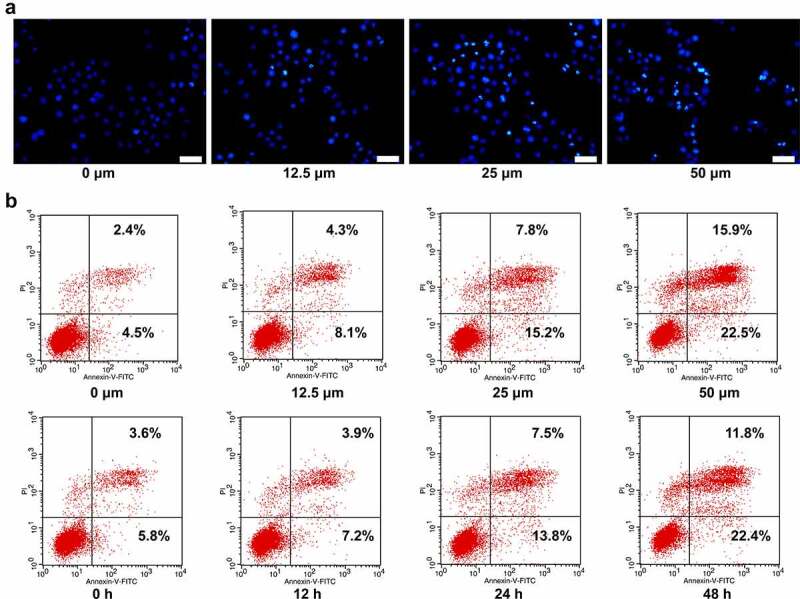
Ginkgetin induced apoptosis of HepG2 cells. (a) Hoechst 33,258 staining of HepG2 cells incubated with 12.5, 25, and 50 μM ginkgetin for 48 h. (b) Annexin V-PI staining of HepG2 cells incubated with 12.5, 25, and 50 μM ginkgetin for 48 h. (c) Annexin V-PI staining of HepG2 cells incubated with 50 μM ginkgetin for 12, 24, and 48 h.
Ginkgetin-triggered apoptosis was mediated by the mitochondrial pathway
Apoptosis can be triggered by the intrinsic/mitochondrial pathway or the exogenous/death receptor signaling pathway. To assess which pathway mediates ginkgetin-induced apoptosis, we studied the activation of caspase-3 by an intrinsic or extrinsic pathway, activation of caspase-8 by death receptor signal, and the release of cytochrome c. It can be seen from Figure 3(a-c) that when treated with 50 μM ginkgetin, caspase-3 activity and cytochrome c release increased about 3 times (P < 0.01) while the activity of caspase-8 did not change significantly (P > 0.05). These results showed that ginkgetin might mediate cell apoptosis through the mitochondrial pathway. Moreover, treatment with 12.5, 25, and 50 μM ginkgetin increased the protein levels of Bax slightly while decreased Bcl-2 and Bcl-XL protein levels significantly (Figure 3(d)). Furthermore, ginkgetin treatment inhibited the levels of p-SATA3 and SATA3 in a dose-dependent manner (Figure 3(e)).
Figure 3.
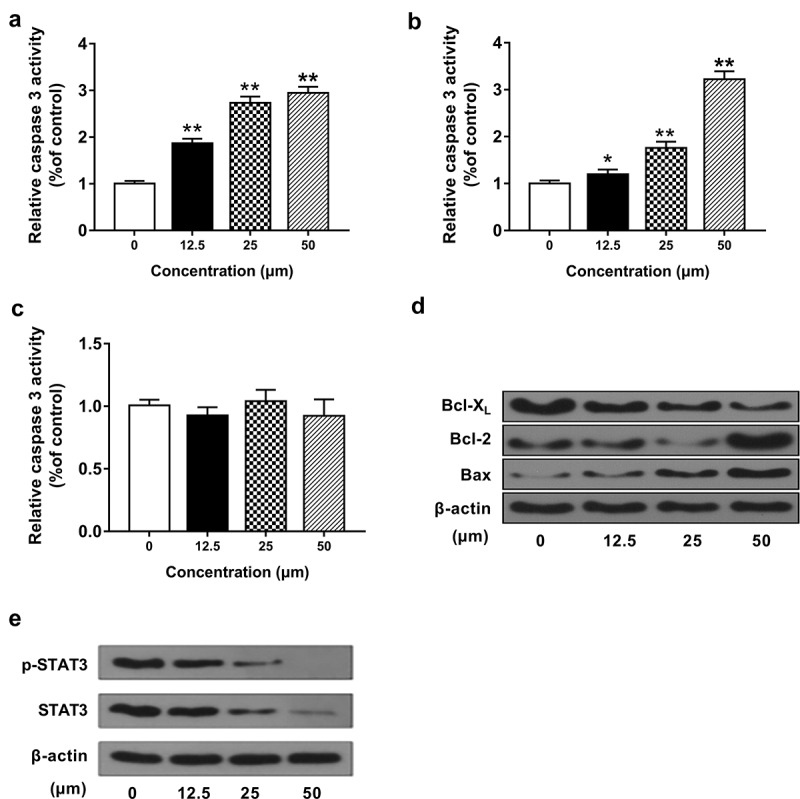
Ginkgetin induced apoptosis of HepG2 cells via the mitochondrial pathway. (a-c) The activity of caspase 3 (a), caspase 8 (c), and cytochrome c release (b). (d-e) The protein levels of Bax, Bcl-2, Bcl-XL, p-SATA3, and SATA3 in HepG2 cells incubated with 12.5, 25, and 50 μM ginkgetin for 48 h. The data are the mean ± SD (n = 3). *P < 0.05, **P < 0.01 compared with the control.
Ginkgetin initiated cell cycle arrest at S phase via influencing cell cycle-related factors
To investigate whether the apoptosis induced by ginkgetin is related to cell cycle changes, the dose and time effects of ginkgetin on cell cycle were examined. With the ginkgetin concentration increased from 0, 12.5, 25 to 50 μM, the population of cells at the G0/G1 phase decreased from 67.4% to 35.8%, that of cells at the S phase increased from 25.5% to 62.7%, and that of cells at G2/M phase decreased from 7.1% to 1.5%. The same trend was also observed after treatment with ginkgetin treatment for 12 h, 24 h, and 48 h. Especially when treated with 50 μM ginkgetin for 48 h, most cells were arrested at the S phase (Figure 4(a)).
Figure 4.
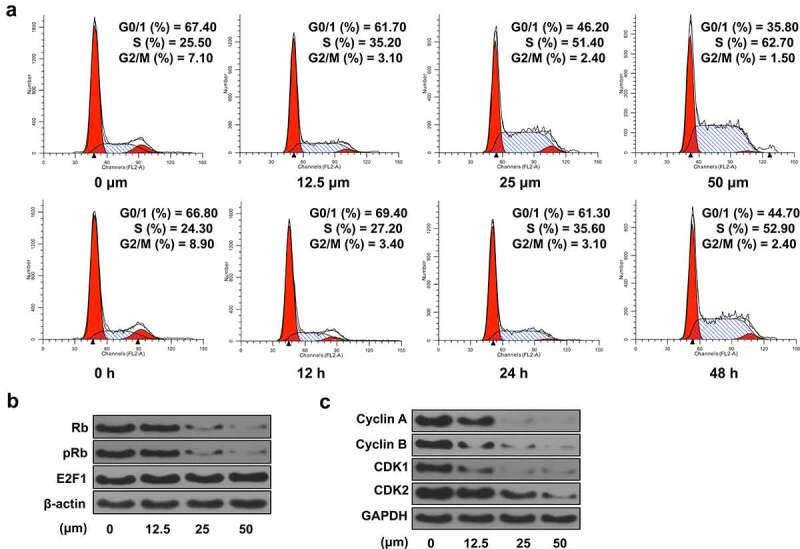
Ginkgetin initiated cell cycle arrest at S phase via cell cycle-related factors in HepG2 cells. (a) The dose- and time-dependent effects of ginkgetin on cell cycle distribution. Shown are the percentages of cells at G0/G1, S, and G2/M phases. (b and c) The protein levels of Rb, pRb, E2F1 (b), and cyclin A, cyclin B, CDK1, and CDK2 (c) in cells treated with 12.5, 25, and 50 nM ginkgetin for 48 h.
To investigate the mechanism by which ginkgetin triggers this cell cycle arrest, the changes in retinoblastoma protein (Rb) were determined. After 48 h of treatment with 12.5–50 μM ginkgetin, total Rb gradually decreased to an almost undetectable level. At the same time, phosphorylated Rb (pRb) level was also decreased significantly. However, total E2F1 protein expression was unchanged. E2F1 protein preferentially binds to pRb in a cell cycle-dependent manner and could mediate cell proliferation and p53-dependent/independent apoptosis at the same time. Thus, the results indicated that a decrease in Rb might contribute to the increased release of free E2F1 to trigger cells entering the S phase (Figure 4(b)). The level of cyclin A/CDK1 or CDK2 complex was also significantly reduced. In addition, a decrease in the expression of cyclin B/CDK1 complex, known as mitotic promoter (MPF), was also observed, indicating a blockade of the G2-M transition (Figure 4(c)). These results suggested that ginkgetin might simultaneously cause more liver cancer cells to pass through the G1 to S phase and stop the S phase output, leading to S phase arrest in the cells.
Anti-tumor effect of ginkgetin in human HCC cells
To clarify whether ginkgetin-induced apoptosis and cell cycle arrest were cell-specific, a correlation assay was performed using human HCC cell line SK-HEP-1. As shown in Figure 5(a), ginkgetin decreased SK-HEP-1 cell viability in a dose- and time-dependent manner, similar to that of HepG2 cells. When ginkgetin dose reached 50 μM, the cell viability did not continue to decrease (P < 0.05, P < 0.01). The results of Hoechst staining and caspase-3 activity assay confirmed that ginkgetin could induce apoptosis of SK-HEP-1 cells (P < 0.05, P < 0.01) (Figure 5(b,c)). In addition, the proportion of SK-HEP-1 cells in the S phase increased from 16.2% to 28.4% in a concentration-dependent manner. These phenomena were further supported by the fact that ginkgetin blocked the S phase of human HCC cells and induced apoptosis without cell line-specific effects (Figure 5(d)). As ginkgetin concentration increasing, the tumor weight became smaller and smaller (P < 0.05, P < 0.01) (Figure 5(e)).
Figure 5.
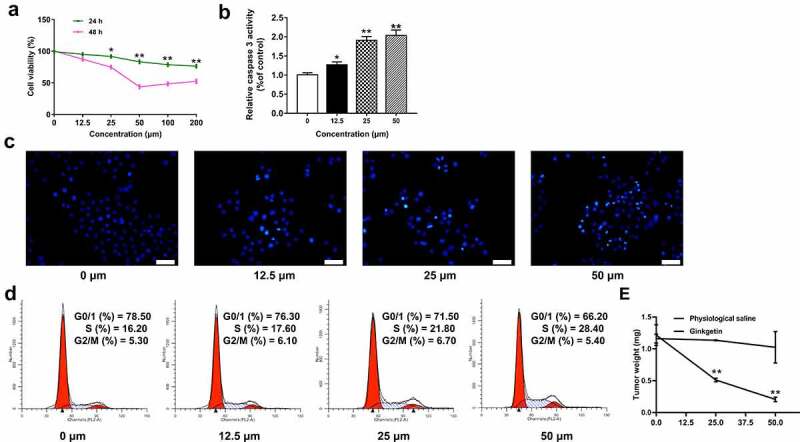
The effects of ginkgetin on human HCC cell line SK-HEP-1. (a and b) The effects of ginkgetin on cell viability (a) and caspase 3 activity (b). The data are the mean ± SD (n = 3). *P < 0.05, **P < 0.01 compared with the control. (c) Hoechst 33,258 staining of SK-HEP-1 cells treated with ginkgetin. (d) The effect of ginkgetin on cell cycle distribution. Cells were treated with 12.5, 25, and 50 μM ginkgetin for 48 h (B, C and D). (e) Mean tumor weight in each group at the end of treatment. Data are presented as the mean ± SD, n = 6. *p < 0.05.
Discussion
Ginkgo biloba leaves contain many different ingredients with different pharmacological effects [29]. Among them, ginkgetin is one of the main active ingredients. Related studies have confirmed that ginkgetin has anti-inflammatory, analgesic resistance, oxidative, hypolipidemic, and other pharmacological activities [30]. Because of its unique pharmacological activity, it has great value in clinical application.
In recent years, related basic medical and clinical medical researches have found that ginkgetin also has certain anti-tumor activity and plays corresponding roles in tumor cell proliferation, metastasis, and apoptosis, such as blocking tumor cell proliferation, reducing cancer cell growth, and promoting liver cancer cell apoptosis [31]. Related studies found that ginkgetin could inhibit prostate cancer (PC-3) cell proliferation, activate caspase-3, and attenuate the expression of survival genes at protein and mRNA levels [32]. In recent years, the anti-tumor effect of ginkgetin has been dementedly explored, and its pharmacological effects have been affirmed in related researches. However, its specific mechanism of action is still not fully understood, and further research is needed. Its main mechanism of action may be related to inhibiting tumor cell proliferation, inducing apoptosis, and interfering with protein kinases in cell signaling pathways [7,33].
This study explored the effects of ginkgetin on human HCC and its underlying mechanisms were explored using different methods. In the experiment, ginkgetin significantly reduced the viability of HepG2 cells. In addition, HepG2 cells treated with ginkgetin showed apoptotic morphological characteristics, such as increased apoptotic bodies and cell contractions with a significant concentration-dependent effect.
Annexin V-FITCTC binds to the exposed phosphatidylserine on apoptotic and necrotic cells to allow PI to enter at the late stage [34,35]. Annexin V-PI double staining found that the apoptosis rate of HCC cells was positively correlated with ginkgetin treatment time and concentration. Apoptosis could be triggered by the intrinsic/mitochondrial pathway or the exogenous/death receptor signaling pathway. Caspase-3 is an intrinsic or extrinsic pathway-activated protein, while caspase-8 is a signal-activated protein of death receptor [36,37]. When endogenous pathways are activated, cytochrome c is secreted from mitochondria to cytosol [38]. We found that caspase-3 activity and cytochrome c release increased with ginkgetin concentration increasing, but caspase-8 activity remained unchanged, suggesting that ginkgetin might induce cell apoptosis through the mitochondrial pathway.
The Bcl-2 protein family is one of the most important regulators of apoptosis and affects the stability of mitochondrial membranes after endogenous or exogenous stimuli. The ratio of pro-apoptotic proteins such as Bax and anti-apoptotic proteins such as Bcl-2 and Bcl-XL plays an important role in determining whether cells are killed or alive [39,40]. Ginkgetin triggered a decrease in the protein levels of Bcl-2 and Bcl-XL. Therefore, we speculated that ginkgetin could increase Bax/Bcl-2 ratio and Bcl-XL expression, leading to mitochondria destruction and cytochrome c release, then activate caspase-3, and ultimately trigger apoptosis. Our experimental results confirm our conjecture. Previous studies have shown that ginkgo flavonoids could induce apoptosis of prostate cancer cells by regulating STAT3 expression [7], and apoptosis of non-small-cell lung cancer cells [9].
Cell growth needs to pass through G1, S, G2 and M4 phases, and cell cycle plays an important role in screening anticancer drugs [41]. When cells enter the S phase from G1, three proteins, cyclin Ds/ CDK4 /6, cyclin E/CDK2, and cyclin A/CDK2, play important roles. Cyclin D1 phosphorylates cell cycle-related protein Rb. Phosphorylated Rb then binds to transcription factor E2F1 at rest, releases activated E2F1, and promotes S phase to initiate the required gene transcription. Enabling cells to enter S phase from G1, also known as a restriction point, is the key to determining whether the cell cycle continues [42,43]. Our study found that ginkgetin could arrest cells at the S phase of the cell cycle and decrease Rb phosphorylation significantly. Any changes in total E2F1 protein suggested that the reduction of Rb might contribute to the increase of free E2F1 release and then trigger cells to enter S phase more quickly. Besides the cyclin A/CDK1 or CDK2 complex, the cyclin B/CDK1 complex was also significantly reduced. Therefore, we can infer that ginkgetin could promote more cells to pass through G1 to S phase by reducing the expression of major proteins in cell cycle, stop exporting from S phase, and ultimately make cells accumulate in the S phase. The results of human HCC cell line SK-HEP-1 showed that ginkgetin could induce nonspecific cell apoptosis and cycle arrest.
As far as we know, this is the first systematic study to show that compared with normal cells, ginkgetin could induce apoptosis and cell cycle arrest of HCC cells. Although our experimental results were only carried out at the cellular level and no clinical trials were carried out, we carried out tumorigenesis experiments in mice and found that ginkgetin did have a certain inhibitory effect on HCC. Our study suggests that ginkgetin may be used as a potential drug for HCC treatment and provides a new direction for HCC treatment in the future.
Conclusion
Ginkgetin could inhibit proliferation and promote apoptosis of human HCC cells and trigger the S phase arrest of the cell cycle. In addition, the effect of ginkgetin was significantly correlated with its concentration. To further explore whether ginkgetin plays an important role in clinical research, the in vivo effect of ginkgetin on liver cancer cells needs to be further explored.
Funding Statement
This work was supported by Hunan Natural Science Foundation, Youth Project (Grant No. 2018JJ3466).
Disclosure statement
No potential conflict of interest was reported by the authors.
Data availability statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Author contribution
LYC: experiment, analysis, manuscript writing; YHN and PHZ: experiment, analysis; WJY: supervision, experiment, analysis, manuscript writing; All authors read and approved the final manuscript.
References
- [1].Yan J, Mao D, Liu X, et al. Isolation and functional characterization of a circadian-regulated CONSTANS homolog (GbCO) from Ginkgo biloba. Plant Cell Rep. 2017;36(9):1387–1399. [DOI] [PubMed] [Google Scholar]
- [2].Zhou YH, Yu JP, Liu YF, et al. Effects of Ginkgo biloba extract on inflammatory mediators (SOD, MDA, TNF-alpha, NF-kappaBp65, IL-6) in TNBS-induced colitis in rats. Mediators Inflamm. 2006;2006(5):92642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Tang Y, Zhou G, Yao L, et al. Protective effect of Ginkgo biloba leaves extract, EGb761, on myocardium injury in ischemia reperfusion rats via regulation of TLR-4/NF-kappaB signaling pathway. Oncotarget. 2017;8(49):86671–86680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Dartigues JF, Grasset L, Helmer C, et al. Ginkgo biloba extract consumption and long-term occurrence of death and dementia. J Prev Alzheimers Dis. 2017;4(1):16–20. [DOI] [PubMed] [Google Scholar]
- [5].Qiu J, Chen X, Netrusov AI, et al. Screening and identifying antioxidative components in Ginkgo biloba Pollen by DPPH-HPLC-PAD coupled with HPLC-ESI-MS2. PLoS One. 2017;12(1):e0170141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Yang X, Zheng T, Hong H, et al. Neuroprotective effects of Ginkgo biloba extract and Ginkgolide B against oxygen-glucose deprivation/reoxygenation and glucose injury in a new in vitro multicellular network model. Front Med. 2018;12(3):307–318. [DOI] [PubMed] [Google Scholar]
- [7].Jeon YJ, Jung SN, Yun J, et al. Ginkgetin inhibits the growth of DU-145 prostate cancer cells through inhibition of signal transducer and activator of transcription 3 activity. Cancer Sci. 2015;106(4):413–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Sun CM, Syu WJ, Huang YT, et al. Selective cytotoxicity of ginkgetin from Selaginella moellendorffii. J Nat Prod. 1997;60(4):382–384. [DOI] [PubMed] [Google Scholar]
- [9].Lou J-S, Zhao L-P, Huang Z-H, et al. Ginkgetin derived from Ginkgo biloba leaves enhances the therapeutic effect of cisplatin via ferroptosis-mediated disruption of the Nrf2/HO-1 axis in EGFR wild-type non-small-cell lung cancer. Phytomedicine. 2021;80:153370. [DOI] [PubMed] [Google Scholar]
- [10].Baek SH, Lee JH, Ko JH, et al. Ginkgetin blocks constitutive STAT3 activation and induces apoptosis through induction of SHP-1 and PTEN tyrosine phosphatases. Phytother Res. 2016;30(4):567–576. [DOI] [PubMed] [Google Scholar]
- [11].Hayashi K, Hayashi T, Morita N.. Mechanism of action of the antiherpesvirus biflavone ginkgetin. Antimicrob Agents Chemother. 1992;36(9):1890–1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Balasegaram M. Complete hepatic dearterialization for primary carcinoma of the liver. Report of twenty-four patients. Am J Surg. 1972;124(3):340–345. [DOI] [PubMed] [Google Scholar]
- [13].Maharajan K, Hey HWD, Tham I, et al. Solitary vertebral metastasis of primary clear cell carcinoma of the liver: a case report and review of literature. J Spine Surg. 2017;3(2):287–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Hu H, Zhu W, Qin J, et al. Acetylation of PGK1 promotes liver cancer cell proliferation and tumorigenesis. Hepatology. 2017;65(2):515–528. [DOI] [PubMed] [Google Scholar]
- [15].Zheng C, Zheng L, Yoo JK, et al. Landscape of infiltrating T Cells in liver cancer revealed by single-cell sequencing. Cell. 2017;169(7):1342–1356. e1316. [DOI] [PubMed] [Google Scholar]
- [16].Zucman-Rossi J, Villanueva A, Nault JC, et al. Genetic landscape and biomarkers of hepatocellular Carcinoma. Gastroenterology. 2015;149(5):1226–1239.e1224. [DOI] [PubMed] [Google Scholar]
- [17].Forner A, Reig M, Bruix J. Hepatocellular carcinoma. Lancet. 2018;391(10127):1301–1314. [DOI] [PubMed] [Google Scholar]
- [18].Li WF, Dai H, Ou Q, et al. Overexpression of microRNA-30a-5p inhibits liver cancer cell proliferation and induces apoptosis by targeting MTDH/PTEN/AKT pathway. Tumour Biol. 2016;37(5):5885–5895. [DOI] [PubMed] [Google Scholar]
- [19].Liang L, Wong CM, Ying Q, et al. MicroRNA-125b suppressesed human liver cancer cell proliferation and metastasis by directly targeting oncogene LIN28B2. Hepatology. 2010;52(5):1731–1740. [DOI] [PubMed] [Google Scholar]
- [20].Liao YH, Lin CC, Lai HC, et al. Adjunctive traditional Chinese medicine therapy improves survival of liver cancer patients. Liver Int. 2015;35(12):2595–2602. [DOI] [PubMed] [Google Scholar]
- [21].Liao YH, Lin CC, Li TC, et al. Utilization pattern of traditional Chinese medicine for liver cancer patients in Taiwan. BMC Complement Altern Med. 2012;12:146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Kim KS, Rhee KH, Yoon JH, et al. Ginkgo biloba extract (EGb 761) induces apoptosis by the activation of caspase-3 in oral cavity cancer cells. Oral Oncol. 2005;41(4):383–389. [DOI] [PubMed] [Google Scholar]
- [23].Mu J, Liu T, Jiang L, et al. The traditional Chinese medicine baicalein potently inhibits gastric cancer cells. J Cancer. 2016;7(4):453–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Xu AH, Chen HS, Sun BC, et al. Therapeutic mechanism of ginkgo biloba exocarp polysaccharides on gastric cancer. World J Gastroenterol. 2003;9(11):2424–2427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Xu A, Jia X, Chen H, et al. Studies of ginkgo biloba testa polysaccharides in inhibiting liver cancer and inducing apoptosis of the liver cancer cell in mice. Trad Chine Drug Res Clin Pharmacol. 2001;12(5):340–341. [Google Scholar]
- [26].Dong S, Su SB. Advances in mesenchymal stem cells combined with traditional Chinese medicine therapy for liver fibrosis. J Integr Med. 2014;12(3):147–155. [DOI] [PubMed] [Google Scholar]
- [27].Wang H, Wu X, Lezmi S, et al. Extract of Ginkgo biloba exacerbates liver metastasis in a mouse colon cancer Xenograft model. BMC Complement Altern Med. 2017;17(1):516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Lou JS, Bi WC, Chan GKL, et al. Ginkgetin induces autophagic cell death through p62/SQSTM1-mediated autolysosome formation and redox setting in non-small cell lung cancer. Oncotarget. 2017;8(54):93131–93148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Van Beek TA, Scheeren HA, Rantio T, et al. Determination of ginkgolides and bilobalide in Ginkgo biloba leaves and phytopharmaceuticals. J Chromatogr A. 1991;543:375–387. [Google Scholar]
- [30].Byoung-Mog K, Lee Y-J. Ginkgetin inhibits the growth of cancer cells via the cell cycle arrest at G2-phase. [abstract]. Proceedings of the 105th Annual Meeting of the American Association for Cancer Research; 2014; San Diego, CA. [Google Scholar]
- [31].Pan LL, Wu WJ, Zheng GF, et al. Ginkgetin inhibits proliferation of human leukemia cells via the TNF-alpha signaling pathway. Z Naturforsch C J Biosci. 2017;72(11–12):441–447. [DOI] [PubMed] [Google Scholar]
- [32].You OH, Kim SH, Kim B, et al. Ginkgetin induces apoptosis via activation of caspase and inhibition of survival genes in PC-3 prostate cancer cells. Bioorg Med Chem Lett. 2013;23(9):2692–2695. [DOI] [PubMed] [Google Scholar]
- [33].Lee YJ, Kang YR, Lee SY, et al. Ginkgetin induces G2-phase arrest in HCT116 colon cancer cells through the modulation of bMyb and miRNA34a expression. Int J Oncol. 2017;51(4):1331–1342. [DOI] [PubMed] [Google Scholar]
- [34].Lee J, Lee S, Kim SL, et al. Corn silk maysin induces apoptotic cell death in PC-3 prostate cancer cells via mitochondria-dependent pathway. Life Sci. 2014;119(1–2):47–55. [DOI] [PubMed] [Google Scholar]
- [35].Ma J, Liu J, Lu C, et al. Pachymic acid induces apoptosis via activating ROS-dependent JNK and ER stress pathways in lung cancer cells. Cancer Cell Int. 2015;15:78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Fianco G, Cenci C, Barila D. Caspase-8 expression and its Src-dependent phosphorylation on Tyr380 promote cancer cell neoplastic transformation and resistance to anoikis. Exp Cell Res. 2016;347(1):114–122. [DOI] [PubMed] [Google Scholar]
- [37].Zhao X, Wang D, Zhao Z, et al. Caspase-3-dependent activation of calcium-independent phospholipase A2 enhances cell migration in non-apoptotic ovarian cancer cells. J Biol Chem. 2006;281(39):29357–29368. [DOI] [PubMed] [Google Scholar]
- [38].Wolf BB, Schuler M, Li W, et al. Defective cytochrome c-dependent caspase activation in ovarian cancer cell lines due to diminished or absent apoptotic protease activating factor-1 activity. J Biol Chem. 2001;276(36):34244–34251. [DOI] [PubMed] [Google Scholar]
- [39].Danial NN. BCL-2 family proteins: critical checkpoints of apoptotic cell death. Clin Cancer Res. 2007;13(24):7254–7263. [DOI] [PubMed] [Google Scholar]
- [40].Reed JC, Miyashita T, Takayama S, et al. BCL-2 family proteins: regulators of cell death involved in the pathogenesis of cancer and resistance to therapy. J Cell Biochem. 1996;60(1):23–32. [DOI] [PubMed] [Google Scholar]
- [41].Gamet-Payrastre L, Li P, Lumeau S, et al. Sulforaphane, a naturally occurring isothiocyanate, induces cell cycle arrest and apoptosis in HT29 human colon cancer cells. Cancer Res. 2000;60(5):1426–1433. [PubMed] [Google Scholar]
- [42].Lees E, Faha B, Dulic V, et al. Cyclin E/cdk2 and cyclin A/cdk2 kinases associate with p107 and E2F in a temporally distinct manner. Genes Dev. 1992;6(10):1874–1885. [DOI] [PubMed] [Google Scholar]
- [43].Taya Y, Jung HK, and Ikeda M, et al. Cdk4-cyclin D1 and Cdk2-cyclin E/A phosphorylate different sites in the RB protein. In: Mihich E, and Hartwell L, editors. Genomic Instability and Immortality in Cancer. Pezcoller Foundation Symposia. Vol 8 (pp. 229-231). Boston (MA): Springer; 1997. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.