

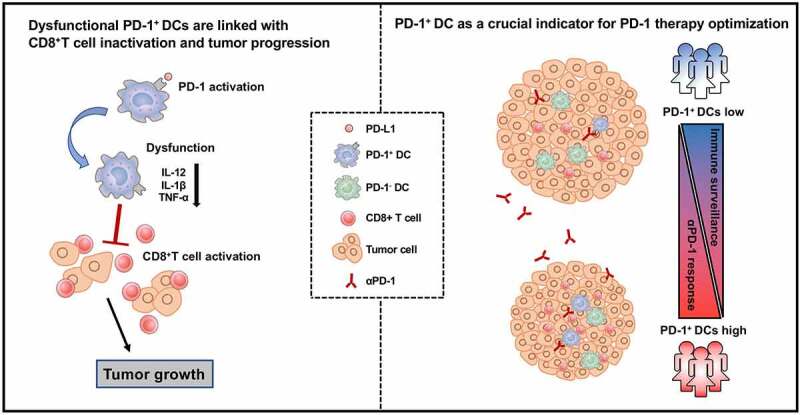
ABSTRACT
Various predictive biomarkers are needed to select candidates for optimal and individualized treatments. Tumor‐infiltrating immune cells have gained increasing interest in cancer research for the prediction of therapeutic response and survival. However, the role of dendritic cells (DCs) in PD-1 blockade immunotherapy remains unclear. In this study, we identified a population of PD-1+ DCs in the tumor microenvironment (TME) of cervical cancer (CC). The accumulation of PD-1+ DCs in cervical tumors was correlated with advanced stages, elevated preoperative squamous cell carcinoma antigen levels and lymph-vascular space invasion. PD-1 expression was induced on activated tumor-associated DCs (TADCs) in vitro compared with their resting counterparts. This PD-1+ DC population was characterized by reduced secretion of cytokines (IL-12, TNF-α, and IL-1β) and dysfunctional induction of T cell proliferation and cytotoxic reaction. PD-1 blockade significantly reinvigorated PD-1+ DCs to release IL-12, TNF-α, and IL-1β compared with PD-1- DCs. TILs from samples with higher PD-1+ DC infiltration could be induced to achieve a greater killing effect of PD-1 blockade treatment. Our findings suggested a role for PD-1+ DCs in immune surveillance dysfunction and CC progression. PD-1+ DC density in the TME may serve as a diagnostic factor for predicting the optimal beneficiaries of PD-1/PD-L1 blockade immunotherapy in CC.
KEYWORDS: Dendritic cell, cervical cancer, immunotherapy, PD-1, predictive marker
Graphical abstract
1. Introduction
Despite improvements in screening and prophylactic vaccination, cervical cancer (CC) remains the fourth most common malignancy and third leading cause of cancer-related mortality among women worldwide.1 It has been widely accepted that persistent high-risk human papillomavirus (HPV) infection is the major risk factor for CC.2 In recent years, a better understanding of HPV tumor–host immune system interactions supporting the immunogenicity of CC, and the development of new therapeutics targeting immune checkpoints have generated interest in the use of immunotherapy in advanced cervical cancer.3,4 However, it is desirable to explore predictive biomarkers to select candidates for optimal treatment and design individualized therapy.
Tumor‐infiltrating immune cells have gained increasing interest in cancer research given that these cells predict the response to therapy and survival.5 Intratumoral immune cells exhibit potential as biomarkers in different cancer types; however, each subtype has its own unique role in antitumor immunity.6 In the CC tumor microenvironment (TME), cell-mediated immunity is impaired in general. Decreased accumulation of tumor-infiltrating CD4 + T cells and a reduced CD4+/CD8 + T cell ratio correlate with rapid tumor growth and lymph node metastasis.7 Moreover, higher T cell accumulation (including CD3+, and CD4 + T cells) and an increased CD4+/CD8 + T cell ratio are associated with a decreased risk of relapse and better prognosis.8,9 The correlation of CD8 + T cell infiltration and lymph node metastasis or survival differs in studies.7,8,10 Regulatory T cells (Tregs), a population of immunosuppressive immune cells, were significantly associated with unfavorable outcomes in squamous cell carcinoma (SCC) but not in adenocarcinoma.11,12
As inducers of the T cell response, dendritic cells (DCs) are the foundation of antitumor immunity and are critical for responses to cytotoxic and targeted agents.13 The density of DCs in tumor tissue has been reported to correlate with prognosis in certain human carcinomas.14–16 In CC, it has been reported that the number of mature DCs and the cDC2 proportion are significantly decreased in the TME, while the pDC proportion is increased.7,17 However, the clinical impact and biological properties of intratumoral DCs are not completely understood. Here, we report the role of dysfunctional PD-1+ DCs in immune surveillance and CC progression. In addition, the density of PD-1+ DCs in the TME may serve as a diagnostic factor for predicting the optimal beneficiaries of PD-1/PD-L1 blockade immunotherapy.
2. Materials and methods
2.1. Patients and specimens
A total of 40 patients pathologically diagnosed with CC at the Obstetrics and Gynecology Hospital of Fudan University from December 2019 to September 2021 were enrolled in this study. At least two hospital pathologists histologically assessed the tumor specimens. Fresh tumor tissue specimens from each patient were obtained after surgical resection, and each tumor was bisected for subsequent phenotypic analysis and treatment assays. Blood samples were obtained from two healthy donors to generate monocyte-derived DCs. Patients and clinical data were collected through chart review and electronic patient records. HPV infection and SCC antigen levels were determined by preoperative examination reports. All procedures using human tissues conformed to the guidelines of the Declaration of Helsinki and Tokyo for human experimentation and were approved by the Ethics Committee of the Obstetrics and Gynecology Hospital of Fudan University (2019–123). Informed consent was obtained from all subjects.
2.2. Tissue dissociation
Collected fresh patient tumor samples were minced into 1-mm3 pieces with a scalpel and then digested in RPMI 1640 medium supplemented with 0.1% collagenase type IV, 0.01% hyaluronidase and 0.002% DNase I (Sigma-Aldrich, St. Louis, MO, USA) for 2 hours at 37°C in 5% CO2. Then, the single-cell suspensions derived from tumors were filtered through a 100-μm strainer and washed twice with phosphate-buffered saline (PBS).
2.3. Cell culture
The TC-1 and HeLa cervical cancer cell lines were maintained in RPMI 1640 medium (Thermo Fisher Scientific, Waltham, MA, USA) supplemented with penicillin-streptomycin (Sigma-Aldrich) and 10% fetal bovine serum (FBS) (Thermo Fisher Scientific, Waltham, MA, USA). The B16 and B16-OVA cells were maintained in DMEM (Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 1% penicillin-streptomycin and 10% FBS. To prepare tumor-conditioned medium (TCM), 5 × 106 TC-1 and HeLa cells were kept in 10 mL complete medium containing 10% FBS in a 100 mm cell culture dish for 48 hours. Supernatants were harvested and stored at −80°C for later use.
Bone marrow-derived DCs (BMDCs) and tumor-associated DCs (TADCs) were generated from 6- to 8-week-old C57BL/6 mice as previously described.18 In brief, single bone marrow cells were cultured in RPMI 1640 medium containing 10% FBS, 20 ng/mL mouse granulocyte macrophage colony stimulating factor (mGM-CSF), 10 ng/ml mouse interleukin 4 (mIL-4) (PeproTech, Rocky Hill, NJ, USA), 100 IU/mL penicillin G, and 100 mg/mL streptomycin sulfate with or without the addition of TC-1 TCM (20% by volume). PMBCs from healthy donors were isolated by Ficoll and monocytes were isolated from PBMCs via plastic adherence in tissue-culture flasks. Monocyte-derived DCs (Mo-DCs) were generated in X–VIVO 15 supplemented with 100 ng/ml of rhIL-4 and 100 ng/ml rhGM-CSF (PeproTech, Rocky Hill, NJ, USA), 100 IU/mL penicillin G, and 100 mg/mL streptomycin sulfate with or without the addition of HeLa TCM (20% by volume). After 6 days, 1 μg/mL lipopolysaccharide (LPS; PeproTech, NY, USA) was added to the culture medium for further stimulation.
2.4. DC endocytosis assay
DCs were incubated with 0.5 g/ml FITC-dextran (40,000 Mr) (Sigma, USA, FD40S-100 MG) for 1 hour at 37°C and then analyzed by flow cytometry for FITC fluorescence expression. Cells incubated in FITC-dextran solution at 4°C were used as a negative control.
2.5. Allogenic mixed lymphocyte reaction (MLR)
TADCs were generated from the bone marrow of C57BL/6 mice, and Mo-DCs were generated from PBMCs. Then, PD-1+ and PD-1- DCs were selected by incubation with an APC-conjugated anti-mouse PD-1 antibody followed by incubation with magnetic anti-APC nanobeads (Biolegend, 480071) and isolation using a magnetic separator according to the manufacturer’s instructions (Biolegend). These cells were then washed three times with ice-cold PBS and plated into 96-well U-bottom culture plates (Corning, NY, USA) at 1 × 104 cells/well as stimulators for T cells.
For MLR with TADCs, CD3 + T cells were obtained from splenocytes of allogeneic Balb/c mice purified with anti-CD3 nanobeads (Biolegend, 480099). For MLR with Mo-DCs, allogenic lymphocytes were obtained from PBMCs of other healthy donors isolated by plastic inadhesion in tissue-culture flasks. Purified mouse CD3 + T cells or human lymphocytes (responder cells) were labeled with arboxyfluorescein succinimidyl ester (CFSE; Dojindo, Kumamoto, Japan) and then added to 96-well U-bottom culture plates at 1 × 105 cells/well. The plates were incubated at 37°C in 5% CO2 for 72 hours. T cell proliferation was measured by evaluating CFSE dilution. The production of interferon gamma (IFN-γ) by T cells was measured using flow cytometry.
2.6. Tumor specific antigen T cell assay
PD-1+ and PD-1-TADCs were sorted and pulsed with OVA peptide 257–264 (MedChemExpress, NJ, USA, HY-P1489) at 40 μg/ml for 24 hours. CD8 + T cells were purified from OT-I mice spleen with anti-CD8a nanobeads (Biolegend, 480135) and stimulated with OVA-loaded PD-1+ or PD-1- TADCs for 72 hours at 37°C to generate antigen-specific OT-I T cells. The proliferation of OT-I T cells was measured by CFSE dilution as described in the MLR reaction. For the cytotoxic T lymphocyte (CTL) assay, antigen-specific OT-I T cells (effector cells) were harvested and then cultured together with 2000 B16 or B16-OVA cells (target cells) in 96-well plates at effector:target cell ratios of 100:1 for 18 hours. The suspensions were collected to evaluate the lactate dehydrogenase (LDH) leakage level using a Cytotoxicity LDH Assay Kit-WST (Dojindo, Kumamoto, Japan) according to the manufacturer’s protocol. The percentage of specific lysis was calculated as follows: % specific lysis = [(experimental release – E spontaneous release – T spontaneous release)/(T maximum release -T spontaneous release)] Χ 100%.
2.7. Ex vivo stimulations
Isolated single cells derived from tumors were cultured in X–VIVO 15 medium (Lonza, Switzerland) supplemented with 2% human serum (Biological Industries, USA) at 37°C and 5% CO2. Cells were treated with an anti-PD-1 antibody (pembrolizumab, 20 μg/ml) for 24 hours and then subjected to phenotypic analysis by flow cytometry.
2.8. Flow cytometry
For characterization of in vitro-cultured DCs, CC tissue-isolated cells and ex vivo-stimulated cells, cells were blocked with Fc receptor (Human TruStain FcX™, 422302, Biolegend), and incubated for 30 min in the dark with antibodies recognizing specific surface proteins. An Annexin V apoptosis detection kit with propidium iodide (PI; BioLegend, 640932) was used to analyze cell apoptosis. For the detection of intracellular proteins, cells were stimulated for 6 hours with 10 μg/ml brefeldin A and 2 μM ionomycin (Absin) before staining. Cells were then fixed and permeabilized using fixation/permeabilization wash buffer (BioLegend) and stained with antibodies for 30 min in the dark. For Ki67 staining, cells were incubated in 70% ethanol for 1 hour at −20°C and then stained with an anti-Ki67 antibody (350519; BioLegend). All antibodies used in this study are listed in Supplementary Table 1. All samples were run on a CytoFLEX platform (Beckman Coulter, Brea, CA, USA) and analyzed using FlowJo version 10.8 software (BD Biosciences).
2.9. Statistical analysis
All data are expressed as the means ± SDs of a minimum of three independent experiments. Student’s t-tests were used to compare two groups, one-way analysis of variance (ANOVA) with least significant difference (LSD) tests was used to compare multiple groups, and linear regression was used to identify the association between two indicators. Additionally, two-tailed paired t-tests were used for paired comparisons. P-values <0.05 were considered statistically significant.
3. Results
3.1. Identification of enriched intratumoral PD-1+ DCs in advanced CC
To assess DC infiltration, 40 resected fresh CC specimens were dissociated into single-cell suspensions and analyzed by flow cytometry. Due to the scarcity of DC-exclusive markers, we used a depletion strategy as in a previous study to identify total DCs.19 A series of lineage markers whose expression was low or absent in DCs including CD3, CD19, CD56, CD14, and CD16 (collectively defined lineage markers) were used to exclude contaminant cells. Total DCs were selected by CD45 positive expression, HLA-DR high expression, and lineage-negative expression (Supplementary Figure 1a).
The clinical characteristics of the patient cohort used in this analysis are shown in Supplementary Table 2. We examined the relationship between DC infiltration and disease progression in patients with different stages ranging from FIGO (International Federation of Gynecology and Obstetrics) stages I to III. No significant correlation was observed between the percentage of DCs in the CD45+ leukocyte population and clinical stage (Figure 1a), which is consistent with a previous report.20 Recently, PD-1 was found to be expressed in innate cells, including macrophages, MDSCs, and DCs.21 We analyzed PD-1 expression on intratumoral total DCs and found that higher grade tumor stages were correlated with an increased percentage of PD-1+ DCs (Figure 1b). Considering that the infiltration number of CD45+ cells varied significantly (Supplementary Figure 2a), the absolute number of DCs or PD-1+ DCs was also calculated, and the results consistently showed that the accumulation of PD-1+ DCs increased in the higher stage (Supplementary Figure 2b). These findings suggested that abundant PD-1+ DCs were associated with malignant progression. The relationships between intratumoral PD-1+ DCs and various clinicopathological features were further assessed. PD-1+ DC accumulation in the tumor bed appeared to be increased in patients with elevated preoperative SCC antigen levels and lymph-vascular space invasion (LVSI) (Figure 1c,d, Supplementary Figure 2c,d). However, the differences in the percentage or absolute number of total DCs did not reach statistical significance (Figure 1c,d, Supplementary Figure 2c,d). In our experiments, we found that HPV infection was associated with a higher percentage of PD-1+ cells in DCs (Supplementary Figure 2e) and an increased absolute number of total DCs (Supplementary Figure 2f). However, samples without HPV infection accounted for only 4 of the 40 total cases, and we could not determine whether the significant difference was due to random error. In addition, no significant differences in age, histological type, tumor size, or stromal invasion depth were noted for either total DC infiltration or PD-1+ DC accumulation. Overall, these data suggest a potential role for PD-1+ DCs rather than total DCs in CC progression and poor clinical outcomes.
Figure 1.
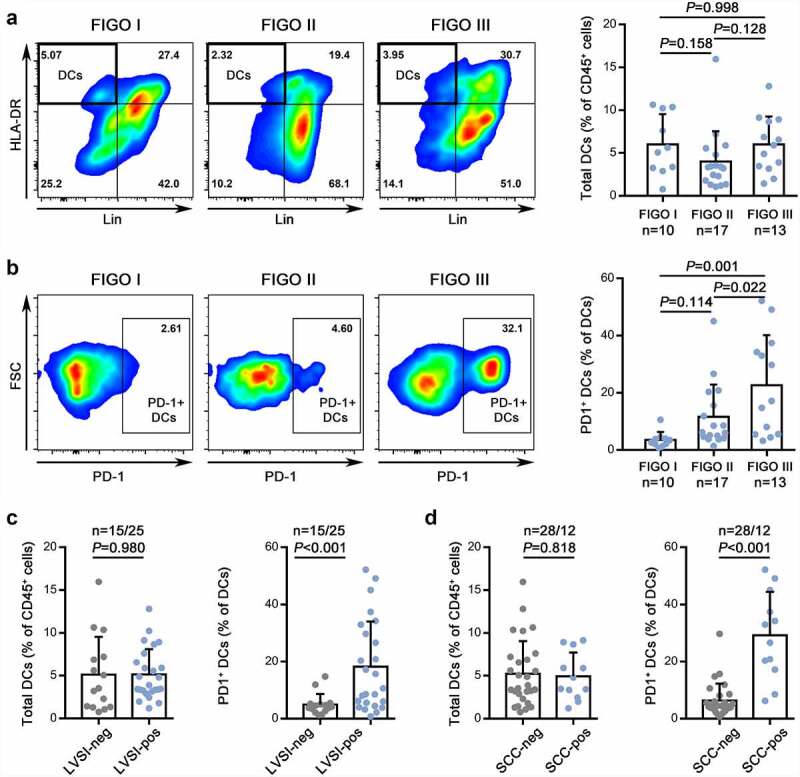
Identification of enriched intratumoral PD-1+ DCs in advanced CC.
(a,b) Left: The gating strategies for DCs (A) and PD-1+ DCs (B) in CC at different stages. Right: The percentages of DCs (A) and PD-1+ DCs (B) quantified in FIGO stages I, II, and III of CC.(c,d) The percentage of DCs (left) and PD-1+ DCs (right) in CC with or without lymph-vascular space invasion (C) or elevated preoperative SCC antigen levels (D).
3.2. Intratumoral PD-1+ DCs with highly expressed costimulated markers exhibit dysfunctional properties
DCs are a heterogeneous population composed of several distinct subsets. To precisely define these PD-1+ DCs, different DC subgroups were discriminated by the expression of CD11c, CD1c, CD141, CD123, CD16, and CCR7 and then measured with PD-1 levels (Supplementary Figure 1a) in 10 samples. PD-1 expression was partly positive in all 5 subsets including cDC1, cDC2, pDC, CD16+ DC and undefined DCs. For different samples, the proportion of each DC subset varied distinctly, and the fractions of each subset in PD-1+ DCs were accordingly different (Figure 2a). In general, nonmigratory cDC2s, which occupied the highest proportion of DCs, derived most PD-1+ DCs (34%–62.7%). Since PD-1 positive expression was found in all subtypes, total DC was analyzed in subsequent experiments.
Figure 2.
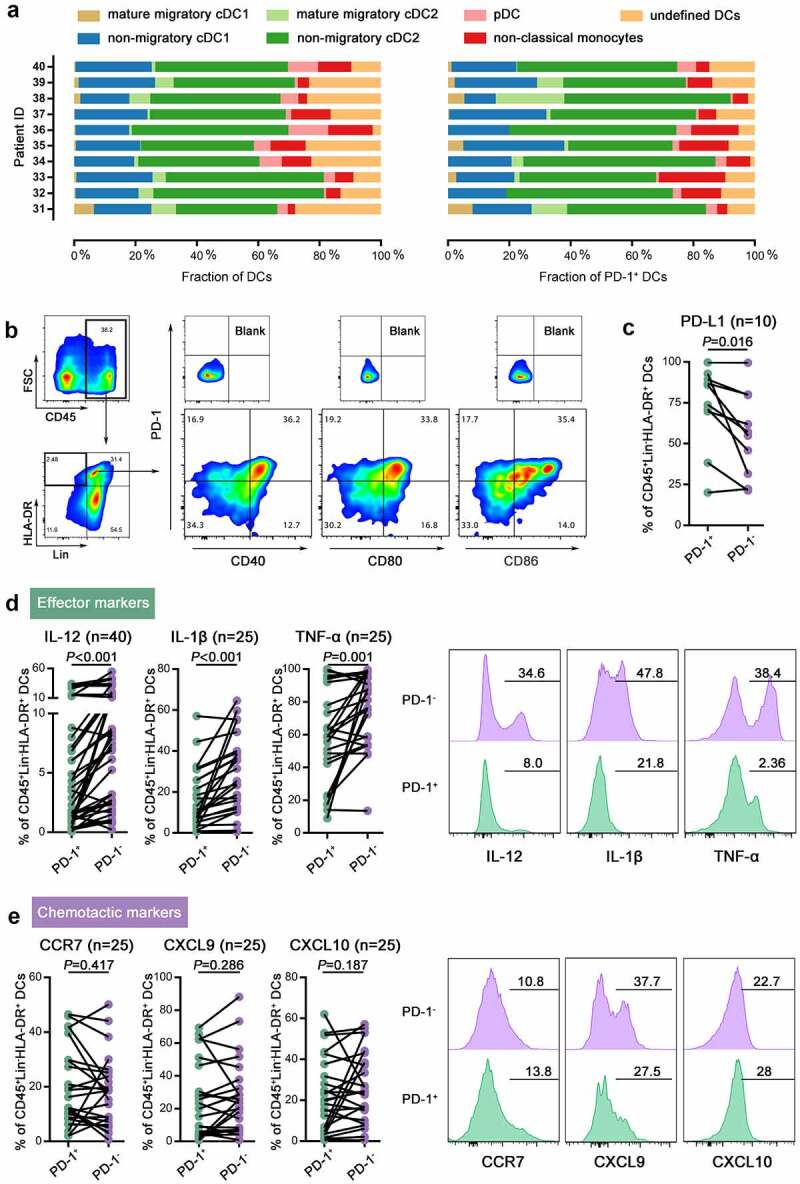
Intratumoral PD-1+ DCs with high expression of costimulatory markers exhibited dysfunctional properties.
(a) The fractions of different subgroups among DCs (left) and PD-1+ DCs (right) in each patient. (b) The gating strategy for CD40, CD80, CD86, and PD-1 expression on DCs. (c) Quantification of PD-L1 expression on PD-1+ and PD-1- DCs in CC. (d,f) Left: Quantification of the expression of effector markers (IL-12, IL-1β, and TNF-α, D) or chemotactic markers (CCR7, CXCL9, and CXCL10, E) on PD-1+ and PD-1- DCs in CC. Right: Representative flow cytometric histograms of the expression of effector markers (IL-12, IL-1β, and TNF-α, D) or chemotactic markers (CCR7, CXCL9, and CXCL10, E) on PD-1+ and PD-1- DCs in CC.
To investigate whether PD-1 plays a role in the function of DCs in cervical tumors, we first examined costimulatory, effector and chemotactic markers in intratumoral PD-1+ DCs from 40 specimens. We found that PD-1 was mainly expressed on CD40+ CD80+ CD86+ DCs, a subset with an activated phenotype (Figure 2b). Moreover, higher PD-L1 expression were observed on PD-1+ DCs than PD-1- DCs (Figure c). DCs are an abundant source of IL-12, TNF-α, and IL-1β, which are crucial in eliciting immune responses and eliminating cancer cells. PD-1+ DCs secreted lower levels of these cytokines than their PD-1- counterparts (Figure 2d). CCR7, a key receptor in DC homing to the lymph nodes, showed no difference in expression between PD-1+ DCs and PD-1- DCs (Figure 2e). The levels of two chemokines that mediate T cell recruitment to the tumor, CXCL9 and CXCL10, also displayed no difference in PD-1+ DCs and PD-1- DCs (Figure 2e).
3.3. PD-1+ DCs are induced upon activation in tumor conditions and deficient in eliciting an adaptive response
To examine the induction of PD-1+ DCs, we generated BMDCs and TADCs from C57BL/6 mice to observe PD-1 expression during DC maturation in vitro. On day 6 of culture, LPS was added to the culture medium to stimulate mature DCs. After stimulation for 6, 12, 24, or 48 hours, the percentage of PD-1+ cells gradually increased with maturation, especially for TADCs activated by LPS (Figure 3a). BMDCs cultured under normal conditions exhibited slight PD-1 expression after LPS stimulation, which was significantly lower than that of TADCs stimulated with LPS (Figure 3a). The PD-1 level strongly correlated with the percentage of CD40+ CD80+ CD86+ activated DCs (Figure 3b). We examined the activation status of PD-1+ TADCs based on CD40, CD80 and CD86 expression, and the results showed consistency with the CC specimens in which PD-1 was mainly induced on activated DCs (Figure 3c). We observed that LPS-treated DCs (either under normal or tumor conditions) lost their capacity for antigen phagocytosis compared to immature BMDCs. However, we found no difference in phagocytic capacity between PD-1+ DCs and PD-1- DCs (Figure 3d). Moreover, PD-1+ DCs exhibited lower IL-12 and TNF-α secretion than PD-1- DCs, which showed a consistent trend in both BMDCs and TADCs (Figure 3e). The secretion level of cytokines is comparable between BMDCs and TADCs, indicating the dysfunction of TADCs, especially those expressing PD-1.
Figure 3.
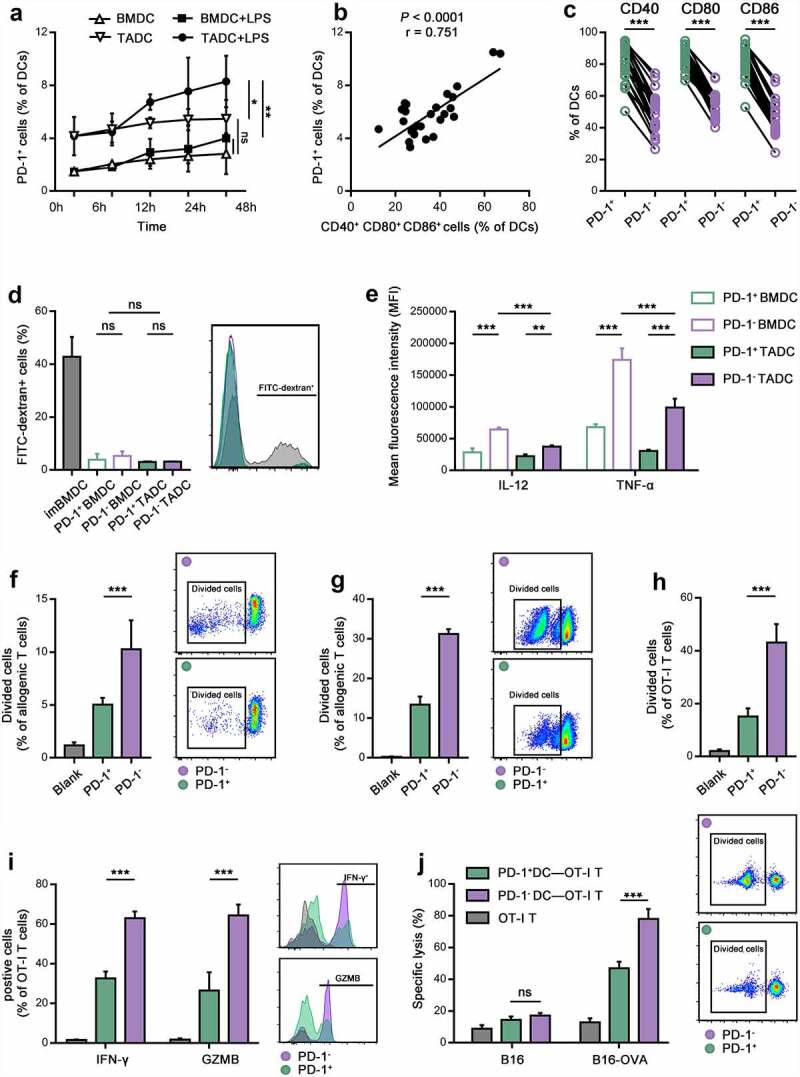
TCM contributed to PD-1 expression on activated DCs with impaired function in vitro.
(a) Quantification of PD-1+ DCs under normal and tumor conditions after treatment with or without LPS for 0, 6, 12, 24, and 48 hours. (b) The percentage of PD-1+ DCs correlated with CD40+ CD80+ CD86+ DCs cultured under tumor conditions after treatment with LPS for 6 hours. (c) The percentage of CD40+, CD80+, and CD86+ cells in PD-1+ or PD-1- DCs cultured under tumor conditions. (d) The percentage of FITC-dextran endocytosis in immature BMDCs and PD-1+ and PD-1- BM/TADCs. (e) IL-12 and TNF-α expression in PD-1+ and PD-1- TADCs as calculated by the MFI. (f,g) The division of allogeneic mouse T cells (f) or allogeneic human lymphocytes (g) after stimulation in an MLR with PD-1+ or PD-1- DCs isolated from TADCs or Mo-DCs for 72 hours. (h,i) The division (h) and IFN-γ, and GZMB production (i) of antigen-specific OT-I T cells after stimulation with OVA-loaded PD-1+ or PD-1- TADCs for 72 hours. (j) The CTL response of antigen-specific OT-I T cells (effector cells) stimulated with OVA-loaded PD-1+ or PD-1- TADCs to kill B16-OVA or B16 cells (target cells) measured by LDH release. T cells without treated with DCs were control.
To understand the function of PD-1+ DCs in priming the antitumor response, we performed antigen nonspecific and antigen-specific assays. In an allogenic MLR, proliferation of the T cells, as measured by the decrease in CFSE by flow cytometry, was significantly inhibited when cocultured with the PD-1+ DCs compared to PD-1- DCs (Figure 3f). Meanwhile, a parallel MLR was conducted using human Mo-DCs and allogenic lymphocytes. We generated Mo-DCs under tumor conditions by the addition of human HeLa cells and isolated PD-1+ and PD-1- DCs. The results were consistent with those in mice which the ability of DCs to stimulate T cell proliferation was significantly decreased for those expressing PD-1 (Figure 3g). To evaluate the effect of PD-1+ DCs on the cytotoxicity of antigen-specific T cells, OVA model antigen and OT-I T cells were used. As shown in Figure 3h,i, OVA-loaded PD-1+ DCs showed dampened effects on the proliferation, activation, and degranulation of antigen-specific OT-I T cells as indicated by decreases in division, IFN-γ and GZMB release. Moreover, the OT-I T cell cells stimulated by OVA-loaded PD-1+ DCs had a relatively low efficiency in the immune killing of B16-OVA cells compared to OVA-loaded PD-1- DCs (Figure 3j). Taken together, the results support a role for PD-1 on DCs in tumor conditions to shape their dysfunction properties.
3.4. Intratumoral PD-1+ DC infiltration is associated with CD8 + T cell inactivation
To further investigate the role of PD-1+ DC infiltration in the tumor immune microenvironment, we classified 40 CC patients into 2 subgroups on the basis of the median frequency of PD-1-expressing tumor-infiltrating DCs. The median percentage of PD-1+ tumor-infiltrating DCs was 6% (range, 0.78%–52.13%). Patients with greater than 6% of their intratumoral DCs expressing PD-1 were termed high PD-1+ DC expressers, whereas those with less than 6% PD-1+ DCs were termed low PD-1+ DC expressers. Our cohort comprised 50% high PD-1+ DC expressers and 50% low PD-1+ DC expressers.
We next investigated the density and effector cytokines of tumor-infiltrating CD4 + T cells, CD8 + T cells and natural killer (NK) cells using flow cytometry to dissect the tumor-killing functions of the immune system. We observed that the proliferation of these cells remained unaffected, as the percentages of Ki67+ cells in the CD4 + T cell, CD8 + T cell and NK cell populations were not different between the two patient groups (Figure 4a). Our results revealed that high and low PD-1+ DC expressers showed no differences in the infiltration density of total CD45+ cells, NK cells or CD4 + T cells (Figure 4b). The cytotoxic functions of NK cells and CD4 + T cells were also similar between high and low PD-1+ DC expressers, as indicated by similar cytolytic markers, including CD107a, GZMB and perforin, and effector cytokines IL-2, IFN-γ and TNF-α (Figure 4a). Strikingly, high PD-1+ DC expressers had a significantly higher percentage of CD8 + T cells among tumor-infiltrating leukocytes and a significantly higher CD8 + T cell density in the tumor bed (Figure 4c). Interestingly, although naïve T cells typically circulate through blood and lymphoid organs,22 a small proportion of CD8 + T cells infiltrating the in tumor were CD45RA+CCR7+ naïve (Figure 4a,d). This indicated that in addition to cytotoxic effector cell recruitment from lymph nodes, there was also local T cell activation in the tumor tissue, and this primary activation was stronger in high PD-1+ DC expressers (Figure 4d). However, feature profiles of tumor-infiltrating CD8 + T cells revealed that the levels of Eomes, PD-1, and Tim-3, which are well-established markers of exhausted T cells,23 were increased on CD8 + T cells in high PD-1+ DC expressers. Additionally, the tumor-infiltrating CD8 + T cells of high PD-1+ DC expressers exhibited significantly lower capacities to produce CD107a and GZMB and release IFN-γ than those of low PD-1+ DC expressers (Figure 4d). Collectively, these data indicated that intratumoral PD-1+ DC infiltration is relevant to CD8 + T cell dysfunction.
Figure 4.
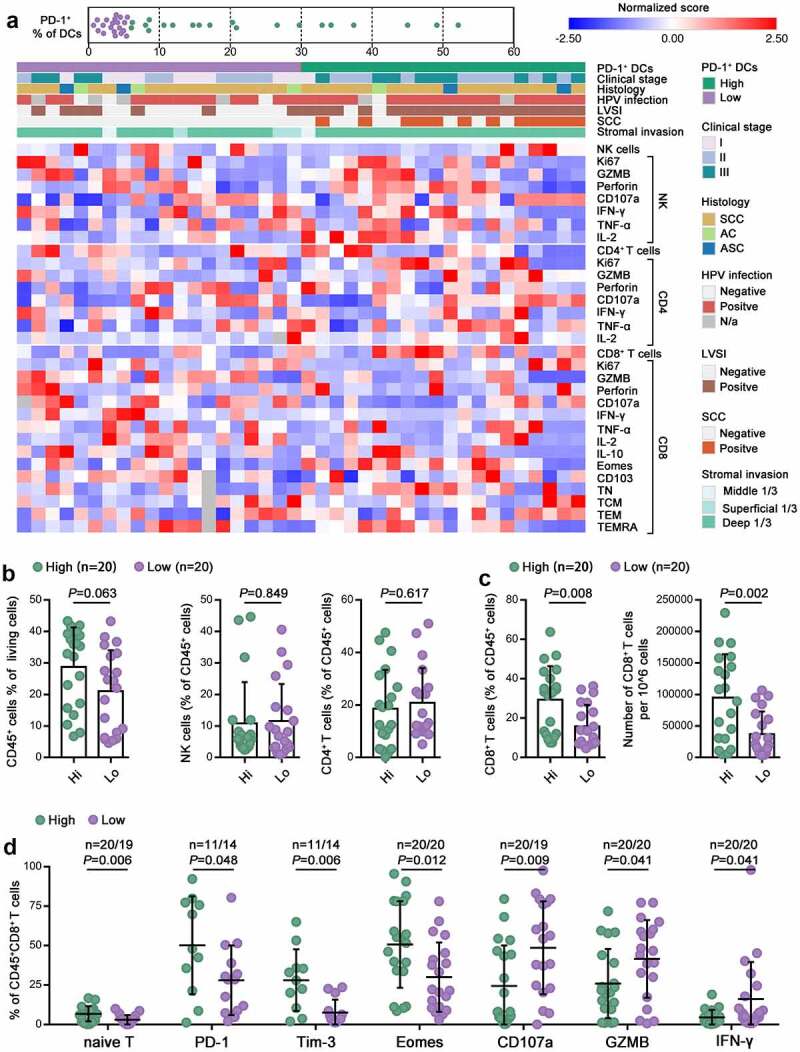
Intratumoral PD-1+ DC infiltration was associated with CD8 + T cell inactivation.
(a) Heatmap showing the frequencies of CD4 + T, CD8 + T and NK cells and the percentage of the indicated marker-positive cells in the CD4 + T, CD8 + T and NK cell populations within tumors. Clinical parameters and the frequency of PD-1+ cells are depicted on top. Patients were clustered into high and low groups based on PD-1+ cell frequency. (b,c) The percentage of intratumoral CD45+ cells in total living cells, the percentage of intratumoral NK cells, CD4 + T cells, and CD8 + T cells in CD45+ cells, and the absolute number of CD8 + T cells in high and low PD-1+ DC expressers. (d) Quantification of naïve T cells and the percentages of PD-1+, Tim-3+, Eomes+, CD107a+, GZMB+, and IFN-γ+ cells in the CD8 + T cell population in high and low PD-1+ DC expressers.
3.5. The accumulation of intratumoral PD-1+ DCs is correlated with a favorable response to PD-1 blockade
Furthermore, we evaluated the clinical significance of PD-1+ DC infiltration for PD-1 blockade using the same 40 CC specimens. We added pembrolizumab, a Food and Drug Administration (FDA)-approved anti-PD-1 monoclonal antibody, to suspensions of freshly resected CC tissue and detected PD-1+ DC function, CD8 + T cell cytotoxicity and tumor cell apoptosis after 24 hours of treatment. Interestingly, we found that dysfunctional PD-1+ DCs were restored by blockade of PD-1 and exhibited significantly elevated secretion of IL-12, IL-1β and TNF-α, whereas the functions of PD-1- DCs were not further enhanced (Figure 5a,b). Moreover, ex vivo cytolytic assays demonstrated that the cell killing efficacy of pembrolizumab was remarkable in samples with high PD-1+ DC infiltration but not obvious in those with low PD-1+ DC infiltration (Figure 5c). More importantly, PD-1 blockade further enhanced CD107a, GZMB, and IFN-γ production by CD8+ tumor-infiltrating lymphocytes (TILs) from high PD-1+ DC expressers but did not have a similar impact on low PD-1+ DC expressers (Figure 4d). In CD8 + T cells, the proliferation marker Ki67 seemed unaffected by pembrolizumab in both high and low PD-1+ DC expressers. Together, these results suggested that PD-1 blockade enhanced PD-1+ DC function and might achieve more effective functional restoration of CD8+ TILs from high PD-1+ DC expressers.
Figure 5.
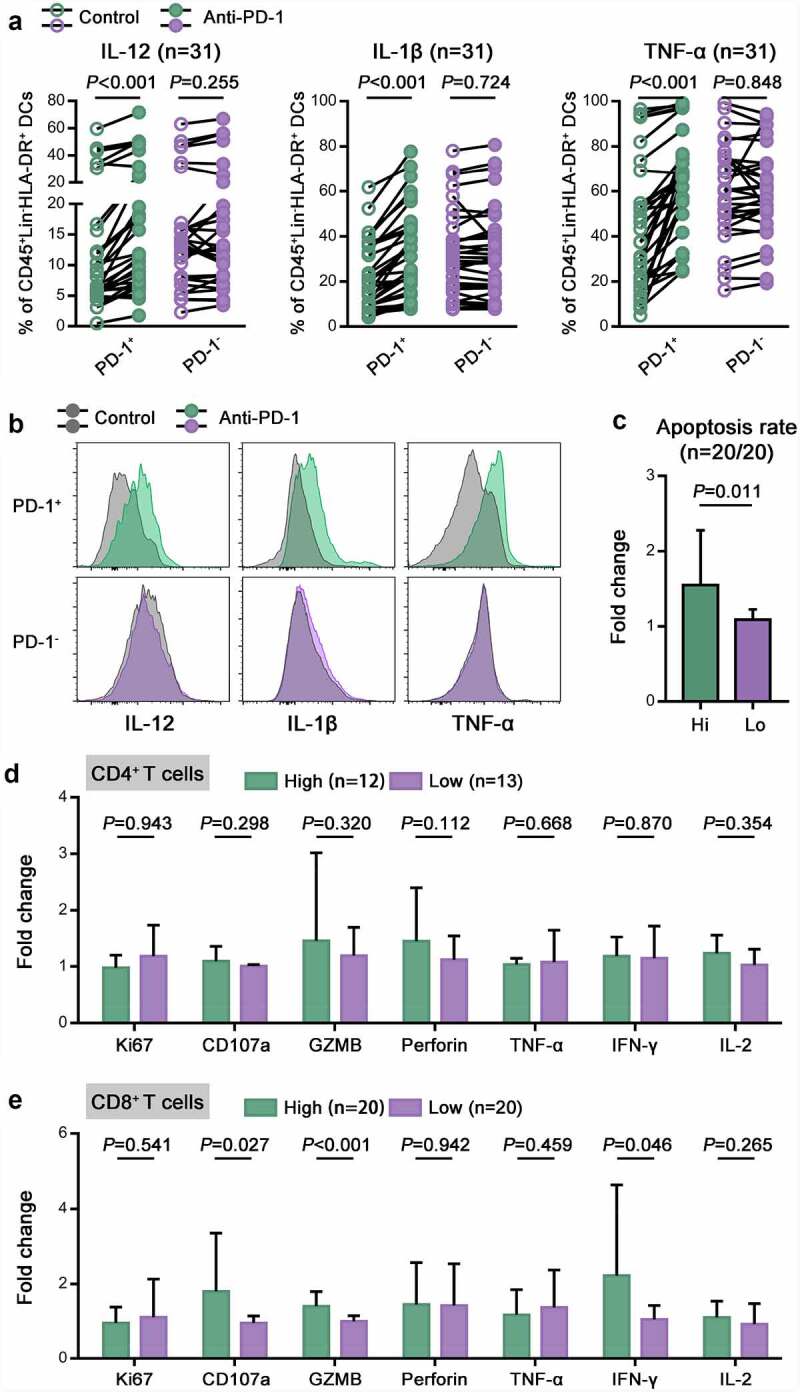
The accumulation of intratumoral PD-1+ DCs was correlated with a favorable response to PD-1 blockade.
(a) The percentages of IL-12+, IL-1β+ and TNF-α+ cells in PD-1+ or PD-1- DCs from CC tumors before and after treatment with pembrolizumab. (b) Representative flow cytometric histograms showing IL-12, IL-1β and TNF-α expression on PD-1+ or PD-1- DCs in CC before and after treatment with pembrolizumab. (c) The apoptosis rate of tumor cells from high and low PD-1+ DC expressers after treatment, as determined by Annexin V-PI staining. The fold change was calculated as the ratio between the pembrolizumab group and the control group. (d,e) The percentages of Ki67+, CD107a+, GZMB+, perforin+, TNF-α+, IFN-γ+ and IL-2+ cells in CD4 + T cells (D) or CD8 + T cells (E) from high and low PD-1+ DC expressers. The fold change was calculated as the ratio between the pembrolizumab group and the control group.
4. Discussion
As orchestrators of the immune system bridging innate and adaptive immunity, DCs are key players in directing antitumor immunity. Generally, after sufficient activation and antigen recognition, DCs subsequently activate the adaptive immune response. However, the association of activated DC infiltration with clinical outcome suggested differences between cancers. In pancreatic adenocarcinoma and skin cutaneous melanoma, higher intratumoral activated DCs were associated with good prognosis, while the opposite relationship occurred in breast invasive carcinoma.24 In this context, activated DCs may shift to a tolerogenic or immunosuppressive state through undetermined negatively regulated signals. Here, we identified a population of activated DCs in the TME that express the inhibitory molecule PD-1. Although the signaling pathways may be multifaceted, our experiments may shed light on the role of PD-1 in DC dysfunction in the CC TME.
To date, mechanistic studies of PD-1 have mainly focused on T cells. PD-1 is inducibly expressed on T cells by T cell antigen receptor (TCR) signaling and then acts as a molecular brake to negatively control T cell activity. Upon interaction with the ligand PD-L1, a pair of tyrosines within the cytoplasmic tail of PD-1 is phosphorylated and recruits the protein tyrosine phosphatases SHP2 and SHP1, which then dephosphorylate both the TCR and CD28 costimulatory molecules.25,26 These biochemical events ultimately lead to the attenuation of T cell proliferation, cytokine production, and cytolytic activities.27 Despite the widely accepted notion that PD-1 primarily functions as a T cell inhibitory receptor, accumulating evidence suggests that PD-1 is also expressed on other types of immune cells (including macrophages, NKs, DCs, and B cells).28–31 As shown in both our in vitro experiments and CC samples, we found that PD-1 was induced on DCs concomitant with the activation process, indicating that PD-1 may serve as a marker of high tumor-responsive DCs. Moreover, the induction of PD-1 was much more apparent under tumor conditions than under normal conditions, whereas DC activation was inhibited under tumor conditions. These observations indicated that the TME may exert extra induction pressure, which is weak in a normal environment, to induce PD-1 expression on activated DCs, which deserves further exploration.
Of note, those PD-1+ matured DC exhibited dampened ability in producing proinflammtory cytokines and eliciting adaptive T immune response. Although the biochemical mechanism underlying PD-1-mediated inhibition of DCs has yet to be clarified, some reported studies are helpful to clarify our results. Studies in tumor associated macrophages (TAMs) showed that PD-1 engagement by PD-L1 actively recruited SHP-2 to the PD-1 cytoplasmic tail, leading to the inhibited glycolysis, M1 type cytokine production and phagocytic potency. Blockade of PD-1/PD-L1 signaling with either PD-L1 removal or PD-1 mAb rescued the TAM function by reversing immune metabolic dysfunctions as well as promoting M1 polarization.28,32,33 Similarly, PD-1 signaling in DCs suppresses intracellular canonical NF-κB pathway in an SHP-2-dependent manner and sequentially inhibits self-function.34 DC apoptosis also starts to increase after PD-1 upregulation.35 The PD-L1/PD-1 interaction has been intensively studied as trans interaction between PD-1 on T cells and PD-L1 on tumor cells or tumor infiltrating immune cells. Interestingly, a recent report showed that PD-1 interacts in cis with both PD-L1 and PD-L2 on cell membranes.36 Coexpression of PD-L1 and PD-1 on DCs observed in CC specimens raises the possibility that the PD-1/PD-L1 cis-interactions may deliver inhibitory signal and thus contribute to shaping dysfunction of PD-1+ DCs. These findings thus reveal the role of PD-1 signaling in promoting tumor escape by regulating the function of DCs.
Generally, tumor-infiltrated DCs (TIDCs) take up antigen, become mature, and migrate to tumor-draining LNs (tdLNs) in a CCR7-dependent mechanism.37,38 In tdLNs, DCs prime naïve tumor antigen-specific T cells to activate, and effector T cells are then recruited to the TME mediated by CXCL9/10 produced by TIDCs.38,39 However, we found that most of the cDCs in the CC TME were nonmigratory. This is consistent with previous studies showing that only a small fraction of tumor cDCs will end up migrating to the lymph nodes, possibly related to controlled expression of CCR7 by tumor derived suppressive molecules.40,41 It is possible that nonmigratory TIDCs are similarly important in maintaining the in situ antitumor immune response. In support of this, intratumoral cDCs are required for tumor regression after adoptive T cell transfer irrespective of the migration of T cells to tdLNs.42 Moreover, local T cell priming and activation within tumors were not impacted in mice that lacked LN or when T cell recirculation was blocked.43,44 Considering the nonmigratory properties of TIDCs, the lack of differences of chemokines and receptors raised the possibility that the impact of PD-1+ DCs on adaptive immunity may occur either through direct antigen presentation or through establishment of an unfavorable cytokine milieu. As a population of DCs expressing high costimulatory signals (CD40, CD80, CD86), PD-1+ DCs may be able to induce the proliferation of naïve T cells. This was confirmed by the observation of a higher ratio of naïve T cells infiltrating in tumor bed. However, without enough stimulation of proinflammation cytokines (IL-12, IL-1β, and TNF-α), the survival of proliferating naïve T cells will be compromised, and most T cells will develop anergic or apoptotic cells. This was confirmed by the results that dysfunction and exhaustion of CD8 + T cells increased in PD-1+ DCs with higher expression. Our findings reveal potential suppressive mechanisms of PD-1 affecting DC-mediated adaptive immunity: inhibition of proinflammatory cytokine secretion and disruption of the ability to promote antitumor T cell proliferation and activation in the local TME.
DCs are fundamental for the initiation and maintenance of immune responses against malignant cells. As key antigen-presenting cells, DCs are capable of presenting tumor antigens to T lymphocytes and promoting innate immunity via NK cells.45,46 As a population of DCs exhibiting dysfunctional properties, the role of PD-1+ DCs in antitumor immunity was assessed. We found that PD-1+ DCs in tumors were inclined to influence the function of CD8 + T cells rather than that of CD4 + T cells or NK cells. In our results, CC with a higher proportion of PD-1+ DCs was infiltrated with more CD8 + T cells. Infiltration of T cells into tumors is a prerequisite for an inflamed tumor phenotype.47 Common characteristics of responding inflamed tumors include dense CD8 + T cell infiltrates, a broad chemokine profile, PD-L1 expression on immune cells, a type 1 interferon (IFN) signature, and elevated expression of IFN-γ–induced genes.46,48 Therefore, inflamed tumors are thought to have a preexisting CD8 + T-cell response to their tumor. Although CC appears to present more commonly as inflamed, individuals may present with cancer that is inflamed or noninflamed (excluded or immune deserts).48 Those high PD-1+ DC expressers featured increased accumulation of CD8 + T cells, indicating a preexisting CD8 + T-cell response. However, the characteristics of inflamed tumors ultimately induce the adaptive upregulation of the PD-1/PD-L1 pathway on neighboring tumor-infiltrating immune cells and on tumor cells.49,50 The establishment of durable and effective anticancer immunity is kept in check by intratumoral PD-1/PD-L1 expression. Consistent with this negative feedback mechanism, we confirmed that the tumor-infiltrating CD8 + T cells of high PD-1+ DC expressers exhibited inactivation and deficient cytotoxicity as well as a tendency to express an exhausted phenotype. Moreover, a high level of intratumoral PD-1+ DCs was more likely to be detected in advanced CC and lymph-vascular space invasion-positive specimens. Preliminarily, we classified high PD-1+ DC expressers with a median cutoff value, which may require larger samples or prospective multicenter clinical trials to be verified. Our findings identified this dysfunctional PD-1+ DC subpopulation as a contributor to CD8 + T cell dysfunction and tumor progression in CC.
PD-1/PD-L1 blockade immunotherapy has shown clinical efficacy in multiple cancers, including CC, by rescuing exhausted T cells by targeting the PD-1/PD-L1 pathway. Most of the existing research has focused on the reactivation of T cells by PD-1 blockade. However, it has been found that the role of PD-1 blockade in potentiating antitumor immunity is not limited to T cells. Therefore, it is possible that blocking PD-1/PD-L1 exerts antitumor functions by acting synergistically on a variety of immune cells synergistically rather than only T cells. As expressers of PD-l, intratumoral DCs may also respond to PD-1/PD-L1 blockade. In our study, we blocked the PD-1/PD-L1 pathway with pembrolizumab ex vivo and found that PD-1/PD-L1 blockade could restore the function of PD-1+ DCs to a greater extent than that of PD-1- DCs, suggesting that PD-1/PD-L1 blockade was efficient in PD-1+ DCs. PD-L1 expression on tumor cells, mismatch repair and the presence of TILs and cytokines in tumor samples have been used to identify patients who might respond to PD-1/PD-L1 blockade immunotherapy. However, accumulating evidence suggests that these markers are not sufficient to accurately predict the response status. Therefore, additional diagnostic approaches are needed to predict who might respond to PD-1/PD-L1 blockade immunotherapy. We found a subset of patients with high intratumoral PD-1+ DC levels who had a greater response to pembrolizumab, exhibiting restored DC function, enhanced CD8+ TIL cytotoxicity and elevated tumor cell apoptosis. Therefore, we can make a preliminary conclusion that PD-1+ DCs are an important response subgroup toward PD-1 blockade and have a positive impact on the recovery of the downstream immune response. Although pembrolizumab can directly affect CD8 + T cells in our ex vivo treatment model – this is a limitation in our studies that deserves further exploration by developing novel ev vivo treatment platforms – the reactivation of PD-1+ DCs indeed participates in the recovery of CD8 + T function. Our findings suggested that the different frequencies of preexisting intratumoral PD-1+ DCs can serve as an additional diagnostic marker for predicting the response to PD-1/PD-L1 blockade immunotherapy.
In conclusion, our study identified a distinctly dysfunctional status of DCs determined by PD-1 expression. The PD-1+ DC subset in CC was correlated with inactivation of CD8+ TILs and progression of CC. Our results also described a distinct subgroup of CC patients with a large population of intratumoral PD-1+ DCs whose PD-1+ DCs and CD8+ TILs could be reinvigorated by PD-1/PD-L1 blockade. This information may provide a rationale and evidence for establishing optimal strategies for PD-1/PD-L1 blockade in patients with CC.
Supplementary Material
Acknowledgments
The authors would like to thank Dr. Xiao-yang Liu (Laboratory animal center of Nantong university) for OT-I mice.
Funding Statement
This work was supported by funding from the National Natural Science Foundation of China [No. 81971361; to Jun-jun Qiu], the Natural Science Foundation of Shanghai Science and Technology [No. 19ZR1406900; to Jun-jun Qiu], the Shanghai “Rising Stars of Medical Talent” Youth Development Program [No. AB83030002019004; to Jun-jun Qiu], the Clinical Research Plan of SHDC [No.SHDC2020CR4087; to Jun-jun Qiu], Shanghai Municipal Health Commission [No. 202040498; to Jun-jun Qiu], the Research and Innovation Project of the Shanghai Municipal Education Commission [No. 2019-01-07-00-07-E00050; to Ke-qin Hua] and the Clinical Research Plan of SHDC [No. SHDC2020CR1045B; to Ke-qin Hua], and the National Natural Science Foundation of China [No. 81873124; to Gui-ling Li].
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website
References
- 1.Siegel RL, Miller KD, Jemal A.. Cancer statistics, 2020. CA Cancer J Clin. 2020;70(1):7–13. doi: 10.3322/caac.21590. [DOI] [PubMed] [Google Scholar]
- 2.Nobbenhuis MA, Walboomers JM, Helmerhorst TJ, Rozendaal L, Remmink AJ, Risse EK, van der Linden HC, Voorhorst FJ, Kenemans P, Meijer CJ, et al. Relation of human papillomavirus status to cervical lesions and consequences for cervical-cancer screening: a prospective study. Lancet. 1999;354(9172):20–25. doi: 10.1016/s0140-6736(98)12490-x. [DOI] [PubMed] [Google Scholar]
- 3.Attademo L, Tuninetti V, Pisano C, Cecere SC, Di Napoli M, Tambaro R, Valabrega G, Musacchio L, Setola SV, Piccirillo P, et al. Immunotherapy in cervix cancer. Cancer Treat Rev. 2020;90:102088. doi: 10.1016/j.ctrv.2020.102088. [DOI] [PubMed] [Google Scholar]
- 4.Ferrall L, Lin KA-O, Roden RBS, Hung CF, Wu TC. Cervical cancer immunotherapy: facts and hopes. (1557-3265 (Electronic)). doi: 10.1158/1078-0432.CCR-20-2833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Berraondo P, Minute L, Ajona D, Corrales L, Melero I, Pio R. Innate immune mediators in cancer: between defense and resistance. Immunol Rev. 2016;274(1):290–306. doi: 10.1111/imr.12464. [DOI] [PubMed] [Google Scholar]
- 6.Iannello A, Thompson TW, Ardolino M, Marcus A, Raulet DH. Immunosurveillance and immunotherapy of tumors by innate immune cells. Curr Opin Immunol. 2016;38:52–58. doi: 10.1016/j.coi.2015.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wu Y, Ye S, Goswami S, Pei X, Xiang L, Zhang X, Yang H. Clinical significance of peripheral blood and tumor tissue lymphocyte subsets in cervical cancer patients. BMC Cancer. 2020;20(1):173. doi: 10.1186/s12885-020-6633-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nedergaard BS, Ladekarl M, Thomsen HF, Nyengaard JR, Nielsen K. Low density of CD3+, CD4+ and CD8+ cells is associated with increased risk of relapse in squamous cell cervical cancer. Br J Cancer. 2007;97(8):1135–1138. doi: 10.1038/sj.bjc.6604001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sheu BC, Hsu SM, Ho HN, Lin RH, Torng PL, Huang SC. Reversed CD4/CD8 ratios of tumor-infiltrating lymphocytes are correlated with the progression of human cervical carcinoma. Cancer. 1999;86(8):1537–1543. doi:. [DOI] [PubMed] [Google Scholar]
- 10.Piersma SJ, Jordanova ES, Van Poelgeest MI, Kwappenberg KM, Van Der Hulst JM, Drijfhout JW, Melief CJ, Kenter GG, Fleuren GJ, Offringa R, et al. High number of intraepithelial CD8+ tumor-infiltrating lymphocytes is associated with the absence of lymph node metastases in patients with large early-stage cervical cancer. Cancer Res. 2007;67(1):354–361. doi: 10.1158/0008-5472.CAN-06-3388. [DOI] [PubMed] [Google Scholar]
- 11.Yang H, Ye S, Goswami S, Li T, Wu J, Cao C, Ma J, Lu B, Pei X, Chen Y, et al. Highly immunosuppressive HLADRhi regulatory T cells are associated with unfavorable outcomes in cervical squamous cell carcinoma. Int J Cancer. 2020;146(7):1993–2006. doi: 10.1002/ijc.32782. [DOI] [PubMed] [Google Scholar]
- 12.Punt S, van Vliet ME, Spaans VM, de Kroon CD, Fleuren GJ, Gorter A, Jordanova ES. FoxP3(+) and IL-17(+) cells are correlated with improved prognosis in cervical adenocarcinoma. Cancer Immunol Immunother: CII. 2015;64(6):745–753. doi: 10.1007/s00262-015-1678-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sabado RL, Balan S, Bhardwaj N. Dendritic cell-based immunotherapy. Cell Res. 2017;27(1):74–95. doi: 10.1038/cr.2016.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Inoshima N, Nakanishi Y, Minami T, Izumi M, Takayama K, Yoshino I, Hara N. The influence of dendritic cell infiltration and vascular endothelial growth factor expression on the prognosis of non-small cell lung cancer. Clinical Cancer Res. 2002;8:3480. [PubMed] [Google Scholar]
- 15.Hegde S, Krisnawan VE, Herzog BH, Zuo C, Breden MA, Knolhoff BL, Hogg GD, Tang JP, Baer JM, Mpoy C, et al. Dendritic cell paucity leads to dysfunctional immune surveillance in pancreatic cancer. Cancer Cell. 2020;37(3):289–307.e9. doi: 10.1016/j.ccell.2020.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jensen TO, Schmidt H, Møller HJ, Donskov F, Høyer M, Sjoegren P, Christensen IJ, Steiniche T. Intratumoral neutrophils and plasmacytoid dendritic cells indicate poor prognosis and are associated with pSTAT3 expression in AJCC stage I/II melanoma. Cancer. 2012;118(9):2476–2485. doi: 10.1002/cncr.26511. [DOI] [PubMed] [Google Scholar]
- 17.Hayati AR, Zulkarnaen M. An immunohistochemical study of CD1a and CD83-positive infiltrating dendritic cell density in cervical neoplasia. Int J Gynecol Pathol. 2007;26(1):83–88. doi: 10.1097/01.pgp.0000225850.90115.bc. [DOI] [PubMed] [Google Scholar]
- 18.Luo Z, Wang C, Yi H, Li P, Pan H, Liu L, Cai L, Ma Y. Nanovaccine loaded with poly I:C and STAT3 siRNA robustly elicits anti-tumor immune responses through modulating tumor-associated dendritic cells in vivo. Biomaterials. 2015;38:50–60. doi: 10.1016/j.biomaterials.2014.10.050. [DOI] [PubMed] [Google Scholar]
- 19.Solana, A , Lucia, P. 2016. Purification of Human Dendritic Cell Subsets from Peripheral Blood. doi: 10.1007/978-1-4939-3606-9. Dendritic Cell Protocols 3 1423 Elodie, Segura, and Nobuyuki, Onai. Methods in Molecular Biology. New York, NY: Humana Press. XI, 322 978-1-4939-3604-5 [DOI] [Google Scholar]
- 20.Qin Kai CY, Huang L, and Yuan X. Study on the relationship between immune cell infiltration pattern and clinical characteristics and prognosis of cervical cancer based on TCGA database. J Hainan Med Univ. 2021;27(11):1–9. doi: 10.13210/j.cnki.jhmu.20200623.002. [DOI] [Google Scholar]
- 21.Patsoukis N, Wang Q, Strauss L, Boussiotis VA. Revisiting the PD-1 pathway. Sci Adv. 2020;6(38). doi: 10.1126/sciadv.abd2712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.van der Leun AM, Thommen DS, Schumacher TN. CD8(+) T cell states in human cancer: insights from single-cell analysis. Nat Rev Cancer. 2020. doi: 10.1038/s41568-019-0235-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Miller BC, Sen DR, Al Abosy R, Bi K, Virkud YV, LaFleur MW, Yates KB, Lako A, Felt K, Naik GS, et al. Subsets of exhausted CD8+ T cells differentially mediate tumor control and respond to checkpoint blockade. Nat Immunol. 2019;20(3):326–336. doi: 10.1038/s41590-019-0312-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Charoentong P, Finotello F, Angelova M, Mayer C, Efremova M, Rieder D, Hackl H, Trajanoski Z. Pan-cancer immunogenomic analyses reveal genotype-immunophenotype relationships and predictors of response to checkpoint blockade. Cell Rep. 2017;18(1):248–262. doi: 10.1016/j.celrep.2016.12.019. [DOI] [PubMed] [Google Scholar]
- 25.Yokosuka T, Takamatsu M, Kobayashi-Imanishi W, Hashimoto-Tane A, Azuma M, Saito T. Programmed cell death 1 forms negative costimulatory microclusters that directly inhibit T cell receptor signaling by recruiting phosphatase SHP2. J Exp Med. 2012;209(6):1201–1217. doi: 10.1084/jem.20112741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hui E, Cheung J, Zhu J, Su X, Taylor MJ, Wallweber HA, Sasmal DK, Huang J, Kim JM, Mellman I, et al. T cell costimulatory receptor CD28 is a primary target for PD-1-mediated inhibition. Science. 2017;355(6332):1428–1433. doi: 10.1126/science.aaf1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Francisco LM, Sage PT, Sharpe AH. The PD-1 pathway in tolerance and autoimmunity. Immunol Rev. 2010;236:219–242. doi: 10.1111/j.1600-065X.2010.00923.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gordon SR, Maute RL, Dulken BW, Hutter G, George BM, McCracken MN, Gupta R, Tsai JM, Sinha R, Corey D, et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature. 2017;545(7655):495–499. doi: 10.1038/nature22396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hsu J, Hodgins JJ, Marathe M, Nicolai CJ, Bourgeois-Daigneault MC, Trevino TN, Azimi CS, Scheer AK, Randolph HE, Thompson TW, et al. Contribution of NK cells to immunotherapy mediated by PD-1/PD-L1 blockade. J Clin Invest. 2018;128(10):4654–4668. doi: 10.1172/jci99317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yao S, Wang S, Zhu Y, Luo L, Zhu G, Flies S, Xu H, Ruff W, Broadwater M, Choi I-H, et al. PD-1 on dendritic cells impedes innate immunity against bacterial infection. Blood. 2009;113(23):5811–5818. doi: 10.1182/blood-2009-02-203141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xiao X, Lao XM, Chen MM, Liu RX, Wei Y, Ouyang FZ, Chen D-P, Zhao X-Y, Zhao Q, Li X-F, et al. PD-1 hi identifies a novel regulatory B-cell population in human hepatoma that promotes disease progression. Cancer Discov. 2016;6(5):546–559. doi: 10.1158/2159-8290.Cd-15-1408. [DOI] [PubMed] [Google Scholar]
- 32.Chen W, Wang J, Jia L, Liu J, Tian Y. Attenuation of the programmed cell death-1 pathway increases the M1 polarization of macrophages induced by zymosan. Cell Death Dis. 2016;7(2):e2115. doi: 10.1038/cddis.2016.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Qorraj M, Bruns H, Böttcher M, Weigand L, Saul D, Mackensen A, Jitschin R, Mougiakakos D. The PD-1/PD-L1 axis contributes to immune metabolic dysfunctions of monocytes in chronic lymphocytic leukemia. Leukemia. 2017;31(2):470–478. doi: 10.1038/leu.2016.214. [DOI] [PubMed] [Google Scholar]
- 34.Karyampudi L, Lamichhane P, Krempski J, Kalli KR, Behrens MD, Vargas DM, Hartmann LC, Janco JMT, Dong H, Hedin KE, et al. PD-1 blunts the function of ovarian tumor-infiltrating dendritic cells by inactivating NF-κB. Cancer Res. 2016;76(2):239–250. doi: 10.1158/0008-5472.Can-15-0748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Park SJ, Namkoong H, Doh J, Choi JC, Yang BG, Park Y, Chul Sung Y. Negative role of inducible PD-1 on survival of activated dendritic cells. J Leukoc Biol. 2014;95(4):621–629. doi: 10.1189/jlb.0813443. [DOI] [PubMed] [Google Scholar]
- 36.Zhao Y, Harrison DL, Song Y, Ji J, Huang J, Hui E. Antigen-Presenting Cell-Intrinsic PD-1 Neutralizes PD-L1 in cis to Attenuate PD-1 Signaling in T Cells. Cell Rep. 2018;24(2):379–90.e6. doi: 10.1016/j.celrep.2018.06.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fu C, Jiang A. Dendritic cells and CD8 T cell immunity in tumor microenvironment. Front Immunol. 2018;9:3059. doi: 10.3389/fimmu.2018.03059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gardner A, Ruffell B. Dendritic cells and cancer immunity. Trends Immunol. 2016;37(12):855–865. doi: 10.1016/j.it.2016.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Veglia F, Gabrilovich DI. Dendritic cells in cancer: the role revisited. Curr Opin Immunol. 2017;45:43–51. doi: 10.1016/j.coi.2017.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Roberts EW, Broz ML, Binnewies M, Headley MB, Nelson AE, Wolf DM, Kaisho T, Bogunovic D, Bhardwaj N, Krummel MF, et al. Critical role for CD103(+)/CD141(+) dendritic cells bearing CCR7 for tumor antigen trafficking and priming of T cell immunity in melanoma. Cancer Cell. 2016;30(2):324–336. doi: 10.1016/j.ccell.2016.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Villablanca EJ, Raccosta L, Zhou D, Fontana R, Maggioni D, Negro A, Sanvito F, Ponzoni M, Valentinis B, Bregni M, et al. Tumor-mediated liver X receptor-alpha activation inhibits CC chemokine receptor-7 expression on dendritic cells and dampens antitumor responses. Nat Med. 2010;16(1):98–105. doi: 10.1038/nm.2074. [DOI] [PubMed] [Google Scholar]
- 42.Broz ML, Binnewies M, Boldajipour B, Nelson AE, Pollack JL, Erle DJ, Barczak A, Rosenblum M, Daud A, Barber D, et al. Dissecting the tumor myeloid compartment reveals rare activating antigen-presenting cells critical for T cell immunity. Cancer Cell. 2014;26(6):938. doi: 10.1016/j.ccell.2014.11.010. [DOI] [PubMed] [Google Scholar]
- 43.Ma Y, Adjemian S, Mattarollo SR, Yamazaki T, Aymeric L, Yang H, Portela Catani J, Hannani D, Duret H, Steegh K, et al. Anticancer chemotherapy-induced intratumoral recruitment and differentiation of antigen-presenting cells. Immunity. 2013;38(4):729–741. doi: 10.1016/j.immuni.2013.03.003. [DOI] [PubMed] [Google Scholar]
- 44.Thompson ED, Enriquez HL, Fu YX, Engelhard VH. Tumor masses support naive T cell infiltration, activation, and differentiation into effectors. J Exp Med. 2010;207(8):1791–1804. doi: 10.1084/jem.20092454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lion E, Smits EL, Berneman ZN, Van Tendeloo VF. NK cells: key to success of DC-based cancer vaccines? Oncologist. 2012;17(10):1256–1270. doi: 10.1634/theoncologist.2011-0122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gajewski TF, Schreiber H, Fu YX. Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol. 2013;14(10):1014–1022. doi: 10.1038/ni.2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.van der Woude LL, Gorris MAJ, Halilovic A, Figdor CG, de Vries IJM. Migrating into the tumor: a roadmap for T cells. Trends Cancer. 2017;3(11):797–808. doi: 10.1016/j.trecan.2017.09.006. [DOI] [PubMed] [Google Scholar]
- 48.Hegde PS, Karanikas V, Evers S. The where, the when, and the how of immune monitoring for cancer immunotherapies in the era of checkpoint inhibition. Clinical Cancer Res. 2016;22(8):1865–1874. doi: 10.1158/1078-0432.Ccr-15-1507. [DOI] [PubMed] [Google Scholar]
- 49.Taube JM, Anders RA, Young GD, Xu H, Sharma R, McMiller TL, Chen S, Klein AP, Pardoll DM, Topalian SL, et al. Colocalization of inflammatory response with B7-h1 expression in human melanocytic lesions supports an adaptive resistance mechanism of immune escape. Sci Transl Med. 2012;4(127):127ra37. doi: 10.1126/scitranslmed.3003689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gurusamy D, Clever D, Eil R, Restifo NP. Novel “elements” of immune suppression within the tumor microenvironment. Cancer Immunol Res. 2017;5(6):426–433. doi: 10.1158/2326-6066.Cir-17-0117. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.