

ABSTRACT
DNA damage by genotoxic drugs such as gemcitabine and 5-fluorouracil (5-FU) activates the ataxia telangiectasia, mutated (ATM)-Chk pathway and induces the expression of NKG2D ligands such as the MHC class I-related chain A and B (MICA/B). The mechanisms underlying this remain incompletely understood. Here we report that xanthine oxidoreductase (XOR), a rate-limiting enzyme that produces uric acid in the purine catabolism pathway, promotes DNA damage-induced MICA/B expression. Inhibition of the ATM-Chk pathway blocks genotoxic drug-induced uric acid production, TGF-β-activated kinase 1 (TAK1) activation, ERK phosphorylation, and MICA/B expression. Inhibition of uric acid production by the XOR inhibitor allopurinol blocks DNA damage-induced TAK1 activation and MICA/B expression in genotoxic drug-treated cells. Exogenous uric acid activates TAK1, NF-κB, and the MAP kinase pathway. TAK1 inhibition blocks gemcitabine- and uric acid-induced MAP kinase activation and MICA/B expression. Exogenous uric acid in its salt form, monosodium urate (MSU), induces MICA/B expression and sensitizes tumor cells to NK cell killing. MSU immunization with irradiated murine breast cancer cell line RCAS-Neu retards breast cancer growth in syngeneic breast cancer models and delays breast cancer development in a somatic breast cancer model. Our study suggests that uric acid accumulation plays an important role in activating TAK1, inducing DNA damage-induced MICA/B expression, and enhancing antitumor immunity.
KEYWORDS: ATM, MICA/B, XOR, uric acid, TAK1, MAP kinase
Introduction
The NKG2D ligand family includes several members of the retinoic acid early transcript 1 (RAET1) (Rae 1, H60, and Mult1 in rodents, four UL-16 binding proteins in humans) and the MHC class I-related chain A and B (MICA/B), two glycosylated, polymorphic, and non-classical MHC class I transmembrane proteins1. NKG2D ligand expression is undetectable or low in normal epithelial cells but is overexpressed in a variety of tumors such as melanoma, breast, colon, hepatocellular, prostate cancer, and leukemia.2,3 Activation of the MAP kinase pathway due to RAS or BRAF gene mutations and the promoter DNA demethylation contribute to increased MICA/B expression.4–6 NKG2D ligands bind to their receptor expressed on natural killer cells, γδ T cells, and CD8+ T cells and play an important role in activating immune cells and antitumor immunity.1,7
Genotoxic drugs such as 5-fluorouracil (5-FU) or gemcitabine are thought to exert their tumoricidal effects by inhibiting DNA replication and cell proliferation and by inducing apoptosis. Van der Meer et al.8 recently reported that gemcitabine increases the sensitivity of ovarian cancer cells to CD34+ progenitor-derived NK cells in vitro and in vivo in NOD/SCID/IL2RgNull mice. However, it has been increasingly recognized that genotoxic drugs mediate their antitumor activity in part by eliciting antitumor immune response. In particular, genotoxic drugs stimulate MICA/B expression on fast proliferating fibroblasts and tumor cells by activating the ATM-Chk pathway.9 This pathway is primarily responsible for repairing DNA double-strand breaks and for phosphorylating p53 and CDC25A to halt cell cycle progression. Interestingly, activation of the ATM-Chk pathway also leads to up-regulation of MICA/B expression.9–12 Several studies suggest that NF-κB activation is involved in DNA damage-induced MICA/B expression.13,14
The expression of xanthine oxidoreductase (XOR), a rate-limiting enzyme in the purine catabolism pathway, is down-regulated in several types of malignancy including breast, colon, gastric, and lung cancers.15 XOR converts purine nucleotides to uric acid and reactive oxygen species.15,16 Uric acid is a novel “danger signal” that activates innate immunity.17 Uric acid produced in apoptotic or necrotic cells can induce the expression of the co-stimulatory molecules CD80 and CD86 in dendritic cells and promote dendritic cell maturation.18 Our prior study suggests that XOR and uric acid accumulation play an important role in activating the MAP kinase and inducing MICA/B expression in DNA-damaged tumor cells.19 However, whether the ATM-Chk pathway regulates MICA/B expression through uric acid production is not clear. How uric acid activates the MAP kinase pathway and induces MICA/B expression remains unknown.
TAK1 is a serine/threonine kinase that phosphorylates and activates a variety of kinases including ERK, p38, c-Jun terminal kinase (JNK), AMP-activated kinase (AMPK), and the I-kappa B kinase complex (IKK).20 TAK1 plays a crucial role in cell survival, differentiation, apoptosis, autophagy, and the inflammatory response.20,21 TAK1 is activated by various receptors such as the IL-1 and TGF-β receptors, the Toll-like receptors, and the B cell receptor.20,21 Several prior studies have shown that DNA damage activates TAK1, NK-κB, and the MAP kinase pathways.20 Singh et al. recently reported that uric acid is capable of locking TAK1 in an active-state conformation, resulting in sustained TAK1 kinase activation.22 We hypothesize that activation of the ATM-Chk pathway in genotoxic-stressed cells may induce NKG2D ligand expression by uric acid-activated TAK1. Here we report that uric acid production indeed plays a crucial role in activating TAK1 and inducing MICA/B expression in genotoxic drug-treated cells; Inhibition of TAK1 blocks DNA damage- and uric acid-induced MICA/B expression. Our study unveils a previously unrecognized pathway that regulates the expression of NKG2D ligands in DNA-damaged cells.
Materials and methods
Reagents
5Z-7-oxozeaenol (5Z), allopurinol (AP), uric acid, and 5-FU were purchased from Sigma Aldrich, Inc. (St. Louis, MO, USA). MSU crystals were prepared as previously described.23,24 2,7-dichlorodihydrofluorescin diacetate (DCFH-DA) was purchased from Molecular Probes, Inc. (Eugene, OR, USA). KU-55933 (S1092) was purchased from Selleck Chemicals LLC (Shanghai, China). Gemcitabine (Gemzar, Eli Lilly, Indianapolis, IN, USA) was purchased as a lyophilized powder and reconstituted in saline at 130 µM. Antibodies against TAK1 (5206S), TAK1S412 (9339S), ATM (2873S), Chk1 (2360S), p65S539 (3033S), ERKS202/204 (3510S), ERK (4685S), p38T180/182 (9211S), p38 (8690S), JNK (9252S), JNKT183/185 (4668S), and Chk2 (6334S) were purchased from Cell Signaling Technology (Danvers, MA, USA). Antibodies against p65 (sc-372), β-Actin (sc-47778), and glyceraldehyde 3-phosphate dehydrogenase (GAPDH), and debromohymenialdisine (DBH) (sc-202127) were obtained from Santa Cruz Biotechnology, Inc. (San Diego, CA, USA). The XOR expression vector and an anti-XOR rabbit antiserum were kindly provided by Dr. Richard Wright (The University of Colorado, CO, USA). Antibodies against MICA/B mAb (Clones 6D4, #320902), FITC-conjugated anti-human CD80 (Clone 2D10, #305205), and PE-conjugated anti-human CD86 (Clone BU63, #374205) were purchased from BioLegend (San Diego, CA, USA). Recombinant human IL-1β (#201-LB), recombinant human TNF-α (#210-TA), PGE2 (#2296), and FITC-conjugated anti-Pan-Rae1 antibody (#18107) were purchased from R&D Systems (Minneapolis, MN, USA). The anti-NKG2D neutralizing antibody (clone 2D11, #H00135250-M01) was purchased from Novus Biologicals, Inc. (Centennial, CO, USA). TAK1 siRNA (6317S), ATM siRNA (6328S), Chk1 siRNA (6241S), and Chk2 siRNA (6276S) were purchased from Cell Signaling Technology, Inc. (Danvers, MA, USA). The scrambled control siRNA, TurboFect transfection reagent, and siRNA transfection reagent Lipofectamine RNAiMAX were purchased from Life Technologies (Invitrogen Life Technologies, Inc., Grand Island, NY, USA).
Cells
MCF-7 (#HTB-22), HT-29 (#HTB-38), HeLa (#CCL-2), CT26 (#CRL-238), and B16-F10 (#CRL-475) cells were purchased from the American Tissue Culture Collection (Manassas, VA, USA). MCF-7 cells were cultured in MEM medium containing 10% fetal bovine serum and human recombinant insulin (10 µg/ml). RCAS-Neu cells are a murine breast cancer cell line that is derived from a breast cancer induced in TVA transgenic mice through intraductal injection of an avian retroviral vector encoding a mutant rat Neu oncogene.25 RCAS-Neu, CT26, and HT-29 cells were grown in complete RPMI 1640 medium containing 10% fetal bovine serum. HeLa and B16-F10 cells were grown in DMEM with 10% fetal bovine serum. NK-92 cells, a human natural killer cell line kindly provided Dr. Hans-G. Klingemann (Tufts-New England Medical Center, Boston, USA), were grown in MyoloCult H5100 medium (StemCell Technologies, Vancouver, BC, Canada) supplemented with 100 units IL-2/ml.
MICA/B expression in dendritic cells
(DCs). DCs were prepared by culturing human peripheral blood monocytes for 3 days in the presence of GM-CSF (200 IU/ml) and IL-4 (200 IU/ml). The cells were then left unstimulated or stimulated with MSU (200 µg/ml) or TNF-α(20 ng/ml), PGE2 (1 µM), and IL-1β (10 ng/ml) for 16 hr. Cells were analyzed for CD80 and CD86 expression by double staining with anti-CD80-FITC and anti-CD86-PE and FACS. Mouse IgG antibodies conjugated with PE or FITC were included as isotype controls. MICA/B expression was analyzed by staining with a FITC-conjugated anti-MICA/B antibody.
TAK1, ATM, Chk1, and Chk2 knockdown
HeLa cells seeded in 6-well plates were transfected with scrambled control siRNA, TAK1 siRNA, ATM siRNA, Chk1 siRNA, or Chk2 siRNA by using a Lipofectamine RNAiMAX transfection kit according to the manufacturer’s instruction. After incubation for 24 hr, the cells were left untreated or treated with 5-FU (10 µM) or gemcitabine (2 µM) for various lengths of time. Single-cell suspensions were prepared and analyzed for MICA/B expression by flow cytometry. Cell lysates were prepared and analyzed for protein phosphorylation and total protein levels by Western blot.
Flow cytometric analysis
MICA/B expression was stained as reported in our prior publications19,26 and analyzed in a fluorescence-activated cell-sorter scanner (Becton Dickinson, Palo Alto, CA, USA) with Cell Quest software or in a Beckman Coulter flow cytometer (Model CyAn ADP) with FlowJo software.
Quantification of uric acid
HeLa cells were lysed in the buffer containing Tris-HCl, 50 mM, pH 8.0; 2 mM EDTA, 1% Triton X-100 and followed by homogenization for 10 seconds. The lysates were incubated on ice for 30 min and spun down at 2000 g for 15 min. The supernatants were collected and analyzed for uric acid concentration by using a uric acid kit (BioAssay Systems, Hayward, CA, USA).
Reactive oxygen species (ROS) analysis
MCF-7 cells were transfected with pcDNA3.1 or the pCMV-XOR expression vector. After incubation for 24 hr, 5-FU (10 μM) or gemcitabine (2 μM) were added and then incubated for 4 hr. Single-cell suspensions were prepared and then loaded with fluorescein dye DCFH-DA (20 nM) at 37°C for 15 min. Intracellular ROS levels were assayed in a Beckman Coulter flow cytometer (Model CyAn ADP) with FlowJo software.
Western blot
MCF-7 cells were first transiently transfected with the pcDNA3.1 expression vector or the vector encoding a human XOR by using the TurboFect transfection reagent following the manufacturer’s instruction. MCF-7 and HeLa cells were treated with 5-FU (10 µM), gemcitabine (2 µM), or the various concentrations of MSU and then incubated for the indicated time points. Cell lysates were first analyzed for protein phosphorylation by Western blot. Membranes were then stripped and re-probed with antibodies against their total proteins.
Chromium release assay
Cells were grown in T-25 flasks. Upon 90% confluence, the cell monolayers were washed and treated with Cell Dissociation Solution (Sigma, St. Louis, MO). Single-cell suspensions were labeled with 51Cr (50/µCi per 1 × 106 cells) at 37°C for 1 hr. Cells were washed three times with Hank’s balanced salt solution. The cells (5000 per well) were added in triplicate in a 96-well U-bottom plate in the absence or presence of NK-92 cells at the ratio as indicated. Cells were incubated at 37°C in a humidified CO2 incubator for 4 hr. The supernatants were collected and transferred to a Ready-Cap. The radioactivity was read in a gamma counter (Becton Dickinson, Palo Alto, CA, USA). To test whether NK-92 cell-mediated cytotoxic activity on HT-29 and HeLa cells was mediated by MICA/NKG2D interaction, anti-MICA/B mAb (6D4, 1 µg/15,000 tumor cells) and anti-NKG2D mAb (2 µg/75,000 NK-92 cells) were premixed with tumor cells and NK-92 cells, respectively, and incubated for 30 min.
Animal experiments
All animal experiments were conducted in accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocol was approved by the Institutional Animal Care and Use Committee of Rush University Medical Center. Female FVB mice (6-7-wks-old; 6 mice/group) were immunized with MSU (200 µg/mouse) minus or plus irradiated RCAS-Neu breast cancer cells (5x106/mouse) by subcutaneous injection. Irradiation was conducted by exposing cells to a 137 Cs resource in a CIS Biointernational generator at a rate of 1.45 Gy per minute. Two weeks after immunization, RCAS-Neu cells (2x106/mouse) were injected into the fat pad. Tumor volumes were measured using a caliper, twice a week, and calculated based on the formula of length x width2 x π ÷ 6. Mice were sacrificed on day 42 after tumor implantation.
An orthotopic breast cancer model was also used to evaluate the antitumor activity of MSU. Briefly, female FVB mice (6-7-wks-old; 6 mice/group) were immunized with irradiated RCAS-Neu cells (5x106 cells/mouse) minus or plus MSU (200 µg/mouse) by subcutaneous injection. Two weeks after immunization, RCAS-Neu cells (5x104 cells in 10 µl/gland/mouse) were given by intraductal injection. Two weeks after tumor cell implantation, mice were immunized with irradiated RCAS-Neu cells (5x106 cells/mouse) minus or plus MSU (200 µg/mouse) again. Mice were observed weekly for tumor development.
MSU-mediated antitumor immunity was further investigated on a somatic breast cancer model as previously reported.27,28 Briefly, female TVA-transgenic mice (10 mice/group), which express the receptor for an avian retrovirus vector (RCAS) on mammary epithelial cells, were infected with RCAS-Neu virus (1x106 virions/gland/mouse in 10 µl) by intraductal injection. MSU were administered weekly by subcutaneous injection (200 µg/mouse) from the 3rd week for 5 weeks consecutively. Mice were monitored for tumor occurrence by palpation for 7 months after tumor induction.
Statistical analysis
Differences in the density of Western blot bands, uric acid levels, the cytotoxicity of HT-29 and HeLa cells between different treatment groups were statistically analyzed by using an unpaired Student’s t test or Analysis of Variance (ANOVA) when the data set has three or more independent groups. The differences in tumor volumes were statistically analyzed by using the one-way repeated measure ANOVA. The difference of tumor occurrence between the untreated and MSU-treated groups was statistically analyzed by using a Log-Rank test. A p value of <0.05 was considered statistically significant. All statistics were performed with SigmaPlot 11 software (Systat Software, Inc, San Jose, CA, USA).
Results
Genotoxic drugs induce MICA/B expression in XOR-transfected MCF-7
Previous studies showed that MCF-7 cells do not express XOR.29,30 Our prior study suggests that uric acid produced by XOR plays an important role in activating the MAP kinase pathway and subsequently inducing MICA/B expression in DNA-damaged cells.19 Here we examined the effect of XOR overexpression on the levels of ERK, ROS, uric acid production, and MICA/B in MCF-7 cells treated with 5-FU and gemcitabine. As shown in Figure 1a, XOR was overexpressed in MCF-7 cells transiently transfected with an expression vector encoding a human XOR gene (pCMV-XOR). The levels of XOR were very low in MCF-7 cells transfected with pcDNA3.1 (Figure 1a). 5-FU (10 µM) and gemcitabine (2 µM) induced ERK phosphorylation in XOR-transfected MCF-7 cells but only did little in MCF-7 cells transfected the pcDNA3.1 vector (Figure 1a). Of note, the concentrations of 5-FU and gemcitabine used in these studies were based on our prior study19 and many other publications. Consistently, the basal levels of uric acid (Figure 1b) and ROS (Figure 1c) were significantly higher in MCF-7 cells transfected with pCMV/XOR than pcDNA3.1. The levels of uric acid were significantly higher in XOR-transfected cells treated with 5-FU or gemcitabine than in their corresponding pcDNA3.1-transfected controls (Figure 1b). 5-FU and gemcitabine did not significantly increase ROS levels in pcDNA-transfected MCF-7 but significantly increased ROS levels in XOR-transfected cells (Figure 1c). The basal levels of MICA/B expression were significantly higher in XOR-transfected MCF-7 cells than in pcDNA-transfected MCF-7 cells (Figure 1d). 5-FU and gemcitabine further increased MICA/B expression in XOR-transfected but not pcDNA3.1-transfected cells (Figure 1d).
Figure 1.
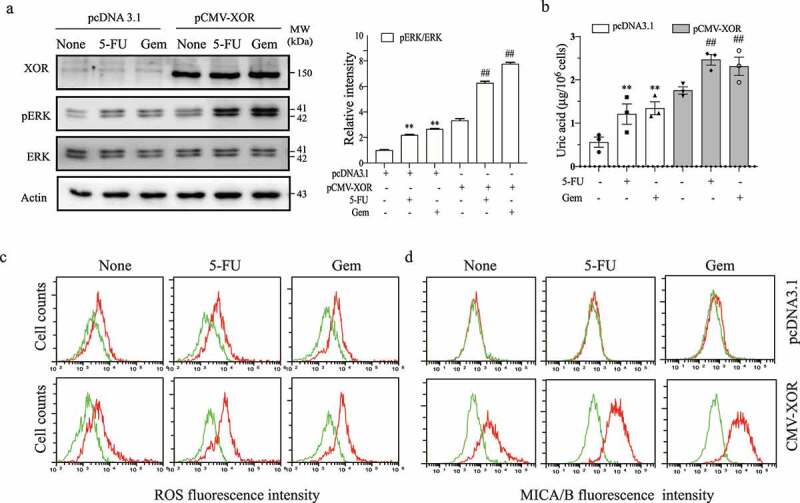
XOR overexpression enhances genotoxic drug-induced MAP kinase activation, ROS production, uric acid production, and MICA/B expression. MCF-7 cells were transiently transfected with the pcDNA3.1 vector or pCMV/XOR. After incubation for 24 hr, the cells were left untreated or treated with 5-FU (10 μM) or gemcitabine (2 μM) for 24 hr. Cell lysates were prepared and analyzed for ERK phosphorylation by Western blot (a) and for uric acid concentrations by using a uric acid assay kit (b). Relative phosphorylation levels were analyzed by quantifying the density of phosphorylated ERK bands normalized by the density of their total protein bands with NIH Image-J software and presented in a bar graph. Data are the mean ± standard deviation (SD) of three experiments. **p < .01, compared to the untreated control. ##p < .01, compared to the corresponding pcDNA3.1-transfected controls. Gem, gemcitabine. For assaying the levels of ROS and MICA/B, MCF-7 cells were treated as above and incubated for 4 or 24 hr, respectively. Single-cell suspensions were prepared and pulsed with the DCFH-DA fluorescent dye (c) or stained with a PE-conjugated anti-MICA/B antibody (d), followed by analysis with flow cytometry. (c) Green line, unstained; Red line, ROS; (d) Green line, isotype control; Red line, MICA/B. The data represent one of three independent experiments with similar results.
ATM-Chk inhibitors block uric acid production, MAP kinase activation, and MICA/B expression
It is well documented that activation of the ATM-Chk pathway leads to NKG2D ligand up-regulation.14 To determine if ATM-Chk activation regulates MICA/B expression through urid acid, we first tested if ATM-Chk activation was also responsible for increased uric acid production and for increased ERK phosphorylation. As shown in Figure 2a, KU-55933 (5 µM), an ATM inhibitor, and DBH (10 µM), a Chk inhibitor, did not affect the basal levels of TAK1 and ERK phosphorylation but blocked 5-FU- and gemcitabine-induced TAK1 and ERK phosphorylation in HeLa cells. Consistently, these inhibitors had no effect on the basal uric acid levels (Figure 2b). 5-FU (10 µM) and gemcitabine (2 µM) significantly increased uric acid levels, which was blocked by the inhibitors of ATM and Chk (Figure 2b). These inhibitors also blocked 5-FU- and gemcitabine-induced MICA/B expression in HeLa cells (Figure 2c). Similar observations were made with HT-29 cells (data not shown).
Figure 2.
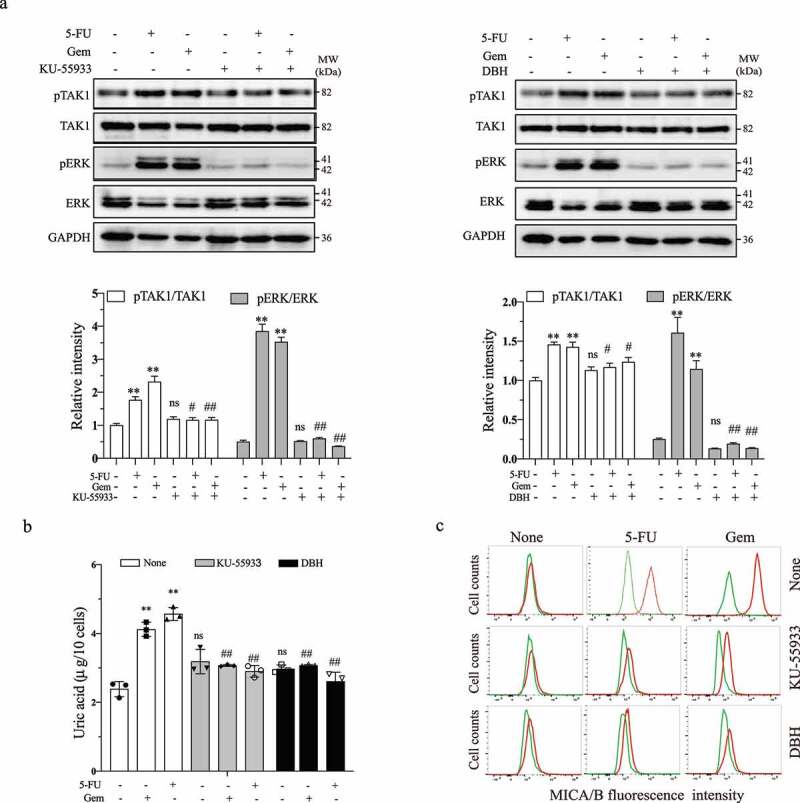
ATM and Chk1/2 inhibition blocks MAP kinase activation, uric acid production, and MICA/B expression. HeLa cells grown in 6-well plates were incubated for 24 hr in the absence or presence of 5-FU (10 μM) or gemcitabine (2 μM) minus or plus KU-55933 (5 μM) or DBH (10 μM). Cell lysates were prepared and analyzed for TAK1 and ERK phosphorylation (a) by Western blot and uric acid levels by using a uric acid assay kit (b). Relative phosphorylation levels were analyzed by quantifying the density of phosphorylated TAK1 and ERK bands normalized by the density of their corresponding total protein bands with NIH Image-J software and presented as bar graphs. Data in (a) and (b) are the mean ± SD of three experiments. **p < .01, compared to the untreated control. #p < .05, ##p < .01; ns, not significant, compared to their corresponding no-inhibitor controls. For assaying the levels of MICA/B, HeLa cells were treated as above and incubated for 24 hr, single-cell suspensions were stained with a PE-conjugated anti-MICA/B antibody (c). The levels of MICA/B expression were analyzed by flow cytometry. Green line, isotype control; Red line, MICA/B. The data represent one of three independent experiments with similar results.
ATM and Chk knockdown blocks DNA damage-induced MICA/B expression
We next tested if inhibition of ATM and Chk activity by siRNA had the same effects as ATM and Chk inhibitors. ATM, Chk1, and Chk2 expression was effectively down-regulated by their siRNA (Figure 3a). 5-FU (10 µM) and Gem (2 µM) significantly increased TAK1 and ERK phosphorylation in control siRNA-transfected cells but not in HeLa cells transfected with ATM, Chk1, and Chk2 siRNA (Figure 3a). 5-FU and gemcitabine increased the levels of uric acid (Figure 3b) and MICA/B (Figure 3c) in HeLa cells transfected with control siRNA but not in those transfected with ATM, Chk1, or Chk2 siRNA. These observations collectively suggest that ATM-Chk activation leads to increased uric acid production.
Figure 3.
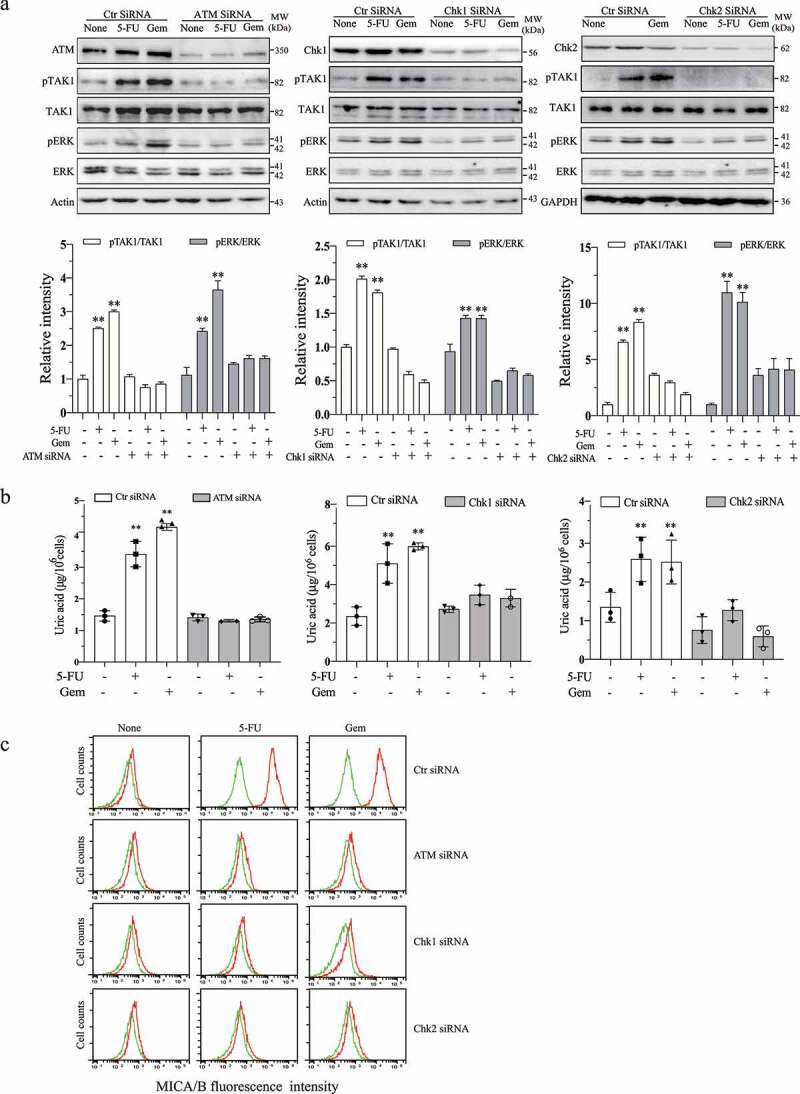
ATM and Chk silencing blocks genotoxic drug-induced ERK phosphorylation, uric acid production, and MICA/B expression. HeLa cells were transfected with scrambled control siRNA, ATM siRNA, Chk1 siRNA, or Chk2 siRNA. After incubation for 24 hr, the cells were left untreated or treated with 5-FU (10 μM) or gemcitabine (2 μM) for another 24 hr. Cell lysates were prepared and analyzed for TAK1 and ERK phosphorylation by Western blot (a) and uric acid levels by using a uric acid assay kit (b). Relative phosphorylation levels were analyzed by quantifying the density of phosphorylated TAK1 and ERK bands normalized by the density of their corresponding total protein bands with NIH Image-J software and presented as bar graphs. Data are the mean ± SD of three experiments. **p < .01, compared to the untreated control. ##p < .01; ns, not significant, compared to the corresponding control siRNA-transfected controls. Single-cell suspensions were prepared and analyzed for the levels of MICA/B expression (c) by flow cytometry. Green line, isotype control; Red line, MICA/B. The data represent one of three independent experiments with similar results.
Gemcitabine induces ERK phosphorylation and MICA/B expression by activating TAK1
Singh et al.22 recently reported that uric acid activates TAK1, a serine/threonine kinase that activates multiple signaling pathways such as ERK and NF-κB. Our prior study showed that gemcitabine induces ERK phosphorylation and MICA/B expression.19 Here we tested if genotoxic drug-induced ERK phosphorylation and MICA/B expression was mediated by uric acid-activated TAK1. As shown in Figure 4a, gemcitabine increased the levels of TAK1, JNK, p38, and p65 phosphorylation in HeLa cells in a time- and dose-dependent manner. 5Z (5 µM), a TAK1-specific inhibitor, blocked gemcitabine-induced TAK1, ERK, JNK, p38, and p65 phosphorylation (Figure 4b). Flow cytometry revealed that gemcitabine-induced MICA/B expression was also blocked by 5Z (5 µM) (Figure 4c). Similar observations were made with 5-FU in HeLa cells and with gemcitabine in HT-29 cells (data not shown).
Figure 4.
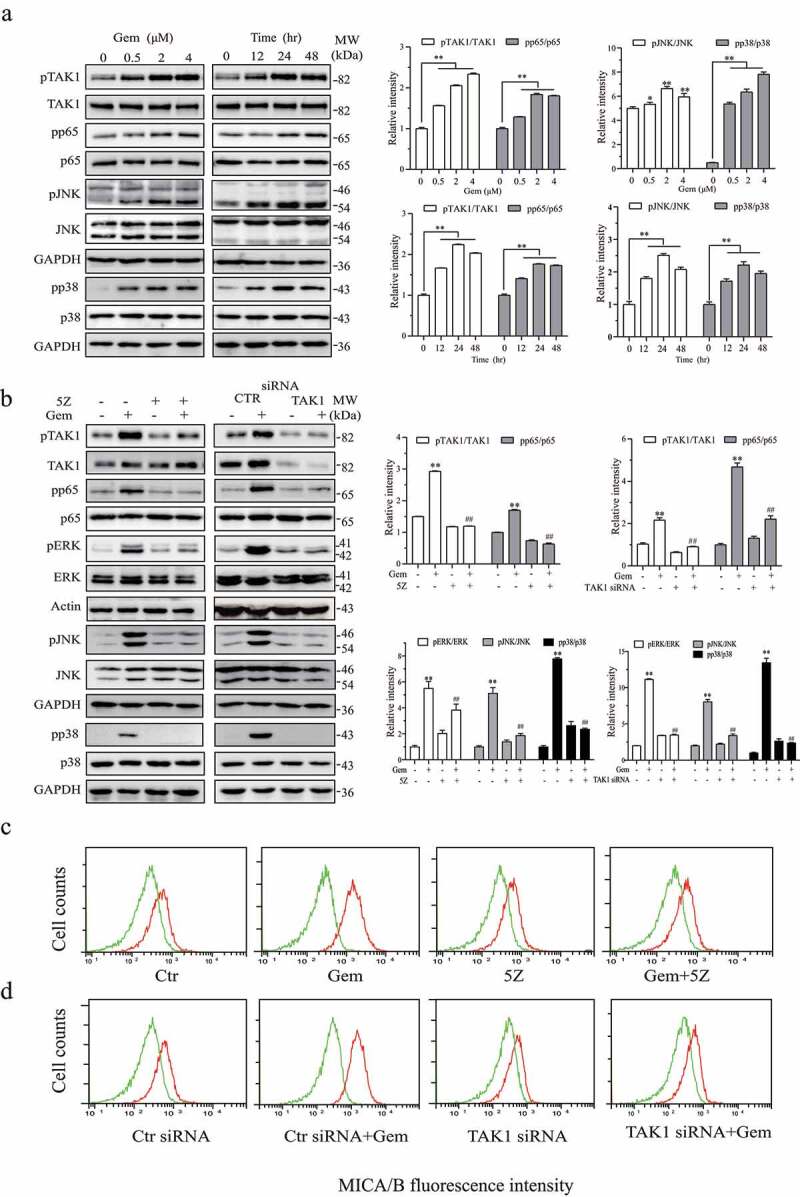
Gemcitabine induces ERK phosphorylation and MICA/B expression by activating TAK1. (a) HeLa cells were treated with the indicated concentrations of gemcitabine for 24 hr or treated with gemcitabine (2 μM) for the indicated lengths of time. Cell lysates were prepared and analyzed for TAK1, ERK, JNK, p38, and p65 phosphorylation as well as their total proteins by Western blot. (b) HeLa cells were incubated for 24 hr in the absence or presence of gemcitabine (2 μM) minus or plus 5Z (5 μM). Alternatively, HeLa cells were transfected with a scrambled control siRNA or TAK1 siRNA. After incubation for 24 hr, the cells were left untreated or treated with gemcitabine (2 μM) for another 24 hr. Cell lysates were prepared and analyzed for TAK1, ERK, JNK, p38, and p65 phosphorylation and their total proteins by Western blot. Relative phosphorylation levels were analyzed by quantifying the density of phosphorylated protein bands normalized by the density of their corresponding total protein bands with NIH Image-J software and presented as bar graphs. Data are the mean ± SD of three experiments. **p < .01, compared to the untreated control. ##p < .01, compared to the corresponding no-inhibitor or control siRNA-transfected controls. (c & d) HeLa cells were treated with gemcitabine minus or plus 5Z (c) or transfected with TAK1 siRNA and treated with or without gemcitabine as described above (d). Single-cell suspensions were prepared and analyzed for MICA/B expression by flow cytometry. Green line, isotype control; Red line, MICA/B. The data represent one of three independent experiments with similar results.
The role of TAK1 in mediating genotoxic drug-induced ERK phosphorylation and MICA/B expression was further confirmed by TAK1 knockdown. As shown in Figure 4b, TAK1 was effectively silenced in HeLa cells transfected with TAK1 siRNA, compared to that transfected with scrambled siRNA as a control. Gemcitabine (2 µM) was able to induce TAK1, ERK, JNK, p38, and p65 phosphorylation in control siRNA-transfected but not TAK1 siRNA-transfected HeLa cells. TAK1 knockdown also blocked gemcitabine-induced MICA/B expression in HeLa cells (Figure 4d).
Genotoxic drugs activate TAK1 by uric acid accumulation
Allopurinol (AP) is an XOR-specific inhibitor that inhibits uric acid production and has been used as an anti-uric acid drug for treating gout.31 Our prior study demonstrated that AP blocks genotoxic drug-induced uric acid and ROS production, ERK phosphorylation, and MICA/B expression.19 Here we tested if inhibition of uric acid production by AP also blocked genotoxic drug-induced activation of TAK1 and its downstream molecules. As shown in Figure 5a, AP (200 µg/ml) indeed blocked TAK1, JNK, ERK, p38, and p65 phosphorylation (Figure 5a) in HeLa cells treated with gemcitabine (2 µM). XOR overexpression enhanced the basal levels of JNK, p38, and p65 phosphorylation in MCF-7 cells, compared to that in pcDNA3.1-treated cells (Figure 5b). 5-FU (10 µM) and gemcitabine (2 µM) weakly or moderately increased TAK1, p38, JNK, and p65 phosphorylation in pcDNA3.1- and XOR-transfected MCF-7, compared to that in the untreated control (Figure 5b).
Figure 5.
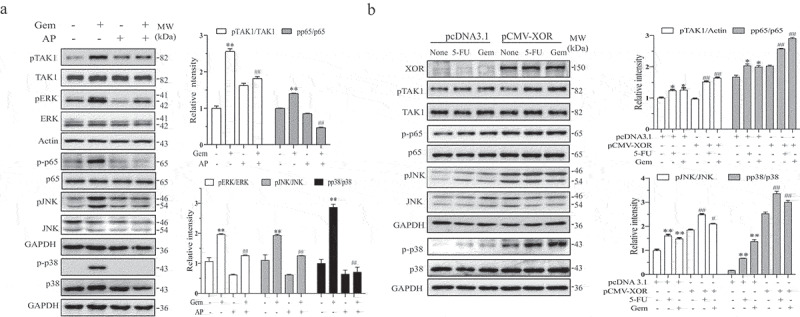
Genotoxic drugs activate TAK1 by uric acid production. (a) HeLa cells were incubated in the absence or presence of gemcitabine (2 μM) and allopurinol (AP) (200 μg/ml). After incubation for 24 hr, cell lysates were prepared and analyzed for TAK1, ERK, JNK, p38, and p65 phosphorylation and their total proteins by Western blot. (b) XOR overexpression enhances genotoxic drug-induced TAK1 activation. MCF-7 cells were transfected with pcDNA3.1 or pCMV/XOR. After incubation for 24 hr, the cells were left untreated or treated with gemcitabine (2 μM) or 5-FU (10 μM) for another 24 hr. The cells were harvested and analyzed for the levels of TAK1, JNK, p38, and p65 phosphorylation and their total proteins by Western blot. Relative phosphorylation levels were analyzed by quantifying the density of phosphorylated protein bands normalized by the density of their corresponding total protein bands with NIH Image-J software and presented as bar graphs. Data are the mean ± SD of three experiments. **p < .01, compared to the untreated control. #p < .05, ##p < .01, compared to the corresponding gemcitabine or pcDNA3.1-transfected controls.
Uric acid activates TAK1 to induce ERK phosphorylation and MICA/B expression
Having shown that uric acid produced by XOR played an important role in DNA damage-induced TAK1 phosphorylation and MICA/B expression, we next tested whether uric acid was able to directly activate TAK1. As shown in Figure 6a, uric acid in its salt form, MSU, increased TAK1, ERK1, JNK, p38, and p65 phosphorylation (Figure 6a) in HeLa cells in a time- and dose-dependent manner. We next tested if MSU-induced ERK and TAK1 phosphorylation and MICA/B expression was indeed mediated by TAK1. We found that 5Z blocked MSU-induced TAK1, ERK, JNK, p38 and p65 phosphorylation (Figure 6b) and MICA/B expression (Figure 6c) in HeLa cells. Consistently, TAK1 siRNA blocked MSU-induced TAK1. ERK, JNK, p38, and p65 phosphorylation (Figure 6b) and MICA/B expression (Figure 6d) in HeLa cells. Similar observations were made with 5-FU in HeLa cells and with gemcitabine in HT29 cells (data not shown).
Figure 6.
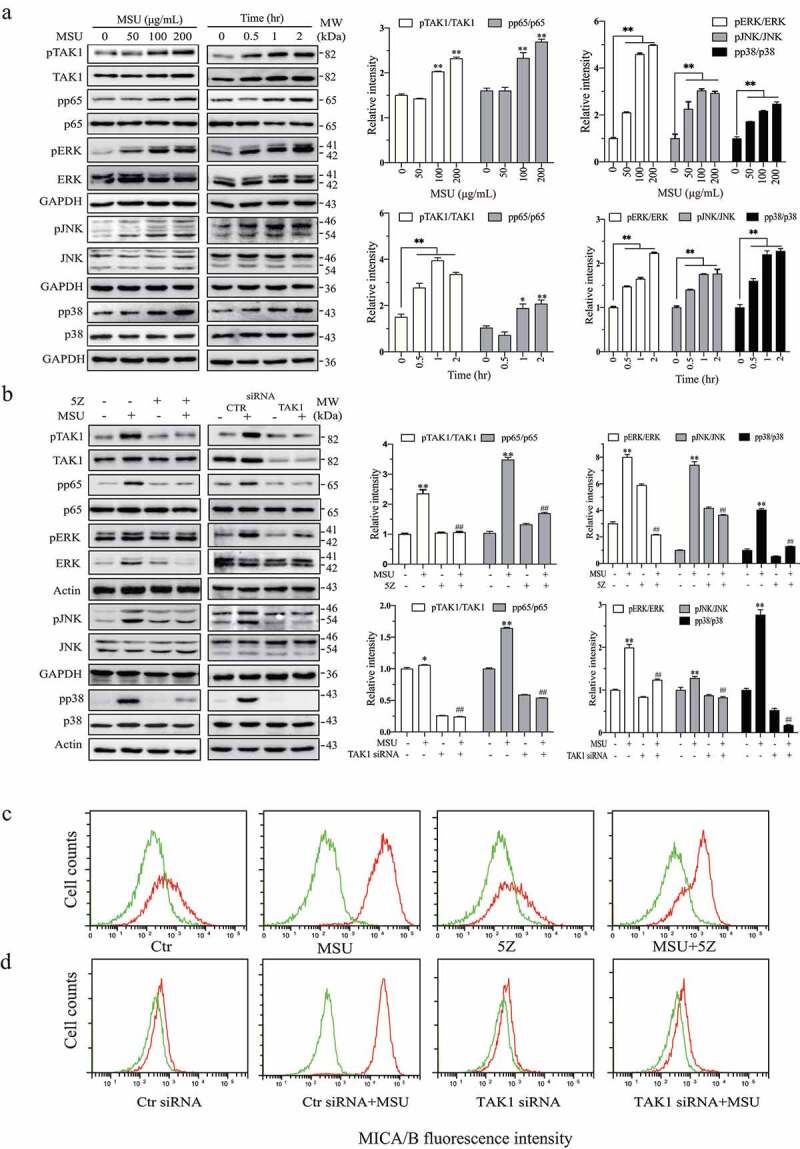
Uric acid activates TAK1 to induce ERK phosphorylation and MICA/B expression. (a) HeLa cells were incubated in the absence or presence of the indicated concentrations of MSU for 2 hr or incubated with MSU (200 μg/mL) for the indicated lengths of time. Cell lysates were prepared and analyzed for TAK1, ERK, JNK, p38, and p65 phosphorylation and their total proteins by Western blot. (b) MSU induces ERK phosphorylation by activating TAK1. HeLa cells were incubated in the absence or presence of MSU (200 μg/mL) and 5Z (5 μM) for 2 hr (b). Alternatively, HeLa cells were transfected with a scrambled control siRNA or TAK1 siRNA. After incubation for 48 hr, the cells were left untreated or treated with MSU (200 μg/ml) for another 2 hr. The cells were harvested and analyzed for TAK1, ERK, JNK, p38, and p65 phosphorylation and their total proteins by Western blot. Relative phosphorylation levels were analyzed by quantifying the density of phosphorylated protein bands normalized by the density of their corresponding total protein bands with NIH Image-J software and presented as bar graphs. Data are the mean ± SD of three experiments. *p < .05, **p < .01, compared to the untreated control. ##p < .01, compared to their corresponding MSU or control siRNA-transfected controls. (c & d) HeLa cells were incubated in the absence or presence of MSU (200 μg/mL) and 5Z (5 μM) for 24 hr. Alternatively, HeLa cells were transfected with a scrambled control siRNA or TAK1 siRNA. After incubation for 24 hr, the cells were left untreated or treated with MSU (200 μg/ml) for another 24 hr. Single-cell suspensions were prepared and stained for MICA/B expression followed by flow cytometry. Green line, isotype control; Red line, MICA/B. The data represent one of three independent experiments with similar results.
MSU induces NKG2D ligand expression and sensitizes tumor cells to NK cell killing
Uric acid has been considered a novel “danger” signal that can alert the immune system.17,23,32 Uric acid has been shown to be able to activate dendritic cells (DCs).23 Here we first determined if MSU could induce MICA/B expression in human DCs. As shown in Figure 7a, MSU increased the levels of CD80/CD86 and MICA/B equivalent to that in DCs activated by TNF-α, IL-1β, and PGE. We next determined the ability of genotoxic drugs and MSU to induced NKG2D ligand expression in murine tumor cells. As shown in Figure 7b, MSU induced Rae I expression in B16-F10 (a melanoma cell line), CT26 (a colon cancer cell line), and RCAS-Neu (a murine breast cancer cell line) cells. As previously reported,19 MSU also induced MICA/B expression in HeLa and HT-29 cells (Figure 7c). We next investigated if increased MICA/B expression in tumor cells would render them more sensitive to NK cell killing. As shown in Figure 7d, NK92 cells, a human NK cell line, killed HeLa and HT-29 cells pretreated with MSU more potently than in untreated controls and those pretreated with allopurinol as a negative control. Increased killing by NK cells was mediated by increased MICA/B expression since an anti-MICA/B and anti-NKG2D neutralizing antibody alone or in combination eliminated NK9 2 cell-mediated cytotoxicity against MSU-pretreated HeLa and HT-29 cells (Figure 7e).
Figure 7.
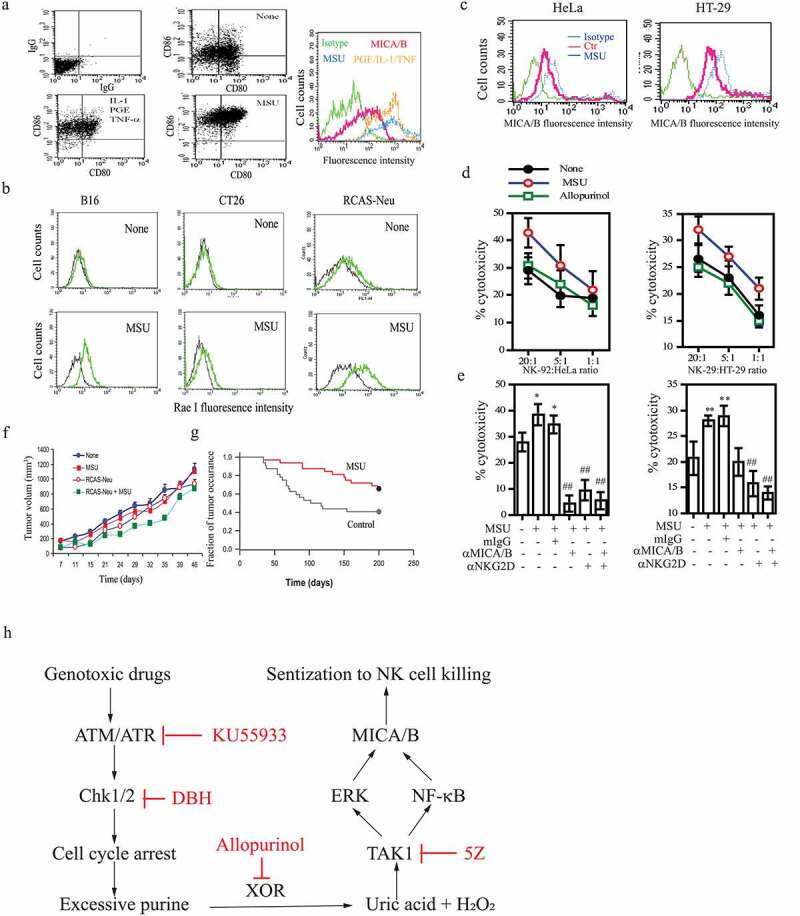
The in vitro and in vivo antitumor activity of MSU. (a) DCs were left unstimulated or stimulated with MSU (200 μg/ml) or a cocktail of the inflammatory cytokines (TNF-α, PGE2, and IL-1β) for 16 hr. Single-cell suspensions were analyzed for CD80 and CD86 expression by double staining with anti-CD80-FITC and anti-CD86-PE by FACS. Mouse IgG antibodies conjugated with PE or FITC were included as isotype controls. MICA/B expression was analyzed by staining with a FITC-conjugated anti-MICA/B antibody. Green line, isotype control; Red, Orange, and blue lines denote MICA/B in DCs left untreated or treated with the cytokine cocktail or MSU, respectively. (b) MSU induces NKG2D ligand expression in murine tumor cell lines. B16-F10, CT26, and RCAS-Neu cells were incubated with MSU (200 µg/ml) for 20 hr, single-cell suspensions were analyzed for Rae I expression by flow cytometry. (c) HeLa and HT-29 cells were pre-incubated with MSU (200 µg/ml) for 20 hr, the cells were harvested and analyzed for MICA/B expression by flow cytometry. (d) MSU-treated cells (20,000 cells/sample) were labeled with 51Cr for cytotoxicity assay and were mixed with NK-92 cells with the indicated ratios. After incubation for 4 hr, the supernatants were collected and assayed for 51Cr release. (e) HeLa and HT-29 cells were pretreated with MSU for 20 hr and then labeled with 51Cr. Tumor cells (20,000 cells/sample) and NK-92 cells (1x106/sample) were preincubated at 37°C for 30 min with an anti-MICA/B mAb (6D4) and anti-NKG2D mAb (clone 1D11), respectively. Mouse IgG was used as a negative control. Cells were mixed at the effector:target ratio of 1:10 and then incubated for 4 hr. The supernatants were collected and analyzed for the release of 51Cr in a scintillation counter. Data are the mean ± SD of three independent experiments. *p < .05, **p < .01, compared to the untreated control. ##p < .01, compared to mIgG control treatment. (f) Uric acid induces the antitumor immunity. RCAS-Neu cells were injected into the fat pad of FVB mice (6 mice/group) pre-immunized with MSU and/or irradiated RCAS-Neu. Tumor volumes were monitored twice weekly and plotted. (g) TVA transgenic mice infected with RCAS-Neu virus (1x106 virion/mouse) by intraductal injection were treated with saline or MSU. Mice were monitored for tumor occurrence for 7 months after tumor induction by palpation. Fraction of tumor-free mammary were plotted. MSU versus control, p < .01. (h) The schematic model of DNA damage-induced NKG2D ligand expression. Activation of the ATM-Chk pathway leads to cell cycle arrest. Excessive purine nucleotides are catalyzed by XOR to produce ROS and uric acid. The latter binds to and activates TAK1, a serine/threonine kinase that activates NK-κB and the MAP kinase pathway. Activation of NF-κB and the MAP kinase-activated AP1 transcription factor leads to transcriptional up-regulation of NKG2D ligand gene expression
MSU induces antitumor immunity
Having shown that MSU was able to induce NKG2D ligand expression in both DCs and tumor cells and to sensitize tumor cells to NK cell killing, we then investigated if MSU could induce antitumor immunity in vivo. We first used a syngeneic murine xenograft model to determine if MSU co-immunization with irradiated RACS-Neu cells could slow down or prevent tumor growth. As shown in figure 7f, immunization once with irradiated RCAS-Neu plus MSU was able to weakly but significantly retard the growth of RCAS-Neu tumors implanted in the fat pad of FVB mice. Immunization with MSU or irradiated RCAS-Neu cells alone did not affect RCAS-Neu tumor growth. We next determined if MSU plus RCAS-Neu immunization twice could elicit antitumor activity in a syngeneic orthotopic breast cancer model. As shown Table 1, twice immunization with MSU plus RCAS-Neu almost completely prevented the development of RCAS-Neu breast cancer in a syngeneic orthotopic model. In contrast, unimmunized mice or mice immunized with RCAS-Neu cells alone developed breast cancer in all FVB mice (Table 1). The adjuvant activity of MSU was further investigated in a somatic breast cancer model in which breast cancer was induced in TVA transgenic mice by intraductal injection of RCAS-Neu virus, an avian retroviral vector encoding a truncated rat Neu oncogene. As shown in Figure 7g, MSU treatment (200 µg/mouse) significantly delayed the formation of breast cancer, compared to those in saline-treated control group. These observations collectively suggest that uric acid crystals elicit a strong anti-tumor immunity.
Table 1.
Control of breast cancer by immunization with RCAS-Neu cells plus MSU*.
| Immunization | Tumor incidence | Tumor Weight | p value |
|---|---|---|---|
| None | 8/8 | 0.755 ± 0.376 | |
| RCAS-Neu | 8/8 | 0.771 ± 0.352 | n.s. |
| RCAS-Neu + MSU | 1/8 | 0.1 | < 0.01 |
*FVB mice were left unimmunized or immunized with irradiated RCAS-Neu breast cancer cells (5x106/mouse) minus or plus MSU (200 μg/mouse) by subcutaneous injection. Two weeks later, mice were inoculated with RCAS-Neu cells (5x104/gland/mouse in 10 μl PBS) by intraductal injection. Mice were immunized once again two weeks after tumor implantation and monitored for tumor occurrence for 3 months.
Discussion
It is well established that activation of the ATM-Chk pathway induces NKG2D ligand expression in cells undergoing genotoxic stress.33 Our present study focuses on the mechanisms of DNA damage-induced MICA/B expression. We provide several lines of evidence that uric acid accumulation and subsequent TAK1 activation play an important role in DNA damage-induced MICA/B expression: 1) Two genotoxic drugs were able to induce uric acid and ROS production, ERK phosphorylation, and MICA/B expression in MCF-7 cells overexpressing XOR; 2) Inhibition of the ATM-Chk pathway by their specific inhibitors or by siRNA blocked 5-FU- and gemcitabine-induced uric acid production, MAP kinase activation, and MICA/B expression; 3) Exogenous uric acid activated TAK1 and several downstream molecules such as p38, JNK, and NF-κB; 4) Inhibition of XOR activity by allopurinol blocked genotoxic drug-induced TAK1 activation and MICA/B expression. These observations collectively suggest that TAK1 is activated by uric acid and plays a central role in DNA damage-induced MICA/B expression. Our study has delineated a previously unrecognized pathway that regulates NKG2D ligand expression in DNA-damaged cells (Figure 7h).
XOR is a rate-limiting enzyme that converts hypoxanthine to xanthine, and xanthine to uric acid.15,16 Although XOR is down-regulated in several types of malignancy including breast, colon, gastric, and lung cancer, some patients with lymphoma or breast cancer treated with anticancer drugs develop the “tumor lysis” syndrome, a disorder characterized by dramatic increase of serum uric acid levels.15 Since the PI-3 and MAP kinase pathways are frequently mutated in a variety of cancer and promote purine and pyrimidine nucleotide biosynthesis, cancer cells are addicted to produce abundant pyrimidine and purine nucleotides.34,35 When the cell cycle is halted due to the activation of the ATM-Chk pathway, the buildup of excessive nucleotides in cancer cells is then converted into uric acid by XOR. Our present study shows that uric acid levels were significantly increased in XOR-transfected MCF-7 cells treated with two genotoxic drugs but not in pcDNA3.1-transfected cells (Figure 1b). Inhibition of the ATM-Chk pathway by their specific inhibitors or siRNA blocked genotoxic drug-induced uric acid production (Figure 2b & 3b). This is consistent with our prior study showing that DNA damage by genotoxic drugs or irradiation increases uric acid production in HeLa and HT29 cells, and that inhibition of XOR activity by allopurinol blocks DNA damage-induced uric acid accumulation.19,36 These observations collectively suggest that activation of the ATM-Chk pathway in cells undergoing genotoxic stress leads to uric acid accumulation.
Several prior studies have shown that DNA damage activates TAK1 through the retention of ATM in the cytoplasm and the formation of a complex containing ATM, NEMO, Ubc13, and XIAP or TRAF6.20 XIAP or TRAF6 functions as a ubiquitin ligase to ubiquitinate RIPK1, ELKS, or NEMO and recruit TAK1 for its activation. Hinz et al. reported that the cytoplasmic ATM-TRAF6-cIAP1 module ubiquitinates NEMO to activate TAK1 and NF-κB in irradiated HeLa cells.37 Wu et al. reported that ubiquitination of ELKS by the ubiquitin ligase XIAP recruits TAK1 to its ubiquitin chain and activates it,38 whereas Yang et al. reported that the cytosolic ATM/NEMO/RIPK1 complex recruits TAK1 into the polyubiquitin chain of RIPK1, leading to the activation of TAK1 and its downstream molecule, NF-κB and p38 in doxorubicin-treated murine embryonic fibroblast cells.39 Our present study shows that TAK1 and several downstream signaling pathways were activated in HeLa cells and in XOR-transfected MCF-7 cells by genotoxic drugs; Genotoxic drug-induced TAK1 activation was abrogated by the XOR inhibitor allopurinol; exogenous uric acid accumulation was able to activate TAK1. These observations suggest that uric acid plays a critical role in mediating DNA damage-induced TAK1 activation. In support of this notion, Singh et al. recently reported that uric acid stabilizes and activates TAK1.22 It appears that ATM may induce the formation of a cytoplasmic complex and provide a platform for initial TAK1 activation, whereas uric acid binds TAK1 and sustain its activation.
The ATM-Chk pathway plays an important role in cell cycle arrest and double-strand DNA break repair. Activation of this pathway also activates the MAP kinase pathway.40,41 For example, etoposide, a genotoxic drug, is unable to induce ERK kinase phosphorylation in ATM-deficient GM05823 fibroblast cells.42 How the ATM-Chk pathway cross-activates the MAP kinase pathway is not fully understood.43 Our present study shows that inhibition of the ATM-Chk pathway by their specific inhibitors or siRNA blocked genotoxic drug-induced TAK1 and ERK phosphorylation; Inhibition of TAK1 activity by 5Z or siRNA blocked genotoxic drug-induced ERK phosphorylation. In addition, we and others have shown that uric acid activates the MAP kinase pathway.19,44,45 We conclude that activation of the MAP kinase pathway through the ATM-Chk pathway is mediated through uric acid-activated TAK1 (Figure 7h).
Uric acid is the end-product of the purine catabolism pathway in humans and has been long recognized as a “danger” signal to alert immune system.17,32 Our prior study showed that uric acid accumulation leads to increased ERK phosphorylation and MICA/B expression in HeLa and HT29 cells.19 Exogenous uric acid induces ERK phosphorylation. Inhibition of the MAP kinase pathway abrogates uric acid-induced MICA/B expression.19 Moreover, genotoxic drugs lose their ability to induce NKG2D expression in XOR-silenced HeLa cells.19 Complementary to these observations, our present study demonstrates that genotoxic drugs induced uric acid accumulation, ERK phosphorylation, and MICA/B expression in XOR-transfected MCF-7 cells (Figure 1). Based on the observations that activation of the MAP kinase pathway in tumor cells with RAS or BRAF gene mutations up-regulates MICA/B expression through the AP1 transcription factor,26 we propose that uric acid induces MICA/B expression largely by activating the MAP kinase pathway (Figure 7h).
It should be noted that several prior studies have shown that activation of NF-κB and IRF3 is responsible for DNA damage-induced NKG2D ligand expression.33 For example, Hu et al. reported that NKG2D ligand expression is increased in tumor cells by CD8 T-cell-mediated engagement and activation of NF-κB and the p300/CBP-associated factor.46 Lam et al. reported that DNA damage activates the cGAS-STING pathway and its downstream IRF3 transcription factor to induce Rae I and ULBP expression in murine lymphoma cell lines.11 NF-κB activation is involved in regulating MICA/B expression in DNA-damaged endothelial cells33,47 and activated T cells.48,49 Our present study shows that TAK1 inhibition largely blocked genotoxic drug- and uric acid-induced p65 phosphorylation. This suggests that genotoxic drugs activate NF-κB in part by uric acid-activated TAK1. Since both NF-κB and AP1 are involved in MICA/B transcriptional regulation, we propose that DNA damage induces MICA/B expression through collaborative action of these two transcription factors (Figure 7h). However, our present study focused only on the MAP kinase pathway since we reported earlier that MAP kinase activation by uric acid crystals or by BRAF gene mutation leads to increased MICA/B expression.19,26
Uric acid has been long implicated in playing an important role in antitumor immunity. For example, Hu et al.50 reported that inhibition of uric acid production by allopurinol delays the rejection of immunogenic EL-4 tumors, which express the high levels of chicken ovalbumin; MSU administration alone induces the rejection of non-regressing EL-4 tumors, which express chicken ovalbumin at low levels. Behren et al.51 reported that immunization with MSU plus the murine MMC breast cancer cell line induces the production of anti-Neu antibody and prevents the growth of subcutaneously implanted MMC tumors. Endo et al.33 reported that uric acid production in adenovirus-infected tumor cells stimulates DCs to produce IFN-γ and IL-12 and induces an anticancer immune response. We reported earlier that inhibition of uric acid production in RCAS-Neu cells by XOR knockdown alleviates gemcitabine-mediated antitumor activity in a murine breast cancer model.19 Consistent with these observations, our present study shows that immunization with MSU plus irradiated RCAS-Neu retarded RCAS-Neu tumor growth (Figure 7e) and abrogated breast cancer development in an orthotopic syngeneic breast cancer model (Table 1). Furthermore, MSU administration alone delayed the development of the Neu oncogene-induced breast cancer in a somatic breast cancer model (Figure 7g), which recapitulates the oncogenesis of human breast cancer. MSU-mediated antitumor activity was much stronger in the orthotopic and somatic breast cancer models (Table 1 & Figure 7g) than the syngeneic model (figure 7f). While we did not characterize the nature of MSU-induced antitumor activity, we speculate that MSU may elicit its antitumor immunity by activating DCs and inducing NKG2D ligand expression in DCs and tumor cells. Nevertheless, our findings collectively suggest that uric acid produced in DNA-damaged tumor cells and exogenous MSU function as an adjuvant to enhance the antitumor immunity. It should be noted that, while uric acid production and subsequent NKG2D ligand expression could sensitize tumor cells to immune cell killing, TAK1 and NF-κB activation may render tumor cells more resistant to drug-induced apoptosis. Thus, whether the status of XOR can be uses as a marker for the prognosis of chemoimmunotherapy remains unclear.
In summary, our present study demonstrates that activation of the ATM-Chk pathway in tumor cells undergoing genotoxic stress leads to uric acid accumulation and increased MICA/B expression. Uric acid accumulation and exogenous uric acid activates TAK1 and its downstream signaling pathway. Inhibition of TAK1 by 5Z or siRNA blocks genotoxic drug- and uridine-induced MAP kinase activation and MICA/B expression. Uric acid crystals sensitize tumor cells to NK cell killing. Our study highlights the role of XOR and uric acid in mediating the ATM-Chk pathway-induced TAK1 activation, NKG2D ligand expression, and antitumor immunity in DNA-damaged tumor cells.
Acknowledgments
We thank Dr. Hans-G. Klingemann (Tufts-New England Medical Center, Boston) for kindly providing the NK-92 cell line, Dr. Yi Li (Baylor College of Medicine, Houston, TX, USA) for kindly providing the RCAS-Neu cell line, RCAS-Neu plasmid, and TVA-transgenic mice, and Dr. Richard Wright (The University of Colorado, CO) for kindling providing the XOR expression vector and an anti-XOR rabbit antiserum.
Correction Statement
This article has been republished with minor changes. These changes do not impact the academic content of the article.
Funding Statement
This work was supported in part by the National Natural Science Foundation of China (81672643) and the Priority Academic Program Development of Jiangsu Higher Education Institutions to Xiulong Xu.
Disclosure statement
The authors declare that there are no conflicts of interest
Author Contributions
Investigation, methodology, data curation: W.J., K.L., T.X., P.L.; Editing, R.A.P.; Conceptualization, validation, supervision, administration, writing, review, and editing: X.X.
References
- 1.Lanier LL. NKG2D Receptor and its ligands in host defense. Cancer Immunol Res. 2015;3(6):575–14. doi: 10.1158/2326-6066.CIR-15-0098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Liu H, Wang S, Xin J, Wang J, Yao C, Zhang Z. Role of NKG2D and its ligands in cancer immunotherapy. Am J Cancer Res. 2019;9:2064–2078. [PMC free article] [PubMed] [Google Scholar]
- 3.Mantovani S, Oliviero B, Varchetta S, Mele D, Mondelli MU. Natural killer cell responses in hepatocellular carcinoma: implications for novel immunotherapeutic approaches. Cancers (Basel). 2020;12(4):926. doi: 10.3390/cancers12040926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu H XV, Ss TJJ, Kamran N, Gasser S, Gasser S. Ras activation induces expression of raet1 family NK receptor ligands. J Immunol. 2012;189(4):1826–1834. doi: 10.4049/jimmunol.1200965. [DOI] [PubMed] [Google Scholar]
- 5.Ho SS, Gasser S. NKG2D ligands link oncogenic RAS to innate immunity. Oncoimmunology. 2013;2(1):e22244. doi: 10.4161/onci.22244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schmiedel D, Mandelboim O. NKG2D ligands-critical targets for cancer immune escape and therapy. Front Immunol. 2018;9:2040. doi: 10.3389/fimmu.2018.02040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lopez-Soto A, Huergo-Zapico L, Acebes-Huerta A, Villa-Alvarez M, Gonzalez S. NKG2D signaling in cancer immunosurveillance. Int J Cancer. 2015;136(8):1741–1750. doi: 10.1002/ijc.28775. [DOI] [PubMed] [Google Scholar]
- 8.Van der Meer JMR, de Jonge P, van der Waart AB, Geerlings AC, Moonen JP, Brummelman J, de Klein J, Vermeulen MC, Maas RJA, Schaap NPM, et al. CD34 + progenitor-derived NK cell and gemcitabine combination therapy increases killing of ovarian cancer cells in NOD/SCID/IL2Rg null mice. Oncoimmunology. 2021;10(1):1981049. doi: 10.1080/2162402X.2021.1981049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gasser S, Orsulic S, Brown EJ, Raulet DH. The DNA damage pathway regulates innate immune system ligands of the NKG2D receptor. Nature. 2005;436(7054):1186–1190. doi: 10.1038/nature03884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Soriani A, Iannitto ML, Ricci B, Fionda C, Malgarini G, Morrone S, Peruzzi G, Ricciardi MR, Petrucci MT, Cippitelli M, et al. Reactive oxygen species- and DNA damage response-dependent NK cell activating ligand upregulation occurs at transcriptional levels and requires the transcriptional factor E2F1. J Immunol. 2014;193(2):950–960. doi: 10.4049/jimmunol.1400271. [DOI] [PubMed] [Google Scholar]
- 11.Lam AR, Bert NL, Ho SS, Shen YJ, Tang MLF, Xiong GM, Croxford JL, Koo CX, Ishii KJ, Akira S, et al. RAE1 ligands for the NKG2D receptor are regulated by STING-dependent DNA sensor pathways in lymphoma. Cancer Res. 2014;74(8):2193–2203. doi: 10.1158/0008-5472.CAN-13-1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Le Bert N, Lam AR, Ho SS, Shen YJ, Liu MM, Gasser S. STING-dependent cytosolic DNA sensor pathways regulate NKG2D ligand expression. Oncoimmunology. 2014;3:e29259. doi: 10.4161/onci.29259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Antonangeli F, Soriani A, Cerboni C, Sciume G, Santoni A. How mucosal epithelia deal with stress: role of NKG2D/NKG2D ligands during inflammation. Front Immunol. 2017;8:1583. doi: 10.3389/fimmu.2017.01583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Raulet DH, Marcus A, Coscoy L. Dysregulated cellular functions and cell stress pathways provide critical cues for activating and targeting natural killer cells to transformed and infected cells. Immunol Rev. 2017;280(1):93–101. doi: 10.1111/imr.12600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Battelli MG, Polito L, Bortolotti M, Bolognesi A. Xanthine oxidoreductase in cancer: more than a differentiation marker. Cancer Med. 2015;5:546–547. doi: 10.1002/cam4.601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bortolotti M, Polito L, Battelli MG, Bolognesi A. Xanthine oxidoreductase: one enzyme for multiple physiological tasks. Redox Biol. 2021;41:101882. doi: 10.1016/j.redox.2021.101882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Martinon F. Mechanisms of uric acid crystal-mediated autoinflammation. Immunol Rev. 2010;233(1):218–232. doi: 10.1111/j.0105-2896.2009.00860.x. [DOI] [PubMed] [Google Scholar]
- 18.Shi Y, Mucsi AD, Ng G. Monosodium urate crystals in inflammation and immunity. Immunol Rev. 2010;233(1):203–217. doi: 10.1111/j.0105-2896.2009.00851.x. [DOI] [PubMed] [Google Scholar]
- 19.Xu X, Rao G, Li Y. Xanthine oxidoreductase is required for genotoxic stress-induced NKG2D ligand expression and gemcitabine-mediated antitumor activity. Oncotarget. 2016. 7(37):59220-59235. doi:18632/oncotarget.11042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mukhopadhyay H, Lee NY. Multifaceted roles of TAK1 signaling in cancer. Oncogene. 2020;39(7):1402–1413. doi: 10.1038/s41388-019-1088-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mihaly SR, Ninomiya-Tsuji J, Morioka S. TAK1 control of cell death. Cell Death Differ. 2014;21(11):1667–1676. doi: 10.1038/cdd.2014.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Singh AK, Haque M, O’Sullivan K, Chourasia M, Ouseph MM, Ahmed S. Suppression of monosodium urate crystal-induced inflammation by inhibiting TGF-beta-activated kinase 1-dependent signaling: role of the ubiquitin proteasome system. Cell Mol Immunol. 2019;18:162–170. doi: 10.1038/s41423-019-0284-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shi Y, Evans JE, Rock KL. Molecular identification of a danger signal that alerts the immune system to dying cells. Nature. 2003;425(6957):516–521. doi: 10.1038/nature01991. [DOI] [PubMed] [Google Scholar]
- 24.Shi Y, Parhar RS, Zou M, Baitei E, Kessie G, Farid NR, Alzahrani A, Al-Mohanna FA. Gene therapy of anaplastic thyroid carcinoma with a single-chain interleukin-12 fusion protein. Hum Gene Ther. 2003;14(18):1741–1751. doi: 10.1089/104303403322611755. [DOI] [PubMed] [Google Scholar]
- 25.Du Z, Li Y. RCAS-TVA in the mammary gland: an in vivo oncogene screen and a high fidelity model for breast transformation? Cell Cycle. 2007;6(7):823–826. doi: 10.4161/cc.6.7.4074. [DOI] [PubMed] [Google Scholar]
- 26.Xu X, Rao G, Gaffud MJ, Ding HG, Maki G, Klingemann H-G, Groh V, Spies T, Caillat-Zucman S, Gattuso P, et al. Clinicopathologic significance of major histocompatibility complex class I-related chain A and B (MICA/B) expression in thyroid cancer. J Clin Endocrinol Metab. 2006;91:2704–2712. doi: 10.1210/jc.2006-0492. [DOI] [PubMed] [Google Scholar]
- 27.Usha L, Rao G, Christopherson Ii K, Xu X, Li Y. Mesenchymal stem cells develop tumor tropism but do not accelerate breast cancer tumorigenesis in a somatic mouse breast cancer model. PLoS One. 2013;8(9):e67895. doi: 10.1371/journal.pone.0067895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Du Z, Podsypanina K, Huang S, McGrath A, Toneff MJ, Bogoslovskaia E, Zhang X, Moraes RC, Fluck M, Allred DC, et al. Introduction of oncogenes into mammary glands in vivo with an avian retroviral vector initiates and promotes carcinogenesis in mouse models. Proc Natl Acad Sci U S A. 2006;103(46):17396–17401. doi: 10.1073/pnas.0608607103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Linder N, Lundin J, Isola J, Lundin M, Raivio KO, Joensuu H. Down-regulated xanthine oxidoreductase is a feature of aggressive breast cancer. Clin Cancer Res. 2005;11(12):4372–4381. doi: 10.1158/1078-0432.CCR-04-2280. [DOI] [PubMed] [Google Scholar]
- 30.Taibi G, Carruba G, Miceli V, Cocciadiferro L, Nicotra CM. Estradiol decreases xanthine dehydrogenase enzyme activity and protein expression in non-tumorigenic and malignant human mammary epithelial cells. J Cell Biochem. 2009;108(3):688–692. doi: 10.1002/jcb.22305. [DOI] [PubMed] [Google Scholar]
- 31.Stamp LK, Day RO, Yun J. Allopurinol hypersensitivity: investigating the cause and minimizing the risk. Nat Rev Rheumatol. 2016;12(4):235–242. doi: 10.1038/nrrheum.2015.132. [DOI] [PubMed] [Google Scholar]
- 32.Rock KL, Kataoka H, Lai JJ. Uric acid as a danger signal in gout and its comorbidities. Nat Rev Rheumatol. 2013;9(1):13–23. doi: 10.1038/nrrheum.2012.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Endo Y, Sakai R, Ouchi M, Onimatsu H, Hioki M, Kagawa S, Uno F, Watanabe Y, Urata Y, Tanaka N, et al. Virus-mediated oncolysis induces danger signal and stimulates cytotoxic T-lymphocyte activity via proteasome activator upregulation. Oncogene. 2008;27(17):2375–2381. doi: 10.1038/sj.onc.1210884. [DOI] [PubMed] [Google Scholar]
- 34.Ali ES, Sahu U, Villa E, O’Hara BP, Gao P, Beaudet C, Wood AW, Asara JM, Ben-Sahra I. ERK2 phosphorylates PFAS to mediate posttranslational control of de novo purine synthesis. Mol Cell. 2020;78(6):1178–91 e6. doi: 10.1016/j.molcel.2020.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ben-Sahra I, Howell JJ, Asara JM, Manning BD. Stimulation of de novo pyrimidine synthesis by growth signaling through mTOR and S6K1. Science. 2013;339(6125):1323–1328. doi: 10.1126/science.1228792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xu X, Rao GS, Groh V, Spies T, Gattuso P, Kaufman HL, Plate J, Prinz RA. Major histocompatibility complex class I-related chain A/B (MICA/B) expression in tumor tissue and serum of pancreatic cancer: role of uric acid accumulation in gemcitabine-induced MICA/B expression. BMC Cancer. 2011;11:194. doi: 10.1186/1471-2407-11-194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hinz M, Stilmann M, Arslan SC, Khanna KK, Dittmar G, Scheidereit C. A cytoplasmic ATM-TRAF6-cIAP1 module links nuclear DNA damage signaling to ubiquitin-mediated NF-kappaB activation. Mol Cell. 2010;40(1):63–74. doi: 10.1016/j.molcel.2010.09.008. [DOI] [PubMed] [Google Scholar]
- 38.Wu ZH, Wong ET, Shi Y, Niu J, Chen Z, Miyamoto S, Tergaonkar V. ATM- and NEMO-dependent ELKS ubiquitination coordinates TAK1-mediated IKK activation in response to genotoxic stress. Mol Cell. 2010;40(1):75–86. doi: 10.1016/j.molcel.2010.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yang Y, Xia F, Hermance N, Mabb A, Simonson S, Morrissey S, Gandhi P, Munson M, Miyamoto S, Kelliher MA, et al. A cytosolic ATM/NEMO/RIP1 complex recruits TAK1 to mediate the NF-kappaB and p38 mitogen-activated protein kinase (MAPK)/MAPK-activated protein 2 responses to DNA damage. Mol Cell Biol. 2011;31(14):2774–2786. doi: 10.1128/MCB.01139-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Blackford AN, Jackson SPATM,ATR, Dna-pk. The trinity at the heart of the DNA damage response. Mol Cell. 2017;66(6):801–817. doi: 10.1016/j.molcel.2017.05.015. [DOI] [PubMed] [Google Scholar]
- 41.Rezatabar S, Karimian A, Rameshknia V, Parsian H, Majidinia M, Kopi TA, Bishayee A, Sadeghinia A, Yousefi M, Monirialamdari M, et al. RAS/MAPK signaling functions in oxidative stress, DNA damage response and cancer progression. J Cell Physiol. 2019;234:14951–14965. doi: 10.1002/jcp.28334. [DOI] [PubMed] [Google Scholar]
- 42.Tang D, Wu D, Hirao A, Lahti JM, Liu L, Mazza B, Kidd VJ, Mak TW, Ingram AJ. ERK activation mediates cell cycle arrest and apoptosis after DNA damage independently of p53. J Biol Chem. 2002;277(15):12710–12717. doi: 10.1074/jbc.M111598200. [DOI] [PubMed] [Google Scholar]
- 43.Hawkins AJ, Golding SE, Khalil A, Valerie K. DNA double-strand break - induced pro-survival signaling. Radiother Oncol. 2011;101(1):13–17. doi: 10.1016/j.radonc.2011.05.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tao M, Shi Y, Tang L, Wang Y, Fang L, Jiang W, Lin T, Qiu A, Zhuang S, Liu N, et al. Blockade of ERK1/2 by U0126 alleviates uric acid-induced EMT and tubular cell injury in rats with hyperuricemic nephropathy. Am J Physiol Renal Physiol. 2019;316(4):F660–F73. doi: 10.1152/ajprenal.00480.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cheng TH, Lin JW, Chao HH, Chen Y-L, Chen C-H, Chan P, Liu J-C. Uric acid activates extracellular signal-regulated kinases and thereafter endothelin-1 expression in rat cardiac fibroblasts. Int J Cardiol. 2010;139(1):42–49. doi: 10.1016/j.ijcard.2008.09.004. [DOI] [PubMed] [Google Scholar]
- 46.Hu J, Xia X, Gorlick R, Li S. Induction of NKG2D ligand expression on tumor cells by CD8(+) T-cell engagement-mediated activation of nuclear factor-kappa B and p300/CBP-associated factor. Oncogene. 2019;38(49):7433–7446. doi: 10.1038/s41388-019-0960-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cerboni C, Fionda C, Soriani A, Zingoni A, Doria M, Cippitelli M, Santoni A. The DNA damage response: a common pathway in the regulation of NKG2D and DNAM-1 ligand expression in normal, infected, and cancer cells. Front Immunol. 2014;4:508. doi: 10.3389/fimmu.2013.00508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lin D, Lavender H, Soilleux EJ, O’Callaghan CA. NF-kappaB regulates MICA gene transcription in endothelial cell through a genetically inhibitable control site. J Biol Chem. 2012;287(6):4299–4310. doi: 10.1074/jbc.M111.282152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cerboni C, Zingoni A, Cippitelli M, Piccoli M, Frati L, Santoni A. Antigen-activated human T lymphocytes express cell-surface NKG2D ligands via an ATM/ATR-dependent mechanism and become susceptible to autologous NK- cell lysis. Blood. 2007;110(2):606–615. doi: 10.1182/blood-2006-10-052720. [DOI] [PubMed] [Google Scholar]
- 50.Hu DE, Moore AM, Thomsen LL, Brindle KM. Uric acid promotes tumor immune rejection. Cancer Res. 2004;64(15):5059–5062. doi: 10.1158/0008-5472.CAN-04-1586. [DOI] [PubMed] [Google Scholar]
- 51.Behrens MD, Wagner WM, Krco CJ, Erskine CL, Kalli KR, Krempski J, Gad EA, Disis ML, Knutson KL. The endogenous danger signal, crystalline uric acid, signals for enhanced antibody immunity. Blood. 2008;111(3):1472–1479. doi: 10.1182/blood-2007-10-117184. [DOI] [PMC free article] [PubMed] [Google Scholar]