

ABSTRACT
The main biological function of the tumor suppressor p53 is to control cell cycle arrest and apoptosis. Among the p53 target genes, p21 has been identified as a key player in p53-mediated G1 arrest, while Killin, via its high DNA binding affinity, has been implicated in S and G2/M arrest. However, whether Killin is involved in G1 arrest remains unclear. This research aimed to explore the role of Killin in p53-mediated G1 arrest. Knockout of killin in human colorectal cells led to a dramatic decrease in p53-mediated G1 arrest upon DNA damage. Moreover, double knockout of killin and p21 completely abolished G1 arrest, similar to that of p53 knockout cells. We further showed that Killin could upregulate p21 protein expression independent of p53 via ubiquitination pathways. Immunoprecipitation studies indicated that Killin may directly bind to proteasome subunits, thereby disrupting proteasomal degradation of p21. Together, these results demonstrate that Killin is involved in multiple cell cycle checkpoint controls, including p53-mediated G1 arrest.
KEYWORDS: Killin, p53, p21, proteasome, G1 arrest
Introduction
p53 is one of the major tumor suppressors and the most frequently mutated gene in human cancer [1–3]. Under cellular stress, such as DNA damage, p53 is activated as a transcription factor that leads to cell cycle arrest, allowing a cell to either repair its DNA or undergo apoptosis [4]. p53 induces G1 arrest primarily through activation of its best-characterized downstream gene, p21, which encodes a cyclin-dependent kinase (CDK) inhibitor [5]. G2/M arrest mediated by p53 activation has been linked to the induction of GADD45 and 14-3-3δ [6]. Among the known p53 target genes involved in apoptosis are BCL2 family members such as Noxa and PUMA [7].
p21 is one of the best-known downstream genes of p53, induces G1 cell cycle arrest and inhibits cell apoptosis [2,5,8]. p21 is a cyclin-dependent kinase (CDK) inhibitor via its interaction with the N-terminal CDK kinase motif and causes G1 arrest [9]. Another important role of p21 is to inhibit apoptosis; p21 binds to and inhibits procaspase-3 and apoptosis signal-regulating kinase 1 (ASK1) in the cytoplasm, thereby blocking the apoptotic pathway [10,11]. Although p21 is mainly regulated at the transcriptional level by p53 upon DNA damage, post-translational modifications also play a critical role in the regulation of p21 through the ubiquitin-mediated proteasome degradation system [12,13].
killin is another p53 target gene discovered by differential display that encodes a 178 amino acid nuclear protein [14]. killin is located on human chromosome 10 at a locus extremely close to the pTEN tumor suppressor gene [14]. Many diseases or cancers that are associated with PTEN have also been linked to Killin [15–17]. Genetic screening and biochemical analysis have demonstrated that Killin binds to S phase DNA in a nonoverlapping pattern to the PCNA replication fork protein and inhibits DNA synthesis, which leads to S phase arrest [14,18]. In contrast, in interphase cells, Killin is localized in the nucleolus and has been implicated in the inhibition of rRNA synthesis [18]. Germline and somatic killin variants dysregulate G2/M cell cycle arrest [19]. Killin causes stalled replication forks, which in turn leads to upregulated p53 expression, thus forming a positive feedback loop for p53 activation [16,20,21]. In conclusion, as a tumor suppressor, the main functions of Killin are closely associated with p53-mediated S and G2/M phase arrest, and the distribution of Killin protein changes with cell cycle progression [18]. Based on the correlation between Killin and p53-mediated cell cycle arrest, we hypothesized that Killin is also involved in p53-mediated G1 arrest.
In this study, via a somatic gene knockout strategy, we showed that p53-mediated G1 cell cycle arrest upon DNA damage requires the induction of both p21 and Killin in human colorectal cells. Further analysis revealed that full induction of p21 requires Killin, which functions as a proteasome inhibitor that prevents p21 from ubiquitin-dependent proteolytic degradation. Together, these results suggest that Killin is involved in p53-mediated G1 arrest and provide a potential molecular basis for Killin in G1 arrest.
Results
Killin induces G1 arrest in HCT116 cells
To elucidate the physiological functions of Killin, we adopted CRISPR/Cas9 technology to selectively knock out killin in the HCT116 cell line containing the wild-type p53 gene, activating p53 in response to genotoxic stress, such as doxorubicin treatment [5]. Compared with the parental control cells, the killin knockout cells showed neither the complete killin gene in the genome (Figure 1a, Figure S1B) nor expression at the mRNA and protein levels after DOX treatment (Figure 1b,c, Figure S2B). Under normal conditions, the killin knockout cells showed little difference in cell growth (Figure 1d), morphology and cell cycle profile (Figure 1e,f) compared to their parental control cells.
Figure 1.
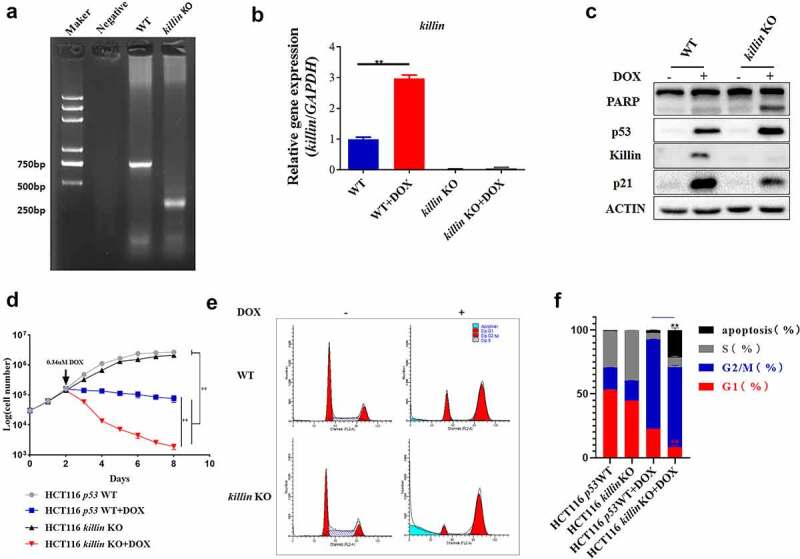
killin KO disrupts p53 induced G1 arrest under DNA damage.
(a) PCR amplification of killin gene with primer killin-check-F and killin-check-R to identify killin knocked out in HCT116. Size of the resulting PCR products was ~750bp (killin WT) or ~350bp (killin KO). (b) qPCR analysis identified that the efficiency of killin KO under DNA damage in HCT116. Mean ± SD of 3 replicates is shown. (c) killin KO accelerated p53-induced PARP cleavage and reduced p53-induced p21 expression. HCT116 parental cells, killin KO cells were treated or mock-treated with DOX (0.34 μM) for 24h. The induction of p53, p21, cleavage PARP and Killin were confirmed by Western blot analysis with ACTIN as a control for equal sample loading. (d) killin KO broke p53-mediated growth arrest under DNA damage in HCT116. (e, f) Cell cycle analysis showed killin KO disruption of p53-mediated G1 arrest following DOX (0.34 μM) treatment, the different phases of the cell cycle are presented (e) and the quantitative measurement of cell cycle phase (f). The killin gene was knocked out (KO) by CRISPR/Cas9 in HCT116 cells.
However, upon DNA damage, the levels of p53 protein and apoptosis as indicated by PARP cleavage were increased in the killin knockout cells compared to the parental HCT116 (p53 WT) cells, as shown by Western blot analysis (Figure 1c, Figure 2a, Figure S2A, D, E, H), which was consistent with a previous report [20]. Viable cell number markedly dropped for the killin knockout cells, while parental HCT116 (p53 WT) cells instead showed growth inhibition under DNA damage condition (Figure 1d). The proportion of G1 arrest was also reduced in the killin knockout cells, as shown by FACS analysis (Figure 1e,f). These phenotypes resemble those of p21 knockout cells [22], which show enhanced cell apoptosis and decreased G1 arrest compared to HCT116 (p53 WT) cells under DNA damage conditions. Interestingly, disruption of Killin expression reduced the protein level of p53-induced p21 (Figure 1c, Figure 2a, Figure S2C, G), which suggested a connection between Killin and p21.
Figure 2.
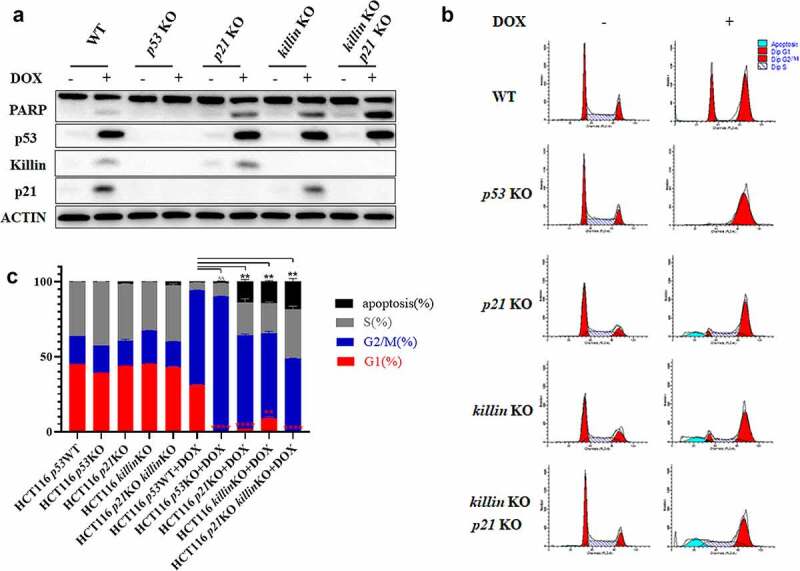
Killin and p21 completed the p53-mediated G1 arrest.
(a) killin and p21 double KO synergistically promote p53-induced PARP cleavage. HCT116 parental cells, p53 KO, p21 KO, killin KO and killin /p21 KO cells were treated or mock-treated with DOX (0.34 μM) for 24h. The induction of p53, p21, cleavage PARP and Killin were confirmed by Western blots analysis with ACTIN as a control for equal sample loading. (b, c) Cell cycle analysis reveals killin and p21 double KO blocking p53-mediated G1 arrest after DOX (0.34 μM) treatment, the different phases of the cell cycle are showed (b) and the quantitative measurement of cell cycle phase (c). The killin, p21, p53, and killin/p21 were knocked out (KO) by CRISPR/Cas9 in HCT116 cells.
Complete G1 arrest by p53 requires both p21 and Killin
To determine the relationship between Killin and p21 in p53-mediated G1 arrest, we knocked out p21, p53 and p21/killin in HCT116 (p53 WT) cells using the CRISPR/Cas9 system. After DOX treatment for 24h, both killin and p21 knockout cells showed a reduced proportion in G1 phase arrest, as demonstrated by FACS analysis (Figure 2b,c), and accelerated cell apoptosis to a similar extent, as shown by PARP cleavage and FACS analysis (Figure 2a, b,c, Figure S2F, H). Cells with double knockout of killin/p21 cells showed little G1 arrest, similar to p53 knockout cells, and an additive effect on apoptosis compared with p21 or killin knockout cells (Figure 2a, b,c, Figure S2F, H). In addition, we performed cell cycle analysis by FACS with 0.34 µM DOX treatment for 48h and 72h. The proportion of G1 phase arrest was almost nonexistent, and apoptotic cells were also increased in the p21 KO, killin KO and p21/killin KO cells (Figure S3). Although p21 has a predominant role in p53-mediated G1 arrest, our results showed that killin knockout can also decrease G1 arrest, similar to the disruption of p21. There is little G1 arrest in double knockout of killin/p21 cells after DNA damage, similar to p53 knockout cells; in other words, p53-mediated G1 phase arrest was completed by p21 and Killin.
Killin upregulates p21 expression independent of p53
p21, as one of the major target genes of p53, mainly induces G1 arrest and inhibits apoptosis. Surprisingly, we found that p21 expression was suppressed in the killin knockout cells under the stimulation of DNA damage (Figure 1c, 2a, Figure S2C, G); these results suggested Killin may directly regulate expression of p21. To further elucidate the relationship between Killin and p21, we overexpressed GFP-Killin via the TET-OFF system in DLD-1 cells. Removal of tetracycline induced either GFP or GFP-Killin at different time points (0h, 24h, 48h, 72h), and the p21 and p53 levels were upregulated with the expression of GFP-Killin as expected (Figure 3a, Figure S4A, B, C). However, there was no significant increase in cleaved PARP (Figure 3a, Figure S4D), which once again showed that the main function of Killin was not to induce cell apoptosis. p21 expression is usually controlled at the transcriptional level in a p53-dependent way [23], and we knocked out p53 in DLD-1 cells to induce GFP-Killin expression. Notably, the results showed that the ability of GFP-Killin to upregulate p21 expression was not affected (Figure 3b, Figure S4E, F, G), which confirms that Killin upregulated p21 expression independent of p53.
Figure 3.
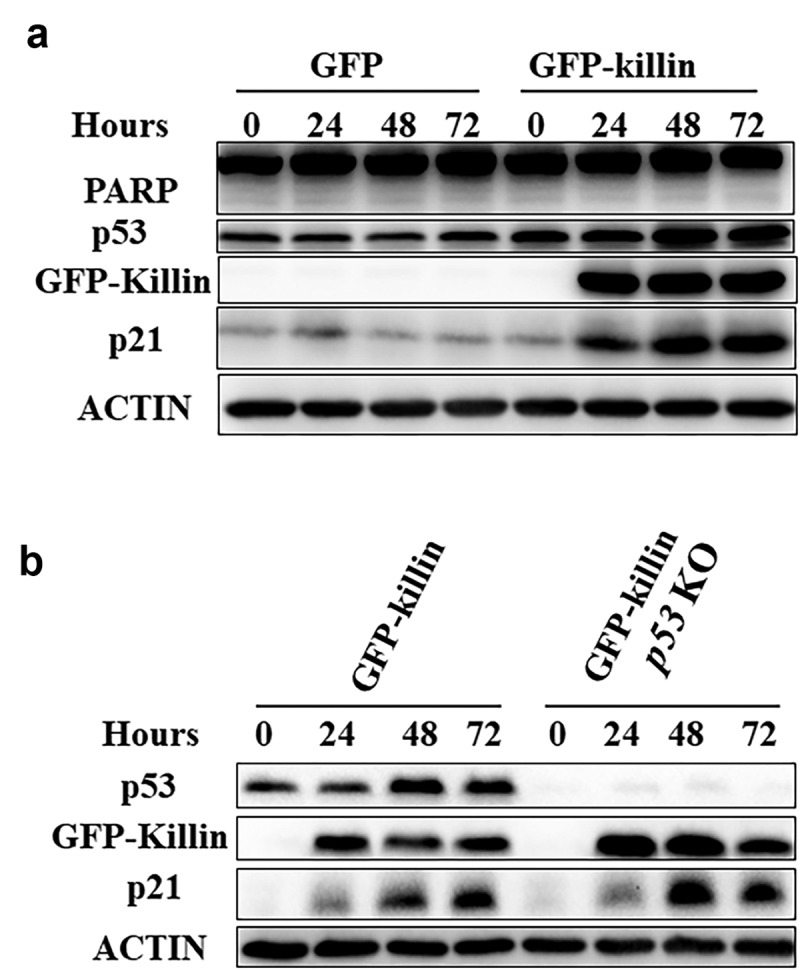
Killin upregulates p21 independent of p53.
(a) Western blots analysis of DLD-1 that the induction of GFP-Killin led to increase in p53 as well as its major target gene products, such as p21 compared with GFP at different time points (0h,24h,48h,72h). The cleavage PARP was not significantly increased after overexpression of GFP-Killin. (b) Knockout of p53 did not affect the induction of p21 by overexpression of GFP-Killin. The p53 KO and parental DLD-1 were induced GFP-Killin expression at different time points (0h,24h,48h,72h) before Western blots analysis. Though CRISPR/Cas9 knocked out (KO) p53 in DLD-1 cells with the ability of GFP-Killin inducing expression. GFP-Killin and GFP are induced to express by withdrawing tetracycline.
Killin stabilizes p21 through the proteasome pathway
To identify critical mechanisms involved in the Killin upregulation of p21 expression, we profiled the transcriptome of DLD-1 cells under the induction of GFP and GFP-Killin expression for 48h. Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis of genes related to both p21 and killin showed that these genes were predominantly distributed in pathways related to the proteasome, protein processing in the endoplasmic reticulum, and amyotrophic lateral sclerosis (Figure 4a). Among the genes involved in the proteasome pathway were multiple proteasome subunit proteins (Supplementary Table 3). These results suggested that Killin might upregulate p21 expression via proteasome pathways.
Figure 4.
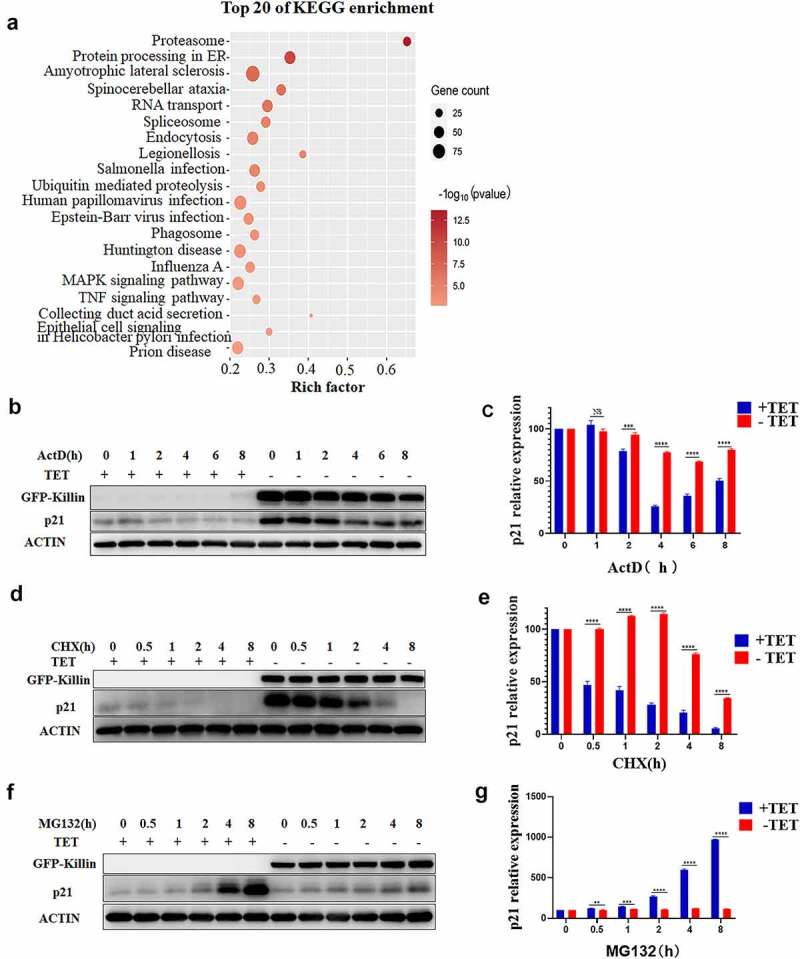
Killin via proteasome pathway upregulates p21.
(a) Pathway enrichment analysis of genes associated with p21 and Killin in DLD-1 cells with GFP-Killin expression. The x-axis shows the enrichment factor, the y-axis corresponds to the pathway, and the color of the dot represents the p-value, and the size of the dot indicates the number of genes mapped to the reference pathways. (b, d) GFP-Killin overexpression in DLD-1 cells increased the stability of p21. After the induction of GFP-Killin for 24h, 5 μg/mL actinomycin D (ActD) was added to suppress transcription (b) or 100 μg/mL cycloheximide (CHX) was added to block translation (d), and aliquots were isolated at the indicated times. Total proteins were extracted, and the level of p21 protein was detected by Western blots. (c, e) The expression of p21 protein relative to ACTIN was quantified (n = 3). (f) Disruption of proteasome pathway did not markedly upregulate the expression of p21 in DLD-1 cell with GFP-Killin expression. After the induction of GFP-Killin for 24h, 5 μM MG132 was added to inhibit proteasomal activity, and aliquots were isolated and analyzed at the indicated times by Western blot. (g) The expression of p21 protein relative to ACTIN was quantified (n = 3) in DLD-1 cell after MG132 treatment.
Therefore, to determine whether Killin participates in the proteasome pathway, we tested the protein stability using actinomycin D (ActD) to inhibit RNA synthesis and cycloheximide (CHX) to block the translational elongation, and the assay results indicated that the half-life of p21 was significantly prolonged in the GFP-Killin-expressing cells compared to the GFP-expressing cells (Figure 4b, c, d, e). Furthermore, we treated DLD-1 cells with GFP or GFP-Killin using MG132, a proteasome inhibitor, which blocks the proteasome pathway and upregulates p21 expression [24]. The results showed that the expression of p21 was greatly upregulated in the GFP-expressing cells after treatment of MG132, while the GFP-Killin-expressing cells only showed slight upregulation of p21 expression (figure 4f, g). Under normal conditions, p21 is degraded by the ubiquitin proteasome system, and blocking the proteasome degradation pathway increases p21 expression [25]. However, overexpression of Killin obviously diminished the upregulation of p21 expression by MG132, which could be because Killin completely inactivated the function of the proteasome pathways. These results support Killin upregulation of p21 expression through inhibition of proteasome pathways.
Killin upregulates p21 protein expression by inhibiting its ubiquitination
The ubiquitination of proteins plays a key role in protein degradation via the proteasome pathway. To determine whether Killin has effects on the ubiquitination of protein, we detected ubiquitination of whole protein and p21 by Western blots. Although the overexpression of GFP-Killin improved the ubiquitination of the whole protein (Figure 5a), polyubiquitination of p21 was significantly reduced in the DLD-1 cells with GFP-Killin (Figure 5b). Then, after treating DLD-1 cells with GFP or GFP-Killin using MG132, we found that polyubiquitination of p21 in the DLD-1 cells with GFP-Killin was also significantly lower than that in the GFP-expressing cells (Figure 5c,d). Killin inhibited the polyubiquitination of p21, which decreased the degradation of p21 by the proteasome. To further elucidate how Killin inhibits the polyubiquitination of p21, we detected Killin coimmunoprecipitated proteins by Western blots, and the results showed that Killin could bind to p21 (figure 5f). The direct binding of proteins inhibits ubiquitination and proteasomal degradation [26], and our results suggest that Killin might also directly bind to p21 to disrupt its ubiquitination and proteasomal degradation.
Figure 5.
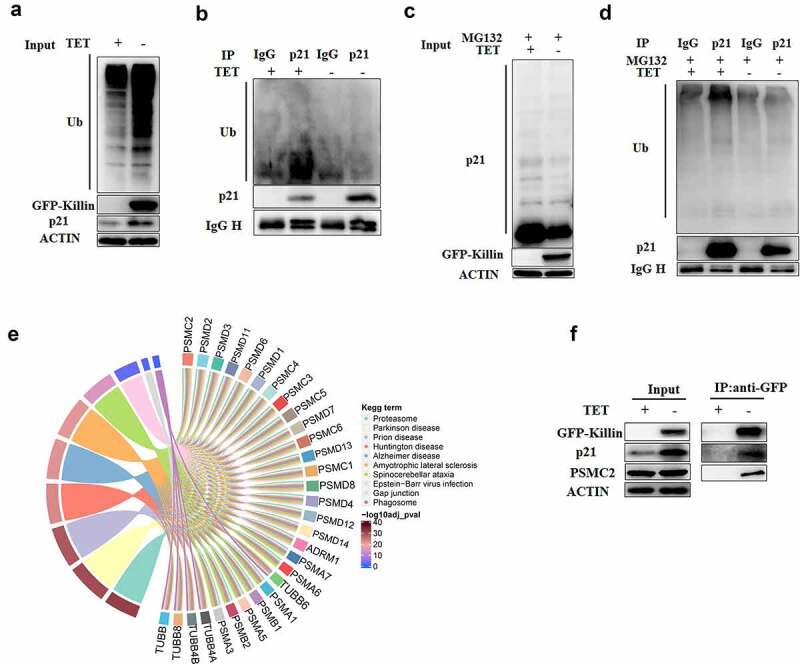
Killin reduces the degradation of p21 though suppression its ubiquitination.
(a, b, c, d) GFP-Killin inhibited the ubiquitination level of p21. Cell lysates from with (-TET) and without (+TET) GFP-Killin expressing for 48h were subjected to immunoprecipitation with anti-p21 antibody or immunoglobulin G control. The cell lysates (a) or immunoprecipitants (b) were detected using the indicated antibodies by Western blots. (c) After inducing GFP-Killin expression for 24h, 5 μM MG132 was added to inhibit proteasomal activity for 24h. The extracted of total proteins were subjected to immunoprecipitation with anti-p21 antibody or immunoglobulin G control. The cell lysates (c) or immunoprecipitants (d) were detected with the indicated antibodies by Western blots. (e) Pathway enrichment analysis of protein interaction with Killin. The colored small squares in the figure represent the proteins of interaction with Killin, linking each protein to the processes to which it is related. The color of the small squares in the figure represents the p value of the pathways. (f) GFP-Killin interacted with p21 and PSMC2. Cell lysates from with (-TET) and without (+TET) GFP-Killin expressing for 48h were subjected to immunoprecipitation with anti-GFP beads. The cell lysates (left) or immunoprecipitants (right) were detected using the indicated antibodies by Western blots.
Killin directly binds proteasome subunits and inhibits proteasomal activity
To further shed light on the molecular mechanism of Killin in the proteasome pathway, we performed immunoprecipitation coupled to mass spectrometry (IP-MS). Potential Killin-interacting proteins were co-pulled down with anti-GFP antibody-conjugated beads and subsequently applied to MS analysis. Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis of the proteins coimmunoprecipitated with Killin showed that these genes were predominantly distributed in pathways related to the proteasome, Parkinson’s disease, and prion disease, etc (Figure 5e). Among the genes involved in the proteasome, multiple proteasome subunit proteins were identified (Figure 5e, Supplementary Table 4). Input and Killin coprecipitated proteins were detected by immunoblotting. Although GFP-Killin had no effect on the expression of the proteasome 26S subunit protein PSMC2, the results of the co-IP assay with anti-GFP showed that Killin forms a complex with PSMC2 (figure 5f). Killin combined with proteasome 26S subunits, suggesting that Killin may inhibit proteasomal activity by directly binding proteasome subunits.
Discussion
The interpretation of the network of p53 transcriptionally regulated target genes is vital to comprehensively understand the complexity of p53-dependent tumor suppression [1]. p53 is known as the “guardian of the genome” and one of its main functions is regulating the cell cycle [2]. Through a nonbiased and systematic screening conducted using saturation fluorescent differential display technology, we identified the p53 target gene Killin, which is involved in p53-mediated S- and G2/M-phase arrest [14,18,19]. In this study, we showed that Killin induces G1 arrest and inhibits cell apoptosis. Overexpression of Killin did not immediately lead to cell apoptosis and mainly caused cell growth arrest (Figure 3a). The main function of Killin may be induction of cell cycle arrest, which allows time for the activation of the DNA repair pathway in response to DNA damage. All cell cycle processes including G1, G2/M, and S phase arrest are related to Killin, which suggests Killin may be a generalist involved in p53-mediated cell cycle arrest upon DNA damage.
p21 is perhaps one of the most characteristic downstream target genes of p53, which mainly induces G1 arrest and inhibits apoptosis [9]. p21 knockout cells did not completely abolish G1 arrest, as p53 knockout cells did upon DNA damage [5]; in other words, there are other genes participating in p53-mediated G1 arrest. As target genes of p53, both Killin and p21 can induce G1 arrest. We showed that double knockout of killin/p21 completely blocked p53-mediated G1 arrest and that Killin may be another gene involved in p53-mediated G1 arrest. In response to DNA damage, p53-induced Killin and p21 act as growth and death inhibitors, allowing enough time for DNA repair to maintain genomic stability [2]. Killin can also maintain chromosomal stability by preserving H3K9 methyltransferase activity and H3K9 trimethylation [27]. Maintaining genomic stability may be one of the important physiological functions of Killin. Upon DNA damage, knockdown of killin leads to an increase in genomic instability [20]. Our results also showed that upregulation of p53 expression was more obvious in the killin knockout cells compared to the parental HCT116 (p53 WT) cells under DNA damage conditions (Figure 1c,2a, Figure S2A, E). This is consistent with knockout of killin increased genomic instability, which in term leads to p53 upregulation. Killin upregulates p53 through replication fork arrest and direct binding to the p53ʹ promoter in normal condition [17,21].
During analysis of the mechanism of Killin-induced G1 phase arrest, we confirmed the upregulation of p21 expression by Killin. One of the major functions of Killin is as a component of a positive feedback loop to upregulate p53 expression [21], and the expression of p21 is tightly regulated by p53 at the transcriptional level [5]. Unexpectedly, our results showed that upregulation of p21 expression by Killin is not affected by p53 knockout. We profiled the transcriptome of the DLD-1 cells expressing GFP and GFP-Killin, and the proteasome pathway was enriched. Overexpression of Killin significantly extended the half-life of p21, similar to proteasomal inhibitors. Inhibition of the proteasome by MG132 can not obviously upregulate p21 expression in the DLD-1 cells with GFP-Killin expression, which may be due to disruption of the proteasome pathway by Killin.
The ubiquitination of proteins is the first step in the proteasomal degradation pathway, which plays a vital role in regulating protein half-life [25]. The direct binding of proteins can inhibit ubiquitination and proteasomal degradation [26]. We showed that Killin forms a complex with p21 that may inhibit the ubiquitination of p21. In addition, cytoplasmic Killin may affect E3 ubiquitin ligase activity [28], which can also decrease the ubiquitination of p21. Ubiquitination of p21 was inhibited in the DLD-1 cells with GFP-Killin overexpression, which resulted in increased stability of p21. Furthermore, Killin may directly bind proteasome subunits and impact proteasomal degradation. Killin stabilizes p21 by inhibiting ubiquitination and proteasome activity. However, the specific mechanism needs to be further studied in the future. Killin with different cell localizations has different functions; cytoplasmic Killin directly affects the proteasomal pathway-mediated upregulation of p21 expression, while nuclear Killin directly binds DNA/RNA and inhibits DNA/RNA synthesis [18]. Proteins and RNA are extensively synthesized during G1 phase, and Killin can also bind DNA/RNA and inhibit RNA synthesis, which restrains cell growth and leads to G1 arrest. Thus, Killin is involved in p53-mediated G1 arrest via both upregulation of p21 expression and inhibition of RNA synthesis. Analysis of the crystal structure of the Killin protein is important to understand the pattern of Killin binding to proteins and RNA/DNA, but the expression of Killin protein is hindered by its toxicity [14,29]. We may confirm the binding domain by truncation or point mutations of the Killin protein following bioinformatics analysis in the future, which will shed light on the definite role of Killin in proteasomal degradation.
Taken together, these results strongly support an important role of Killin in p53-mediated G1 arrest. Mechanistically Killin can cause G1 arrest either via upregulation of p21 expression, or by directly binding to DNA/and RNA during different cycle phases, thereby inhibiting their synthesis. We have provided a schematic diagram to illustrate the multifunctional roles of Killin in p53-mediated cell cycle arrest and apoptosis (Figure 6). The most important and rather surprising revelation of this study has been that complete G1 arrest of colorectal cancer cells with WT p53 requires both killin and p21, with Killin being able to inhibit p21 degradation via proteasome pathway. Absence of either of these p53 target genes would sensitize the cancer cells to p53-dependent apoptosis upon DNA damage.
Figure 6.
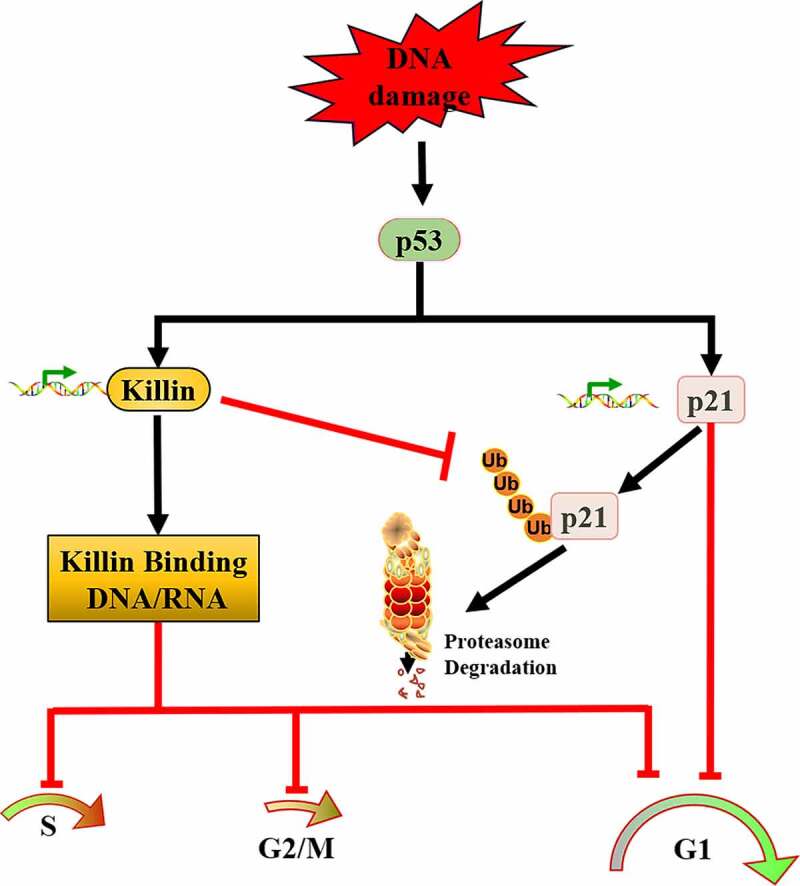
Schematic diagram depicting the pathway.
DNA damage activates p53, which induces its targets Killin and p21 expression to lead to p53-mediated G1 arrest. Though inhibition of p21 ubiquitination, Killin upregulates p21 expression. Killin can also induce G1, G2/M, S arrest via directly binding and inhibiting DNA/RNA synthesis.
Materials and methods
Cell culture, cell transfection, and drug treatments
HCT116 cells were propagated in Dulbecco’s Modified Eagles Medium (DMEM) (Hyclone) with 10% fetal bovine serum (HyClone) and 1% penicillin/streptomycin (Hyclone). Inducible Killin cell lines were cultured as described [14], in brief, the cells were washed three times with PBS and then cultured in Tetracycline – free medium. All plasmids were transfected into HCT116 and DLD-1 cell lines using FUGENE 6 (Promega, USA). MG-132 (S2619), Doxorubicin (S1208), and Tetracycline (S2574) were purchased from Selleck. Cells were treated with an indicated chemical compound at 80% confluence for the indicated time.
Antibodies and Western blots
Antibodies used for this study were as follows: p53 (sc-126) (Santa Cruz Bio), ACTIN (TA-09) (ZSbio), p21 (#2947), PARP (#9532), and Ub (#3936) (CST). The polyclonal Killin antibody was produced with the C terminus of Killin (amino acids 124–178) fused to GST (pGEX4T-1) as an antigen by Covance [14]. Cellular protein extraction was carried out by using the standard protocol as described [14]. Cellular proteins were separated using 10–12% SDS-PAGE gels, then transferred to PVDF membranes (Millipore). The membranes were blocked with 5% BSA (sigma) and hybridized to an appropriate primary antibody and HRP-conjugated secondary antibody in 5% BSA. Western lightning ECL reagent (Millipore) was used for signal detection.
Somatic gene knock out
Gene knockout was carried out essentially by CRISPR/Cas9 as described [30]. In short, the sgRNA primers were designed through website: https://zlab.bio/guide-design-resources. All sgRNA primer sequences are in Supplementary Table 1 in this study. The annealed sgRNA template was cloned into BbsI (NEB) sites of PX459 (addgene) and confirmed by DNA sequencing. After plasmid transfected into HCT116 p53WT or DLD-1, puromycin (Selleck, S9631) or hygromycin (Selleck, S2908) was used for selecting to obtain clones by limiting dilution. Gene knockout was verified by Western blots in pool and single clone cells.
Quantitative real-time PCR
Total RNA was extracted from treatment and control cells using TRIZOL (Invitrogen). cDNA was synthesized from 1 μg of total RNA as a template by Revert Aid First Strand cDNA Synthesis Kit (Thermo Fisher Scientific), according to the manufacturer’s protocol. Each reaction contained total RNA sample (1ug), RNase free water, Oligo (dT)18 primer. Then, samples were denatured at 65 ℃ for 5 minutes and then chill on ice. Next, 10 mM dNTPs mix, RiboLock RNase Inhibitor, reaction buffer and RevertAid M-MuLV RT were added to the reaction and samples were incubated at 42 ℃ for 60 min. Finally, the reaction was terminated by heating at 70°C for 5 min.
Quantitative PCR was performed using Universal SYBR® Green Super mix (Bio-Rad), according to the manufacturer's protocol. The qPCR reactions were performed in CFX-960 Real-time PCR System (Bio-Rad) and using Universal SYBR® Green Super mix (Bio-Rad) according to the manufacturer’s instructions. The thermal cycle conditions included heating to 95°C for 5 min, followed by 40 cycles at 95°C for 15 s, 55°C for 30 s, and 72°C for 30 s. The procedure ended by a melt-curve ramping from 55 to 95°C to check the PCR specificity at 0.1°C/s. Using the comparative Ct (2−ΔΔCt) method [31], relative expression levels of Killin mRNA were calculated for each sample after normalization against GAPDH. Primers used for qPCR are listed in Supplementary Table 2.
FACS analysis
FACS analysis was essentially as described [14]. After DOX treatment, all cells were harvested and washed with ice-cold PBS at the same time, then fixed within 70% ethanol overnight at −20°C, and stained with the DNA binding dye Propidium Iodide (PI) (50 μg/mL) and RNase (50 μg/mL) (Sigma) for 30 min at 37°C, cell suspension was immediately analyzed the cycling characteristics of cell populations with flow cytometry (FACS) (BD FACScalibur).
Co-immunoprecipitation (Co-IP)
Cells were collected, washed with cold PBS, and resuspended in RIPA (50mM Tris-HCl, pH 7.4, 150 mM NaCl, 0.25% deoxycholic acid, 1% NP-40, 1 mM EDTA) with Protease Inhibitor Cocktail (Bimake, B14001) and Phosphatase Inhibitor Cocktail (Bimake, B15001). For Co-IP of Killin, crude cell lysates were diluted to 1 mg/mL, 1 mg of protein per sample was incubated with 60ul of anti-GFP beads (Kangti-life) at 4°C overnight. For Co-IP of p21, cell lysates were incubated with 2ug of anti-p21 antibody or IgG control at room temperature for 4 h followed by incubation with protein A-agarose beads at 4°C overnight. Beads were then washed five times. All samples were stored at −80°C freezer for Western blotting or mass spectrometry (MS) analysis.
RNA extraction, sequencing, and Data analysis
In brief, total RNA was extracted from DLD-1 with GFP and GFP-Killin expression using TRIZOL (Invitrogen) following the manufacturer’s recommendations. Purification of mRNA using poly-T oligo-attached magnetic beads was fragmented with divalent cations. After constructing cDNA libraries, NGS sequencing was performed on Illumine Hiseq platform. Sequencing reads were aligned to human reference genome GRCh38 (ftp://ftp.ensembl.org), performed quality control on the Raw fastq file to get clean data and quantified by Salmon v0.81. To explore the function of genes, cluster profiler package was used for Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis.
Statistical analysis
All experiments were performed in triplicate. All data were shown as mean values ± SD (shown as error bars). Statistical analysis and comparisons were analyzed by two-tailed Student’s t-tests. The significance of the result is based on the calculated p-value, where the p-value less than 0.05 is considered statistically significant and where *p < 0.05, **p < 0.01 or***p < 0.001.
Supplementary Material
Acknowledgments
This work was founded by the grants from the National Natural Science Foundation of China (31801143), the general Project of Sichuan Education Department (18ZB0150), the general project of Natural Science Foundation of Chengdu Medical College (CYZ16-09).
Disclosure statement
No potential conflict of interest was reported by the author(s).
Author contributions
P.L. and D.L. conceived the ideas and supervised the study; P.L. and D.L. designed experiments; D.L., C.Y., C.S., S.L. carried out experiments; J.Y. performed bioinformatics analysis; P.L., D.L. wrote the manuscript.
Data availability statement
All data generated or analyzed during this study are included in this published article and its supplementary information files.
Supplementary material
Supplemental data for this article can be accessed here
References
- [1].Tokino T, Idogawa M, Sasaki Y.. P53 pathway and cancer: from bench to clinic. Pers Med Universe. 2015;4:1–3. [Google Scholar]
- [2].Boutelle AM, Attardi LD. p53 and tumor suppression: it takes a network. Trends Cell Biol. 2021;31(4):298–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Vogelstein B, Papadopoulos N, Velculescu VE, et al. Cancer genome landscapes. Science. 2013;339(6127):1546–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Pflaum J, Schlosser S, Mã¼ller M. p53 family and cellular stress responses in cancer. Front Oncol. 2014;4(4). DOI: 10.3389/fonc.2014.00285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Waldman T, Kinzler KW, Vogelstein B. p21 is necessary for the p53-mediated G1 arrest in human cancer cells. Cancer Res. 1995;55(22):5187–5190. [PubMed] [Google Scholar]
- [6].Giono LE, Manfredi JJ. The p53 tumor suppressor participates in multiple cell cycle checkpoints. J Cell Physiol. 2006;209(1):13–20. [DOI] [PubMed] [Google Scholar]
- [7].Pitolli C, Wang Y, Candi E, et al. p53-mediated tumor suppression: DNA-damage response and alternative mechanisms. Cancers (Basel). 2019;11(12):1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Schwab M. P21. 2012.
- [9].Xiao BD, Zhao Y-J, Jia X-Y, et al. Multifaceted p21 in carcinogenesis, stemness of tumor and tumor therapy. World J Stem Cells. 2020;12(6):481–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Zhan J, Easton JB, Huang S, et al. Negative regulation of ASK1 by p21Cip1 involves a small domain that includes serine 98 that is phosphorylated by ASK1 in vivo. Mol Cell Biol. 2007;27(9):3530–3541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Suzuki A, Tsutomi Y, Akahane K, et al. Resistance to Fas-mediated apoptosis: activation of caspase 3 is regulated by cell cycle regulator p21WAF1 and IAP gene family ILP. Oncogene. 1998;17(8):931–939. [DOI] [PubMed] [Google Scholar]
- [12].Starostina NG, Kipreos ET. Multiple degradation pathways regulate versatile CIP/KIP CDK inhibitors. Trends Cell Biol. 2012;22(1):33–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Li Y, Huang J, Zeng B, et al. PSMD2 regulates breast cancer cell proliferation and cell cycle progression by modulating p21 and p27 proteasomal degradation. Cancer Lett. 2018;430:109–122. [DOI] [PubMed] [Google Scholar]
- [14].Cho YJ, Liang P. Killin is a p53-regulated nuclear inhibitor of DNA synthesis. Proc Natl Acad Sci U S A. 2008;105(14):5396–5401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Bennett KL, Mester J, Eng C. Germline epigenetic regulation of KILLIN in Cowden and Cowden-like syndrome. JAMA. 2010;304(24):2724–2731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Wang Y, Radhakrishnan D, He X, et al. Transcription factor KLLN inhibits tumor growth by AR suppression, induces apoptosis by TP53/TP73 stimulation in prostate carcinomas, and correlates with cellular differentiation. J Clin Endocrinol Metab. 2013;98(3):E586–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Wang Y, He X, Yu Q, et al. Androgen receptor-induced tumor suppressor, KLLN, inhibits breast cancer growth and transcriptionally activates p53/p73-mediated apoptosis in breast carcinomas. Hum Mol Genet. 2013;22(11):2263–2272. [DOI] [PubMed] [Google Scholar]
- [18].Qiao M, Luo D, Kuang Y, et al. Cell cycle specific distribution of killin: evidence for negative regulation of both DNA and RNA synthesis. Cell Cycle. 2015;14(12):1823–1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Nizialek EA, Peterson C, Mester JL, et al. Germline and somatic KLLN alterations in breast cancer dysregulate G2 arrest. Hum Mol Genet. 2013;22(12):2451–2461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Sankunny M, Eng C. KLLN-mediated DNA damage-induced apoptosis is associated with regulation of p53 phosphorylation and acetylation in breast cancer cells. Cell Death Discov. 2018;4:92–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Cho Y-J, Liang P. S-phase-coupled apoptosis in tumor suppression. Cell Mol Life Sci. 2011;68(11):1883–1896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Polyak K, Waldman T, He TC, et al. Genetic determinants of p53-induced apoptosis and growth arrest. Genes Dev. 1996;10(15):1945–1952. [DOI] [PubMed] [Google Scholar]
- [23].Gartel AL, Tyner AL. Transcriptional regulation of the p21 (WAF1/CIP1) gene. Exp Cell Res. 1999;246(2):280–289. [DOI] [PubMed] [Google Scholar]
- [24].Zhang L, Hu JJ, Gong F. MG132 inhibition of proteasome blocks apoptosis induced by severe DNA damage. Cell Cycle. 2011;10(20):3515–3518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Dang F, Nie L, Wei W. Ubiquitin signaling in cell cycle control and tumorigenesis. Cell Death Differ. 2020;28(2):427–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Egunsola AT, Bae Y, Jiang -M-M, et al. Loss of DDRGK1 modulates SOX9 ubiquitination in spondyloepimetaphyseal dysplasia. J Clin Invest. 2017;127(4):1475–1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Nizialek EA, Sankunny M, Niazi F, et al. Cancer-predisposition gene KLLN maintains pericentric H3K9 trimethylation protecting genomic stability. Nucleic Acids Res. 2016;44(8):3586–3594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Sankunny M, Eng C. Identification of nuclear export signal in KLLN suggests potential role in proteasomal degradation in cancer cells. Oncotarget. 2020;11(50):4625–4636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Ma Z, Luo D, Huang A, et al. pKILLIN: a versatile positive-selection cloning vector based on the toxicity of Killin in Escherichia coli. Gene. 2014;544(2):228–235. [DOI] [PubMed] [Google Scholar]
- [30].Wang H, Yang H, Shivalila CS, et al. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell. 2013;153(4):910–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc. 2008;3(6):1101–1108. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated or analyzed during this study are included in this published article and its supplementary information files.