

ABSTRACT
Retargeting of T lymphocytes toward cancer cells by bispecific antibodies has demonstrated its therapeutic potential, with one such antibody approved for the treatment of acute lymphoblastic leukemia (blinatumomab) and several other in clinical trials. However, improvement of their efficacy and selectivity for solid tumors is still required. Here, we describe a novel tandem T-cell recruiting trispecific antibody for the treatment of colorectal cancer (CRC). This construct, termed trispecific T-cell engager (TriTE), consists of a CD3-specific single-chain Fv (scFv) flanked by anti-epidermal growth factor receptor (EGFR) and anti-epithelial cell adhesion molecule (EpCAM) single-domain VHH antibodies. The TriTE was well expressed in mammalian and yeast cells, bound the cognate antigens of the three parental antibodies, and enabled the specific cytolysis of EGFR- and/or EpCAM-expressing cancer cells, without inducing T cell activation and cytoxicity against double-negative (EGFR−EpCAM−) cancer cells. Bivalent bispecific targeting of double-positive HCT116 cells by TriTE improved in vitro potency up to 100-fold compared to single-positive cells and significantly prolonged survival in vivo. In addition, it was less efficient at killing single-positive target cells than the corresponding bispecific controls, leading to potentially enhanced tumor specificity. Moreover, dual targeting of two tumor-associated antigens may contribute toward preventing the tumor escape by antigen loss caused by selective pressures from conventional single-targeting T-cell engagers, and may help to overcome antigenic heterogeneity.
KEYWORDS: Trispecific antibodies, single-domain antibodies, scFv, tandem antibodies, cancer immunotherapy, colorectal cancer
Introduction
Recombinant DNA technology has allowed the development of a wide variety of multispecific and multivalent antibodies with potentially enhanced anti-tumoral activity and reduced Fc-associated toxicity. At present, single-chain variable fragments (scFv), consisting of VH and VL domains connected by a flexible linker peptide, and the variable domain of heavy-chain only antibodies (VHH), are the main building blocks used to generate recombinant Fc-free antibodies.1 Linking several of these building blocks with different specificities allows the design of bispecific (BsAbs) and trispecific (TsAbs) antibodies that are able to recognize one or two tumor-associated antigens (TAA) and one activating or costimulatory receptor in effector cells, thereby redirecting the immune response specifically toward TAA-expressing cancer cells. In this context, the anti-CD19 x anti-CD3 blinatumomab was the first tandem scFv, known as ‘bispecific T-cell engager’ (BiTE), approved by FDA (2014) and EMA (2015) for the treatment of B-cell acute lymphoblastic leukemia (B-ALL).2 Since then, an overwhelming number of BsAbs formats has been developed and dozens of them are currently under evaluation in early phases of clinical trials.3 BsAb design has evolved from the simple BiTEs to complex platforms such as trimerbodies, which allows the generation of trimeric and hexavalent BsAbs.4
The next challenge to further enhance the effector functions of immune cells is the generation of trifunctional or trispecific antibodies (TsAb). A recent example is the trifunctional natural killer (NK) cell engager (NKCE), targeting the activating receptors NKp46 and CD16 on NK cells and a TAA on cancer cells.5 Similarly, a TsAb that interacts with CD38, CD3 and CD28 enhances both T cell activation and tumor targeting.6 These two constructs are Fab-based and contain Fc regions, with a molecular weight well above 150 kDa. Interestingly, the CD38 trispecific antibody incorporated a Fc mutation for the ablation of FcγR binding in order to prevent side effects derived from off-target T cell activation by FcγR-expressing cells.7
An alternative for the generation of smaller TsAb formats with improved tumor penetration is the exclusive use of scFv and VHH as building blocks. The first checkpoint inhibitory T cell–engaging (CiTE) antibody described consisted of an anti-CD33 scFv fused to an anti-CD3 scFv and the extracellular domain of PD-1 in a single polypeptide chain.8 Another similar concept was the triplebody, with three scFv fused in tandem recognizing two different TAA and CD169–11 or CD3.12,13 In addition, trispecific killer engagers (TriKE) comprise an anti-CD16 scFv or VHH and an anti-TAA scFv crosslinked by the human interleukin-15 moiety.14–16 Last but not least, the TriTAC format incorporates an anti-albumin VHH for extended serum half-life.17
Overall, the great majority of fragment-based TsAb are focused on NK cell activation in order to treat hematological malignancies. Here, we propose a TsAb-based strategy to specifically activate T cells against two different TAA in solid tumor cells, using a format that we have named TriTE (Trispecific T-cell Engager). Dual TAA targeting may provide additional benefits, such as decreasing the risk of immune escape by antigen loss or decreasing on-target off-tumor side effects by improving tumor selectivity. The modularity of our TsAb design allows binding domains to be shuffled, to accommodate the phenotype of different tumors. In this study, we have designed, expressed and characterized an anti-EpCAM x anti-CD3 x anti-EGFR TsAb in the TriTE format.
In antibody-based therapies for colorectal cancer (CRC), EGFR is the most commonly targeted TAA. However, the efficacy of anti-EGFR mAbs cetuximab and panitumumab is limited due to primary and acquired resistance.18 In addition, high levels of expression of EpCAM have been shown in most of carcinomas and is associated with adhesion, proliferation, migration, and invasion of tumor cells.19 In fact, the EpCAM x CD3 IgG catumaxomab was the first BsAb approved by EMA (2009), although it was withdrawn in 2017 for commercial reasons. Catumaxomab was admininistered i.p. since i.v administration was not feasible due to hepatotoxicity, which was attributed to Fc-mediated, off-target T cell activation in the liver.20
In this proof-of-concept study, we demonstrate that TriTE antibodies were expressed in a functional state, simultaneously binding to EGFR and EpCAM to improve CD3 clustering on T cells and their cyototoxic effect. Moreover, the TriTE antibody showed an enhanced therapeutic effect in vivo compared to that of a control CD3 x EGFR BsAb.
Results
Design and expression of a trispecific T-cell engager (TriTE)
In this study, we generated a trispecific tandem VHH-scFv-VHH protein (AxOxE) by fusing the anti-human EpCAM A2 VHH21 and the anti-human EGFR Ega1 VHH22 to the N- and C-terminus, respectively, of the anti-human CD3 OKT3 scFv23 using flexible five-amino acid (G4S) linkers (Figure 1a). This new format was named trispecific T-cell engager (TriTE). For controls, two bispecific light T-cell engagers (LiTEs)24 were also designed: EpCAM x CD3 (AxO) and CD3 x EGFR (OxE). The three constructs were efficiently produced by transiently transfected human cells. Western blot analysis under reducing conditions of conditioned media (CM) showed a migration pattern consistent with the molecular weights calculated from their amino acid sequence (59 kDa for the AxOxE TriTE, 45 kDa for OxE LiTE and 44 kDa AxO LiTE) and size-exclusion chromatography (SEC) analysis of the AxOxE TriTE in CM showed a peak corresponding to the monomer (Suppl. Figure 1a-b). Next, we demonstrated by ELISA that CM from 293 TTriTE and 293 TLiTE (AxO/OxE) specifically recognized immobilized EpCAM and/or EGFR (Suppl. Figure 1c). Their ability to detect antigens in a cellular context was also studied by flow cytometry. Fluorescence staining was observed after incubation of CM from 293TTriTE and 293TLiTE OxE cells with the CRC cell lines HCT116 (EGFR+, EpCAM+) and CT26EGFR, the latter transduced with retrovirus encoding human EGFR25 (Suppl. Figure 1d). In addition, both AxOxE TriTE and AxO LiTE recognized EpCAM on HCT116 and SW620 cells (EGFR−, EpCAM+). Furthemore, the three antibodies detected CD3 on the surface of human Jurkat T cells (Suppl. Figure 1d) and were able to activate them in the presence of immobilized target antigens (Suppl. Figure 1e). The monoclonal antibodies cetuximab (anti-EGFR), OKT3 (anti-CD3) and Ber-EP4 (anti-EpCAM) were used as positive controls.
Figure 1.
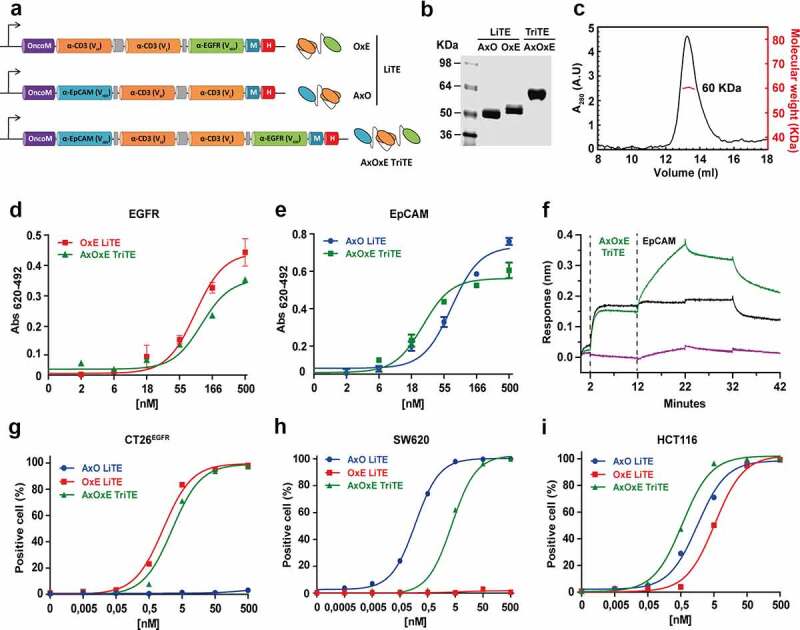
Schematic representation and characterization of trispecific T-cell engager (TriTE) and corresponding light T-cell engagers (LiTEs). (a) Genetic structure of the tandem VHH-scFv AxO LiTE formed by fusing the anti-EpCAM A2 VHH (blue box) N-terminally to the CD3-specific OKT3 scFv (Orange box); the scFv-VHH OxE LiTE comprising the anti-EGFR EGa1 VHH (green box) fused C-terminally to the OKT3 scFv; and the VHH-scFv-VHH AxOxE TriTE with anti-EpCAM and anti-EGFR VHH fused to the N- and C-terminus of OKT3 scFv, respectively. The Oncostatin M signal peptide (purple box) is used to direct secretion of recombinant antibody, and the myc/6xHis tags (dark blue and red boxes) were appended for immunodetection and affinity purification, respectively. Schematic representations showing arrangement of VHH and scFv in each construct are shown on the right. (b) Reducing SDS-PAGE of the three constructs and (c) SEC analysis of the purified AxOxE TriTE with the indicated molecular weight measured at the center of the chromatography peak (red line). (d-e) Titration ELISA against plastic-immobilized EGFR and EpCAM. Experiments were performed at least twice in duplicates. Mean ± SD are shown at each concentration. (f) Biolayer interferometry (BLI)-derived sensorgrams for the interaction between immobilized EGFR and AxOxE TriTE in the presence (green) or not (black) of soluble EpCAM. Note that the TriTE was present during association with EpCAM to prevent dissociation of the TriTE from immobilized EGFR. (g-i) FACS on CT26EGFR (EGFR+EpCAM−), SW620 (EGFR−EpCAM+) and HCT116 cells (EGFR+EpCAM+). Percentages of positive cells are shown at each concentration. AxO = A2 (anti-EpCAM VHH) x OKT3 (anti-CD3 scFv); OxE = OKT3 x Ega1 (anti-EGFR VHH); AxOxE = A2 x OKT3 x Ega1.
Purification and characterization of the EpCAMxCD3xEGFR TriTE
For upscaled production, the three antibodies were expressed in P. pastoris cells after 24 hours of methanol induction and purified by IMAC, with a yield of roughly 5 mg/L for OxE LiTE and 2 mg/L for AxO LiTE and AxOxE TriTE. Coomasie-stained SDS-PAGE analysis of the purified proteins revealed single bands (>95% pure) with molecular weight slightly higher than the observed in the CM of 293 cells, as previously reported for yeast-produced proteins26 (Figure 1b, Suppl. Figure 1a). Purified AxOxE TriTE mainly behaved as a globular protein in solution of about 60 kDa as determined by SEC analysis (Figure 1c). Purified AxOxE TriTE and LiTEs showed similar dose-dependent binding curves to plastic-immobilized EGFR or EpCAM, with AxOxE TriTE displaying a slightly lower signal at the highest concentration (Figure 1d-e).
We next studied whether the binding sites of the AxOxE TriTE can bind concurrently to both EGFR and EpCAM. Biolayer interferometry (BLI)-derived sensorgrams showed binding of AxOxE TriTE to EGFR-coated biosensors, giving additional binding curves upon addition of EpCAM (Figure 1f). This experiment demonstrated that the AxOxE TriTE can simultaneously bind to EGFR and EpCAM, and that these interactions are therefore not sterically incompatible.
Then, the ability of AxOxE TriTE to detect the three cognate antigens as cell surface proteins was studied by flow cytometry (Figure 1g-i, Suppl. Figure 2). All three constructs recognized CD3 on the surface of Jurkat T cells. The AxO LiTE stained EpCAM on the surface of SW620 and HCT116 cells, and the OxE LiTE detected EGFR on CT26EGFR and HCT116 cells. Indeed, the AxO LiTE performed better than TriTE on SW620 cells. However, only the AxOxE TriTE was able to stain the three CRC cell lines. The AxO and OxE LiTE did not stain single-positive CT26EGFR or SW620 cells, respectively. Neither of the three recombinant antibodies bound to the triple negative CT26mock cell line (hereafter referred to as CT26).
It has been described that apparent affinity increases when a bivalent antibody binds to the second target following its binding to the first receptor on the same cell.27,28 This phenomenon is also observed in the case of AxOxE TriTE: apparent affinity is 4.5 nM and 3.94 nM for SW620 and CT26EGFR cells, respectively, but changes to 0.51 nM in double-positive HCT116 (approximately sevenfold). These differences cannot be attributed to higher expression of target antigens in HCT116 cells. Indeed, EpCAM expression levels on the surface of SW620 and HCT116 cells are practically identical (MFI = 633 and 640, respectively) (Suppl. Figure 3a). Moreover, CT26EGFR cells exhibited slightly higher EGFR levels than HCT116 (MFI = 550 vs 505).
Next, we analyzed the long-term stability in serum of the three constructs. For this purpose, purified proteins were incubated in 60% mouse (Figure 2a-b) or human (Figure 2c-d) serum for 0 (control) to 4 days at 37°C. The purified AxOxE TriTE was very stable with 80% EGFR and EpCAM binding activity after 96 hours of incubation, comparable to those of OxE and AxO LITEs.
Figure 2.
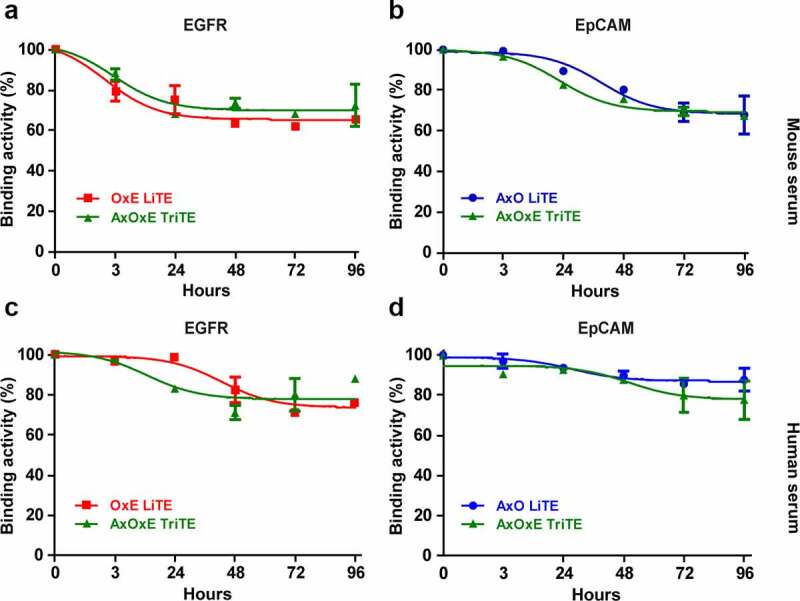
Serum stability of purified EpCAMxCD3xEGFR TriTE and EpCAMxCD3 and CD3xEGFR LiTEs. AxOxE TriTE and LiTEs were incubated in mouse (a-b) or human (c-d) serum at 37°C for 96 hours and their functional activity was analyzed by ELISA against plastic-immobilized EGFR (left) or EpCAM (right). Experiments were performed twice in duplicates. Mean ± SD are shown at each time point. AxO = A2 (anti-EpCAM VHH) x OKT3 (anti-CD3 scFv); OxE = OKT3 x Ega1 (anti-EGFR VHH); AxOxE = A2 x OKT3 x Ega1.
Inhibition of cell proliferation and EGFR phosphorylation
It has been described that Ega1 VHH is able to block the activity of EGFR by preventing the conformational change of the receptor and thus, its dimerization.29 To assess the functionality of the Ega1 moiety in the AxOxE TriTE and OxE LiTE, we studied their ability to inhibit proliferation and block EGFR phosphorylation in A431 cells. Notably, the EGFR-dependent A431 cells also express EpCAM.30 Whereas AxOxE TriTE inhibited A431 proliferation by a 20%, compared with OKT3 (P = .005), the OxE LiTE had no significant effect on proliferation, as previously described31 (Suppl. Figure 4a).
To assess the effect on EGFR phosphorylation status, A431 cells were stimulated with 25 ng/mL of human EGF, after incubation with serial dilutions of AxOxE TriTE or OxE LiTE at the highest concentration. Cetuximab was used as a positive control and untreated cell as negative control. Interestingly, AxOxE TriTE was able to decrease pEGFR in a dose-dependent manner, whereas the OxE LiTE had no effect, in accordance with proliferation results (Suppl. Figure 4b).
EpCAMxCD3xEGFR TriTE specifically activated T cells and triggered the assembly of canonical immunological synapses
We next assayed the purified AxOxE TriTE and LiTEs for their ability to activate T cells in vitro. As shown in Figure 3, LiTEs increased CD69 expression in a dose-dependent manner and more efficiently than AxOxE TriTE when Jurkat cells or PBMCs were co-cultured with single-positive EGFR+EpCAM− CT26EGFR (Figure 3b,f) or EGFR−EpCAM+ SW620 (Figure 3c,g) tumor cells. However, when PBMCs were cocultured with double-positive EGFR+EpCAM+ HCT116 tumor cells in the presence of AxOxE TriTE, the induction of CD69 expression was stronger compared to that of AxO (P = .015) and OxE (P = .002) LiTEs (Figure 3d,h). In fact, AxOxE TriTE reached nearly full activation of PBMCs at 2 nM, whereas CD69 expression was almost basal at the same equimolar concentration of both LiTEs. CD69 expression was not induced when T cells were cocultured with double-negative EGFR−EpCAM− CT26 cells in the presence of AxOxE TriTE or LiTEs (Figure 3a,e).
Figure 3.
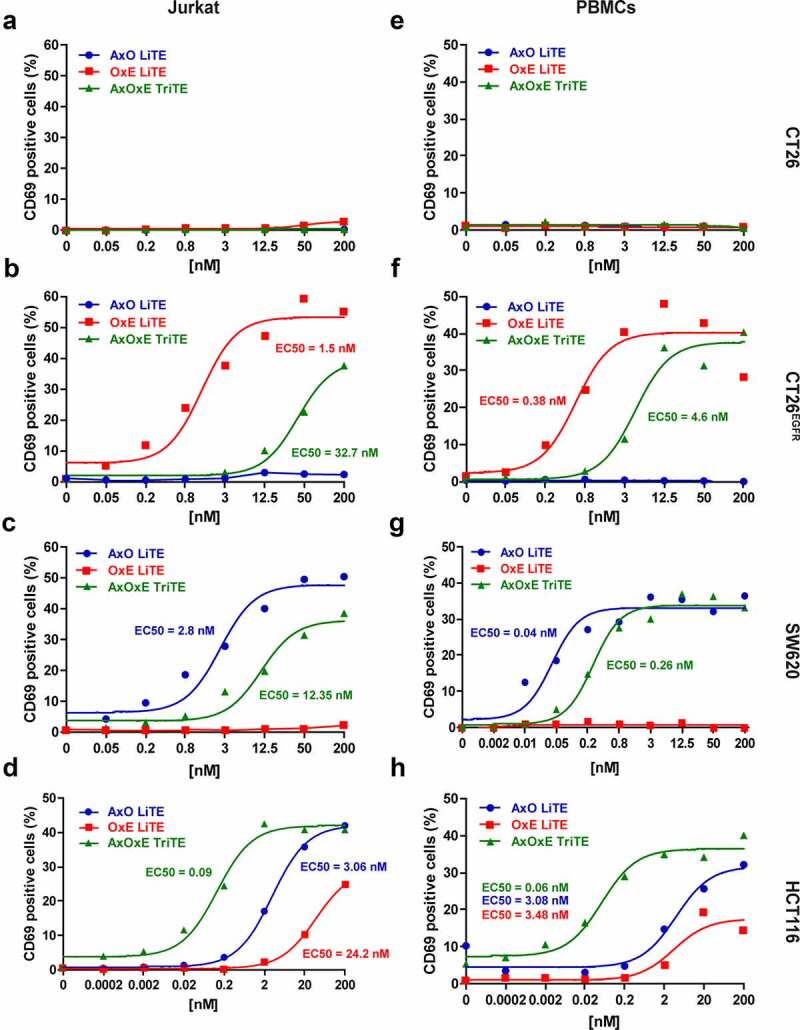
Induction of T cell activation by purified EpCAMxCD3xEGFR TriTE and EpCAMxCD3 and CD3xEGFR LiTEs. (a,e) CT26 cells (EGFR−EpCAM−); (b,f) CT26EGFR cells (EGFR+EpCAM−); (c,g) SW620 cells (EGFR−EpCAM+) or (d,h) HCT116 cells (EGFR+EpCAM+) were cocultured with Jurkat cells (left) or PBMCs (right) at the effector/target (E/T) ratio of 5:1 in the presence of different concentrations of purified AxOxE TriTE and LiTEs. After 24 hours, the surface expression of T cell activation marker CD69 was determined by FACS analysis. EC50 values are provided according to the color code. Experiments were performed three times, one representative experiment is shown. AxO = A2 (anti-EpCAM VHH) x OKT3 (anti-CD3 scFv); OxE = OKT3 x Ega1 (anti-EGFR VHH); AxOxE = A2 x OKT3 x Ega1.
To check if TriTE activity could be improved by swapping VHH domains, the construct ExOxA was generated. In both TriTE, the VHH in C-ter position was less efficient recognizing single-positive cells or activating T cells cocultured with them (Suppl. Figure 5). However, ExOxA TriTE effect in T cell activation was similar to that of AxOxE in the presence of double-positive HCT116 cells, and superior in both cases to the observed with single-positive cells.
In addition, AxOxE TriTE and LiTEs promoted the formation of the immunological synapse (IS) between Jurkat cells and EGFR+EpCAM+ HCT116 cells as assessed by CD3ε and F-actin accumulation at the T cell:target cell contact surface (Figure 4a), while the IS was not assembled in the absence of AxOxE TriTE or LiTEs (Suppl. Figure 6). F-actin polarization at the IS was more efficient in the presence of AxOxE TriTE when compared with both LiTEs (P < .0001) (Figure 4b), consistent with a higher capability of AxOxE TriTE to activate T cells in comparison with LiTEs (Figure 3d,h). 3D confocal microscopy showed that AxOxE TriTE assembled a canonical IS with peripheral and central distribution of F-actin and CD3ε, respectively (Figure 4a), whereas this organization was less distinct when LiTEs were used.
Figure 4.
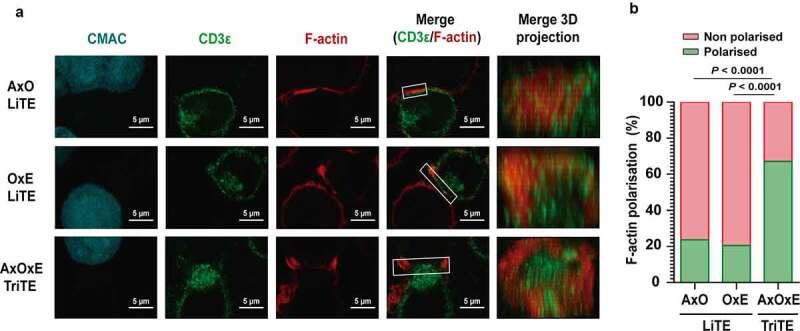
Immunological synapse formation is triggered by EpCAMxCD3xEGFR TriTE. (a) Images of immunological synapse (IS) assembly by Jurkat cells co-cultured with EpCAM+EGFR+ HCT116 cells (cyan) in the presence of AxOxE TriTE or LiTEs. The green (CD3ε) and red (F-actin) channels, as well as the merged images, are shown. The IS topology obtained from the 3D reconstructions of regions of interest in confocal stacks (white square) containing the red and the green channels is shown on the right. Experiments were performed three times; results of one representative experiment are shown. Scale bar 5 µm. (b) Percentages of T cells showing F-actin polarization at the IS in each condition are shown. Statistical differences were examined by two-coiled chi-square test. AxO = A2 (anti-EpCAM VHH) x OKT3 (anti-CD3 scFv); OxE = OKT3 x Ega1 (anti-EGFR VHH); AxOxE = A2 x OKT3 x Ega1.
EpCAMxCD3xEGFR TriTE promoted preferential lysis of EpCAM+EGFR+ cancer cells in vitro
We next assessed the ability and specificity of the AxOxE TriTE to elicit cytotoxic responses in vitro. PBMCs of three healthy donors were cocultured with CT26Luc, CT26EGFR-Luc, SW620Luc or HCT116Luc cells at a 5:1 effector to target (E:T) ratio in the presence of different concentrations of purified antibodies. According to activation data, the three constructs were able to induce dose-dependent killing of EpCAM+ and/or EGFR+ cells (Figure 5a-c), which was strictly antigen-specific since it spared EGFR−EpCAM− CT26 cells (Suppl. Figure 7A). On EGFR+EpCAM+ HCT116Luc cells, AxOxE TriTE exhibited a considerably higher cytotoxic ability than LiTEs, with an EC50 value of 4 pM, compared with EC50 values of 0.26 nM and 27 nM for AxO LiTE (P = .02) and OxE LiTE (P = .01), respectively (Figure 5c). Moreover, AxOxE TriTE preferentially killed double-positive tumor cells over single-positive ones (EC50 of 400 pM for CT26EGFR-Luc cells and 90 pM in SW620Luc cells). This difference could not be attributed to cell line-intrinsic factors other than antigen expression, since cytotoxicity experiments with single antigen-expressing cell lines on the same HCT116 background (KO EGFR Luc and KO EpCAM Luc) rendered results comparable to those obtained with SW620Luc and CT26EGFR-Luc cells, respectively, in the presence of AxOxE TriTE (Figure 5d). Expression profiles by FACS of both KO cell lines are shown in Suppl. Figure 3b.
Figure 5.
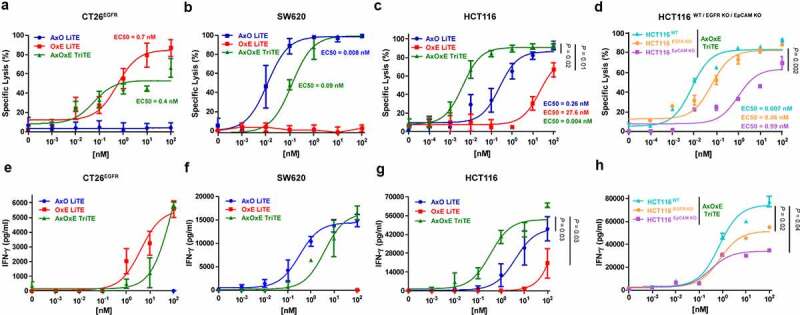
Specific cytotoxicity and IFN-γ secretion elicited by purified EpCAMxCD3xEGFR TriTE and EpCAMxCD3 and CD3xEGFR LiTEs. CT26EGFR-Luc cells (a,e), SW620Luc cells (b,f) or HCT116Luc (c,g) cells were cocultured in 96-well plates with PBMCs at the effector/target (E/T) ratio of 5:1 in the presence of different concentrations of purified AxOxE TriTE and LiTEs. In additional experiments, HCT116Luc were compared with the corresponding EGFR and EpCAM knockout cell lines in the presence of TriTE serial dilutions (d,h). After 72 hours, specific cytolysis of tumor cells were measured by bioluminescence assay (upper) and IFN-γ production was determined in CM by ELISA (lower). Percent specific lysis was calculated relative to an equal number of tumor cells cultured with PBMCs in the absence of purified antibodies. EC50 values are provided according to the color code. PBMCs were obtained from 3 different donors, and experiments were performed in triplicate. Statistical differences were examined by unpaired Student’s t-test assuming a normal distribution. Results are expressed as a mean ± SD. AxO = A2 (anti-EpCAM VHH) x OKT3 (anti-CD3 scFv); OxE = OKT3 x Ega1 (anti-EGFR VHH); AxOxE = A2 x OKT3 x Ega1.
EpCAMxCD3xEGFR TriTE induced strong IFN-γ secretion in vitro
Next, we analyzed IFN-γ secretion by PBMCs that were co-cultured 72 h with tumor cells in the presence of the three antibodies. IFN-γ secretion elicited by AxOxE TriTE was significantly higher than that of AxO and OxE LiTEs (P = .03 and P = .036, respectively) in the cocultures with HCT116Luc cells (Figure 5g), whereas there were no statistically significant differences between AxOxE TriTE and LiTEs in the induction of IFN-γ in the cocultures with CT26EGFR-Luc (Figure 5e) or SW620Luc (Figure 5f). In accordance with cytotoxicity results, AxOxE TriTE promoted higher IFN-γ secretion in cocultures with HCT116 wild type than in the presence of HCT116 EGFR or EpCAM KO (Figure 5h).
Importantly, in the presence of CT26 target cells, analyses of CM revealed no increase of IFN-γ secretion even at 100 nM, the highest concentration of antibodies used (Suppl. Figure 7B).
Antitumor effect of EpCAMxCD3xEGFR TriTE
To study the therapeutic effect of AxOxE TriTE in vivo, HCT116 cells were implanted in Hsd:Athymic Nude-Foxn1nu mice. When the tumors reached average diameters of 0.2 cm (day 4), mice were randomized and human PBMCs were administered intraperitoneally. On day 5, treatment with OxE LiTE or AxOxE TriTE was initiated. Equimolar doses of the antibodies (3 mg/kg for OxE LiTE and 4 mg/kg for AxOxE TriTE) were administered i.p. daily for 10 days (Figure 6a). As shown in Figure 6b, HCT116 tumor-bearing mice treated with AxOxE TriTE showed a delay in tumor growth when compared to their counterparts treated with OxE LiTE. Moreover, 2 out of 4 mice in the TriTE group controlled tumor growth at least until day 21 (Suppl. Figure 8). By day 27 after HCT116 inoculation, all control mice treated with PBS had been euthanized, whereas 100% of mice in the AxOxE TriTE group were alive, vs. 25% in the group receiving OxE LiTE. In fact, only AxOxE TriTE-treated mice showed a statistically significant increased survival in comparison to the PBS group (P = .006) (Figure 6c). No significant differences in body weight were observed before and after the treatment (Figure 6d).
Figure 6.
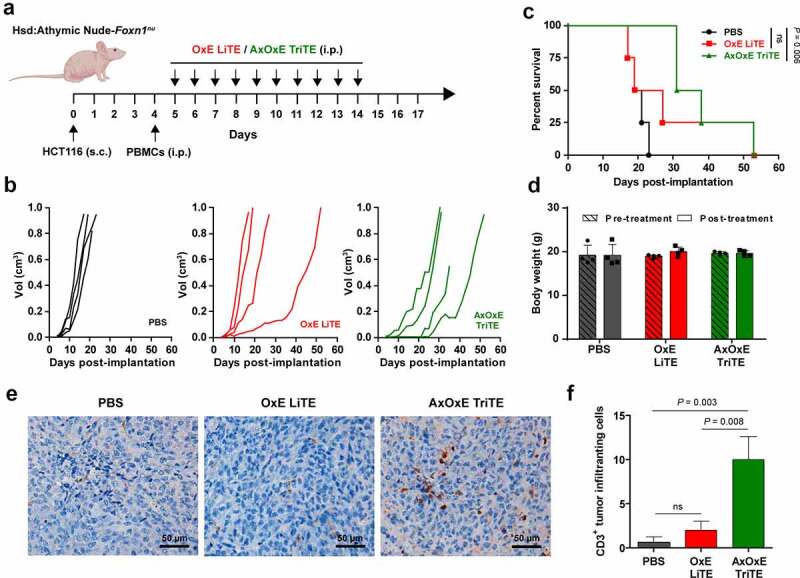
In vivo therapeutic effect of EpCAMxCD3xEGFR TriTE and CD3xEGFR LiTE. (a) Hsd:Athymic Nude-Foxn1nu mice were inoculated subcutaneously with 2 × 106 HCT116 tumor cells. Mice were randomized into groups (n = 4/group) when tumors reached 0.2 cm in diameter and injected intraperitoneally with 107 freshly isolated PBMCs. Then, mice were treated with intraperitoneally injections of PBS, OxE LiTE or AxOxE TriTE daily. (b) Tumor volume growth curves for individual mice are represented. (c) Kaplan-Meier survival curves of AxOxE TriTE- and OxE LiTE-treated mice, log-rank (Mantel-Cox) test. (d) Mice weight before and after treatment are shown. (e) Representative images of CD3+ TIL immunostaining in tissue sections from HCT116 tumors treated with PBS, OxE LiTE or AxOxE TriTE. Tumors were resected at termination of the experiment shown above. Error bar = 50 µm. (f) Quantification of CD3+ TILs in three independent fields/tissue section. Statistical differences were examined by unpaired Student’s t-test assuming a normal distribution. Results are expressed as a mean ± SD. OxE = OKT3 (anti-CD3 scFv) x Ega1 (anti-EGFR VHH), AxOxE = A2 (anti-EpCAM VHH) x OKT3 x Ega1.
In order to assess the ability of the antibodies to promote immune infiltration, resected tumors were analyzed by immunohistochemistry. Numbers of infiltrating CD3+ T cells were significantly higher in AxOxE TriTE-treated mice than in PBS- (P = .003) or OxE LiTE-treated groups (P = .008) (Figure 6e-f).
Discussion
Intratumor heterogeneity has been associated with poor outcome and decreased response to therapy in a variety of human cancer types, suggesting a universal role in therapeutic resistance. Preexisting heterogeneity increases the risk of at least some tumor cells surviving therapy-induced elimination, while ongoing diversification of tumor cell phenotypes during treatment enables tumor cells to adapt to therapeutic selective pressure, leading to de novo resistance.32 For example, carcinoembryonic antigen (CEA) expression heterogeneity and plasticity contribute to resistance to the anti-CEA T-cell bispecific antibody cibisatamab (CEA-CD3-TCB) in patient-derived CRC organoids through CEA antigen loss.33 In patients treated with CD19-directed immunotherapies, such as chimeric antigen receptor (CAR)-engineered T-cells (CAR-T) and the BiTE blinatumomab, up to 60% and 30% of relapses, respectively, are associated with the loss of CD19, rendering the malignant cells invisible to CD19-specific immunotherapies.34
Another escape strategy is the acquisition of mutations that prevent the recognition by the targeting domain, as observed in CRC patients treated with the anti-EGFR mAb cetuximab. Emergence of mutations in the EGFR ectodomain, located in the region of interaction with cetuximab, may disturb this interface and confer resistance to the treatment.35,36 Interestingly, a subset of EGFR mutations preventing binding to cetuximab are still permissive for interaction with panitumumab, the second anti-EGFR mAb approved for CRC treatment.37,38
While effective, single-targeted T-cell engagers such as BiTE and TCB exert selective pressure against a unique target antigen which may lead to tumor relapse. Therefore, simultaneous dual targeted T-cell-redirecting therapies may improve tumor specificity while limiting the risk of immune escape.39 For example, T cells expressing both anti-CD19 and anti-CD123 CARs simultaneously provided superior in vivo activity against B-ALL compared with single-expressing CAR-T-cells.34 Recently, a first-in-human trial of bispecific anti-CD20, anti-CD19 CAR-T-cells for relapsed, refractory B cell malignancies has been reported,40 demonstrating their potential to overcome the antigen loss observed for single-targeted CD19 CAR-T cells.
Despite promising results with dual targeted CAR-T-cells, a similar approach has not been pursued with T-cell redirecting antibodies. Here, we present a novel trispecific T-cell engager (TriTE) as an evolution of the BiTE and LiTE formats. This TriTE was generated by the fusion of an anti-CD3 scFv to single-domain anti-EpCAM and anti-EGFR VHH, for dual targeting of CRC cells. The AxOxE TriTE was secreted as soluble and functional protein by transfected mammalian and yeast cells, recognized the three cognate antigens, and selectively activated and recruited cytolytic human T cells to kill EpCAM+ and/or EGFR+ cancer cells in vitro. As VHH lack the hydrophobic interface mediating interaction with VL,41,42 chain mispairing within the TriTE can be ruled out. Importantly, AxOxE TriTE had no effect when human T cells were cultured with double-negative EpCAM−EGFR− cells.
Another issue to take into account is that most antigens targeted by therapeutic antibodies are tumor-associated, but not unique to tumor cells: that is to say, they are also expressed at lower levels in normal tissues. EpCAM expression on healthy epithelia of the gastrointestinal tract has limited the therapeutic window of EpCAM-directed therapies due to on-target/off tumor side effects.19 The mAb edrecolomab, the first anti-EpCAM mAb approved for the treatment of CRC, was subsequently withdrawn when larger studies showed no benefit compared with standard chemotherapy.43 While edrecolomab efficacy may have been impaired by low binding affinity, the mAbs ING-1 and 3622W94 displayed such a high affinity that they no longer discriminated between normal and malignant cells, and risk of pancreatitis precluded further studies as monotherapy.44 A moderate binding affinity could account for the larger therapeutic window observed in patients treated with the anti-EpCAM mAb adecatumumab.45
In order to avoid systemic toxicity, it has been proposed that a dual-targeted anti-tumor BsAb should preferentially bind to malignant cells rather than normal cells if the affinity of the individual binding domains are sufficiently low as to require the presence of both target antigens for efficient binding (through the avidity effect),46 whereas high-affinity binding domains may efficiently bind normal cells expressing only a single antigen and thereby induce off-tumor toxicity. In some circumstances, dual targeting alone may not be sufficient to guarantee selective tumor-targeting, and affinity fine-tuning of one of the binding domains may be required.47 Affinity-reduced BsAb variants have been shown to mediate a greater degree of tumor selectivity, while the overall therapeutic effect was not ameliorated.48,49 Interestingly, AxOxE TriTE is less efficient in activating T cells (as assessed by CD69 expression and IFN-γ secretion) and triggering their cytotoxic effect on single positive cells than the corresponding LiTEs. This may be a consequence of its engineering in a single-chain polypeptide that may partially interfere at certain extent with antigen recognition by individual binding domains. In fact, AxOxE TriTE EC50 is 100 and 22.5 times higher in CT26EGFR and SW620 than in HCT116, respectively. The anti-EGFR VHH seems to be especially impaired in the C-terminal position of AxOxE TriTE, since its capacity to kill CT26EGFR cells is considerably reduced in comparison with the corresponding anti-EGFR LiTE. The positioning effect on binding affinity of anti-EGFR VHH placed in C-terminal end has been previously described.17 Although this reduction was not deliberate in our case, we can speculate that it may render TriTE able to discriminate to some extent between double-positive and single positive cells, or cells with high vs. low antigen density. Indeed, AxOxE TriTE performs much better in cytotoxicity assays with double-positive HCT116 cells than LiTEs, with EC50 65 and 675 times lower than that of AxO LiTE and OxE LiTEs, respectively, suggesting a potentially more favorable safety profile, while preserving its therapeutic effect in vivo. Discrimination could be further enhanced generating AxOxE TriTE variants through alanine mutagenesis of targeted residues in the CDR3 of anti-EpCAM binding domain as previously described,49 in order to completely abrogate binding to single positive cells, while preserving recognition of double-positive ones. Affinity-reduced variants could also distinguish between high antigen density malignant cells and normal cells with low antigen density.
Interestingly, AxOxE TriTE partially recapitulated the effect of bivalent cetuximab in the inhibition of EGFR-mediated signaling. It had been previously reported that monovalent binding to EGFR by a LiTE had no effect on proliferation or receptor phosphorylation status of A431 cells.31 Although EGFR binding by AxOxE TriTE is also monovalent, this difference may be attributed to EpCAM expression in A431 cells30 and thus the higher avidity of AxOxE TriTE. This could theoretically allow the AxOxE TriTE to function as a dual mechanism therapeutic, redirecting T cells toward CRC cells while simultaneously inhibiting mitogenic signaling from EGFR in the CRC cells.
In summary, we demonstrate the potential of the tandem trispecific T-cell engager format, using anti-EpCAM and anti-EGFR binding domains for proof of concept. Moreover, a wide selection of TAA-specific VHH antibodies are available, and their combination with well-characterized anti-CD3 or anti-CD16 binding domains could easily provide a therapeutic arsenal of TriTEs and TriKEs aimed toward treatment of diverse cancers, with potentially enhanced efficacy, increased tumor selectivity and reduced risk of clonal escape.
Materials and methods
General reagents and antibodies
The human EGFR-Fc (cat#344-ER) and EpCAM-Fc (cat#960-EP) chimeras were from R&D Systems (Minneapolis, MN) and bovine serum albumin (cat#A9647, BSA) was from Sigma–Aldrich (St. Louis, MO). The mAbs used included: mouse anti-c-myc clone 9E10 (cat#ab206486, Abcam, Cambridge, UK), mouse anti-human CD3ε clone OKT3 (Ortho Biotech, Bridgewater, NJ), chimeric anti-human epidermal growth factor receptor (EGFR) cetuximab (Merck KGaA, Darmstadt, Germany), mouse anti-human epithelial antigen clone Ber-EP4 (cat#M0804, Dako), phycoerytrin (PE)-conjugated anti-human CD69 clone FN50 (cat#555531, BD Biosciences, San Jose, CA), fluorescein isothiocyanate (FITC)-conjugated anti-human CD3 clone OKT3 (cat#566783, BD Biosciences), the rabbit anti-human phospho-EGFR (Tyr1068) clone D7A5 (cat#3777, Cell Signaling Technology Inc) and anti-β-actin mouse clone 8226 (cat#ab8226, Abcam). The polyclonal antibodies included: PE-conjugated goat F(ab’)2 fragment anti-mouse IgG, Fc specific, (cat#115-116-071, Jackson Immuno Research, Newmarket, UK); PE-conjugated goat F(ab’)2 fragment anti-human IgG (H&L) (cat#109-116-170, Jackson Immuno Research, Newmarket, UK), horseradish peroxidase (HRP)-conjugated goat anti-mouse IgG (cat#A5278, Sigma-Aldrich), HRP-conjugated goat anti-human IgG (cat#A-0170, Sigma-Aldrich), IRDye800CW-donkey anti-rabbit (cat#925-32213, LI-COR Biosciences) and IRDye680RD-donkey anti-mouse (cat#925-68072, LI-COR Biosciences).
The following reagents were also used: human EGF (cat#130-093-825, Miltenyi Biotec), D-luciferine (cat#E160C, Promega, Madison, WI, USA), CellTiter-Glo luminescent assay (cat#G7571, Promega), glycerol (cat#56-81-5, VWR LifeScience), methanol (cat#K977, Amresco), yeast extract (cat#1702.00, Condalab), peptone (cat#1616.00, Condalab), YNB (cat#1545.00, Condalab) and dextrose (cat#X997.2, Roth). Cetuximab was obtained from the pharmacy at Hospital Puerta de Hierro.
Cells and culture conditions
Human 293T (CRL-3216), SW620 (CCL-227), HCT116 (CCL-247) and Jurkat (TIB-152) cells were obtained from ATCC (American Type Culture Collection, Rockville, MD, USA). Mouse CT26 cells (CRL-2638) infected with p-BABE-puro-EGFR expressing human EGFR (CT26EGFR) or infected with the empty vector retrovirus (CT26mock) were provided by M. Rescigno.25 EGFR knockout (ab281597) and EpCAM knockout (ab281596) HCT116 cell lines were purchased from Abcam. Production of lentiviral vectors for the generation of HCT116 (WT and KO), SW620, CT26 mock and CT26EGFR cells expressing the firefly luciferase (Luc) gene has been described previously.50 The epidermoid carcinoma cell-line A431, carrying an amplification of the EGFR gene, was obtained from the ATCC (CRL-1555) and FreeStyleTM 293 F cells were provided by Invitrogen (R790-07). PBMCs from healthy donors were provided by the Biobank of Hospital Universitario Puerta de Hierro Majadahonda (HUPHM)/Instituto de Investigación Sanitaria Puerta de Hierro-Segovia de Arana (IDIPHISA) (PT17/0015/0020, Spanish National Biobank Network), with the appropriate approval of the Ethics Committee and based on informed consent. Adherent cells were cultured in DMEM medium (Lonza, Walkersville, MD) supplemented with 10% FCS (Sigma-Aldrich, St. Louis, MO) and 1% pen-strep-glutamine (Gibco, Thermo Fisher Scientific, Waltham, MA). Jurkat cells and PBMCs were maintained in RPMI-1640 (Lonza) supplemented with with 10% FCS and 1% pen-strep-glutamine. FreeStyleTM 293 F cells were cultured in FreeStyleTM 293 expression medium (Invitrogen). All cells were routinely screened for mycoplasma contamination by PCR (Biotools, Madrid, Spain) at the Tissue Culture Core Facility, Biomedical Research Institute Puerta de Hierro-Segovia de Arana and were authenticated at the Universidad Complutense de Madrid Genomics Unit using the AmpFLSTR Identifiler PCR Amplification kit (Applied Biosystems, Thermo Fisher Scientific). Pichia pastoris KM71H strain was provided by Dr. Javier Lacadena Gallo (UCM) and cells were cultured with YPD (1% yeast extract, 2% peptone, 2% dextrose), BMXY (1% yeast extract, 2% peptone, 100 mM potassium phosphate buffer pH 6.0, 4 × 10–5 biotin) supplemented with 1% glycerol (BMGY) or 0,5% methanol (BMMY).
Construction of expression vectors
To generate the EpCAMxCD3 (AxO) and CD3xEGFR (OxE) LiTEs expression vectors, DNA fragments encoding the anti-EpCAM A2 (sequence kindly provided by Patrick Chames) and anti-EGFR Ega1 VHH29 were synthesized by Geneart AG (Thermo Fisher Scientific), digested with ClaI/NotI or XhoI/EcoRI, respectively, and ligated into the pCR3.1-OKT3 plasmid. To generate the AxOxE TriTE construct, the PCR fragment Ega1 cleaved with XhoI/EcoRI was ligated into a pCR3.1 vector containing the coding sequence of OKT3, and A2 VHH was cloned via ClaI/NotI, resulting in pCR3.1- AxOxE TriTE expression vector. Finally, in order to obtain the ExOxA construct, PCR fragments Ega1 and A2 were digested with ClaI/NotI or XhoI/EcoRI, respectively, and ligated into the same vector. Individual binding domains in each construct were connected by short linkers (G4S). For medium-scale protein production, the three constructs were subcloned into the ClaI/XbaI digested pPiczα vector (Invitrogen), to obtain pPiczα-AxO, pPiczα-OxE LiTEs and pPiczα-AxOxE TriTE. All plasmids were amplified in chemically competent Escherichia coli TOP10 and purified using Qiagen plasmid Midi kit. Final sequences were verified using F-CMV and R-BGH or 5’-AOX1 and 3’-AOX1.
Expression in mammalian cells
293T cells were transiently transfected with pCR3.1 vectors encoding the three antibodies using calcium phosphate and CM were collected after 48 h. FreeStyleTM 293 F cells (10 x 107) were transfected with a ratio 1:1 of pCR3.1 – AxOxE TriTE (100 μg) and PEI (100 μg) in 100 ml of FreeStyleTM expression medium. Antibody expression was analyzed using ELISA and Western blotting.
Expression in P. pastoris and purification of recombinant antibodies
Electrocompetent P. pastoris KM71H strain cells were electroporated with 10 μg of appropriate linearized pPiczα plasmids after digestion with PmeI, using Bio-Rad Gene pulser apparatus (Bio-Rad, Hercules, CA, USA). The yeast cells containing the integrated sequences were selected in YPD plates with 100, 400 or 750 μg/mL of zeocin. Different clones were tested to select those with the highest yield. For this purpose, individual clones were grown in BMGY (buffered media for yeast containing glycerol) for 24 hours at 30°C and 200 rpm and protein production was induced with BMMY (buffered media for yeast containing methanol) at 15°C and 200 rpm. Methanol 0,5% (v/v) was added every 24 hours and samples of CM were collected to analyze the expression profile by SDS-PAGE and Western blot. Colonies with the highest production of each construction were selected for medium-scale production, grown in baffled erlenmeyer flasks with 2–4 L of BMGY medium at 30°C, 200 rpm for 24 hours and induced with 200–400 mL of BMMY medium at 15°C, 200 rpm for 24 hours. Collected cell-free CM were dialyzed against PBS 1x at 4°C for 24 hours and recombinant antibodies were purified by affinity chromatography with HisTrapTM HP columns (GE Healthcare) using an ÄKTA Prime plus system (GE Healthcare, Uppsala, Sweden). Endotoxin levels were <0.25 EU/ml as determined by the LAL Endotoxin Kit (Pierce).
Size exclusion chromatography
A sample of 200 μL of CM from transfected 293 cells was injected into a Superdex 200 Increase 10/300 GL column (Cytiva, MA, US) on an ÄKTA GO chromatography system (Cytiva) at room temperature, while monitoring light absorbance at 280 nm. The column was equilibrated in phosphate buffered saline pH 7.4 plus 150 mM NaCl and run in the same buffer at 0.5 mL per minute. The column was previously calibrated with a set of Gel Filtration Standards (Biorad, from 1.4 to 670 kDa). The fractions containing monomeric protein were concentrated and reinjected under the same conditions to assess that the monomeric protein was stable and did not aggregate. To check the molecular size of purified AxOxE TriTE in solution, SEC was also performed. An aliquot corresponding to the elution volume of the AxOxE TriTE was re-cromatographed to assess its monomeric state.
Western blot
Samples of cell-free CM or purified proteins were analyzed under reducing conditions on 12% Tris-glycine gels and transferred to nitrocellulose membranes using iBlot system (Life Technologies). After incubation with LI-COR blocking solution (LI-COR, Lincoln, NE, USA), proteins were detected with 1 μg/mL mouse anti-c-myc mAb (9E10, cat#ab206486, Abcam), followed by DyLight800-conjugated goat anti-mouse IgG (cat#610-145-121, Rockland Immunochemicals) diluted 1:5000. Visualization of protein bands was performed with the Odyssey system (LI-COR Biosciences).
Enzyme-linked immunosorbent assay
Human EGFR-Fc or huEpCAM-Fc chimeras were immobilized (5 μg/mL) on Maxisorp plates (NUNC Brand Products) overnight at 4°C. After washing and blocking with Odyssey blocking buffer (Li-Cor Biosciences), CM or increasing amounts of purified antibodies were added for 1 hour at room temperature. The wells were washed and incubated with 1 μg/mL anti-c-myc mAb for 1 hour at room temperature. After washing, proteins were detected with HRP-conjugated goat-anti-mouse IgG (1:1000 dilution) (cat#A5278, Sigma-Aldrich) for 45 minutes at room temperature. Finally, the plates were developed using O-phenylenediamine dihydrochloride (OPD) in citrate phosphate buffer and the reaction was stopped using sulfuric acid 1 M. Cetuximab (5 μg/mL) (pharmacy at Hospital Puerta de Hierro) and Ber-EP4 (1:200) were used as positive controls and detected with HRP-conjugated goat-anti-human IgG (1:1000 dilution) or HRP-conjugated goat-anti-mouse IgG (1:1000 dilution), respectively.
Biolayer interferometry
The simultaneous binding of AxOxE TriTE to immobilized huEGFR-Fc (R&D Systems) and huEpCAM-Fc (R&D Systems) in solution was measured using biolayer interferometry on an Octet RED96 system (Fortebio). Prior to the experiment, anti-hFc capture biosensors (Fortebio) were incubated with 40 nM of huEGFR-Fc for 20 minutes in HEPES-buffered saline (HBS, 20 mM HEPES, 150 mM NaCl, pH 7.4). The antibody was loaded onto the immobilized huEGFR-Fc at 100 nM for 10 minutes in HBS. One biosensor was then moved into a solution still containing 100 nM of the AxOxE TriTE, and another into a solution with both 100 nM of the antibody and 300 nM of EpCAM. A control biosensor which was loaded with huEGFR-Fc but not the AxOxE TriTE also was also incubated with 300 nM of EpCAM. After monitoring the association with EpCAM for 10 minutes, the biosensors were moved back into solutions containing only 100 nM of the antibody to measure EpCAM dissociation for 10 minutes, and then moved into HBS only to monitor antibody dissociation for an additional 10 minutes.
Flow cytometry
CT26, CT26EGFR, SW620, HCT116 or Jurkat cells were incubated with CM or purified antibodies for 1 hour on ice. In case of titration experiments, the antibodies were used in tenfold dilution series spanning the concentration range from 500 nM down to 0.5 pM. After washing, 1 μg/mL anti-c-myc mAb was added for 1 hour at 4°C and detected using a phycoerithrin (PE)-conjugated goat-anti-mouse antibody (1:200 dilution). Cetuximab, Ber-EP4 and OKT3 mAbs were used as positive controls. Cells incubated without primary antibody were used as negative controls. Samples were acquired on a MACSQuant Analyzer 10 (Miltenyi Biotec, Bergisch Gladbach, Germany) and analyzed using FlowJo (BD Biosciences, Franklin Lakes, NJ, USA) at the Flow Cytometry Core Facility, Biomedical Research Institute Puerta de Hierro-Segovia de Arana.
Serum stability
Purified antibodies (6 ug) were incubated in PBS 60% human and mouse serum at 37°C for 5 days. Samples were collected at 3 and every 24 hours and their binding activities were tested by ELISA, representing the sample at 0 hours 100% of functionality.
T cell Activation assays
Microtiter 96-well plates were seeded with tumor cells (2x104/well) 24 hours before. Consecutively, wells were incubated with CM or increasing amounts of purified antibodies for 30 min at 37°C. After washing, Jurkat cells or human PBMCs, isolated from healthy volunteers by density-gradient centrifugation, were added at 5:1 effector:target (E:T) ratio. After 24 hours, the expression profile of activation marker CD69 was determined by FACS using a PE-conjugated anti-CD69 mAb and FITC-conjugated anti-CD3 mAb incubated for 30 minutes on ice. Samples were analyzed with a MACSQuant Analyzer 10 (Miltenyi Biotec GmbH).
Immunological synapse formation
HCT116 cells were labeled with 1 μM cell tracker dye 7-amino-4-chloromethylcoumarin (CMAC; Life Technologies) and incubated with 5 nM AxOxE TriTE or LiTEs for 30 minutes. After washing, an equal number of Jurkat and HCT116 cells were co-incubated for 15 minutes on poly-L-lysine-coated coverslips at 37°C in a humidified atmosphere with 5% CO2. Samples were fixed with 4% paraformaldehyde for 5 minutes at room temperature and permeabilised with TBS-Triton 0.1% for 5 minutes at room temperature. After blocking for 20 minutes with 10 μg/ml human gamma globulin (Sigma-Aldrich), samples were stained with mouse α-human CD3ɛ antiserum (kindly provided by Francisco Sanchez-Madrid, Hospital Universitario de la Princesa, Madrid, Spain) diluted 1/2 in TNB buffer (Roche Diagnostics) for 1 hour at room temperature. After washing with TBS, cells were incubated with Alexa Fluor 488-conjugated donkey anti-rabbit antibody (1:500; Life Technologies) and phalloidin-Alexa-647 (1:200; Thermo Scientific) for 30 minutes at room temperature. Finally, after washing, samples were embedded in mowiol (Sigma Aldrich) and allowed to dry at RT. Confocal microscopy analysis was performed in a Leica SP8 microscope with a 63X oil objective (Leica Microsystems, Germany) using 405 nm (for CMAC), 488 nm (for Alexa-488) and 647 nm (for Alexa-647) excitation lines. Confocal sections were acquired every 0.25 μm along the z axis and 3D reconstructions were obtained with Image J software (National Institutes of Health). Graphs and statistics were made using PRISM 6 (GraphPad Software, USA).
Cytotoxicity assay
CT26Luc, CT26EGFR-Luc, SW620Luc or HCT116Luc cells were plated in triplicates in 96-well microtiter plates at 2 × 104 cells/well 24 hours before the assay. Then cells were incubated with CM or increasing amounts of purified antibodies for 30 min at 37°C. After that, Jurkat cells or human PBMCs were added at 5:1 E:T ratio. After 72 hours, specific cytotoxicity was determined adding the D-luciferin substrate (20 μg/mL, Promega) and relative light units (RLU) were measured with the luminescence plate reader Infinite 1200 (Tecan, Männedorf, Switzerland). Wells with target and efector cells in the absence of CM or purified antibodies were set as 100%. CM were also collected after 72 h hours and assayed for IFN-γ secretion by ELISA (cat#851.560, Diaclone).
Inhibition of EGFR-mediated cell proliferation
A431 cells were seeded in triplicates at 2.000 cells/well in 96-well plates in DMEM supplemented with 10% FBS. After 24 h, medium was changed by DMEM containing 1% FBS and equimolar concentrations of purified AxOxE TriTE and OxE LiTE were added. Cetuximab and OKT3 were used as positive and negative controls, respectively. After 72 h, medium was removed and cell proliferation was measured adding CellTiter Glo luminescent assay (Promega, Madison, USA). Bioluminiscence was assessed using a Tecan Infinite F200 plate-reading luminometer.
EGFR signaling inhibition assay
A431 cells were seeded at 100.000 cells/well in 12-well plates in DMEM supplemented with 10% FBS and incubated for 24 hours. Afterward, cells were starved for 16 h with 1% FBS DMEM. Subsequently, cells were incubated with serum-free DMEM containing serial dilutions of AxOxE TriTE (200–0 nM) or OxE LiTE at 200 nM. Cetuximab was used as positive control. Then, cells were stimulated for 5 min with 25 ng/mL of human EGF and lysed in Laemmli-lysis buffer (Bio-Rad, CA, USA) for 10 min, on ice. Samples were analyzed by SDS-PAGE and Western blot using iBlot Dry Blotting System (Invitrogen Life Technologies). Membranes were incubated ON with a rabbit anti-human phospho EGFR Tyr1068 mAb (clone D7A5, Cell Signaling, Leiden, The Netherlands) and a anti β-actin mouse mAb (clone 8226, Abcam, Cambridge, UK), followed by incubation with an IRDye800-conjugated donkey anti-rabbit antibody (Rockland Immunochemicals, Limerick, PA, USA) and IRDye680-conjugated donkey anti-mouse antibody (Rockland Immunochemicals). Odyssey infrared imaging system (LI-COR Biosciences, Lincoln, NE, USA) was used to visualize and analyzed protein bands.
In vivo antitumoral effect
HCT116 (2x106 cells/mouse) in PBS mixed with 30% matrigel (BD Biosciences) were implanted s.c into the right dorsal space of 5-week-old female Hsd:Athymic Nude-Foxn1nu mice. Tumors were measured three times a week with a calliper and their volumes estimated by using the formula: length x width2 x 0.52, where length represents the largest tumor diameter and width represents the perpendicular tumor diameter. At day 4, mice were divided into groups with average diameter of 0.2 cm just before PBMCs administration. Randomization occurred in a blinded fashion. One day after intraperitoneal infusion of fresh PBMCs (1x107 cells/mouse) from a healthy donor (day 5), mice received i.p. injections of PBS, 60 μg/mouse of OxE LiTE or 80 μg/mouse of AxOxE TriTE (equimolar conditions), then treatment continued daily for another 9 days (until day 14). Mice were euthanized when tumor size reached 1 cm3 or at the onset of any sign of distress. All experiments were conducted in compliance with the institutional guidelines provided by the Biomedical Research Institute Hospital Puerta de Hierro Animal Ethics Committee. Procedures were additionally approved by the Animal Welfare Division of the Environmental Affairs Council, Comunidad Autónoma de Madrid (PROEX 066/14).
Histological studies
Mouse CRC xenografts from all mice were routinely formalin-fixed and paraffin-embedded in the Department of Pathology, Hospital Universitario Puerta de Hierro. Sections of 4 μm were stained with hematoxylin and eosin according to standard protocols or processed for immunohistochemistry using the Dako-Omnis automated staining platform. The polyclonal rabbit anti-human CD3 ready-to-use (cat#GA503, Dako-Agilent) was developed using EnVision Flex High pH visualization system. At least two sections (three fields/section) of each tumor were blindly scored by the pathologist.
Statistical analysis
Results were shown as mean ± standard deviation (SD). Data were analyzed by unpaired two-tailed Student’s t-test, assuming a normal distribution, using Prism software v5 (GraphPad, San Diego, CA, USA). Data were considered statistically significant when P < .05.
Supplementary Material
Acknowledgments
The authors wish to thank donors and Biobank of Hospital Universitario Puerta de Hierro Majadahonda for the human specimens used in this study.
Funding Statement
This study was funded by grants from Instituto de Salud Carlos III PI16/00357, PI19/00132), partially supported by the European Regional Development Fund (ERDF), Comunidad Autónoma de Madrid (S2010-BMD-2312), and Ministerio de Economía y Competitividad (RTC-2016-5118-1) to L.S.; and from Ministerio de Ciencia e Innovación (SAF2017-89437-P and PID2020-117323RB-I00), partially supported by ERDF, the Spanish Association Against Cancer (AECC 19084) and the CRIS Cancer Foundation FCRIS-2018-0042, FCRIS-2021-0090 (FCRIS-2018-0042 and FCRIS-2021-0090) to L.A-V. A.T-G. was supported by a predoctoral fellowship from Comunidad Autónoma de Madrid (PEJD-2018-PRE/BMD-8314);Spanish Ministry of Science and Innovation [SAF2017-89437-P, PID2020-117323RB-I00].
Abbreviations:
BsAb, bispecific antibody; BiTE, bispecific T-cell engager; EGFR, epidermal growth factor receptor; EpCAM, epithelial cell adhesion molecule, PBMCs, peripheral blood mononuclear cells; scFv, single-chain variable fragment; TAA, tumor-associated antigen; TriKE, trispecific killer engager; TriTE, trispecific T-cell engager; TsAb, trispecific antibody; VH, immunoglobulin variable heavy chain; VHH, variable domain of heavy-chain only antibodies; VL, immunoglobulin variable light chain.
Consent to Participate
Informed consent was obtained from all participants included in the study.
Disclosure statement
No potential conflicts of interest were disclosed.
Ethics Approval
All procedures involving animals were in accordance with the ethical standards of the corresponding institutional and regional/national committees.
Supplementary Material
Supplemental data for this article can be accessed on the publisher’s website.
References
- 1.Kontermann RE, Brinkmann U.. Bispecific Antibodies. Drug Discov Today. 2015;20(7):838–14. doi: 10.1016/j.drudis.2015.02.008. [DOI] [PubMed] [Google Scholar]
- 2.Przepiorka D, Ko C-W, Deisseroth A, Yancey CL, Candau-Chacon R, Chiu H-J, Gehrke BJ, Gomez-Broughton C, Kane RC, Kirshner S, et al. FDA Approval: blinatumomab. Clin Cancer Res. 2015;21(18):4035–4039. doi: 10.1158/1078-0432.CCR-15-0612. [DOI] [PubMed] [Google Scholar]
- 3.Suurs FV, Lub-de Hooge MN, de Vries EGE, de Groot DJA. A Review of Bispecific Antibodies and Antibody Constructs in Oncology and Clinical Challenges. Pharmacol Ther. 2019;201:103–119. doi: 10.1016/j.pharmthera.2019.04.006. [DOI] [PubMed] [Google Scholar]
- 4.Compte M, Harwood SL, Muñoz IG, Navarro R, Zonca M, Perez-Chacon G, Erce-Llamazares A, Merino N, Tapia-Galisteo A, Cuesta AM, et al. A Tumor-Targeted Trimeric 4-1BB-Agonistic Antibody Induces Potent Anti-Tumor Immunity without Systemic Toxicity. Nat Commun. 2018;9(1):4809. doi: 10.1038/s41467-018-07195-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gauthier L, Morel A, Anceriz N, Rossi B, Blanchard-Alvarez A, Grondin G, Trichard S, Cesari C, Sapet M, Bosco F, et al. Multifunctional Natural Killer Cell Engagers Targeting NKp46 Trigger Protective Tumor Immunity. Cell. 2019;177(7):1701–1713.e16. doi: 10.1016/j.cell.2019.04.041. [DOI] [PubMed] [Google Scholar]
- 6.Wu L, Seung E, Xu L, Rao E, Lord DM, Wei RR, Cortez-Retamozo V, Ospina B, Posternak V, Ulinski G, et al. Trispecific Antibodies Enhance the Therapeutic Efficacy of Tumor-Directed T Cells through T Cell Receptor Co-Stimulation. Nat Cancer. 2020;1(1):86–98. doi: 10.1038/s43018-019-0004-z. [DOI] [PubMed] [Google Scholar]
- 7.Moore GL, Bernett MJ, Rashid R, Pong EW, Nguyen D-HT, Jacinto J, Eivazi A, Nisthal A, Diaz JE, Chu SY, et al. A Robust Heterodimeric Fc Platform Engineered for Efficient Development of Bispecific Antibodies of Multiple Formats. Methods. 2019;154:38–50. doi: 10.1016/j.ymeth.2018.10.006. [DOI] [PubMed] [Google Scholar]
- 8.Herrmann M, Krupka C, Deiser K, Brauchle B, Marcinek A, Ogrinc Wagner A, Rataj F, Mocikat R, Metzeler KH, Spiekermann K, et al. Bifunctional PD-1 × ΑCD3 × ΑCD33 Fusion Protein Reverses Adaptive Immune Escape in Acute Myeloid Leukemia. Blood. 2018;132(23):2484–2494. doi: 10.1182/blood-2018-05-849802. [DOI] [PubMed] [Google Scholar]
- 9.Braciak TA, Roskopf CC, Wildenhain S, Fenn NC, Schiller CB, Schubert IA, Jacob U, Honegger A, Krupka C, Subklewe M, et al. Dual-Targeting Triplebody 33-16-123 (SPM-2) Mediates Effective Redirected Lysis of Primary Blasts from Patients with a Broad Range of AML Subtypes in Combination with Natural Killer Cells. Oncoimmunology. 2018;7(9):e1472195. doi: 10.1080/2162402X.2018.1472195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schubert I, Kellner C, Stein C, Kügler M, Schwenkert M, Saul D, Mentz K, Singer H, Stockmeyer B, Hillen W, et al. A Single-Chain Triplebody with Specificity for CD19 and CD33 Mediates Effective Lysis of Mixed Lineage Leukemia Cells by Dual Targeting. MAbs. 2011;3(1):21–30. doi: 10.4161/mabs.3.1.14057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kügler M, Stein C, Kellner C, Mentz K, Saul D, Schwenkert M, Schubert I, Singer H, Oduncu F, Stockmeyer B, et al. A Recombinant Trispecific Single-Chain Fv Derivative Directed against CD123 and CD33 Mediates Effective Elimination of Acute Myeloid Leukaemia Cells by Dual Targeting. Br J Haematol. 2010;150(5):574–586. doi: 10.1111/j.1365-2141.2010.08300.x. [DOI] [PubMed] [Google Scholar]
- 12.Roskopf CC, Schiller CB, Braciak TA, Kobold S, Schubert IA, Fey GH, Hopfner K-P, Oduncu FS. T Cell-Recruiting Triplebody 19-3-19 Mediates Serial Lysis of Malignant B-Lymphoid Cells by a Single T Cell. Oncotarget. 2014;5(15):6466–6483. doi: 10.18632/oncotarget.2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Roskopf CC, Braciak TA, Fenn NC, Kobold S, Fey GH, Hopfner K-P, Oduncu FS. Dual-Targeting Triplebody 33-3-19 Mediates Selective Lysis of Biphenotypic CD19+ CD33+ Leukemia Cells. Oncotarget. 2016;7(16):22579–22589. doi: 10.18632/oncotarget.8022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Felices M, Kodal B, Hinderlie P, Kaminski MF, Cooley S, Weisdorf DJ, Vallera DA, Miller JS, Bachanova V. Novel CD19-Targeted TriKE Restores NK Cell Function and Proliferative Capacity in CLL. Blood Adv. 2019;3(6):897–907. doi: 10.1182/bloodadvances.2018029371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vallera DA, Felices M, McElmurry R, McCullar V, Zhou X, Schmohl JU, Zhang B, Lenvik AJ, Panoskaltsis-Mortari A, Verneris MR, et al. IL15 Trispecific Killer Engagers (TriKE) Make Natural Killer Cells Specific to CD33+ Targets While Also Inducing Persistence, In Vivo Expansion, and Enhanced Function. Clin Cancer Res. 2016;22(14):3440–3450. doi: 10.1158/1078-0432.CCR-15-2710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Arvindam US, van Hauten PMM, Schirm D, Schaap N, Hobo W, Blazar BR, Vallera DA, Dolstra H, Felices M, Miller JS. A Trispecific Killer Engager Molecule against CLEC12A Effectively Induces NK-Cell Mediated Killing of AML Cells. Leukemia. 2021;35(6):1586–1596. doi: 10.1038/s41375-020-01065-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Austin RJ, Lemon BD, Aaron WH, Barath M, Culp PA, DuBridge RB, Evnin LB, Jones A, Panchal A, Patnaik P, et al. TriTACs, a Novel Class of T-Cell-Engaging Protein Constructs Designed for the Treatment of Solid Tumors. Mol Cancer Ther. 2021;20(1):109–120. doi: 10.1158/1535-7163.MCT-20-0061. [DOI] [PubMed] [Google Scholar]
- 18.Catalano I, Trusolino L. The Stromal and Immune Landscape of Colorectal Cancer Progression during Anti-EGFR Therapy. Cancer Cell. 2019;36(1):1–3. doi: 10.1016/j.ccell.2019.06.001. [DOI] [PubMed] [Google Scholar]
- 19.Gires O, Pan M, Schinke H, Canis M, Baeuerle PA. Expression and Function of Epithelial Cell Adhesion Molecule EpCAM: where Are We after 40 Years? Cancer Metastasis Rev. 2020;39(3):969–987. doi: 10.1007/s10555-020-09898-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Borlak J, Länger F, Spanel R, Schöndorfer G, Dittrich C. Immune-Mediated Liver Injury of the Cancer Therapeutic Antibody Catumaxomab Targeting EpCAM, CD3 and Fcγ Receptors. Oncotarget. 2016;7(19):28059–28074. doi: 10.18632/oncotarget.8574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Even-Desrumeaux K, Nevoltris D, Lavaut MN, Alim K, Borg J-P, Audebert S, Kerfelec B, Baty D, Chames P. Masked Selection: a Straightforward and Flexible Approach for the Selection of Binders against Specific Epitopes and Differentially Expressed Proteins by Phage Display. Mol Cell Proteomics. 2014;13(2):653–665. doi: 10.1074/mcp.O112.025486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hofman EG, Ruonala MO, Bader AN, van den Heuvel D, Voortman J, Roovers RC, Verkleij AJ, Gerritsen HC, van Bergen En Henegouwen PMP. EGF Induces Coalescence of Different Lipid Rafts. J Cell Sci. 2008;121(Pt 15):2519–2528. doi: 10.1242/jcs.028753. [DOI] [PubMed] [Google Scholar]
- 23.Holliger P, Manzke O, Span M, Hawkins R, Fleischmann B, Qinghua L, Wolf J, Diehl V, Cochet O, Winter G, et al. Carcinoembryonic Antigen (CEA)-Specific T-Cell Activation in Colon Carcinoma Induced by Anti-CD3 x Anti-CEA Bispecific Diabodies and B7 x Anti-CEA Bispecific Fusion Proteins. Cancer Res. 1999;59:2909–2916. [PubMed] [Google Scholar]
- 24.Mølgaard K, Harwood SL, Compte M, Merino N, Bonet J, Alvarez-Cienfuegos A, Mikkelsen K, Nuñez-Prado N, Alvarez-Mendez A, Sanz L, et al. Bispecific Light T-Cell Engagers for Gene-Based Immunotherapy of Epidermal Growth Factor Receptor (EGFR)-Positive Malignancies. Cancer Immunol Immunother. 2018;67(8):1251–1260. doi: 10.1007/s00262-018-2181-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pozzi C, Cuomo A, Spadoni I, Magni E, Silvola A, Conte A, Sigismund S, Ravenda PS, Bonaldi T, Zampino MG, et al. The EGFR-Specific Antibody Cetuximab Combined with Chemotherapy Triggers Immunogenic Cell Death. Nat Med. 2016;22(6):624–631. doi: 10.1038/nm.4078. [DOI] [PubMed] [Google Scholar]
- 26.Frenzel A, Hust M, Schirrmann T. Expression of Recombinant Antibodies. Front Immunol. 2013;4:217. doi: 10.3389/fimmu.2013.00217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zheng S, Moores S, Jarantow S, Pardinas J, Chiu M, Zhou H, Wang W. Cross-Arm Binding Efficiency of an EGFR x c-Met Bispecific Antibody. MAbs. 2016;8(3):551–561. doi: 10.1080/19420862.2015.1136762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Harms BD, Kearns JD, Iadevaia S, Lugovskoy AA. Understanding the Role of Cross-Arm Binding Efficiency in the Activity of Monoclonal and Multispecific Therapeutic Antibodies. Methods. 2014;65(1):95–104. doi: 10.1016/j.ymeth.2013.07.017. [DOI] [PubMed] [Google Scholar]
- 29.Schmitz KR, Bagchi A, Roovers RC, van Bergen En Henegouwen PMP, Ferguson KM. Structural Evaluation of EGFR Inhibition Mechanisms for Nanobodies/VHH Domains. Structure. 2013;21(7):1214–1224. doi: 10.1016/j.str.2013.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hristodorov D, Amoury M, Mladenov R, Niesen J, Arens K, Berges N, Hein L, Di Fiore S, Pham A-T, Huhn M, et al. EpCAM-Selective Elimination of Carcinoma Cells by a Novel MAP-Based Cytolytic Fusion Protein. Mol Cancer Ther. 2014;13(9):2194–2202. doi: 10.1158/1535-7163.MCT-13-0781. [DOI] [PubMed] [Google Scholar]
- 31.Harwood SL, Alvarez-Cienfuegos A, Nuñez-Prado N, Compte M, Hernández-Pérez S, Merino N, Bonet J, Navarro R, Van Bergen En Henegouwen PMP, Lykkemark S, et al. ATTACK, a Novel Bispecific T Cell-Recruiting Antibody with Trivalent EGFR Binding and Monovalent CD3 Binding for Cancer Immunotherapy. Oncoimmunology. 2017;7(1):e1377874. doi: 10.1080/2162402X.2017.1377874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Marusyk A, Janiszewska M, Polyak K. Intratumor Heterogeneity: the Rosetta Stone of Therapy Resistance. Cancer Cell. 2020;37(4):471–484. doi: 10.1016/j.ccell.2020.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gonzalez-Exposito R, Semiannikova M, Griffiths B, Khan K, Barber LJ, Woolston A, Spain G, von Loga K, Challoner B, Patel R, et al. CEA Expression Heterogeneity and Plasticity Confer Resistance to the CEA-Targeting Bispecific Immunotherapy Antibody Cibisatamab (CEA-TCB) in Patient-Derived Colorectal Cancer Organoids. J Immunother Cancer. 2019;7(1):101. doi: 10.1186/s40425-019-0575-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ruella M, Barrett DM, Kenderian SS, Shestova O, Hofmann TJ, Perazzelli J, Klichinsky M, Aikawa V, Nazimuddin F, Kozlowski M, et al. Dual CD19 and CD123 Targeting Prevents Antigen-Loss Relapses after CD19-Directed Immunotherapies. J Clin Invest. 2016;126(10):3814–3826. doi: 10.1172/JCI87366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Montagut C, Dalmases A, Bellosillo B, Crespo M, Pairet S, Iglesias M, Salido M, Gallen M, Marsters S, Tsai SP, et al. Identification of a Mutation in the Extracellular Domain of the Epidermal Growth Factor Receptor Conferring Cetuximab Resistance in Colorectal Cancer. Nat Med. 2012;18(2):221–223. doi: 10.1038/nm.2609. [DOI] [PubMed] [Google Scholar]
- 36.Bertotti A, Papp E, Jones S, Adleff V, Anagnostou V, Lupo B, Sausen M, Phallen J, Hruban CA, Tokheim C, et al. The Genomic Landscape of Response to EGFR Blockade in Colorectal Cancer. Nature. 2015;526(7572):263–267. doi: 10.1038/nature14969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Metges J, Ramée JF, Dupuis O, Deguiral P, Boucher E, Cojocarasu O, Ferec M, Porneuf M, Douillard J, Grude F. Panerb Study: which Category of Patients, Suffering from Metastatic Colorectal Cancer, Can Benefit From Panitumumab Treatment After Cetuximab-Based Regimen Failure? Annals of Oncology. 2012;23:ix196. doi: 10.1016/S0923-7534(20)33183-5. [DOI] [Google Scholar]
- 38.Arena S, Bellosillo B, Siravegna G, Martínez A, Cañadas I, Lazzari L, Ferruz N, Russo M, Misale S, González I, et al. Emergence of Multiple EGFR Extracellular Mutations during Cetuximab Treatment in Colorectal Cancer. Clin Cancer Res. 2015;21(9):2157–2166. doi: 10.1158/1078-0432.CCR-14-2821. [DOI] [PubMed] [Google Scholar]
- 39.Goebeler M-E, Bargou RC. T Cell-Engaging Therapies - BiTEs and Beyond. Nat Rev Clin Oncol. 2020;17(7):418–434. doi: 10.1038/s41571-020-0347-5. [DOI] [PubMed] [Google Scholar]
- 40.Shah NN, Johnson BD, Schneider D, Zhu F, Szabo A, Keever-Taylor CA, Krueger W, Worden AA, Kadan MJ, Yim S, et al. Bispecific Anti-CD20, Anti-CD19 CAR T Cells for Relapsed B Cell Malignancies: a Phase 1 Dose Escalation and Expansion Trial. Nat Med. 2020;26(10):1569–1575. doi: 10.1038/s41591-020-1081-3. [DOI] [PubMed] [Google Scholar]
- 41.Rossotti MA, Bélanger K, Henry KA, Tanha J. Immunogenicity and Humanization of Single-Domain Antibodies. FEBS J. 2021. doi: 10.1111/febs.15809. [DOI] [PubMed] [Google Scholar]
- 42.Bannas P, Hambach J, Koch-Nolte F. Nanobodies and Nanobody-Based Human Heavy Chain Antibodies As Antitumor Therapeutics. Front Immunol. 2017;8:1603. doi: 10.3389/fimmu.2017.01603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Punt CJ, Nagy A, Douillard J-Y, Figer A, Skovsgaard T, Monson J, Barone C, Fountzilas G, Riess H, Moylan E, et al. Edrecolomab Alone or in Combination with Fluorouracil and Folinic Acid in the Adjuvant Treatment of Stage III Colon Cancer: a Randomised Study. The Lancet. 2002;360(9334):671–677. doi: 10.1016/S0140-6736(02)09836-7. [DOI] [PubMed] [Google Scholar]
- 44.Goel S, Bauer RJ, Desai K, Bulgaru A, Iqbal T, Strachan B-K, Kim G, Kaubisch A, Vanhove GF, Goldberg G, et al. Pharmacokinetic and Safety Study of Subcutaneously Administered Weekly ING-1, a Human Engineere Monoclonal Antibody Targeting Human EpCAM, in Patients with Advanced Solid Tumors. Ann Oncol. 2007;18(10):1704–1707. doi: 10.1093/annonc/mdm280. [DOI] [PubMed] [Google Scholar]
- 45.Schmidt M, Scheulen ME, Dittrich C, Obrist P, Marschner N, Dirix L, Schmidt M, Rüttinger D, Schuler M, Reinhardt C, et al. An Open-Label, Randomized Phase II Study of Adecatumumab, a Fully Human Anti-EpCAM Antibody, as Monotherapy in Patients with Metastatic Breast Cancer. Ann Oncol. 2010;21(2):275–282. doi: 10.1093/annonc/mdp314. [DOI] [PubMed] [Google Scholar]
- 46.Jarantow SW, Bushey BS, Pardinas JR, Boakye K, Lacy ER, Sanders R, Sepulveda MA, Moores SL, Chiu ML. Impact of Cell-Surface Antigen Expression on Target Engagement and Function of an Epidermal Growth Factor Receptor × c-MET Bispecific Antibody. J Biol Chem. 2015;290(41):24689–24704. doi: 10.1074/jbc.M115.651653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zuckier LS, Berkowitz EZ, Sattenberg RJ, Zhao QH, Deng HF, Scharff MD. Influence of Affinity and Antigen Density on Antibody Localization in a Modifiable Tumor Targeting Model. Cancer Res. 2000;60:7008–7013. [PubMed] [Google Scholar]
- 48.Mazor Y, Oganesyan V, Yang C, Hansen A, Wang J, Liu H, Sachsenmeier K, Carlson M, Gadre DV, Borrok MJ, et al. Improving Target Cell Specificity Using a Novel Monovalent Bispecific IgG Design. MAbs. 2015;7(2):377–389. doi: 10.1080/19420862.2015.1007816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mazor Y, Sachsenmeier KF, Yang C, Hansen A, Filderman J, Mulgrew K, Wu H, Dall’Acqua WF. Enhanced Tumor-Targeting Selectivity by Modulating Bispecific Antibody Binding Affinity and Format Valence. Sci Rep. 2017;7(1):40098. doi: 10.1038/srep40098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Navarro R, Tapia-Galisteo A, Martín-García L, Tarín C, Corbacho C, Gómez-López G, Sánchez-Tirado E, Campuzano S, González-Cortés A, Yáñez-Sedeño P, et al. TGF-β-Induced IGFBP-3 Is a Key Paracrine Factor from Activated Pericytes That Promotes Colorectal Cancer Cell Migration and Invasion. Mol Oncol. 2020;14(10):2609–2628. doi: 10.1002/1878-0261.12779. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.