

Abstract
Ectodysplasin A receptor (EDAR) is a death receptor in the Tumour Necrosis Factor Receptor (TNFR) superfamily with roles in the development of hair follicles, teeth and cutaneous glands. Here we report that human Oestrogen Receptor (ER) negative breast carcinomas which display squamous differentiation express EDAR strongly. Using a mouse model with a high Edar copy number, we show that elevated EDAR signalling results in a high incidence of mammary tumours in breeding female mice. These tumours resemble the EDAR-high human tumours in that they are characterised by a lack of oestrogen receptor expression, contain extensive squamous metaplasia, and display strong β-catenin transcriptional activity. In the mouse model, all of the tumours carry somatic deletions of the third exon of the CTNNB1 gene that encodes β-catenin. Deletion of this exon yields unconstrained β-catenin signalling activity. We also demonstrate that β-catenin activity is required for transformed cell growth, showing that increased EDAR signalling creates an environment in which β-catenin activity can readily promote tumourigenesis. Together, this work identifies a novel death receptor oncogene in breast cancer, whose mechanism of transformation is based on the interaction between the WNT and Ectodysplasin A (EDA) pathways.
Subject terms: Breast cancer, Oncogenes, Growth factor signalling, Disease model, Transdifferentiation
Introduction
Ectodysplasin A receptor (EDAR) is one of eight death domain receptors encoded within the human genome [1]. During development, EDAR signalling is stimulated by its ligand, Ectodysplasin A (EDA), leading to signal transduction through EDAR-associated death domain to the canonical NFκB pathway. Rather than inducing apoptosis, as effected by other death domain receptors such as Tumour Necrosis Factor Receptor 1 (TNFR1) and FAS, this signal is essential for the formation of placodes, the first step in the development of many ectodermal appendages and glands, including hair, teeth and sweat glands [2]. In humans, loss-of function mutations within EDAR pathway components lead to Hypohydrotic Ectodermal Dysplasia (HED), which is characterised by missing or misshapen teeth, sparse hair, a reduced ability to sweat [3, 4] and lack of full mammary development [5, 6]. Similar phenotypes are seen in the mouse strains, downless (EdardlJ) and Tabby (EdaTa/Ta), which carry loss-of-function mutations in the receptor and ligand respectively [4, 7, 8], and when NFκB signalling is reduced in the developing skin [9]. The developmental action of EDAR is tightly associated with WNT/β-catenin signalling, with several levels of interaction between these pathways having been described in early hair follicle development [10, 11].
The role of EDAR signalling in tumour development has received little attention, despite its ability to activate NFκB [12] and the clear association of NFκB signalling with many different cancer types, including breast cancer [13–15]. Furthermore, its signalling in the skin induces the expression of known mammary gland oncogenes [11, 16, 17], including Wnt10b whose overexpression in the developing mammary gland induces precocious alveolar development and focal mammary adenocarcinomas [18]. Receptor Activator of Nuclear Factor κ B (RANK), a closely related member of the TNF receptor superfamily lacking a death domain, also contributes to progestin-induced mammary gland tumours during pregnancy [19].
Thus, based on its signalling function and its role in mammary development, we evaluated EDAR as a novel candidate mammary oncogene. We assessed expression data from human breast carcinomas, identifying a specific subtype of ductal carcinoma with squamous metaplasia that expressed EDAR strongly. To determine whether high EDAR levels are capable of causing breast cancer, we examined mammary tumour formation in a transgenic mouse line expressing high levels of Edar from its endogenous regulatory elements. We found that breeding female mice develop mammary tumours, and, as observed in the EDAR-high human tumours, these tumours contain extensive squamous metaplasia. Our analysis of the tumours indicated that the modestly increased EDAR signalling in the EdarTg951/951 mice is further increased in transformed cells, as is β-catenin transcriptional activity. All tumours carry exon 3 deletions of CTNNB1, conferring WNT-independent activity on β-catenin. This β-catenin activity is required for tumour cell proliferation, highlighting the interplay between EDAR and WNT pathways in tumourigenesis. Together our data identify EDAR as a novel breast oncogene.
Results
EDAR is highly expressed in basal and metaplastic human breast cancers
The action of EDAR in breast tissue, its similarity to the known mammary oncogene RANK, and its ability to activate NFκB, led us to investigate whether EDAR could be a causative factor in human breast cancer. We first analysed expression of EDAR in a meta-dataset of 1107 invasive human breast cancers classified into the basal-like, luminal A, luminal B, normal breast-like, and HER2 molecular subtypes [20, 21]. Within this dataset, there was a small group of tumours that expressed very high levels of EDAR, which almost invariably belonged to the basal-like subgroup (Fig. 1A). These basal-like EDAR-high tumours have low ESR expression, which encodes the oestrogen receptor. There was also a positive correlation between high EDAR and expression of the skin-specific genes LORICRIN (LOR), KERATIN1 (KRT1) and INVOLUCRIN (IVL), suggesting the occurrence of squamous metaplasia within the EDAR-high cancers (Fig. 1A).
Fig. 1. EDAR is highly expressed in basal and metaplastic human breast cancers.
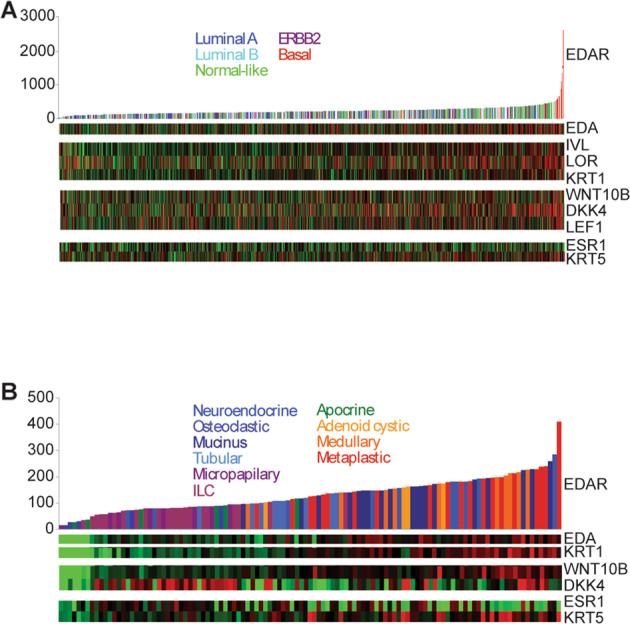
Expression of EDAR pathway components, skin markers and EDAR target genes in a meta-analysis of 1107 primary tumours (A), and 113 breast cancers of special histological type (B). Tumours were ordered left to right by increasing EDAR expression in both datasets and coloured by molecular (A) or histological subtype (B); white bars represent tumours that were not clearly assigned to any subtype. The heat bars show high expression (red) of EDA, skin markers (IVL,KRT1), and EDAR target genes, WNT10B and DKK4 in EDAR-high tumours. Increased LEF1 expression indicated increased WNT/β-catenin signalling in the EDAR-high tumours. Low expression (green) of ESR and high expression of KRT5 was seen in the EDAR-high tumours confirming they were ER-ve tumours with a basal molecular subtype.
In light of the correlation between EDAR and the expression of epidermal marker genes, we also examined EDAR expression in a second dataset of 113 histologically special type tumours, which include metaplastic cancers that can display extensive squamous metaplasia (Fig. 1B) [22]. The tumours expressing the highest levels of EDAR were the adenocystic, medullary and metaplastic cancers, which are those most closely related to the basal molecular subtype of invasive breast cancers of no specific type [23, 24]. These tumours also showed high levels of KRT1 and IVL expression, indicating squamous differentiation (Fig. 1B). Thus elevated EDAR expression is associated with squamous metaplasia in human breast cancers.
High Edar copy number in the EdarTg951 line leads to elevated expression and signalling in the mammary gland
To assess whether elevated Edar expression can be a causative factor in mammary tumourigenesis, we employed the EdarTg951 transgenic mouse line; homozygous EdarTg951/951animals carry approximately 32 additional copies of the Edar locus [25]. We determined Edar expression levels in non-transgenic wild-type and homozygous EdarTg951/951 mammary tissue at 4 and 8 weeks of age, finding increased Edar expression in the transgenic line (Fig. 2A). We assessed the location of Edar expression by in situ hybridisation, detecting the transcript in luminal cells of the epithelial ducts of both non-transgenic wild-type and EdarTg951/951 mice (Fig. 2B). Increased Edar expression in the EdarTg951/951 line is accompanied by elevated expression of the EDAR target genes Wnt10b and Dickkopf4 (Dkk4) (Fig. 2C), demonstrating that signal transduction through this pathway is amplified in the transgenic mammary glands. To determine whether increased Edar expression can cause elevated signal transduction in mammary epithelial cells, we transfected C57MG mouse mammary epithelial cells with an Edar expression vector and assessed NFκB luciferase activity. Transfection and high-level Edar expression in this cell line did lead to increased NFκB activity (Fig. 2D) demonstrating the capacity of mammary epithelial cells to respond to high-level EDAR expression by activating NFκB.
Fig. 2. Increased Edar expression and function in the EdarTg951 mouse line.

A qRT-PCR analysis of Edar expression in nulliparous mouse mammary glands of wild-type (WT) and EdarTg951/951 at 4 and 8 weeks of age (wv), normalised to Keratin18 (**P < 0.01, *P < 0.05, N = 4). B In situ hybridisation to detect Edar and dapB negative control probesets on mammary gland sections from EdarTg951/951 and WT female mice at 6 weeks of age. Edar expression was detected in the ductal luminal cells. Scale bar = 50 µm. C qRT-PCR analysis comparing expression of Wnt10b and Dkk4 in WT versus normal EdarTg951/951 mammary tissue at 4 weeks of age. Expression was normalised to Keratin18. Wnt10b and Dkk4 expression were elevated in the EdarTg951/951 mammary gland relative to WT (**P < 0.01, *P < 0.05, N ≥ 5). D Luciferase reporter assay detecting NFκB activity in C57MG cells in response to transfection of an Edar expression vector (*P < 0.05, N = 2). Error bars indicate SEM.
Elevated EDAR signalling leads to mammary tumourigenesis in breeding female mice
Having defined increased Edar expression and signalling activity in the EdarTg951/951 mammary gland, we next assessed the tumour incidence in these mice. We followed cohorts of female nulliparous and continuously breeding EdarTg951/951, along with wild-type mice, over a 1-year period. The continuously breeding EdarTg951/951 mice developed solitary mammary gland tumours from as early as 3 months of age and with an average latency of approximately 7 months (Fig. 3A, B). In contrast, no tumours occurred in the continuously breeding WT mice and only one of the nulliparous EdarTg951/951 mice developed a tumour (Fig. 3B), as did a single male EdarTg951/951 mouse in the colony. We also observed tumours in 2 out of the 3 EdarTg951/+ breeding female mice that we have had in the colony. Histological analysis of the EDAR-induced tumours showed that all were adenocarcinomas (Fig. 3C). No tumours were noted in any other organ in the course of the study.
Fig. 3. Elevated EDAR signalling leads to pregnancy-dependent mammary tumours with squamous metaplasia.

A Gross image of a tumour from a breeding 6-month-old EdarTg951/951 mouse. Dotted line shows the outline of the mammary tumour. Scale bar = 50 mm. B Mammary tumour incidence in continuously breeding and nulliparous cohorts of EdarTg951/951 mice, compared to continuously breeding WT mice. Results are expressed as the percentage of mice remaining tumour free in cohorts of mice as a function of their age (median survival for breeding EdarTg951/951 female mice 215 days, difference in survival curves P < 0.0001, N ≥ 10). C Histological section from an Edar-induced tumour showing the development of adenocarcinoma. Scale bar = 200 µm. D Western analysis of oestrogen receptor (ER) expression in nuclear protein lysates from Edar-induced mammary tumours. Lysates from 8 week nulliparous (8wv) and day 15 pregnant (p15) mammary glands were used as positive controls. Lamin B1 was used as a loading control. Lanes 1–11 represent lysates from 11 different Edar-induced tumours. Immunohistochemical analysis of Ki67 (E) and cleaved Caspase-3 (F) expression in Edar-induced mouse mammary tumours (i); in control sections with secondary antibody only (ii); and in normal mammary gland tissue lying adjacent to the tumour (iii). High levels of Ki67 and cleaved Caspase-3 are seen in the Edar-induced tumours, indicating that the tumours are highly proliferative and contain areas of necrosis. Scale bars = 800 µm in i–ii and 200 µm in iii. G Histological sections from Edar-induced mouse mammary tumours showing areas of (i) transdifferentiation to epidermis and (ii) associated Keratin-like pearls. Scale bar = 200 µm. Immunohistochemical analysis of KERATIN1 (H) and LORICRIN (I) expression in Edar-induced tumour sections (i); in control sections with secondary antibody only (ii); and in WT mammary gland tissue (iii) and skin (iv). KERATIN1 and LORICRIN mark the intermediate and terminally differentiated epidermal cells, respectively, and are absent in the WT mammary gland. Scale bar = 800 µm in i–ii, and 200 µm in iii–iv. J qRT-PCR analysis of Edar and EDAR target genes A20, Wnt10b and Dkk4 in tumours relative to normal EdarTg951/951 mammary tissue, normalised to Keratin18 expression (*P < 0.05, N ≥ 3). K Western analysis of p65 expression in nuclear and cytoplasmic lysates from Edar-induced tumours and WT mammary glands from 8-week nulliparous (8wv) and day 15 pregnant (p15) mice. Lamin B1 was used as a loading control. Nuclear accumulation of p65 indicative of NFκB signalling was observed within the tumours. L Soft agar colony forming assay using EpH4 cells transduced with Edar expression vector (pCDH) (**P < 0.01, N = 3). Error bars indicate SEM.
Analysis of clinical markers of human breast cancer revealed that the tumours arising in the EdarTg951/951 mice lacked Oestrogen Receptor (ER) expression (Fig. 3D) were highly proliferative, with many cells staining positively for the proliferation marker Ki67 (Fig. 3E), and contained large areas of apoptosis, marked by cleaved Caspase-3 staining (Fig. 3F). All tumours also displayed areas of squamous metaplasia, albeit to varying extents, which in some cases led to the formation of Keratin-like pearls (Fig. 3G). This transdifferentiation into skin was verified by expression of KERATIN1 and LORICRIN, markers of intermediate and terminally differentiated layers of the skin, respectively (Figs. 3H, 3I). The areas of transdifferentiation also expressed KERATIN14, a marker of the basal layer of the skin, which is also expressed in myoepithelial cells within the normal mammary gland (Fig. S1). Tumour tissue displayed elevated EDAR target gene expression (Fig. 3J) and an increase in nuclear p65 (Fig. 3K), indicative of ongoing increased NFκB signalling.
To test whether elevation of EDAR signalling in epithelial cells of mature mammary glands is capable of causing transformation alone, we transduced EpH4 cells, a cell line derived from mammary epithelial cells taken from pregnant mice, with an Edar expressing lentiviral vector and determined colony number in a soft agar growth assay. Forced Edar expression led to a statistically significant increase in colony formation (Fig. 3L), demonstrating that short-term stimulation of the EDAR pathway is capable of transforming cells derived from the wild-type adult mammary gland.
EDAR-induced tumours have high levels of β-catenin activity and carry somatic mutations in CTNNB1
Based on the morphological similarities between EdarTg951/951 mammary glands (See Supplementary material and Figs. S2–4) and those developing under conditions of elevated WNT signalling, and on the key role for WNT/β-catenin signalling as a driver of mammary tumorigenesis [26, 27], we assessed β-catenin status and function in Edar-induced tumours. qRT-PCR analysis identified elevated expression of β-catenin target genes Axin2 and Lef1 in EdarTg951/951 tumour tissue (Fig. 4A), as well as elevated Edar transcript abundance when compared to untransformed EdarTg951/951 mammary tissue (Figs. 4A, 4B). Nuclear immunostaining for β-catenin protein was also readily detectable (Fig. 4C), indicating its activated state. It is also interesting to note that within a series of mouse mammary tumour models elevated levels of Edar expression are only seen MMTV-Wnt1 mice (Fig. S5A).
Fig. 4. Edar-induced mammary tumours are characterised by high β-catenin activity.

A qRT-PCR detection of WNT/β-catenin target genes Axin2 and Lef1 in tumours of EdarTg951/951 mice (*P < 0.05, N ≥ 3). B In situ hybridisation detecting Edar expression in tumour epithelial cells. The dapB probeset is a negative control. Scale bar = 50 μm. C Immunodetection of nuclear β-catenin in tumours. Scale bar = 50 μm. D Western analysis of β-catenin protein in tumour homogenates. In addition to the full-length β-catenin, a more intense lower band representing a truncated form is detected in all tumour samples. PCR analysis across exon 3 of Ctnnb1 showing products run on an agarose gel (E) and diagram of different mutations identified (F). G Western analysis of β-catenin protein in distinct tissues from the individual carrying tumour T301113. The short form of β-catenin protein is detected only in the mammary tumour isolate and not in other tissues. H Schematic of β-catenin gene and protein structure, with exon boundaries indicated by dotted lines and number of coding nucleotides per exon indicated below. Amino terminal phosphorylation sites, which induce protein degradation, are indicated by ‘P’. The function of the WNT signalling cascade, leading to suppression of GSK3 activity, is depicted. I Expression of γ-H2AX and nuclear β-catenin in normal EdarTg951/951 gland and in tumour tissue. Scale bars = 50 μm. J Transformation of EpH4 cells by transduction with β-catenin expression constructs lacking the portion encoded by exon 3 (****P < 0.0001, N = 3). Error bars indicate SEM.
We used Western analysis to assess β-catenin protein status in the tumour tissue. Surprisingly, in addition to the ~95 kDa band expected for full-length β-catenin, all tumours also displayed a smaller band with a more intense signal (Fig. 4D). These findings indicated that heterozygosity for a somatic mutation in CTNNB1, encoding β-catenin, could underlie the presence of a smaller form of this protein. We sequenced cDNA and genomic DNA from tumours, identifying in every case a mutation leading to deletion of CTNNB1 exon 3; in one case the deletion encompassed part of exon 4 also (Fig. 4E, F). This shortened form of β-catenin is specific to tumour tissue and is not detected in other tissues, nor in the normal contralateral mammary gland (Fig. 4G and Fig. S5B). These somatic deletions remove the exon encoding the phosphorylation sites required for Glycogen Synthase Kinase 3 (GSK3)-triggered β-catenin degradation while maintaining the open reading frame, permitting WNT-independent β-catenin activity (Fig. 4H). Within the tumours, the nuclear β-catenin corresponded with sites of double-strand DNA breaks, as indicated by phospho-γ-H2AX staining (Fig. 4I). Repair of such double-strand breaks can occur through non-homologous end joining (NHEJ), which often leads to small deletion mutations consistent with those identified that remove the third exon of CTNNB1. The resulting exon 3 deleted forms of β-catenin selected in the tumours are sufficient to drive cellular transformation of EpH4 cells (Fig. 4J). Thus the high-level β-catenin activity observed in all EdarTg951/951 tumours can be explained by their independent, but functionally identical, somatic mutations in CTNNB1.
Involution and DNA damage in EdarTg951/951 mammary glands
Elevated EDAR signalling, together with activation of β-catenin activity through somatic mutation, appears to be a sufficient oncogenic stimulus to explain the high frequency of mammary tumours in the EdarTg951/951 line. However, as it is not clear why continual breeding has such a marked effect on tumour incidence, we assessed Edar expression and function through the mammary gland’s pregnancy/lactation/involution cycle. qRT-PCR analysis detected Edar transcript at all stages of pregnancy, involution and lactation, with the lowest expression observed during lactation (Fig. 5A and Fig. S6A). In situ hybridisation shows that this expression occurs in luminal epithelial cells but is higher in EdarTg951/951 than wild-type mammary glands (Fig. 5B and Fig. S6B).
Fig. 5. Edar expression and induced changes through the pregnancy/lactation cycle.

A qRT-PCR analysis of Edar expression levels through the first pregnancy cycle in wild-type and EdarTg951/951 glands (*P < 0.05, **P < 0.01, **P < 0.0001, N = 4). B In situ hybridisation detecting Edar expression in WT and EdarTg951/951 mammary glands at involution day 2 (I2). Scale bar = 50 μm. C Histological assessment of mammary phenotype in EdarTg951/951 mice at involution day 2 during first and second pregnancy. Scale bar = 50 μm. D Immunodetection of cleaved caspase-3 in mammary glands at involution day 2 during second pregnancy. Scale bar = 20 μm. E, F Quantification of apoptotic frequency in wild-type and EdarTg951/951 mammary epithelia at involution day 2 (***P < 0.001, N ≥ 28). G Histological appearance of mastitis in EdarTg951/951 gland found at involution day 2, accompanied by double-strand breaks identified as immunodetection of γ-H2AX. Scale bar = 50 μm.
Histological analysis of the mammary gland through pregnancy, lactation and involution reveals a similar structure in transgenic and non-transgenic at all stages (Fig. S6C), apart from early involution. At this stage, we observed an increased number of apoptotic cells in the EdarTg951/951 mice (Fig. 5C–F), which was more pronounced after the second pregnancy than the first. However, as the area of epithelial tissue remaining in wild-type and Edartg951/951 mice was not different at involution day 6 (Fig. S6D, E), we conclude that the increase in the number of apoptotic cells seen at involution day 2 in EdarTg951/951 mice arose from a failure to clear apoptotic cells rather than an increased rate of apoptosis. We also noted a high rate of mastitis in breeding female EdarTg951/951 animals (Fig. 5G) that we did not observe in wild-type mice. Mastitis may have arisen when milk ducts and acini became blocked with uncleared apoptotic cells in the transgenic line. Damaged DNA was also detected in mammary epithelial cells as γ-H2AX positive foci, revealing the presence of double-stranded DNA breaks (Fig. 5G). Thus the mastitis observed during mammary involution may provide an environment in which deletion mutations can more readily occur, potentially contributing to the mammary tumour incidence in the EdarTg951/951 line.
β-catenin activity is required for rapid proliferation of EdarTg951/951 tumour cells
The morphological phenotype of the EdarTg951/951 mammary gland is similar in most respects to that obtained by augmentation of WNT signalling [26, 27], and the activation of Wnt10b gene expression by increased Edar in transgenic mammary glands suggests a positive relationship between these pathways. However, if EDAR signalling had only a simple and positive relationship with β-catenin activity, then it is difficult to explain the observed selection for somatic mutations in CTNNB1 that produce an activated β-catenin protein in tumours of the EdarTg951/951 line. To address their signalling relationship, we assessed the short-term effects of EDAR signalling on β-catenin transcriptional regulatory activity by transfecting HEK293T cells with a β-catenin responsive luciferase reporter (pTOP-FLASH), together with an Edar expression construct. We found that introduction of high EDAR signalling in these conditions suppressed β-catenin activity (Fig. S7A). The transfection of a delEx3β-catenin expressing construct, which misses the portion of the protein encoded by the third exon and thus matches the truncated form observed in all EdarTg951/951 tumours, evaded this EDAR driven inhibition and restored reporter gene response in transfected cells (Fig. S7A).
To assess the functional requirement for unrestrained β-catenin activity in EdarTg951/951 tumour cell proliferation, we set out to impair β-catenin activity in these cells. Cells transfected with delEx3β-catenin display increased levels of TOPFLASH reporter activity, which is suppressed using iCRT3, a small molecule inhibitor of β-catenin interaction with TCF class transcription factors, validating its inhibitory action in this signalling context (Fig. 6A). We isolated mammary gland epithelial cells from wild-type, normal EdarTg951/951 glands, and from EdarTg951/951 tumours, and grew these in culture to assess their proliferative characteristics. Tumour cells incorporated the DNA synthesis label EdU at far greater frequency than normal cells, indicating their elevated rate of proliferation. Addition of iCRT3 to the culture medium suppressed proliferation of the tumour cells to levels that were not different from untransformed cells (Fig. 6B), demonstrating that β-catenin activity is required for elevated proliferative index of cells of the EdarTg951/951 tumours; we also observed a decrease in the expression of the Wnt target gene Lef1 in the iCRT3-treated tumour cells (Fig. S7B) but no change in the rate of apoptosis (Fig. S7C).
Fig. 6. The elevated growth rate of EDAR transformed cells is dependent on β-catenin activity.

A Luciferase assay detecting β-catenin reporter activity in cells transfected with delEx3β-catenin encoding constructs, with or without incorporation of iCRT3 to the culture (**P < 0.01, ***P < 0.001, N = 2). B EdU incorporation by cells from EdarTg951/951 and wild-type mammary glands, and from EdarTg951/951 tumours, grown in culture. iCRT3 administration suppresses proliferation of EdarTg951/951 mammary tumour cells (**P < 0.01, ***P < 0.001, N = 3).
Discussion
EDAR represented a potential mammary gland oncogene based on its signalling mode [28] and its role in mammary gland development [8, 29]. Our analyses of expression data from human breast cancer tissue identified a specific subtype of human breast cancer characterised by high EDAR expression. These tumours also displayed high expression levels of epidermal differentiation genes (such as IVL, LOR and KRT1), indicating that these tumours had undergone squamous metaplasia, and an activation of the WNT/β-catenin signalling pathway (expression of WNT10B and LEF1). The latter could explain the overexpression of WNT10B that is seen in some human breast cancers [30].
To test whether elevated EDAR signalling is sufficient to lead to mammary tumourigenesis, we employed the EdarTg951 mouse model which carries a high copy number of Edar expressed from its endogenous regulatory elements [7, 25, 31]. This line has an altered rate of mammary gland development, such that ductal extension and branching is greater in the transgenic during puberty, but this morphological phenomenon is largely corrected by adult life. The high rate of mammary tumours in EdarTg951 mice upon continual mating, which was not observed in wild-type mated or transgenic nulliparous females, demonstrated a driving role for EDAR as an oncogene. In this mouse model, the characteristic squamous metaplasia and expression of epidermal differentiation genes in mammary tumours matches the expression phenotype of the human metaplastic tumours with high EDAR expression. Thus in both human and mouse increased EDAR expression is associated with this distinct mammary tumour type.
Previous work has highlighted a role for NFκB signalling in mammary gland development and tumourigenesis. In particular, RANK signalling has been shown to play an essential role downstream of Progesterone Receptor in the formation of the milk-producing alveoli during pregnancy [32, 33] and to contribute to Progestin-induced mammary gland tumours [19, 34]. However there is some indication within the published literature that NFκB signalling may play a role in pubertal development as well. For example, transplanted mammary tissue, which lacks IκBα, forms outgrowths in the cleared fat pad of a nulliparous animal or matrigel that are substantially more branched than those derived from wild-type tissue [35]. These phenotypes have been attributed to changes in signalling activated by RANK. However given our results here and those of Voutilainen et al. [8], the phenotypes seen could instead be attributed to changes in EDAR-induced NFκB signalling. In fact, the extra branching seen in the ductal tree of virgin animals when RANKL is overexpressed from the mouse mammary tumour virus-long terminal repeat (MMTV-LTR) could be due to RANK/NFκB signalling mimicking endogenous EDAR/NFκB signalling at this stage [36]. However, we do not observe the precocious development of alveoli in the EdarTg951/951 mice that is seen in the MMTV-RankL mice or the ductal hyperplasia observed in the transplanted tissue that lacks IκBα [35, 36]. This most likely reflects differences in the downstream targets of RANK and EDAR signalling. Mammary tumourigenesis has also been reported in the mouse models where RANK/NFκB signalling is activated within the mammary gland [19, 37]. Interestingly, most of the tumours that develop in the MMTV-c-rel mouse strain display squamous metaplasia [37], suggesting that WNT/β-catenin signalling may be activated in these tumours as well.
During development, EDAR signalling is intertwined with that of the WNT/β-catenin pathway [10, 11]. Both positive and negative interactions have been reported and suggested, based on target genes of each pathway. The morphological effect of increased EDAR function on the EdarTg951 gland is consistent with an increase in WNT signalling [18, 38], and is consistent with increased Wnt10b expression, suggesting a stimulatory effect of EDAR on WNT pathway signalling over a sustained period. However, an inhibitory effect of EDAR signalling on β-catenin function is revealed by EDAR overexpression over a 48 h period in cultured cells, potentially through the activation of WNT/β-catenin pathway inhibitors such as DKK4 [10, 11]. Thus, positive and negative impacts of EDAR function on β-catenin signalling are apparent. The negative impact of EDAR overexpression on β-catenin signalling may explain the absence in EdarTg951/951 females of one mammary gland phenotype associated with excess WNT/β-catenin signalling, precocious lobular-alveloar development. This phenotype is seen when the pathway is activated directly by overexpressing a WNT protein or a non-degradable form of β-catenin [39, 40]. In these animals, β-catenin signalling is unregulated. In contrast, the expression of DKK4 or other negative regulators of WNT signalling will limit or control WNT/β-catenin signalling in the EdarTg951/951 mice. If lobular-alveolar development requires a strong WNT/β-catenin signal, then it is reasonable to hypothesise that the signal is simply insufficient in EdarTg951/951 females to drive lobular-alveolar development.
The short-term inhibitory relationship between EDAR and β-catenin is also likely to apply a selective pressure for unrestrained β-catenin activity to drive cell transformation. In tumours, the release of β-catenin activity from this inhibitory EDAR influence is provided by somatic mutation, with cells carrying β-catenin exon 3 deletions growing out to form tumours. The signalling environment caused by EDAR, with very high NFκB activity, likely provides a suitable environment for cells carrying these specific mutations to proliferate rapidly. The occurrence of deletion mutations can be explained by the high frequency of double-strand breaks in the EDAR driven tumours. In non-replicative cells, like the involuting mammary gland, these breaks are repaired by NHEJ which frequently leads to deletions like those observed. It is also interesting to note that mutations in WNT pathway components, although rare in human breast cancer in general, are common in metaplastic breast cancers [41].
The basis for the specificity of squamous metaplasia observed in EDAR driven tumours is unclear. EDAR acts during development of the surface ectoderm to control the patterning and morphogenesis of cutaneous appendages, but does not appear to have a direct influence on epidermal differentiation. Rather, EDAR signalling tends to drive cells away from an epidermal fate and towards that of ectodermal appendage [42]. Thus, it is unexpected, based on its developmental role, to find EDAR promoting growth of a differentiated epidermis within the mammary gland. Alternatively, the squamous metaplasia may simply reflect the operation of EDAR signalling outside its normal developmental context, particularly its operation in conjunction with WNT signalling. In this latter scenario, it is important to remember that squamous metaplasia is seen in many mouse models with unregulated Wnt/β-catenin signalling [26, 27].
Alleles of EDAR encoding a receptor with altered signalling potentials exist in human populations, with East Asian and Native American groups having a non-synonymous polymorphism leading to substantially increased signalling [25, 43]. These alleles may increase the incidence or progression of breast tumours with squamous metaplasia in these populations. A number of recombinant protein and antibody reagents to stimulate and repress the EDAR pathway in vivo have been developed recently [44, 45], with one of the stimulators in clinical trial for early-life correction of HED caused by EDA loss of function [46]. Such agents may represent useful modalities for future diagnosis and treatment of breast cancer with squamous metaplasia.
Materials and methods
Meta-analyses of microarray datasets
A meta-analysis of six Affymetrix gene expression datasets comprising 1107 primary human breast cancers was performed as previously described [21]. Centroid prediction [47] was used to assign the tumours to the five Norway/Stanford subtypes (Basal, Luminal A, Luminal B, ERBB2, and Normal-like) [48]. The dataset of 113 breast tumours representing special histological types [22] was downloaded from Array Express (http://www.ebi.ac.uk/arrayexpress/) experiment number E-NCMF-3. Treeview was used to generate heatmaps (http://rana.lbl.gov/EisenSoftware.htm).
Animals
EdarTg951/951 mice were on an inbred FVB/N genetic background and have been described previously [25]. All procedures carried out under the UK Home Office license. Cohorts of ten nulliparous or constantly breeding EdarTg951/951 and wild-type FVB/N mice were established to analyse tumour formation. For the phenotypic analysis at least three animals were examined for each genotype and stage, specific numbers used are given in the appropriate figure legends. Randomisation and blinding were not used.
Mammary gland whole mount preparation and morphological analysis
Whole mounts of the rostral inguinal (4th) mammary gland were prepared as previously reported [29]. Images of the whole mounts were captured digitally and morphometric analysis was performed using Image J 1.43 software.
Immunohistochemistry
Rostral inguinal mammary glands and shaven back skin were fixed in 4% paraformaldehyde and paraffin-embedded. Antigen retrieval was carried out on mounted sections using a Borg Decloaker (Biocare Medical) and the sections were subsequently stained as described before [42]. Controls were performed using the secondary antibody only. See Supplementary Table 1 for the primary antibodies.
In situ hybridisation
In situ hybridisation was carried out on 8-µm sections from paraffin-embedded mammary tissue using the RNAScope system (Advanced Cell Diagnostics, Hayward, CA), according to the manufacturer’s instructions.
Western blotting
Cell lysates were obtained from powdered frozen mammary tissue samples in 1 ml 2× RIPA lysis buffer (300 mM NaCl, 100 mM Tris pH 7.4, 10 mM EDTA, 2% NP-40, 2% Deoxycholic acid, 0.2% SDS, 10 mM NaF, 1 mM NaVO4 and 1× Protease inhibitor cocktail (Calbiochem)) per gram of tissue and incubating for 15 min on ice. Lysates were then spun at 18,000 g to remove nuclei and genomic DNA. Nuclear-enriched fractions were obtained from powdered frozen mammary tissue samples in 1 ml NP-40 lysis buffer (10% w/v glycerol, 50 mM Tris-HCl pH 7.4, 100 mM NaCl, 1% v/v NP-40, 2 mM MgCl2, with protease/phosphatase inhibitors as above) per gram of tissue and incubating for 15 minutes on ice. Lysates were then spun at 18,000 g for 30 min at 4 °C. The nuclear pellets were resuspended in SDS buffer (10 mM Tris-HCl pH 7.4, 2% SDS) and genomic DNA was sheared using a probe sonicator. Equal quantities of the protein samples were run on SDS/polyacrylamide gels as described [49]. See supplementary Table 1 for all primary antibodies.
Quantitative RT-PCR
RNA was extracted from mammary gland tissue using Trizol (Invitrogen) and treated with RNAse-free DNAse 1 to remove contaminating genomic DNA (New England Biolabs). Two micrograms of total RNA was reverse transcribed using the High Capacity RNA to cDNA kit (Applied Biosystems). The resultant cDNA was diluted tenfold and 2 µl was used as template for subsequent quantitative PCR reactions.
qPCR was carried out in triplicate using the primers listed in supplementary Table 2, the Step One Plus RT-PCR system (Applied Biosystems), and 2× SYBR-green PCR master mix (Applied Biosystems). PCR products were separated on 2% agarose gels to verify their size and the absence of non-specific products. The relative amounts of products were evaluated using the comparative Ct method, and Keratin18 as a normalisation control to account for differences in the amount of ductal epithelium in each sample.
Sequencing
Changes within the sequence of exon 3 of the β-catenin cDNA and genomic locus were screened and sequenced by amplifying the relevant region from either cDNA or the genomic DNA found within the initial RNA extraction prepared from tumours respectively, using the relevant primers listed in supplementary table 2. The amplified bands were then sequenced using nested sequencing primers (supplementary Table 2) using Big Dye Terminator (Applied Biosystems).
FACS analysis
The cell populations present within the adult mammary gland were analysed as described in [50].
Cell lines
The normal mouse mammary epithelial cell lines C57MG and EpH4 were gifts from Dr. Anthony Brown (Weill Medical College of Cornell University, USA) and Prof. Christine Watson (University of Cambridge, UK). Luciferase assays were conducted in HEK293T and EpH4 cells as described in [51], using pNFĸB-Luc and pTopFlash to monitor NFκB and WNT signalling respectively. EpH4 cells stably transduced with a pCDH EF1α-Edar-T2A-GFP or a pCDH EF1α-ΔExon3-β-catenin-T2A-RFP lentivirus were generated as described in [52]. Expression of the proteins was confirmed by western blotting and the expression of downstream target genes confirmed signalling (Fig. S8). Soft agar assays were carried out as described in [49].
Statistical analyses
All data are presented as the mean ± SEM. Statistical significance was evaluated, where appropriate, using unpaired t-tests, one-way ANOVA followed by a Tukey’s test, or log-rank (Mantel–Cox) test.
Supplementary information
Acknowledgements
We would like to thank Rebecca Rock for her help establishing the qRT-PCR assays, and Julia Cheung for training SJ in histology techniques. The work was supported by a BBSRC Ph.D. studentship to SJ and RW, and by grants from the Breast Cancer Campaign (KB, grant numbers 2010NovPR63 and 2011MayPR33), Wellcome Trust (KB, grant number 085073), Breakthrough Breast Cancer (AHS) and BBSRC (DJH).
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Rebecca Williams, Stephanie Jobling.
Contributor Information
Denis J. Headon, Email: denis.headon@roslin.ed.ac.uk
Keith Brennan, Email: keith.brennan@manchester.ac.uk.
Supplementary information
The online version contains supplementary material available at 10.1038/s41388-021-01902-6.
References
- 1.Ashkenazi A, Salvesen G. Regulated cell death: signaling and mechanisms. Annu Rev Cell Dev Biol. 2014;30:337–56. doi: 10.1146/annurev-cellbio-100913-013226. [DOI] [PubMed] [Google Scholar]
- 2.Mikkola ML. Genetic basis of skin appendage development. Semin Cell Dev Biol. 2007;18:225–36. doi: 10.1016/j.semcdb.2007.01.007. [DOI] [PubMed] [Google Scholar]
- 3.Clarke A, Phillips DI, Brown R, Harper PS. Clinical aspects of X-linked hypohidrotic ectodermal dysplasia. Arch Dis Child. 1987;62:989–96. doi: 10.1136/adc.62.10.989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kere J, Srivastava AK, Montonen O, Zonana J, Thomas N, Ferguson B, et al. X-linked anhidrotic (hypohidrotic) ectodermal dysplasia is caused by mutation in a novel transmembrane protein. Nat Genet. 1996;13:409–16. doi: 10.1038/ng0895-409. [DOI] [PubMed] [Google Scholar]
- 5.Al Marzouqi F, Michot C, Dos Santos S, Bonnefont JP, Bodemer C, Hadj-Rabia S. Bilateral amastia in a female with X-linked hypohidrotic ectodermal dysplasia. Br J Dermatol. 2014;171:671–3. doi: 10.1111/bjd.13023. [DOI] [PubMed] [Google Scholar]
- 6.Haghighi A, Nikuei P, Haghighi-Kakhki H, Saleh-Gohari N, Baghestani S, Krawitz PM, et al. Whole-exome sequencing identifies a novel missense mutation in EDAR causing autosomal recessive hypohidrotic ectodermal dysplasia with bilateral amastia and palmoplantar hyperkeratosis. Br J Dermatol. 2013;168:1353–6. doi: 10.1111/bjd.12151. [DOI] [PubMed] [Google Scholar]
- 7.Headon DJ, Overbeek PA. Involvement of a novel Tnf receptor homologue in hair follicle induction. Nat Genet. 1999;22:370–4. doi: 10.1038/11943. [DOI] [PubMed] [Google Scholar]
- 8.Voutilainen M, Lindfors PH, Lefebvre S, Ahtiainen L, Fliniaux I, Rysti E, et al. Ectodysplasin regulates hormone-independent mammary ductal morphogenesis via NF-kappaB. Proc Natl Acad Sci USA. 2012;109:5744–9. doi: 10.1073/pnas.1110627109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schmidt-Ullrich R, Aebischer T, Hulsken J, Birchmeier W, Klemm U, Scheidereit C. Requirement of NF-kappaB/Rel for the development of hair follicles and other epidermal appendices. Development. 2001;128:3843–53. doi: 10.1242/dev.128.19.3843. [DOI] [PubMed] [Google Scholar]
- 10.Fliniaux I, Mikkola ML, Lefebvre S, Thesleff I. Identification of dkk4 as a target of Eda-A1/Edar pathway reveals an unexpected role of ectodysplasin as inhibitor of Wnt signalling in ectodermal placodes. Dev Biol. 2008;320:60–71. doi: 10.1016/j.ydbio.2008.04.023. [DOI] [PubMed] [Google Scholar]
- 11.Zhang Y, Tomann P, Andl T, Gallant NM, Huelsken J, Jerchow B, et al. Reciprocal requirements for EDA/EDAR/NF-kappaB and Wnt/beta-catenin signaling pathways in hair follicle induction. Dev Cell. 2009;17:49–61. doi: 10.1016/j.devcel.2009.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Headon DJ, Emmal SA, Ferguson BM, Tucker AS, Justice MJ, Sharpe PT, et al. Gene defect in ectodermal dysplasia implicates a death domain adapter in development. Nature. 2001;414:913–6. doi: 10.1038/414913a. [DOI] [PubMed] [Google Scholar]
- 13.Biswas DK, Shi Q, Baily S, Strickland I, Ghosh S, Pardee AB, et al. NF-kappa B activation in human breast cancer specimens and its role in cell proliferation and apoptosis. Proc Natl Acad Sci USA. 2004;101:10137–42. doi: 10.1073/pnas.0403621101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kendellen MF, Bradford JW, Lawrence CL, Clark KS, Baldwin AS. Canonical and non-canonical NF-kappaB signaling promotes breast cancer tumor-initiating cells. Oncogene. 2014;33:1297–305. doi: 10.1038/onc.2013.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shostak K, Chariot A. NF-kappaB, stem cells and breast cancer: the links get stronger. Breast Cancer Res. 2011;13:214. doi: 10.1186/bcr2886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pummila M, Fliniaux I, Jaatinen R, James MJ, Laurikkala J, Schneider P, et al. Ectodysplasin has a dual role in ectodermal organogenesis: inhibition of Bmp activity and induction of Shh expression. Development. 2007;134:117–25. doi: 10.1242/dev.02708. [DOI] [PubMed] [Google Scholar]
- 17.Schmidt-Ullrich R, Tobin DJ, Lenhard D, Schneider P, Paus R, Scheidereit C. NF-kappaB transmits Eda A1/EdaR signalling to activate Shh and cyclin D1 expression, and controls post-initiation hair placode down growth. Development. 2006;133:1045–57. doi: 10.1242/dev.02278. [DOI] [PubMed] [Google Scholar]
- 18.Lane TF, Leder P. Wnt-10b directs hypermorphic development and transformation in mammary glands of male and female mice. Oncogene. 1997;15:2133–44. doi: 10.1038/sj.onc.1201593. [DOI] [PubMed] [Google Scholar]
- 19.Gonzalez-Suarez E, Jacob AP, Jones J, Miller R, Roudier-Meyer MP, Erwert R, et al. RANK ligand mediates progestin-induced mammary epithelial proliferation and carcinogenesis. Nature. 2010;468:103–7. doi: 10.1038/nature09495. [DOI] [PubMed] [Google Scholar]
- 20.Perou CM, Sørlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, et al. Molecular portraits of human breast tumours. Nature. 2000;406:747–52. doi: 10.1038/35021093. [DOI] [PubMed] [Google Scholar]
- 21.Sims AH, Smethurst GJ, Hey Y, Okoniewski MJ, Pepper SD, Howell A, et al. The removal of multiplicative, systematic bias allows integration of breast cancer gene expression datasets - improving meta-analysis and prediction of prognosis. BMC Med Genomics. 2008;1:42. doi: 10.1186/1755-8794-1-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Weigelt B, Horlings HM, Kreike B, Hayes MM, Hauptmann M, Wessels LF, et al. Refinement of breast cancer classification by molecular characterization of histological special types. J Pathol. 2008;216:141–50. doi: 10.1002/path.2407. [DOI] [PubMed] [Google Scholar]
- 23.Weigelt B, Kreike B, Reis-Filho JS. Metaplastic breast carcinomas are basal-like breast cancers: a genomic profiling analysis. Breast Cancer Res Treat. 2009;117:273–80. doi: 10.1007/s10549-008-0197-9. [DOI] [PubMed] [Google Scholar]
- 24.Weigelt B, Geyer FC, Reis-Filho JS. Histological types of breast cancer: how special are they? Mol Oncol. 2010;4:192–208. doi: 10.1016/j.molonc.2010.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mou C, Thomason HA, Willan PM, Clowes C, Harris WE, Drew CF, et al. Enhanced ectodysplasin-a receptor (EDAR) signaling alters multiple fiber characteristics to produce the East Asian hair form. Hum Mutat. 2008;29:1405–11. doi: 10.1002/humu.20795. [DOI] [PubMed] [Google Scholar]
- 26.Brennan KR, Brown AM. Wnt proteins in mammary development and cancer. J Mammary Gland Biol Neoplasia. 2004;9:119–31. doi: 10.1023/B:JOMG.0000037157.94207.33. [DOI] [PubMed] [Google Scholar]
- 27.Miyoshi K, Rosner A, Nozawa M, Byrd C, Morgan F, Landesman-Bollag E, et al. Activation of different Wnt/beta-catenin signaling components in mammary epithelium induces transdifferentiation and the formation of pilar tumors. Oncogene. 2002;21:5548–56. doi: 10.1038/sj.onc.1205686. [DOI] [PubMed] [Google Scholar]
- 28.Mikkola ML. The Edar subfamily in hair and exocrine gland development. Adv Exp Med Biol. 2011;691:23–33. doi: 10.1007/978-1-4419-6612-4_3. [DOI] [PubMed] [Google Scholar]
- 29.Chang SH, Jobling S, Brennan K, Headon DJ. Enhanced Edar signalling has pleiotropic effects on craniofacial and cutaneous glands. PLoS ONE. 2009;4:e7591. doi: 10.1371/journal.pone.0007591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bui TD, Rankin J, Smith K, Huguet EL, Ruben S, Strachan T, et al. A novel human Wnt gene, WNT10B, maps to 12q13 and is expressed in human breast carcinomas. Oncogene. 1997;14:1249–53. doi: 10.1038/sj.onc.1200936. [DOI] [PubMed] [Google Scholar]
- 31.Majumder K, Shawlot W, Schuster G, Harrison W, Elder FF, Overbeek PA. YAC rescue of downless locus mutations in mice. Mamm Genome. 1998;9:863–8. doi: 10.1007/s003359900884. [DOI] [PubMed] [Google Scholar]
- 32.Brisken C, Ayyannan A, Nguyen C, Heineman A, Reinhardt F, Tan J, et al. IGF-2 is a mediator of prolactin-induced morphogenesis in the breast. Dev Cell. 2002;3:877–87. doi: 10.1016/s1534-5807(02)00365-9. [DOI] [PubMed] [Google Scholar]
- 33.Fata JE, Kong YY, Li J, Sasaki T, Irie-Sasaki J, Moorehead RA, et al. The osteoclast differentiation factor osteoprotegerin-ligand is essential for mammary gland development. Cell. 2000;103:41–50. doi: 10.1016/s0092-8674(00)00103-3. [DOI] [PubMed] [Google Scholar]
- 34.Schramek D, Leibbrandt A, Sigl V, Kenner L, Pospisilik JA, Lee HJ, et al. Osteoclast differentiation factor RANKL controls development of progestin-driven mammary cancer. Nature. 2010;468:98–102. doi: 10.1038/nature09387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brantley DM, Chen CL, Muraoka RS, Bushdid PB, Bradberry JL, Kittrell F, et al. Nuclear factor-kappaB (NF-kappaB) regulates proliferation and branching in mouse mammary epithelium. Mol Biol Cell. 2001;12:1445–55. doi: 10.1091/mbc.12.5.1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fernandez-Valdivia R, Mukherjee A, Ying Y, Li J, Paquet M, DeMayo FJ, et al. The RANKL signaling axis is sufficient to elicit ductal side-branching and alveologenesis in the mammary gland of the virgin mouse. Dev Biol. 2009;328:127–39. doi: 10.1016/j.ydbio.2009.01.019. [DOI] [PubMed] [Google Scholar]
- 37.Romieu-Mourez R, Kim DW, Shin SM, Demicco EG, Landesman-Bollag E, Seldin DC, et al. Mouse mammary tumor virus c-rel transgenic mice develop mammary tumors. Mol Cell Biol. 2003;23:5738–54. doi: 10.1128/MCB.23.16.5738-5754.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tsukamoto AS, Grosschedl R, Guzman RC, Parslow T, Varmus HE. Expression of the int-1 gene in transgenic mice is associated with mammary gland hyperplasia and adenocarcinomas in male and female mice. Cell. 1988;55:619–25. doi: 10.1016/0092-8674(88)90220-6. [DOI] [PubMed] [Google Scholar]
- 39.Imbert A, Eelkema R, Jordan S, Feiner H, Cowin P. Delta N89 beta-catenin induces precocious development, differentiation, and neoplasia in mammary gland. J Cell Biol. 2001;153:555–68. doi: 10.1083/jcb.153.3.555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Michaelson JS, Leder P. beta-catenin is a downstream effector of Wnt-mediated tumorigenesis in the mammary gland. Oncogene. 2001;20:5093–9. doi: 10.1038/sj.onc.1204586. [DOI] [PubMed] [Google Scholar]
- 41.Hayes MJ, Thomas D, Emmons A, Giordano TJ, Kleer CG. Genetic changes of Wnt pathway genes are common events in metaplastic carcinomas of the breast. Clin Cancer Res. 2008;14:4038–44. doi: 10.1158/1078-0432.CCR-07-4379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mou C, Jackson B, Schneider P, Overbeek PA, Headon DJ. Generation of the primary hair follicle pattern. Proc Natl Acad Sci USA. 2006;103:9075–80. doi: 10.1073/pnas.0600825103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bryk J, Hardouin E, Pugach I, Hughes D, Strotmann R, Stoneking M, et al. Positive selection in East Asians for an EDAR allele that enhances NF-kappaB activation. PLoS ONE. 2008;3:e2209. doi: 10.1371/journal.pone.0002209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kowalczyk-Quintas C, Willen L, Dang AT, Sarrasin H, Tardivel A, Hermes K, et al. Generation and characterization of function-blocking anti-ectodysplasin A (EDA) monoclonal antibodies that induce ectodermal dysplasia. J Biol Chem. 2014;289:4273–85. doi: 10.1074/jbc.M113.535740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kowalczyk C, Dunkel N, Willen L, Casal ML, Mauldin EA, Gaide O, et al. Molecular and therapeutic characterization of anti-ectodysplasin A receptor (EDAR) agonist monoclonal antibodies. J Biol Chem. 2011;286:30769–79. doi: 10.1074/jbc.M111.267997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Huttner K. Future developments in XLHED treatment approaches. Am J Med Genet A. 2014;164A:2433–6. doi: 10.1002/ajmg.a.36499. [DOI] [PubMed] [Google Scholar]
- 47.Calza S, Hall P, Auer G, Bjöhle J, Klaar S, Kronenwett U, et al. Intrinsic molecular signature of breast cancer in a population-based cohort of 412 patients. Breast Cancer Res. 2006;8:R34. doi: 10.1186/bcr1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sorlie T, Tibshirani R, Parker J, Hastie T, Marron JS, Nobel A, et al. Repeated observation of breast tumor subtypes in independent gene expression data sets. Proc Natl Acad Sci USA. 2003;100:8418–23. doi: 10.1073/pnas.0932692100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stylianou S, Clarke RB, Brennan K. Aberrant activation of notch signaling in human breast cancer. Cancer Res. 2006;66:1517–25. doi: 10.1158/0008-5472.CAN-05-3054. [DOI] [PubMed] [Google Scholar]
- 50.Shehata M, Teschendorff A, Sharp G, Novcic N, Russell IA, Avril S, et al. Phenotypic and functional characterization of the luminal cell hierarchy of the mammary gland. Breast Cancer Res. 2012;14:R134. doi: 10.1186/bcr3334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Collu GM, Hidalgo-Sastre A, Acar A, Bayston L, Gildea C, Leverentz MK, et al. Dishevelled limits Notch signalling through inhibition of CSL. Development. 2012;139:4405–15. doi: 10.1242/dev.081885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Meurette O, Stylianou S, Rock R, Collu GM, Gilmore AP, Brennan K. Notch activation induces Akt signaling via an autocrine loop to prevent apoptosis in breast epithelial cells. Cancer Res. 2009;69:5015–22. doi: 10.1158/0008-5472.CAN-08-3478. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.