

Abstract
Ammonia is diffused and transported across all plasma membranes. This entails that hyperammonemia leads to an increase in ammonia in all organs and tissues. It is known that the toxic ramifications of ammonia primarily touch the brain and cause neurological impairment. However, the deleterious effects of ammonia are not specific to the brain, as the direct effect of increased ammonia (change in pH, membrane potential, metabolism) can occur in any type of cell. Therefore, in the setting of chronic liver disease where multi-organ dysfunction is common, the role of ammonia is challenged. This review provides insights and evidence that increased ammonia can disturb many organ and cell types and hence lead to dysfunction.
Effect of ammonia on cell function
Ammonia is used for a number of different metabolic reactions (including the synthesis of non-essential amino acids). However it is primarily a waste product of cellular metabolism and due to its harmful properties at higher concentrations, ammonia is quickly detoxified and transformed into less toxic compounds (Cooper and Plum, 1987; Butterworth, 2002). Under physiological conditions, blood levels of ammonia (in systemic circulation) are carefully maintained at concentrations <50μM in adults, 50–75μM in term neonates and 50–150μM in preterm neonates (Braissant et al., 2013). Here, the liver plays a vital role in regulating the levels of ammonia via the production of urea (a cycle of enzymes which together are solely found in the liver). Ammonia is also removed in extrahepatic organs, including the muscle and brain (astrocytes), through the amidation of glutamate to glutamine via the enzyme glutamine synthesis (GS) (Cooper and Plum, 1987).
The main source of ammonia generation occurs in the intestines; from lysis of blood-borne urea and also from protein digestion/deamination by urease-positive bacteria and microbial deaminase (Jones et al., 1969). A large amount of metabolically generated ammonia is absorbed into the blood and via the portal vein is detoxified through the liver (Cooper and Plum, 1987; Brusilow et al., 2010). The concentration of ammonia in portal vein can be 5–10x higher than in the systemic circulation (Abdo, 2006) and during conditions of liver disease or failure, ammonia is poorly removed (due to impaired ureagenesis from hepatocellular dysfunction and portosystemic shunting) and is liberated into the systemic circulation and exposed to all organs (Rovira et al., 2008). Glutaminase, an enzyme which generates glutamate and ammonia from glutamine and which is found in the intestine, kidney and brain (neurons), also plays a contributing role to the development of hyperammonemia. Diseases causing hyperammonemia include acute liver failure (ALF), chronic liver disease (CLD) and portal-systemic shunting (Felipo and Butterworth, 2002). Hyperammonemia also develops in children with inborn errors of the urea cycle and tend to present with higher blood ammonia concentrations (up to 5 mM) compared to acquired causes (such ALF and CLD) which usually result in hyperammonemia levels between 0.2–1 mM (Ratnakumari et al., 1992; Matkowskyj et al., 1999; Butterworth, 2002; Lichter-Konecki et al., 2008).
Ammonia in solution (i.e blood) is present as NH3 and NH4+ with the ratio NH3/NH4+ depending on the pH as defined by the Henderson-Hasselbach equation. Under physiological conditions with a blood pH of 7.4, more than 98% of ammonia is in NH4+ form (Bromberg et al., 1960). Both forms affect pH, electrolytic, acid-base and ion equilibrium. NH3 is a weak base in gaseous form and uncharged, therefore lipid soluble and capable to cross cell membranes through diffusion. Conversely, NH4+ is a weak acid, water soluble and because of its ionic properties crosses cell membranes at a less rapid rate through various transport channels (Bosoi and Rose, 2009). Due to almost identical hydration radius with K+ (Kikeri et al., 1989), NH4+ can substitute K+ in its channels and transporters (ATPase transporters Na+/K+ and H+/K+ (Moser, 1987) and Na+/K+/Cl− cotransporters (Kelly et al., 2009)). This NH4+ substitution in K+ channels mediates membrane depolarization in vitro (Allert et al., 1998; Norenberg, 1998), but putatively not in situ (Rangroo Thrane et al., 2013). Ammonia can also enter cells through aquaporin-8 channel (Liu et al., 2006; Saparov et al., 2007) and ammonia specific transporters (human non-erythroid Rhesus glycoprotein B and C (Bakouh et al., 2006)). There is also growing evidence that ammonia can cross blood-brain barrier (BBB) preferentially by active transport through ion transporters rather than diffusion (Sørensen, 2013) and it has been shown that ammonia invade paracellular and transcellular passage of different molecules across the BBB (Braissant, 2012; Skowronska and Albrecht, 2012), as well as to a lesser extent by direct diffusion of NH3. Furthermore, ammonia primarily enters astrocytes lining the BBB as these cells have the highest affinity for potassium (Marcaggi et al., 2004; Bosoi and Rose, 2009; Rangroo Thrane et al., 2013).
Ammonia alters both intracellular and extracellular pH and can cause intracellular acidification as well as alkalisation, all depending on ammonium concentration, pH and rate of NH3 vs. NH4+ transport through the cell membrane (Norenberg, 1998; Bosoi and Rose, 2009; Rangroo Thrane et al., 2013). Ammonia has also be shown to cause a rise in intracellular Ca2+ primarily due to cytosolic alkalinisation in vitro (Rose et al., 2005) and through less well characterized mechanisms lead to increased intracellular Ca2+ signalling in vivo (Rangroo Thrane et al., 2013). Ammonia-induced changes in pH, membrane potential as well as alterations in cellular metabolism negatively impacts cell function by influencing signalling transduction pathways, activities of many enzymes, alternation in protein phosphorylation and the state of various other ion channels and transporters (Busa and Nuccitelli, 1984; Norenberg, 1998; Cudalbu, 2013). Furthermore, increased ammonia metabolism will lead to metabolic disturbances. NH4+ detoxification by converting α-ketoglutarate to glutamate and glutamate to glutamine leads to α-ketoglutarate depletion consequently stressing the tricarboxylic acid cycle (Braissant et al., 2013).
Elevated concentrations of ammonia have been shown to generate free radicals (Kosenko et al., 1997; Murthy et al., 2001; Sinke et al., 2008; Norenberg et al., 2009) and leads to excessive production of nitric oxide (NO) by stimulating the citrulline-NO cycle (Braissant et al., 1999; Bachmann et al., 2004; Zielinska et al., 2011). Alteration of NO synthesis and oxidative stress can result in the induction of mitochondrial permeability transition (MPT) (Halestrap et al., 1997; Kowaltowski et al., 2001), alterations of BBB permeability (in some disease models) (Rangroo Thrane et al., 2012; Skowronska and Albrecht, 2012; Braissant et al., 2013) and activation of MAPKs (Cagnon and Braissant, 2009). Ammonia also activates the transcription factor NF-κB, involved in immune and inflammatory reactions, (Sinke et al., 2008) probably as a result of increased oxidative stress, activated MAPKs and induced MPT (Marchetti et al., 1996; Bowie and O’Neill, 2000; Kyriakis and Avruch, 2001).
In addition, liver function impairment independently provokes deviation from physiological values of over 20 different compounds in the circulation (Zieve, 1987) as well as many other factors including inflammation and oxidative stress. In some cases it can be very difficult to distinguish the independent effect of ammonia and therefore other factors (synergistic effects) should be considered (Butterworth, 2008; Bosoi et al., 2012; DeMorrow, 2013; Aldridge et al., 2015).
The effects of ammonia toxicity on the brain
In the brain, ammonia homeostasis is tightly regulated and linked to recycling of the major excitatory and inhibitory neurotransmitters glutamate and γ-aminobutyric acid (GABA) (Paulsen et al., 1987; Bak et al., 2006; Hertz and Kala, 2007). Interestingly, GS in astrocytes has a higher affinity for ammonia than glutamate, indicating that the removal of ammonia may be more important than that of the principle excitatory neurotransmitter (Waniewski, 1992). Ammonia neurotoxicity is a phenomenon that affects all cerebrate species, from fish to humans (Ip and Chew, 2010). Gross neurological dysfunction occurs in conditions that lead to excess blood and therefore brain ammonia, including encephalopathy, seizures, ataxia and coma (Butterworth, 2002).
Toxic levels of ammonia and alterations in pH, electrolyte disturbances, membrane potential depolarization, are thought to lead to neurological dysfunction primarily by causing cellular swelling accompanied by brain edema and metabolic dysfunction (Bosoi and Rose, 2009). Ammonia is likely to be particularly toxic to astrocytes as they are the only cells that possess the enzyme GS responsible for detoxifying ammonia in the brain through condensation with glutamate (Martinez-Hernandez et al., 1977; Hertz and Zielke, 2004). In fact, astrocytes are likely to take up four times more ammonia than any other cell type in the brain (Cooper et al., 1979). Moreover, one of the early pathophysiological findings in hepatic encephalopathy (HE) was the presence of brain edema, and specifically astrocyte swelling in histological specimens and in vitro. Astrocyte swelling is also found in the later stages of HE in most animal models of liver failure and hyperammonemia (Gregorios et al., 1985a, 1985b; Ganz et al., 1989; Willard-Mack et al., 1996; Tanigami et al., 2005; Cagnon and Braissant, 2007; Jayakumar et al., 2008; Butterworth et al., 2009; Rangroo Thrane et al., 2012; Rao et al., 2014). These findings correlated with increased ammonia and glutamine levels and it was therefore hypothesized that ammonia exerted its neurotoxic effects by accumulation of glutamine causing astrocyte swelling (termed osmotic gliopathy). However, astrocyte swelling in the context of ammonia neurotoxicity had until recently not been directly visualized in living tissue. In a hyperacute model of isolated hyperammonemia (urea cycle deficiency), doses of ammonia sufficient to cause stupor, seizures and ataxia did not induce astrocyte swelling in vivo (Rangroo Thrane et al., 2013). Additionally, deleting the main astrocyte water channel, aquaporin 4 (AQP4), did not affect disease outcome. However, lethal doses of parenteral ammonia did cause brain swelling in vivo and astrocyte swelling in situ (Rangroo Thrane et al., 2013), and AQP4 deletion does appear to protect against brain edema in the context of HE (Rao et al., 2014). Therefore, although brain edema is a prominent and widely acknowledged clinical feature of the later stages of HE and contributes to mortality, it does not explain all the neurotoxic effects of ammonia (Joshi et al., 2014).
Several alternate and/or synergistic neurotoxic effects of ammonia on astrocytes have previously been studied. These include impairment of oxidative metabolism; with a consequent increase glycolysis (mainly in astrocytes) and dangerously elevated brain lactate levels (Ott et al., 2005; Dam et al., 2013; Bosoi et al., 2014). Ammonia itself is also widely known to cause pH changes that might affect cellular function due to its ability to act both as a weak acid and base. However, in vivo studies using liver failure models, pH electrodes and NMR show either an overall increase of intracellular pH ranging from 0.1 to 0.4 or no change (Fitzpatrick et al., 1989; Swain et al., 1991; Kanamori and Ross, 1997; Rangroo Thrane et al., 2013). The main limitations of these studies are the indirect measures of pH and the inability to accurately distinguish intra- from extracellular pH. Ammonia has also recently been shown to impair astrocytic calcium signaling in vivo, which is closely linked to many astrocytic housekeeping functions, such as K+ homeostasis (Rose et al., 2005; Wang et al., 2012; Rangroo Thrane et al., 2013). Additionally, ammonia appears to directly disrupt astrocyte potassium buffering in vivo by competing for transport via either the Na+-K+-ATPase, K+ channels or Na+-K+-Cl− cotransporters (Alger and Nicoll, 1983; Brookes and Turner, 1993; Marcaggi et al., 2004; Rangroo Thrane et al., 2013). An acute increase in both extracellular ammonia and potassium has been shown to cause a depolarizing shift in the GABA equilibrium potential (EGABA), thus leading to decreased neuronal inhibition in vivo (Lux, 1971; Benjamin et al., 1978; Rangroo Thrane et al., 2013). This effect likely explains the seizure phenotype seen in models of urea cycle deficiencies (Cagnon and Braissant, 2007). Additionally, ammonia has been found to contribute to seizure generation in epileptic children with normal liver function (Yamamoto et al., 2013), and reactive gliosis seen in temporal lobe epilepsy may induce seizures via an almost identical mechanism (Robel et al., 2015).
More recently, a contribution of other cell types than astrocytes to the pathophysiology of HE and hyperammonemia has gained greater recognition (Butterworth, 2011). Microglia are the resident immune cells in the brain, and known to survey the brain microenvironment for signs of pathogens and inflammation with their fine processes (Nimmerjahn, 2005). When activated, they become more amoeboid in structure, facilitating phagocytosis and release multiple pro-inflammatory mediators (Ransohoff and Cardona, 2010). In the late stages of HE, histological studies have shown that microglia are activated in animal models of CLD, ALF, portocaval shunting and in post mortem tissue from patients (Jiang et al., 2009a, 2009b; Rodrigo et al., 2010; Agusti et al., 2011; Zemtsova et al., 2011). Several studies indicate that this microglial activation is correlated with with BBB opening and brain edema (Jiang et al., 2009a, 2009b; Rangroo Thrane et al., 2012). However, microglia were not found to be activated in a model of portal vein ligation (Brück et al., 2011) and acute isolated hyperammonemic mice (Rangroo Thrane et al., 2012). It is therefore possible that the microglial activation is not a response to ammonia per se, but rather relates to other secondary features of ammonia neurotoxicity such as oxidative stress, cellar distress signals or BBB opening. Finally, ammonia likely has a range of direct and indirect effects on neurons, pericytes and endothelial cells, which have been more extensively reviewed in other publications (Szerb and Butterworth, 1992; Rodrigo and Felipo, 2006; Leke et al., 2011; Thumburu et al., 2012; Shaik et al., 2013).
In summary, the best described deleterious effects of ammonia are associated with the brain as the brain is exquisitely sensitive to even minor increases in the blood ammonia levels. Within the brain, although elevated levels of ammonia are noxious for astrocytes, it also has deleterious effects on all cells of the central nervous system (Figure 1). However, can ammonia be considered solely as a neurotoxin? The toxic effects of ammonia are not specific to the brain and there is evidence depicting ammonia toxicity is diffused to organs/tissues and cells in the body.
Figure 1. Effects of ammonia toxicity in brain.
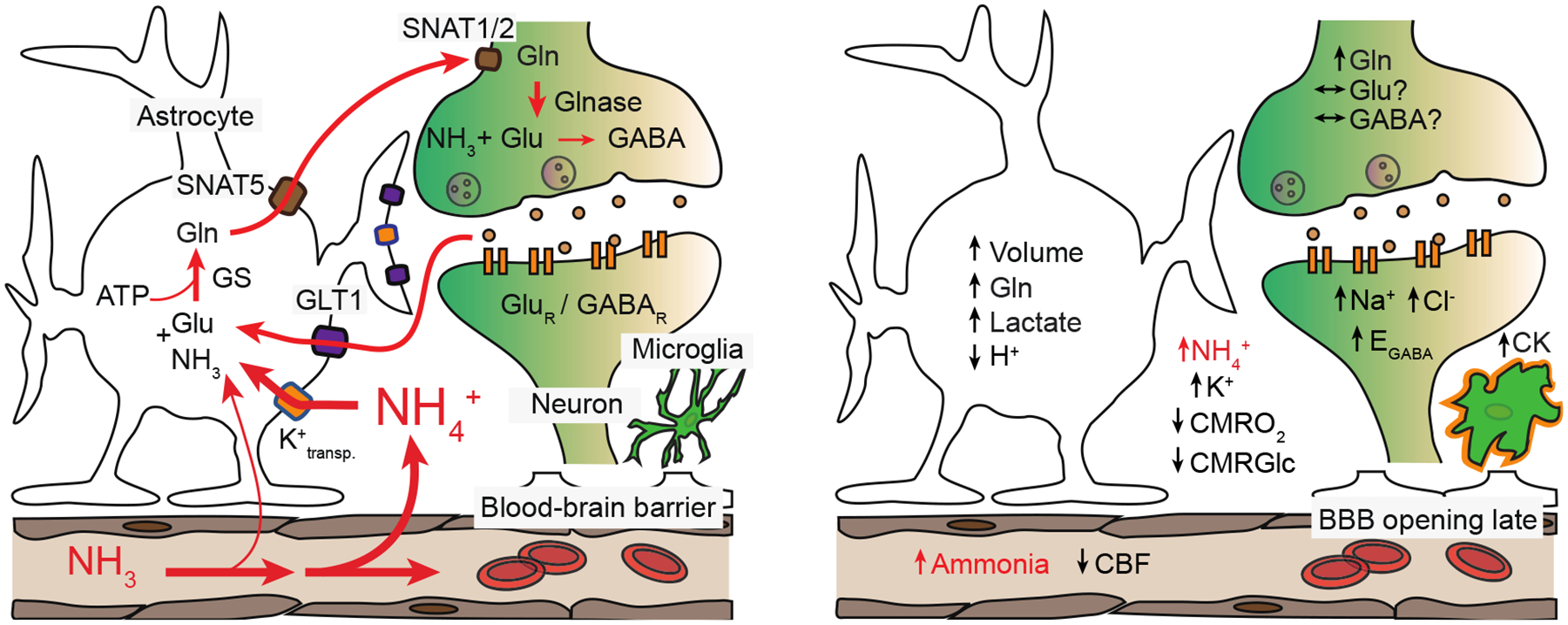
Left, physiological ammonia handling in the brain. Right, some of the proposed neurotoxic effects of excess brain ammonia. Glutamate (Glu), glutamine synthetase (GS), glutamine (Gln), adenosine triphosphate (ATP), sodium-coupled neutral amino acid transporters (SNAT1/2 and 5), glutamate transporter 1 (GLT1), glutaminase (Glnase), glutamate and γ-aminobutyric acid (GABA), glutamate receptor (GluR), GABA receptor (GABAR), cerebral blood flow (CBF), cerebral metabolic rate of oxygen (CMRO2), cerebral metabolic rate of glucose (CMRGlc), GABA reversal potential (EGABA), cytokines (CK).
The effects of ammonia toxicity on muscle
Sarcopenia or loss of skeletal muscle mass is the most frequent and potentially reversible complication in cirrhosis (Dasarathy, 2012). Despite the universally recognized adverse consequences on survival, development of other complications in cirrhosis, quality of life, and post liver transplant outcomes, there are no effective therapies primarily because the mechanisms of sarcopenia are incompletely understood (Dasarathy, 2012). In addition to the lower muscle mass, muscle strength is also reduced in cirrhosis and contributes to adverse clinical outcomes (Jones et al., 2012). Recent evidence strongly suggests that hyperammonemia is a mediator of the liver muscle axis (Qiu et al., 2013). It is also important to emphasize that identifying ammonia as a mediator of the liver-muscle axis is especially relevant because effective therapeutic strategies to reverse hyperammonemia are available (Phongsamran et al., 2010).
Previous studies have reported an increased muscle uptake of ammonia in cirrhosis results in glutamine synthesis that is released into the circulation (Ganda and Ruderman, 1976; Lockwood et al., 1979; Holecek, 2013). Muscle uptake and metabolism of ammonia is however not a benign process since a number of molecular and metabolic perturbations have been reported during muscle hyperammonemia (Qiu et al., 2012, 2013). The consequences of increased muscle uptake of ammonia include both a reduction in muscle mass and muscle strength (Shanely and Coast, 2002; Qiu et al., 2012). Both metabolic and molecular responses to muscle hyperammonemia contribute to muscle loss and weakness.
Metabolic perturbations during hyperammonemia
Since ureagenesis in the liver is impaired and muscle cannot generate urea, ammonia removal by the muscle utilizes a cataplerotic conversion of α-ketoglutarate, a critical tricarboxylic acid cycle intermediate to generate glutamate and glutamine thereby removing 2 moles of ammonia for each mole of α-ketoglutarate. However, the conversion of α-ketoglutarate to glutamate, the first step in this reaction is catalyzed by glutamate dehydrogenase that has a low affinity for ammonia promoting the anaplerotic direction generating α-ketoglutarate from glutamate rather than glutamate from α-ketoglutarate (Wootton, 1983). Under most physiological conditions, since muscle ammonia concentrations do not reach the Km of this enzyme (~1mM), there is not net cataplerosis of α-ketoglutarate. In cirrhosis, we have consistently reported tissue concentrations of ammonia ~4mM that favors cataplerosis of α-ketoglutarate to glutamate. This is consistent with our observations of lower TCA cycle intermediates in the skeletal muscle during hyperammonemia. Impaired muscle mitochondrial function and ATP generation accompany lower TCA cycle intermediates. These observations provide a mechanistic basis for the previously reported impaired skeletal muscle mitochondrial electron chain complex function in cirrhosis. Reduction in muscle ATP also contributes to lower protein synthesis since peptide chain elongation is a high energy-requiring step. Increased autophagy is a homeostatic response and provides amino acids for anaplerotic reactions. Additionally, reduction in ATP generation also contributes to skeletal muscle contractile dysfunction in cirrhosis.
Skeletal muscle molecular abnormalities during hyperammonemia
In addition to skeletal muscle metabolic derangements, ammonia also results in impaired skeletal muscle protein synthesis and increased autophagy. The principal pathway of impaired protein synthesis involves the canonical Akt/mTOR mediated regulation of cap dependent protein synthesis. Myostatin, a TGF-β superfamily member is a potent inhibitor of muscle protein synthesis and inhibits mTOR activation either via an Akt dependent or independent mechanism. Consistently, blocking myostatin increases muscle mass and protein synthesis (Dasarathy et al., 2011).
Hyperammonemia also increases muscle autophagy that in combination with impaired protein synthesis results in loss of muscle mass (Qiu et al., 2012). Both impaired mTOR signaling and activation of AMPK contribute to the increased autophagy. Ammonia also increases protein nitration either directly or by increased generation of reactive oxygen and nitrogen species and autophagy has been shown to contribute to clearance of post translationally modified muscle proteins (unpublished data). Posttranslational modification of proteins can potentially impair the actomyosin interaction during muscle contraction and result in impaired muscle contractile function.
Therapeutic relevance of muscle hyperammonemia.
The metabolic and molecular data support our belief that hyperammonemia induces a state of anabolic resistance because previous strategies to increase muscle mass by nutritional and hormonal strategies have not been consistently been effective (Tsien et al., 2012). Our studies using an integrated molecular-metabolic approach have shown the interaction between signaling abnormalities and metabolic demands during muscle hyperammonemia lay the foundation for novel mechanistic therapeutic strategies. Our model supports the use of long term ammonia lowering, myostatin antagonism, stimulating muscle mTOR, and providing anaplerotic substrates to reverse the adverse consequences of hyperammonemia. Mitochondrial dysfunction and generation of mitochondrial reactive oxygen species and resultant oxidative stress responses are also potential targets to reverse the adverse skeletal muscle responses of hyperammonemia (unpublished data).
Consistently, myostatin blocking using either follistatin or myostatin knockout in mice have been reported to be effective in reversing hyperammonemia mediated impaired muscle protein synthesis and contractile function (Dasarathy et al., 2011; Qiu et al., 2012, 2013). Even though ammonia lowering strategies have been effective in reversing encephalopathy (Phongsamran et al., 2010), there is little data on the impact on the skeletal muscle. One potential reason is that skeletal muscle contractile proteins are long lived and long-term ammonia lowering strategies may be necessary to result in clinically relevant responses.
Interestingly, recent studies have reported that leucine is an anaplerotic substrate generating of α-ketoglutarate (Schachter and Sang, 1997). Additionally, leucine stimulates mTOR directly providing a compelling therapeutic rationale for the use of leucine to reverse muscle hyperammonemia and its consequences. Consistently, human and animal studies in cirrhosis have shown that high doses of leucine have a beneficial effect in reversing impaired skeletal muscle protein synthesis (Tsien et al., 2015). The use of cell permeable esters of α-ketoglutarate also holds promise as a strategy to increase muscle ammonia disposal. Other potential interventions include enhanced non-hepatic ammonia disposal via non-toxic pathways, antioxidants and mitochondrial stabilizers need to be evaluated. On final note, even though hyperammonemia is a mediator of the liver muscle axis, plasma ammonia is elevated in patients with chronic pulmonary disease, severe heart failure and cancers all of which are accompanied by significant skeletal muscle loss (Bessman and Evans, 1955; Chance et al., 1988; Calvert et al., 2010). Therefore, the current approach has broad therapeutic potential across multiple organ dysfunctions.
In conclusion, hyperammonemia occurs in a number of chronic diseases and potentially contributes to sarcopenia and adverse clinical outcomes. Recent advances in our understanding of the molecular and metabolic responses to hyperammonemia in the skeletal muscle provide a mechanistic basis for developing effective therapies (Figure 2).
Figure 2. Skeletal muscle molecular perturbations induced by hyperammonemia.
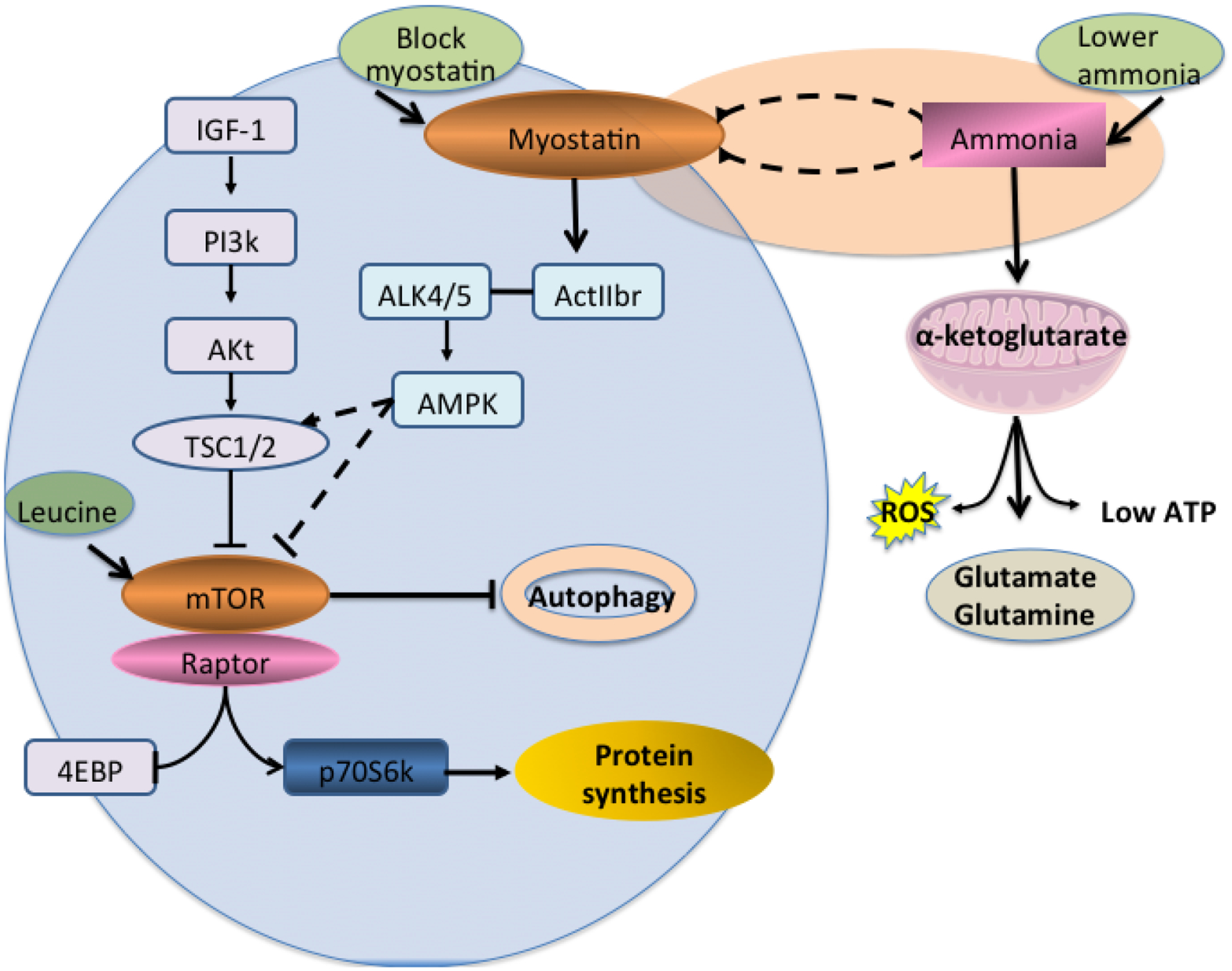
Ammonia transcriptionally up-regulates TGFB superfamily member, myostatin, that inhibits critical regulatory signaling molecule, mTORC1 via AMPK with resultant impaired downstream signaling responses that in turn results in decreased protein synthesis and increased autophagy, both of which contribute to sarcopenia in liver disease. 4EBP1 4E binding protein; ActIIBr activin II b receptor (TGF beta receptor type 2); Akt/PKB protein kinase B;ALK4 activin like kinase 4 (TGFbeta receptor type 1); ALK5 activin like kinase 5 (TGF beta receptor type 1); IGF-1 insulin like growth factor 1, mTORC1 mammalian target of rapamycin 1; PI3K phosphoinositide 3kinase; RiboS6 ribosomal S6 protein, ROS reactive oxygen species, TSC tuberous sclerosis complex.
The effect of ammonia on other organs
Kidney
It has been shown in healthy volunteers that following exposure to hyperammonemia, the kidneys take up net ammonia from the systemic circulation (Owen et al., 1961). However, it has been demonstrated in both cirrhotic patients and hyperammonemic rodents, that the kidney becomes a primary source of ammonia (Owen et al., 1961; Olde Damink et al., 2003; Ytrebø et al., 2006). Elevated systemic ammonia concentrations following CLD may directly interact with glomerular cells and contribute to glomerular injury (Ling et al., 1998). Hyperammonemia also plays a crucial role in tubulointestinal fibrosis (Ling et al., 1998). Gordon et al. displayed evidence ammonia has the capacity to promote tubulointestinal injury due to the interaction of ammonia with the third component of complement, a potent stimulus to the production of reactive oxygen species by polymorphonuclear leukocytes and monocytes (Gordon et al., 1985). In this context, Nath et al. demonstrated that hyperammonemia induces the progression of renal injury, not only through complement cascade, but also through the stimulatory effects of ammonia on renal growth (Nath et al., 1991). In fact, oxidative stress can also cause renal ammoniagenesis which may contribute to progression of renal injury (Dan et al., 2008). By contrast, a very recent study by Satpute et al. observed that subacute exposure to ammonium acetate in rats induces renal necrosis and tubular degeneration, which were not correlated with either oxidative or biochemical changes (Satpute et al., 2014). Indeed, it is apparent that exposure of neutrophils to ammonia impair phagocytosis and promotes increased reactive oxygen species generation that results in oxidative stress at infection sites and collateral damage to host tissue (Shawcross et al., 2008).
During ALF, kidneys are capable of continuous ammonia release into the systemic circulation (Ytrebø et al., 2006). A recent study by Cauli et al. demonstrated in rats with (ALF) that activation of N-methyl D aspartate (NMDA) receptors contribute to hyperammonemia-induced encephalopathy and kidney damage. Moreover, blocking NMDA receptors with MK-801 improved ammonia elimination and glomerular filtration rate and delayed kidney damage (Cauli et al., 2014), confirming the deleterious effects of high ammonia concentration in kidneys. Furthermore, ammonia administered to rats lead to disturbances in renal sodium handling that was preceded by activation of kidney mitogen activated protein kinases/extracellular signal regulated kinases (MAPK/ERKs) signaling pathways, and consequently renal injury (Bento et al., 2005). It has been shown that kidneys perfused with ammonium salts results in cortical and tubular necrosis, with necrotic kidneys demonstrating depressed creatinine clearance (Orvell and Wesson, 1976). Ammonia exposure also inhibits tubular cell proliferation in primary rabbit proximal tubular epithelial cells, therefore affecting cell replication (Rabkin et al., 1993). Collectively, these findings suggest that hyperammonemia does lead to impaired kidney function and sustaining kidney injury.
Liver
As indicated above, whilst astrocytes are major effectors of brain injury in liver failure, it is of interest that they share many similar markers of activation with hepatic stellate cells (HSC), which are implicated in the development of liver fibrosis and also the increase in intra-hepatic resistance with evolving liver injury (Rockey, 1997). Indeed, HSC contraction is one of the mechanisms that is central to the development of portal hypertension, a serious haemodynamic consequence of cirrhosis. HSC have been shown to possess GS (Bode et al., 1998) and recent data shows that in rodent models of cirrhosis, high ammonia concentrations are associated with increased HSC contraction and raised portal pressure and conversely, lowering ammonia decreases HSC activation and ameliorates portal hypertension (Jalan et al., 2016).
In addition to the impact on HSC contraction, elevated ammonia has been shown to promote liver injury through up-regulation of toll-like receptor genes, generation of oxidative stress through activation of resident macrophages and neutrophils and activation of NFκB and iNOS. In addition, ammonia promotes increased hepatocyte apoptosis and changes in cell cycle with higher cyclin D1, thereby further promoting liver injury as liver disease evolves (Jia et al., 2014). It is possible that impaired urea cycle metabolism and reduced liver clearance of ammonia primes the liver and other organs to the impact of acute inflammation and further oxidative stress, as occurs in conditions such as acute-on-chronic liver failure, though this assertion requires mechanistic evaluation.
Lung
Several studies have discussed the detrimental effects of ammonia on lungs following exposure to ammonia gas. Ammonia inhalation also can damage respiratory tract leading to acute lung injury and pulmonary edema, which can ultimately lead to death (Ortiz-Pujols et al., 2014). Furthermore, ammonia inhalation to rats causes increased oxidative stress, decreased antioxidant enzymes and acute lung injury which was attenuated following α-ketoglutarate treatment (Ali et al., 2012). Ammonia induced acute lung injury have also been shown in rabbits associated with an elevated airway pressure and a decrease in PaO2 (Sjöblom et al., 1999) and has been shown to induce lung fibrosis (Ohnuma-Koyama et al., 2013). Ammonia and enzymatically active urease released from parasitic cells of Coccidioides posadasii contribute to host tissue damage and exacerbate the severity of coccidioidal lung infections (Mirbod et al., 2002; Mirbod-Donovan et al., 2005; Wise et al., 2013). In this context, it has been concluded that enzymatically active urease is partly responsible for increased extracellular ammonia concentrations at sites of lung infection, contributing to both localized host tissue damage and exacerbation of the respiratory disease (Lichtenstein et al., 1997).
The effect of ammonia on vascular function
Chronic hyperammonemia is believed to decrease cGMP formation and thereby secondary signaling for NO, one of the main regulators of tonic vasodilatation (Cauli et al., 2007). Moreover, as previously mentioned, the promotion of reactive oxygen species generation by ammonia gives rise to further endothelial dysfunction by decreasing expression of dimethylarginine dimethylamino hydrolase (DDAH1), a cytosolic enzyme that metabolizes asymmetric dimethylarginine (ADMA), a key endogenous inhibitor of NO. High ADMA levels have previously been shown to be associated with impaired cerebral perfusion during hyperammonemia (Balasubramaniyan et al., 2012) and also with high intrahepatic resistance in advanced liver disease (Mookerjee et al., 2007). Similarly, under high oxidative stress conditions, there is increased un-coupling of nitric oxide synthase with less NO bioavailability (Xia et al., 1998) and greater nitration of proteins with the generation of peroxynitrite and nitrotyrosine, further promoting organ injury. Furthermore, important cationic transporters such as the y+ LAT2 system are also stimulated by high ammonia levels, resulting in arginine efflux from endothelial cells in exchange for glutamine uptake (Zielinska et al., 2011), and thus loss of substrate for nitric oxide synthase to generate NO. Thus at multiple levels, ammonia appears to control endothelial function and resistance to vascular flow, whilst also promoting further organ injury. It is perhaps, therefore, not surprising that with progression to advanced cirrhosis and ascites formation and greater ammonia generation, resistive indices for the middle cerebral artery and renal vasculature have been shown to increase, and correlate with severity of hyperdynamic circulatory dysfunction as noted through high renin levels (Guevara et al., 1998).
Conclusion
Increases in ammonia can lead to changes in pH, membrane potential (due to similar properties of NH4+ and K+) and cell metabolism. While ammonia (hyperammonemia) is unequivocally key in the development of neurological impairment, including HE, the toxic effects of ammonia are not specific to the brain and therefore the deleterious consequences of hyperammonemia can impinge on other organs. To date, there is accumulated evidence indicating ammonia is clearly much more than a neuro-toxin and as CLD and ALF are often associated with multi-organ dysfunction, the role of ammonia has been questioned. Evidently, the brain is more sensitive to ammonia toxicity and recognizing ammonia tolerance/intolerance in other organs remains undefined. The severity of ammonia toxicity on all organs and tissues most likely depends on the degree and acuteness of the onset of hyperammonemia, therefore a better understanding on the consequences of inherited and acquired, as well as acute and chronic hyperammonemia merits to be thoroughly investigated. In turn, improved therapeutic strategies for the management of patients with hyperammonemia can be realized.
Footnotes
Disclosures: The authors have no conflicts to disclose
References:
- Abdo AA (2006) An evidence-based update on hepatic encephalopathy. Saudi J Gastroenterol 12:8–15 [PubMed] [Google Scholar]
- Agusti A, Cauli O, Rodrigo R, Llansola M, Hernández-Rabaza V, Felipo V (2011) p38 MAP kinase is a therapeutic target for hepatic encephalopathy in rats with portacaval shunts. Gut 60:1572–1579 [DOI] [PubMed] [Google Scholar]
- Aldridge DR, Tranah EJ, Shawcross DL (2015) Pathogenesis of hepatic encephalopathy: role of ammonia and systemic inflammation. J Clin Exp Hepatol 5:S7–S20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alger BE, Nicoll RA (1983) Ammonia does not selectively block IPSPs in rat hippocampal pyramidal cells. J. Neurophysiol 49:1381–1391 [DOI] [PubMed] [Google Scholar]
- Ali R, Mittal G, Sultana S, Bhatnagar A (2012) Ameliorative potential of alpha-ketoglutaric acid (AKG) on acute lung injuries induced by ammonia inhalation in rats. Experimental Lung Research 38:435–444 [DOI] [PubMed] [Google Scholar]
- Allert N, Köller H, Siebler M (1998) Ammonia-induced depolarization of cultured rat cortical astrocytes. Brain Res 782:261–270 [DOI] [PubMed] [Google Scholar]
- Bachmann C, Braissant O, Villard A-M, Boulat O, Henry H (2004) Ammonia toxicity to the brain and creatine. Molecular Genetics and Metabolism 81 Suppl 1:S52–S57 [DOI] [PubMed] [Google Scholar]
- Bak LK, Schousboe A, Waagepetersen HS (2006) The glutamate/GABA-glutamine cycle: aspects of transport, neurotransmitter homeostasis and ammonia transfer. J Neurochem 98:641–653 [DOI] [PubMed] [Google Scholar]
- Bakouh N, Benjelloun F, Cherif-Zahar B, Planelles G (2006) The challenge of understanding ammonium homeostasis and the role of the Rh glycoproteins. Transfus Clin Biol. 13:139–146 [DOI] [PubMed] [Google Scholar]
- Balasubramaniyan V, Wright G, Sharma V, Davies N a, Sharifi Y, Habtesion A, Mookerjee RP, Jalan R (2012) Ammonia reduction with ornithine phenylacetate restores brain eNOS activity via the DDAH-ADMA pathway in bile duct-ligated cirrhotic rats. Am J Physiol Gastrointest Liver Physiol 302:G145–G152 [DOI] [PubMed] [Google Scholar]
- Benjamin AM, Okamoto K, Quastel JH (1978) Effects of ammonium ions on spontaneous action potentials and on contents of sodium, potassium, ammonium and chloride ions in brain in vitro. J Neurochem 30:131–143 [DOI] [PubMed] [Google Scholar]
- Bento LMA, Carvalheira JBC, Menegon LF, Saad MJA, Gontijo JAR (2005) Effects of NH4Cl intake on renal growth in rats: role of MAPK signalling pathway. Nephrol. Dial. Transplant 20:2654–2660 [DOI] [PubMed] [Google Scholar]
- Bessman AN, Evans JM (1955) The blood ammonia in congestive heart failure. Am Heart J 50:715–719 [DOI] [PubMed] [Google Scholar]
- Bode JG, Peters-Regehr T, Gressner AM, Häussinger D (1998) De novo expression of glutamine synthetase during transformation of hepatic stellate cells into myofibroblast-like cells. Biochem. J 335 (Pt 3):697–700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosoi CR, Rose CF (2009) Identifying the direct effects of ammonia on the brain. Metab Brain Dis 24:95–102 [DOI] [PubMed] [Google Scholar]
- Bosoi CR, Yang X, Huynh J, Parent-Robitaille C, Jiang W, Tremblay M, Rose CF (2012) Systemic oxidative stress is implicated in the pathogenesis of brain edema in rats with chronic liver failure. Free Radic. Biol. Med 52:1228–1235 [DOI] [PubMed] [Google Scholar]
- Bosoi CR, Zwingmann C, Marin H, Parent-Robitaille C, Huynh J, Tremblay M, Rose CF (2014) Increased brain lactate is central to the development of brain edema in rats with chronic liver disease. J Hepatol 60:554–560 [DOI] [PubMed] [Google Scholar]
- Bowie A, O’Neill LA (2000) Oxidative stress and nuclear factor-kappaB activation: a reassessment of the evidence in the light of recent discoveries. Biochemical Pharmacology 59:13–23 [DOI] [PubMed] [Google Scholar]
- Braissant O (2012) Creatine and guanidinoacetate transport at blood-brain and blood-cerebrospinal fluid barriers. Journal of Inherited Metabolic Disease 35:655–664 [DOI] [PubMed] [Google Scholar]
- Braissant O, Honegger P, Loup M, Iwase K, Takiguchi M, Bachmann C (1999) Hyperammonemia: regulation of argininosuccinate synthetase and argininosuccinate lyase genes in aggregating cell cultures of fetal rat brain. Neuroscience Letters 266:89–92 [DOI] [PubMed] [Google Scholar]
- Braissant O, McLin VA, Cudalbu C (2013) Ammonia toxicity to the brain. J Inherit Metab Dis 36:595–612 [DOI] [PubMed] [Google Scholar]
- Bromberg PA, Robin ED, Forkner CEJ (1960) The existence of ammonia in blood in vivo with observations on the significance of the NH4 plus minus NH3 system. J Clin Invest 39:332–341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brookes N, Turner RJ (1993) Extracellular potassium regulates the glutamine content of astrocytes: mediation by intracellular pH. Neurosci Lett 160:73–76 [DOI] [PubMed] [Google Scholar]
- Brück J, Görg B, Bidmon H-J, Zemtsova I, Qvartskhava N, Keitel V, Kircheis G, Häussinger D (2011) Locomotor impairment and cerebrocortical oxidative stress in portal vein ligated rats in vivo. J Hepatol 54:251–257 [DOI] [PubMed] [Google Scholar]
- Brusilow SW, Koehler RC, Traystman RJ, Cooper AJL (2010) Astrocyte glutamine synthetase: importance in hyperammonemic syndromes and potential target for therapy. NeuroRx 7:452–470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busa WB, Nuccitelli R (1984) Metabolic regulation via intracellular pH. The American Journal of Physiology 246:R409–R438 [DOI] [PubMed] [Google Scholar]
- Butterworth RF (2002) Pathophysiology of hepatic encephalopathy: a new look at ammonia. Metab Brain Dis 17:221–227 [DOI] [PubMed] [Google Scholar]
- Butterworth RF (2008) Pathophysiology of hepatic encephalopathy: The concept of synergism. Hepatology Research: The Official Journal of the Japan Society of Hepatology 38:S116–S121 [DOI] [PubMed] [Google Scholar]
- Butterworth RF (2011) Neuroinflammation in acute liver failure: Mechanisms and novel therapeutic targets. Neurochem. Int 59:830–836 [DOI] [PubMed] [Google Scholar]
- Butterworth RF, Norenberg MD, Felipo V, Ferenci P, Albrecht J, Blei AT (2009) Experimental models of hepatic encephalopathy: ISHEN guidelines. Liver Int. 29:783–788 [DOI] [PubMed] [Google Scholar]
- Cagnon L, Braissant O (2007) Hyperammonemia-induced toxicity for the developing central nervous system. Brain Research Reviews 56:183–197 [DOI] [PubMed] [Google Scholar]
- Cagnon L, Braissant O (2009) CNTF protects oligodendrocytes from ammonia toxicity: intracellular signaling pathways involved. Neurobiology of Disease 33:133–142 [DOI] [PubMed] [Google Scholar]
- Calvert LD, Steiner MC, Morgan MD, Singh SJ (2010) Plasma ammonia response to incremental cycling and walking tests in COPD. Resp Med 104:675–681 [DOI] [PubMed] [Google Scholar]
- Cauli O, Llansola M, Agustí A, Rodrigo R, Hernández-Rabaza V, Rodrigues TB, López-Larrubia P, Cerdán S, Felipo V (2014) Cerebral oedema is not responsible for motor or cognitive deficits in rats with hepatic encephalopathy. Liver Int 34:379–387 [DOI] [PubMed] [Google Scholar]
- Cauli O, Rodrigo R, Piedrafita B, Boix J, Felipo V (2007) Inflammation and hepatic encephalopathy: ibuprofen restores learning ability in rats with portacaval shunts. Hepatology 46:514–519 [DOI] [PubMed] [Google Scholar]
- Chance WT, Cao L, Nelson JL, Foley-Nelson T, Fischer JE (1988) Hyperammonemia in anorectic tumor-bearing rats. Life Sciences 43:67–74 [DOI] [PubMed] [Google Scholar]
- Cooper AJ, McDonald JM, Gelbard AS, Gledhill RF, Duffy TE (1979) The metabolic fate of 13N-labeled ammonia in rat brain. The Journal of Biological Chemistry 254:4982–4992 [PubMed] [Google Scholar]
- Cooper JLA, Plum F (1987) Biochemistry and Physiology of Brain Ammonia. Physiological Reviews 67:440–519 [DOI] [PubMed] [Google Scholar]
- Cudalbu C (2013) In vivo studies of brain metabolism in animal models of Hepatic Encephalopathy using 1H Magnetic Resonance Spectroscopy. Metabolic Brain Disease 28:167–174 [DOI] [PubMed] [Google Scholar]
- Dam G, Keiding S, Munk OL, Ott P, Vilstrup H, Bak LK, Waagepetersen HS, Schousboe A, Sørensen M (2013) Hepatic encephalopathy is associated with decreased cerebral oxygen metabolism and blood flow, not increased ammonia uptake. Hepatology 57:258–265 [DOI] [PubMed] [Google Scholar]
- Dan H, Peng R-X, Ao Y, Liu Y-H (2008) Segment-specific proximal tubule injury in tripterygium glycosides intoxicated rats. Journal of Biochemical and Molecular Toxicology 22:422–428 [DOI] [PubMed] [Google Scholar]
- Dasarathy S (2012) Consilience in sarcopenia of cirrhosis. J Cachexia Sarcopenia Muscle 3:225–237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dasarathy S, McCullough AJ, Muc S, Schneyer A, Bennett CD, Dodig M, Kalhan SC (2011) Sarcopenia associated with portosystemic shunting is reversed by follistatin. J. Hepatol 54:915–921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeMorrow S (2013) The Ammonia Hypothesis of Hepatic Encephalopathy should be Revisited. Journal of Cell Science & Therapy 03: [Google Scholar]
- Felipo V, Butterworth RF (2002) Neurobiology of ammonia. Progress in Neurobiology 67:259–279 [DOI] [PubMed] [Google Scholar]
- Fitzpatrick SM, Hetherington HP, Behar KL, Shulman RG (1989) Effects of acute hyperammonemia on cerebral amino acid metabolism and pHi in vivo, measured by 1H and 31P nuclear magnetic resonance. J Neurochem 52:741–749 [DOI] [PubMed] [Google Scholar]
- Ganda OP, Ruderman NB (1976) Muscle nitrogen metabolism in chronic hepatic insufficiency. Metabolism 25:427–435 [DOI] [PubMed] [Google Scholar]
- Ganz R, Swain M, Traber P, DalCanto M, Butterworth RF, Blei AT (1989) Ammonia-induced swelling of rat cerebral cortical slices: implications for the pathogenesis of brain edema in acute hepatic failure. Metab Brain Dis 4:213–223 [DOI] [PubMed] [Google Scholar]
- Gordon DL, Krueger RA, Quie PG, Hostetter MK (1985) Amidation of C3 at the thiolester site: stimulation of chemiluminescence and phagocytosis by a new inflammatory mediator. J. Immunol 134:3339–3345 [PubMed] [Google Scholar]
- Gregorios JB, Mozes LW, Norenberg LO, Norenberg MD (1985a) Morphologic effects of ammonia on primary astrocyte cultures. I. Light microscopic studies. J. Neuropathol. Exp. Neurol 44:397–403 [DOI] [PubMed] [Google Scholar]
- Gregorios JB, Mozes LW, Norenberg MD (1985b) Morphologic effects of ammonia on primary astrocyte cultures. II. Electron microscopic studies. J. Neuropathol. Exp. Neurol 44:404–414 [DOI] [PubMed] [Google Scholar]
- Guevara M, Bru C, Ginès P, Fernández-Esparrach G, Sort P, Bataller R, Jiménez W, Arroyo V, Rodés J (1998) Increased cerebrovascular resistance in cirrhotic patients with ascites. Hepatology (Baltimore, Md.) 28:39–44 [DOI] [PubMed] [Google Scholar]
- Halestrap AP, Woodfield KY, Connern CP (1997) Oxidative stress, thiol reagents, and membrane potential modulate the mitochondrial permeability transition by affecting nucleotide binding to the adenine nucleotide translocase. The Journal of Biological Chemistry 272:3346–3354 [DOI] [PubMed] [Google Scholar]
- Hertz L, Kala G (2007) Energy metabolism in brain cells: effects of elevated ammonia concentrations. Metab Brain Dis 22:199–218 [DOI] [PubMed] [Google Scholar]
- Hertz L, Zielke HR (2004) Astrocytic control of glutamatergic activity: astrocytes as stars of the show. Trends in Neurosciences 27:735–743 [DOI] [PubMed] [Google Scholar]
- Holecek M (2013) Branched-chain amino acids and ammonia metabolism in liver disease: Therapeutic implications. Nutrition 29:1186–1191 [DOI] [PubMed] [Google Scholar]
- Ip YK, Chew SF (2010) Ammonia production, excretion, toxicity, and defense in fish: a review. Front Physiol 1:134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jalan R, De Chiara F, Balasubramaniyan V, Andreola F, Khetan V, Malago M, Pinzani M, Mookerjee RP, Rombouts K (2016) Ammonia produces pathological changes in human hepatic stellate cells and is a target of therapy of portal hypertension. Journal of Hepatology [DOI] [PubMed] [Google Scholar]
- Jayakumar AR, Liu M, Moriyama M, Ramakrishnan R, Forbush B, Reddy PVB, Norenberg MD (2008) Na-K-Cl Cotransporter-1 in the mechanism of ammonia-induced astrocyte swelling. J. Biol. Chem 283:33874–33882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia B, Yu Z-J, Duan Z-F, Lü X-Q, Li J-J, Liu X-R, Sun R, Gao X-J, Wang Y-F, Yan J-Y, et al. (2014) Hyperammonaemia induces hepatic injury with alteration of gene expression profiles. Liver Int 34:748–758 [DOI] [PubMed] [Google Scholar]
- Jiang W, Desjardins P, Butterworth RF (2009a) Cerebral inflammation contributes to encephalopathy and brain edema in acute liver failure: protective effect of minocycline. J Neurochem 109:485–493 [DOI] [PubMed] [Google Scholar]
- Jiang W, Desjardins P, Butterworth RF (2009b) Direct evidence for central proinflammatory mechanisms in rats with experimental acute liver failure: protective effect of hypothermia. J Cereb Blood Flow Metab 29:944–952 [DOI] [PubMed] [Google Scholar]
- Jones EA, Smallwood RA, Craigie A, Rosenoer VM (1969) The enterohepatic circulation of urea nitrogen. Clinical Science 37:825–836 [PubMed] [Google Scholar]
- Jones JC, Coombes JS, Macdonald GA (2012) Exercise capacity and muscle strength in patients with cirrhosis. Liver Transpl 18:146–151 [DOI] [PubMed] [Google Scholar]
- Joshi D, O’Grady J, Patel A, Shawcross D, Connor S, Deasy N, Willars C, Bernal W, Wendon J, Auzinger G (2014) Cerebral oedema is rare in acute-on-chronic liver failure patients presenting with high-grade hepatic encephalopathy. Liver Int. 34:362–366 [DOI] [PubMed] [Google Scholar]
- Kanamori K, Ross BD (1997) Glial alkalinization detected in vivo by 1H-15N heteronuclear multiple-quantum coherence-transfer NMR in severely hyperammonemic rat. J. Neurochem 68:1209–1220 [DOI] [PubMed] [Google Scholar]
- Kelly T, Kafitz KW, Roderigo C, Rose CR (2009) Ammonium-evoked alterations in intracellular sodium and pH reduce glial glutamate transport activity. Glia 57:921–934 [DOI] [PubMed] [Google Scholar]
- Kikeri D, Sun A, Zeidel ML, Hebert SC (1989) Cell membranes impermeable to NH3. Nature 339:478–480 [DOI] [PubMed] [Google Scholar]
- Kosenko E, Kaminsky Y, Kaminsky A, Valencia M, Lee L, Hermenegildo C, Felipo V (1997) Superoxide production and antioxidant enzymes in ammonia intoxication in rats. Free Radical Research 27:637–644 [DOI] [PubMed] [Google Scholar]
- Kowaltowski AJ, Castilho RF, Vercesi AE (2001) Mitochondrial permeability transition and oxidative stress. FEBS Letters 495:12–15 [DOI] [PubMed] [Google Scholar]
- Kyriakis JM, Avruch J (2001) Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiological Reviews 81:807–869 [DOI] [PubMed] [Google Scholar]
- Leke R, Bak LK, Iversen P, Sø rensen M, Keiding S, Vilstrup H, Ott P, Portela LV, Schousboe A, Waagepetersen HS (2011) Synthesis of neurotransmitter GABA via the neuronal tricarboxylic acid cycle is elevated in rats with liver cirrhosis consistent with a high GABAergic tone in chronic hepatic encephalopathy. J Neurochem 117:824–832 [DOI] [PubMed] [Google Scholar]
- Lichtenstein GR, Kaiser LR, Tuchman M, Palevsky HI, Kotloff RM, O\textquotesingleBrien CB, Furth EE, Raps EC, Berry GT (1997) Fatal hyperammonemia following orthotopic lung transplantation. Gastroenterology 112:236–240 [DOI] [PubMed] [Google Scholar]
- Lichter-Konecki U, Mangin JM, Gordish-dressman H, Hoffman EP, Gallo V (2008) Gene Expression Profiling of Astrocytes from Hyperammonemic Mice Reveals Altered Pathways for Water and Potassium Homeostasis In Vivo. Glia 56:365–377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling H, Ardjomand P, Samvakas S, Simm A, Busch GL, Lang F, Sebekova K, Heidland A (1998) Mesangial cell hypertrophy induced by NH4Cl: role of depressed activities of cathepsins due to elevated lysosomal pH. Kidney Int. 53:1706–1712 [DOI] [PubMed] [Google Scholar]
- Liu K, Nagase H, Huang CG, Calamita G, Agre P (2006) Purification and functional characterization of aquaporin-8. Biology of the Cell / under the Auspices of the European Cell Biology Organization 98:153–161 [DOI] [PubMed] [Google Scholar]
- Lockwood AH, McDonald JM, Reiman RE, Gelbard AS, Laughlin JS, Duffy TE, Plum F (1979) The dynamics of ammonia metabolism in man. Effects of liver disease and hyperammonemia. Journal of Clinical Investigation 63:449–460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lux HD (1971) Ammonium and Chloride Extrusion: Hyperpolarizing Synaptic Inhibition in Spinal Motoneurons. Science 173:555–557 [DOI] [PubMed] [Google Scholar]
- Marcaggi P, Jeanne M, Coles JA (2004) Neuron-glial trafficking of NH4+ and K+: separate routes of uptake into glial cells of bee retina. Eur. J. Neurosci 19:966–976 [DOI] [PubMed] [Google Scholar]
- Marchetti P, Castedo M, Susin SA, Zamzami N, Hirsch T, Macho A, Haeffner A, Hirsch F, Geuskens M, Kroemer G (1996) Mitochondrial permeability transition is a central coordinating event of apoptosis. The Journal of Experimental Medicine 184:1155–1160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Hernandez A, Bell KP, Norenberg MD (1977) Glutamine synthetase: glial localization in brain. Science 195:1356–1358 [DOI] [PubMed] [Google Scholar]
- Matkowskyj KA, Marrero JA, Carroll RE, Danilkovich AV, Green RM, Benya RV (1999) Azoxymethane-induced fulminant hepatic failure in C57BL/6J mice: characterization of a new animal model. Am. J. Physiol 277:G455–G462 [DOI] [PubMed] [Google Scholar]
- Mirbod-Donovan F, Schaller R, Hung C-Y, Xue J, Reichard U, Cole GT (2005) Urease Produced by Coccidioides posadasii Contributes to the Virulence of This Respiratory Pathogen. Infection and Immunity 74:504–515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirbod F, Schaller RA, Cole GT (2002) Purification and characterization of urease isolated from the pathogenic fungus Coccidioides immitis. Med Mycol 40:35–44 [DOI] [PubMed] [Google Scholar]
- Mookerjee RP, Stadlbauer V, Lidder S, Wright GAK, Hodges SJ, Davies NA, Jalan R (2007) Neutrophil dysfunction in alcoholic hepatitis superimposed on cirrhosis is reversible and predicts the outcome. Hepatology 46:831–840 [DOI] [PubMed] [Google Scholar]
- Moser H (1987) Electrophysiological evidence for ammonium as a substitute for potassium in activating the sodium pump in a crayfish sensory neuron. Canadian Journal of Physiology and Pharmacology 65:141–145 [DOI] [PubMed] [Google Scholar]
- Murthy CR, Rama Rao KV, Bai G, Norenberg MD (2001) Ammonia-induced production of free radicals in primary cultures of rat astrocytes. Journal of Neuroscience Research 66:282–288 [DOI] [PubMed] [Google Scholar]
- Nath KA, Hostetter MK, Hostetter TH (1991) Increased Ammoniagenesis as a Determinant of Progressive Renal Injury. American Journal of Kidney Diseases 17:654–657 [DOI] [PubMed] [Google Scholar]
- Nimmerjahn A (2005) Resting Microglial Cells Are Highly Dynamic Surveillants of Brain Parenchyma in Vivo. Science 308:1314–1318 [DOI] [PubMed] [Google Scholar]
- Norenberg MD (1998) Astroglial dysfunction in hepatic encephalopathy. Metabolic Brain Disease 13:319–335 [DOI] [PubMed] [Google Scholar]
- Norenberg MD, Rama Rao KV, Jayakumar a R (2009) Signaling factors in the mechanism of ammonia neurotoxicity. Metabolic Brain Disease 24:103–117 [DOI] [PubMed] [Google Scholar]
- Ohnuma-Koyama A, Yoshida T, Tajima-Horiuchi H, Takahashi N, Yamaguchi S, Ohtsuka R, Takeuchi-Kashimoto Y, Kuwahara M, Takeda M, Nakashima N, et al. (2013) Didecyldimethylammonium chloride induces pulmonary fibrosis in association with TGF-beta signaling in mice. Experimental and Toxicologic Pathology 65:1003–1009 [DOI] [PubMed] [Google Scholar]
- Olde Damink SWM, Jalan R, Deutz NEP, Redhead DN, Dejong CHC, Hynd P, Jalan RA, Hayes PC, Soeters PB (2003) The kidney plays a major role in the hyperammonemia seen after simulated or actual GI bleeding in patients with cirrhosis. Hepatology 37:1277–1285 [DOI] [PubMed] [Google Scholar]
- Ortiz-Pujols S, Jones SW, Short KA, Morrell MR, Bermudez CA, Tilley SL, Cairns BA (2014) Management and Sequelae of a 41-Year-Old Jehovah’s Witness With Severe Anhydrous Ammonia Inhalation Injury. Journal of Burn Care & Research 35:e180–e183 [DOI] [PubMed] [Google Scholar]
- Orvell BD, Wesson LG (1976) Some Effects of Ammonium Salts on Renal Histology and Function in the Dog. Nephron 16:42–49 [DOI] [PubMed] [Google Scholar]
- Ott P, Clemmesen O, Larsen FS (2005) Cerebral metabolic disturbances in the brain during acute liver failure: from hyperammonemia to energy failure and proteolysis. Neurochem Int 47:13–18 [DOI] [PubMed] [Google Scholar]
- Owen EE, Johnson JH, Tyor MP (1961) The effect of induced hyperammonemia on renal ammonia metabolism. Journal of Clinical Investigation 40:215–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulsen RE, Contestabile A, Villani L, Fonnum F (1987) An in vivo model for studying function of brain tissue temporarily devoid of glial cell metabolism: the use of fluorocitrate. J. Neurochem 48:1377–1385 [DOI] [PubMed] [Google Scholar]
- Phongsamran PV, Kim JW, Cupo Abbott J, Rosenblatt A (2010) Pharmacotherapy for hepatic encephalopathy. Drugs 70:1131–1148 [DOI] [PubMed] [Google Scholar]
- Qiu J, Thapaliya S, Runkana A, Yang Y, Tsien C, Mohan ML, Narayanan A, Eghtesad B, Mozdziak PE, McDonald C, et al. (2013) Hyperammonemia in cirrhosis induces transcriptional regulation of myostatin by an NF-κB-mediated mechanism. Proc. Natl. Acad. Sci. U.S.A 110:18162–18167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu J, Tsien C, Thapalaya S, Narayanan A, Weihl CC, Ching JK, Eghtesad B, Singh K, Fu X, Dubyak G, et al. (2012) Hyperammonemia-mediated autophagy in skeletal muscle contributes to sarcopenia of cirrhosis. Am. J. Physiol. Endocrinol. Metab 303:E983–E993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabkin R, Palathumpat M, Tsao T (1993) Ammonium chloride alters renal tubular cell growth and protein turnover. Lab. Invest 68:427–438 [PubMed] [Google Scholar]
- Rangroo Thrane V, Thrane AS, Chanag J, Alleluia V, Nagelhus EA, Nedergaard M (2012) Real-time analysis of microglial activation and motility in hepatic and hyperammonemic encephalopathy. Neuroscience 220:247–255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rangroo Thrane V, Thrane AS, Wang F, Cotrina ML, Smith NA, Chen M, Xu Q, Kang N, Fujita T, Nagelhus EA, et al. (2013) Ammonia triggers neuronal disinhibition and seizures by impairing astrocyte potassium buffering. Nat. Med 19:1643–1648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ransohoff RM, Cardona AE (2010) The myeloid cells of the central nervous system parenchyma. Nature 468:253–262 [DOI] [PubMed] [Google Scholar]
- Rao KVR, Verkman AS, Curtis KM, Norenberg MD (2014) Aquaporin-4 deletion in mice reduces encephalopathy and brain edema in experimental acute liver failure. Neurobiol Dis 63:222–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratnakumari L, Qureshi IA, Butterworth RF (1992) Effects of congenital hyperammonemia on the cerebral and hepatic levels of the intermediates of energy metabolism in spf mice. Biochem. Biophys. Res. Commun 184:746–751 [DOI] [PubMed] [Google Scholar]
- Robel S, Buckingham SC, Boni JL, Campbell SL, Danbolt NC, Riedemann T, Sutor B, Sontheimer H (2015) Reactive Astrogliosis Causes the Development of Spontaneous Seizures. J Neurosci 35:3330–3345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rockey DC (1997) New concepts in the pathogenesis of portal hypertension: hepatic wounding and stellate cell contractility. Clin Liver Dis 1:13–29 [DOI] [PubMed] [Google Scholar]
- Rodrigo R, Cauli O, Gomez-Pinedo U, Agusti A, Hernandez-Rabaza V, Garcia-Verdugo J-M, Felipo V (2010) Hyperammonemia induces neuroinflammation that contributes to cognitive impairment in rats with hepatic encephalopathy. Gastroenterology 139:675–684 [DOI] [PubMed] [Google Scholar]
- Rodrigo R, Felipo V (2006) Brain regional alterations in the modulation of the glutamatenitric oxide-cGMP pathway in liver cirrhosis. Role of hyperammonemia and cell types involved. Neurochem Int 48:472–477 [DOI] [PubMed] [Google Scholar]
- Rose C, Kresse W, Kettenmann H (2005) Acute insult of ammonia leads to calcium-dependent glutamate release from cultured astrocytes, an effect of pH. J. Biol. Chem 280:20937–20944 [DOI] [PubMed] [Google Scholar]
- Rovira A, Alonso J, Córdoba J (2008) MR imaging findings in hepatic encephalopathy. AJNR Am J Neuroradiol 29:1612–1621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saparov SM, Liu K, Agre P, Pohl P (2007) Fast and selective ammonia transport by aquaporin-8. The Journal of Biological Chemistry 282:5296–5301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satpute R, Lomash V, Hariharakrishnan J, Rao P, Singh P, Gujar N, Bhattacharya R (2014) Oxidative stress and tissue pathology caused by subacute exposure to ammonium acetate in rats and their response to treatments with alpha-ketoglutarate and N-acetyl cysteine. Toxicology and Industrial Health 30:12–24 [DOI] [PubMed] [Google Scholar]
- Schachter D, Sang JC (1997) Regional differentiation in the rat aorta for a novel signaling pathway: leucine to glutamate. Am. J. Physiol 273:H1484–H1492 [DOI] [PubMed] [Google Scholar]
- Shaik IH, Miah MK, Bickel U, Mehvar R (2013) Effects of short-term portacaval anastomosis on the peripheral and brain disposition of the blood–brain barrier permeability marker sodium fluorescein in rats. Brain Research 1531:84–93 [DOI] [PubMed] [Google Scholar]
- Shanely RA, Coast JR (2002) Effect of Ammonia on in vitro Diaphragmatic Contractility, Fatigue and Recovery. Respiration 69:534–541 [DOI] [PubMed] [Google Scholar]
- Shawcross DL, Wright GAK, Stadlbauer V, Hodges SJ, Davies NA, Wheeler-Jones C, Pitsillides AA, Jalan R (2008) Ammonia impairs neutrophil phagocytic function in liver disease. Hepatology 48:1202–1212 [DOI] [PubMed] [Google Scholar]
- Sinke AP, Jayakumar AR, Panickar KS, Moriyama M, Reddy PVB, Norenberg MD (2008) NFkappaB in the mechanism of ammonia-induced astrocyte swelling in culture. Journal of Neurochemistry 106:2302–2311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjöblom E, Höjer J, Kulling PEJ, Stauffer K, Suneson A, Ludwigs U (1999) A Placebo-Controlled Experimental Study of Steroid Inhalation Therapy in Ammonia-Induced Lung Injury. Journal of Toxicology: Clinical Toxicology 37:59–67 [DOI] [PubMed] [Google Scholar]
- Skowronska M, Albrecht J (2012) Alterations of blood brain barrier function in hyperammonemia: an overview. Neurotoxicity Research 21:236–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sørensen M (2013) Update on cerebral uptake of blood ammonia. Metabolic Brain Disease 28:155–159 [DOI] [PubMed] [Google Scholar]
- Swain MS, Blei AT, Butterworth RF, Kraig RP (1991) Intracellular pH rises and astrocytes swell after portacaval anastomosis in rats. Am. J. Physiol 261:R1491–R1496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szerb JC, Butterworth RF (1992) Effect of ammonium ions on synaptic transmission in the mammalian central nervous system. Prog. Neurobiol 39:135–153 [DOI] [PubMed] [Google Scholar]
- Tanigami H, Rebel A, Martin LJ, Chen T-Y, Brusilow SW, Traystman RJ, Koehler RC (2005) Effect of glutamine synthetase inhibition on astrocyte swelling and altered astroglial protein expression during hyperammonemia in rats. Neuroscience 131:437–449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thumburu KK, Taneja S, Vasishta RK, Dhiman RK (2012) Neuropathology of acute liver failure. Neurochem. Int 60:672–675 [DOI] [PubMed] [Google Scholar]
- Tsien C, Davuluri G, Singh D, Allawy A, Have GAMT, Thapaliya S, Schulze JM, Barnes D, McCullough AJ, Engelen MPKJ, et al. (2015) Metabolic and molecular responses to leucine-enriched branched chain amino acid supplementation in the skeletal muscle of alcoholic cirrhosis. Hepatology 61:2018–2029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsien CD, McCullough AJ, Dasarathy S (2012) Late evening snack: Exploiting a period of anabolic opportunity in cirrhosis. J Gastroenterol Hepatol 27:430–441 [DOI] [PubMed] [Google Scholar]
- Wang F, Smith NA, Xu Q, Fujita T, Baba A, Matsuda T, Takano T, Bekar L, Nedergaard M (2012) Astrocytes modulate neural network activity by Ca2+-dependent uptake of extracellular K+. Sci Signal 5:ra26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waniewski RA (1992) Physiological levels of ammonia regulate glutamine synthesis from extracellular glutamate in astrocyte cultures. J. Neurochem 58:167–174 [DOI] [PubMed] [Google Scholar]
- Willard-Mack CL, Koehler RC, Hirata T, Cork LC, Takahashi H, Traystman RJ, Brusilow SW (1996) Inhibition of glutamine synthetase reduces ammonia-induced astrocyte swelling in rat. Neuroscience 71:589–599 [DOI] [PubMed] [Google Scholar]
- Wise HZ, Hung C-Y, Whiston E, Taylor JW, Cole GT (2013) Extracellular ammonia at sites of pulmonary infection with Coccidioides posadasii contributes to severity of the respiratory disease. Microb. Pathog 59–60:19–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wootton JC (1983) Re-assessment of ammonium-ion affinities of NADP-specific glutamate dehydrogenases. Activation of the Neurospora crassa enzyme by ammonium and rubidium ions. Biochem. J 209:527–531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Y, Tsai AL, Berka V, Zweier JL (1998) Superoxide generation from endothelial nitric-oxide synthase. A Ca2+/calmodulin-dependent and tetrahydrobiopterin regulatory process. J. Biol. Chem 273:25804–25808 [DOI] [PubMed] [Google Scholar]
- Yamamoto Y, Takahashi Y, Imai K, Mishima N, Yazawa R, Inoue K, Itoh K, Kagawa Y, Inoue Y (2013) Risk factors for hyperammonemia in pediatric patients with epilepsy. Epilepsia 54:983–989 [DOI] [PubMed] [Google Scholar]
- Ytrebø LM, Sen S, Rose C, Davies NA, Nedredal GI, Fuskevaag O-M, Have GAMT, Prinzen FW, Williams R, Deutz NEP, et al. (2006) Systemic and regional hemodynamics in pigs with acute liver failure and the effect of albumin dialysis. Scand J Gastroenterol 41:1350–1360 [DOI] [PubMed] [Google Scholar]
- Zemtsova I, Görg B, Keitel V, Bidmon H-J, Schrör K, Häussinger D (2011) Microglia activation in hepatic encephalopathy in rats and humans. Hepatology 54:204–215 [DOI] [PubMed] [Google Scholar]
- Zielinska M, Ruszkiewicz J, Hilgier W, Fręśko I, Albrecht J (2011) Hyperammonemia increases the expression and activity of the glutamine/arginine transporter y+ LAT2 in rat cerebral cortex: implications for the nitric oxide/cGMP pathway. Neurochemistry International 58:190–195 [DOI] [PubMed] [Google Scholar]
- Zieve L (1987) Pathogenesis of hepatic encephalopathy. Metabolic Brain Disease 2:147–165 [DOI] [PubMed] [Google Scholar]