

Abstract
De novo glycosphingolipid (GSL) biosynthesis defects cause severe neurological diseases, including hereditary sensory and autonomic neuropathy type 1A (HSAN1A), GM3 synthase deficiency, and hereditary spastic paraplegia type 26 (HSPG26), each lacking effective treatment. Recombinant adeno-associated virus (AAV)-mediated gene therapy has emerged as a powerful treatment for monogenic diseases and might be particularly suitable for these neurological conditions.
Disorders of Glycosphingolipid Biosynthesis in Humans
Glycolipids contain a carbohydrate moiety attached via a glycosidic bond to fatty acid tails and are integral components of all eukaryotic cell membranes. GSLs, with the attached lipid being a sphingosine, are enriched in both neurons and oligodendrocytes, and represent the predominant glycolipid class of the mammalian central nervous system (CNS) [1]. They mediate both cis (e.g., architecture of membrane microdomains) and trans (e.g., ligand binding) membrane functions. Deficiencies in de novo GSL biosynthesis lead to severe neurological disorders. To date, three disorders have been identified as monogenic: HSAN1A, GM3 synthase deficiency, and HSPG26, caused by mutations in SPTLC1, ST3GAL5, and B4GALNT1, respectively. Treatment for these disorders is directed at accommodating and/or alleviating symptoms. There are currently no disease-modifying therapies that address the underlying deficiency and maldistribution of cerebral GSLs.
Hereditary Sensory Neuropathy Type 1A
Patients with HSAN1A usually begin to experience symptoms when in their teens or later, starting with sensory loss in the feet followed by distal muscle wasting, skin damage, ulcers, and sensorineural hearing loss, and inheritance usually follows an autosomal dominant pattern [2]. The affected locus was mapped to the SPTLC1 gene. Human SPTLC1 contains a 1422-base pair (bp) cDNA, which encodes serine palmitoyltransferase long chain base subunit 1 (SPTLC1), which is widely expressed and catalyzes the formation of 3-ketosphinganine in de novo sphingolipid synthesis. Missense mutations, such as C133W, C133Y, and S331F, induce progressive neurological impairment by inhibiting serine palmitoyltransferase (SPT) activity and gaining toxicity via deoxysphinoid base (DSB) by-products. HSAN1A affects males and females in equal numbers, with an estimated prevalence of 1 in 500 000.
Transgenic Sptlc1C133W mice overexpressing the SPTLC1 C133W mutant show impaired SPT activity accompanied by elevated DSBs [3]. They also develop age-dependent weight loss and sensory impairments at 10 months, which is in line with the disease pathology in patients with HSAN1A.
GM3 Synthase Deficiency
Simpson et al. identified that an infantile-onset symptomatic epilepsy syndrome in the Old Order Amish community was caused by a single premature stop codon mutation, p.R232X, in ST3GAL5 [4]. The 1257-bp human ST3GAL5 cDNA encodes GM3 synthase, the first enzyme in the ganglioside synthesis pathway. The nonsense mutation R232X in ST3GAL5 abolishes GM3 synthase activity, as supported by the complete lack of GM3 and its ganglioside derivatives in patients with this disorder. GM3 synthase deficiency follows an autosomal recessive pattern, with an estimated prevalence of ~1 in 1200 births in the Amish community. All affected children start to have generalized tonic-clonic seizures and developmental milestone failures from 3 months of age. Brain magnetic resonance imaging at older ages shows generalized cerebral atrophy [5].
Homozygous St3gal5-knockout (St3gal5−/−) mice show no abnormalities other than enhanced insulin sensitivity and complete hearing loss [6]. The milder phenotypes appear to be due to compensation by the remaining gangliosides in mice. Indeed, St3gal5/B4galnt1 double knockout mice, which are unable to synthesize any ganglio-series GSL, phenotypically mirror patients with GM3 synthase deficiency [7]. They develop severe epilepsy, developmental delay, and neurodegenerative symptoms, and die soon after weaning. Biochemical analysis of postmortem brain tissue also confirmed the accumulation of lactosylceramide with the absence of all gangliosides.
Hereditary Spastic Paraplegia Type 26
Fishman et al. documented for the first time the accumulation of GM3 and the absence of complex gangliosides (GM1, GD1a, GD1b, and GT1b) in the brain and liver of a patient with spastic paraplegia [8]. This phenotype indicated the loss of function of B4GALNT1, which is responsible for the synthesis of gangliosides GM2, GD2, and asialo GM2. Later on, mutations were identified in B4GALNT1 from 12 independent HSPG26-affected families [9]. HSPG26 is characterized by a slowly progressive spasticity of the lower extremities resulting from axonal degeneration, and mild to moderate intellectual impairment; some male patients exhibit low serum testosterone levels and hypogonadism.
B4galnt1−/− mice from the 129/Sv strain develop deficits in reflexes, strength, and balance starting from 14–16 weeks of age. Progressive gait disorder, tremors, and catalepsy are seen from 12 months, which are reminiscent of clinical features of patients with HSPG26. In addition, B4galnt1−/− male mice have progressive testicular atrophy with the presence of diffuse vacuoles in Sertoli cells and a severe reduction in serum testosterone [10].
Adeno-Associated Virus-Mediated Gene Therapy
AAV-mediated gene therapy has emerged as a powerful strategy for the treatment of monogenic CNS disease. To date, three AAV gene therapy products have been approved by the European Medicines Agency and the US FDA, and 17 AAV gene therapy clinical trials targeting the CNS are in progress (www.clinicaltrials.gov NCT03952637; NCT04273269; NCT04411654; NCT04133454; NCT03634007; NCT04120493; NCT01621581; NCT04167540; NCT03562494; NCT04127578; NCT02926066; NCT02852213; NCT02725580; NCT03770572; NCT03612869; NCT00151216; and NCT01161576).
Wild-type (wt)AAV has an icosahedral protein capsid encapsidating a 4.8-kilobase (kb) single-stranded DNA genome that is flanked by two inverted terminal repeats (ITRs). Recombinant AAV (rAAV) is an engineered AAV that retains the wtAAV capsid, but replaces the wtAAV genome, except for the ITRs, with therapeutic DNA. The rAAV genome can form a circularized episome that is stable and maintains long-term transgene expression in nondividing cells. This characteristic makes rAAV-mediated gene therapy a ‘one-time’ treatment possibility [11].
A successful CNS-directed rAAV-gene therapy requires a suitable delivery method and a well-designed vector genome. Direct CNS injection, such as intraparenchymal and intracerebroventricular (I.C.V.) administration, usually achieves local transduction in the CNS compared with intravenous (I.V.) injection, which delivers the vector systematically. By contrast, some naturally isolated serotypes, such as AAV9 and AAVrh10, are able to cross the blood–brain barrier and have wide-spread delivery throughout the CNS via I.V. injection, which might be advantageous for treating GSL biosynthesis deficiencies [12].
A well-designed vector genome aims to express the therapeutic genetic material at the appropriate level, location, and time. To have a potent and durable expression, self-complementary AAV that has a mutated ITR to help the vector genome bypass second-strand synthesis is favorable in some circumstances. In addition, the promoter strength and codon usage of the transgene can be optimized to meet expression requirements. Additionally, applying tissue- or cell type-specific promoters or inclusion of miRNA binding sites can yield spatial control over transgene expression [13]. Finally, chemically inducible promoters can be switched ON or OFF by the administration of certain drugs to confer temporal control.
Application of AAV-Mediated Gene Therapy to GSL Biosynthesis Disorders
AAV-mediated gene therapy applications toward HSAN1A, GM3 synthase deficiency, and HSPG26 involve two major and distinct gene therapy strategies, namely gene silencing and gene replacement.
Due to the gain-of-function nature of SPTLC1 mutations, gene silencing could be applied for HSAN1A. One strategy is artificial miRNA (amiRNA)-mediated allele-specific mutant SPTLC1 knockdown. amiRNA expressing siRNAs in a natural pri-miRNA backbone reduces target mRNA expression. By silencing mutant SPTLC1 and sparing the normal allele, DSB production would be reduced while maintaining the normal functions of wtSPTLC1. If achieving allele-specific knockdown is difficult, one alternative is to deliver a dual-function rAAV that expresses both amiRNA and a functional SPTLC1 cDNA that function independently (Figure 1A) [14]. The former is recognized by the endogenous miRNA machinery and degrades the endogenous SPTLC1 mRNA, while the amiRNA-resistant SPTLC1 transcript produces wtSPTLC1 protein that executes its normal catalytical functions (Figure 1B).
Figure 1. Dual-Function Recombinant Adeno-Associated Virus (rAAV) Vectors for Treating Hereditary Sensory and Autonomic Neuropathy Type 1A (HSAN1A) Caused by Serine Palmitoyltransferase Long Chain Base Subunit 1 (SPTLC1) Mutations.
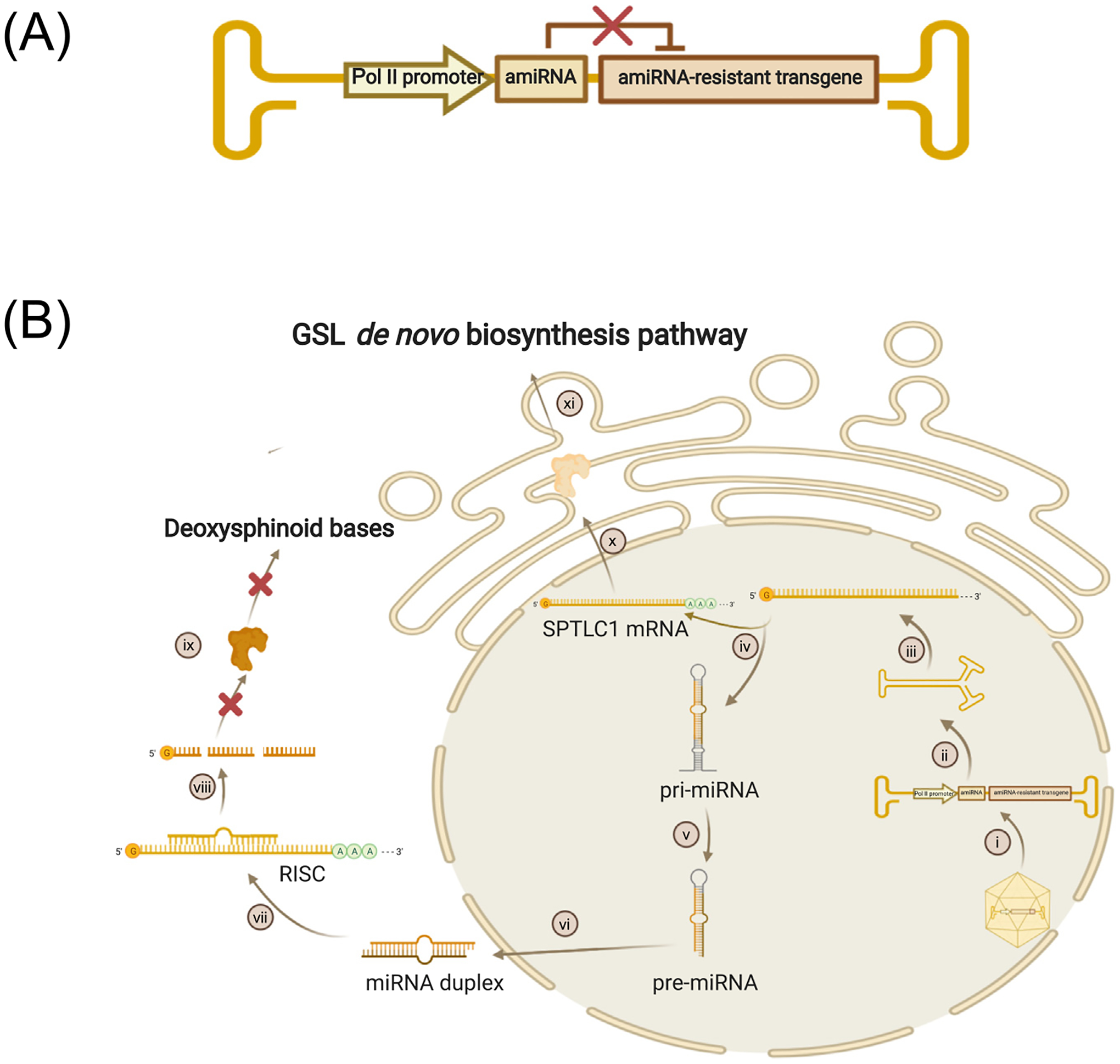
(A) The rAAV genome contains an RNA polymerase II (pol II) promoter driving artificial miRNA (amiRNA) and amiRNA-resistant transgenes simultaneously. amiRNA-resistant transgenes encode the same amino acid sequence as the endogenous normal genes, but carry a different nucleic acid sequence that cannot be recognized by the amiRNA. (B) Dual-function rAAV vectors function in the cell. (i) AAV uncoating in the nucleus; (ii) second-strand synthesis; (iii) transcription and post-transcription modification; (iv) splicing into two independent transcripts: pri-miRNA and amiRNA-resistant SPTLC1 mRNA; (v) cleavage; (vi) export; (vii) formation of RNA-induced silencing complex (RISC) with endogenous SPTLC1 mRNA; (viii) endogenous SPTLC1 mRNA cleavage; (ix) generation of toxic deoxysphinoid bases prevented; (x) wild-type SPTLC1 protein translation; and (xi) SPTLC1 protein involved in normal glycosphingolipid (GSL) de novo synthesis. Created with BioRender.com.
By contrast, loss-of-function mutations in ST3GAL5 and B4GALNT1 could be resolved by rAAV-mediated gene replacement therapy. Due to alternative splicing and multiple start codons, human ST3GAL5 expresses into multiple isoforms. Thus, the appropriate isoform(s) to choose for gene therapy needs to be determined. With the appropriate isoform, the rAAV-borne GM3 synthase is expected to restore the GSL profile and their downstream functions (Figure 2). One potential caveat is that ST3GAL5 overexpression in periphery tissues might cause off-target toxicity, as evidenced by the association between overexpression of ST3GAL5 and cancer or diabetes [15]. Therefore, expressing the exogenous ST3GAL5 in the right cells and to the right extent is pivotal and challenging for gene therapy development. Similarly, the gene replacement therapy strategy and considerations can be applied for delivering B4GALNT1 for HSPG26.
Figure 2. Recombinant Adeno-Associated Virus (rAAV)-Mediated Gene Replacement Therapy for Treating GM3 Synthase Deficiency.
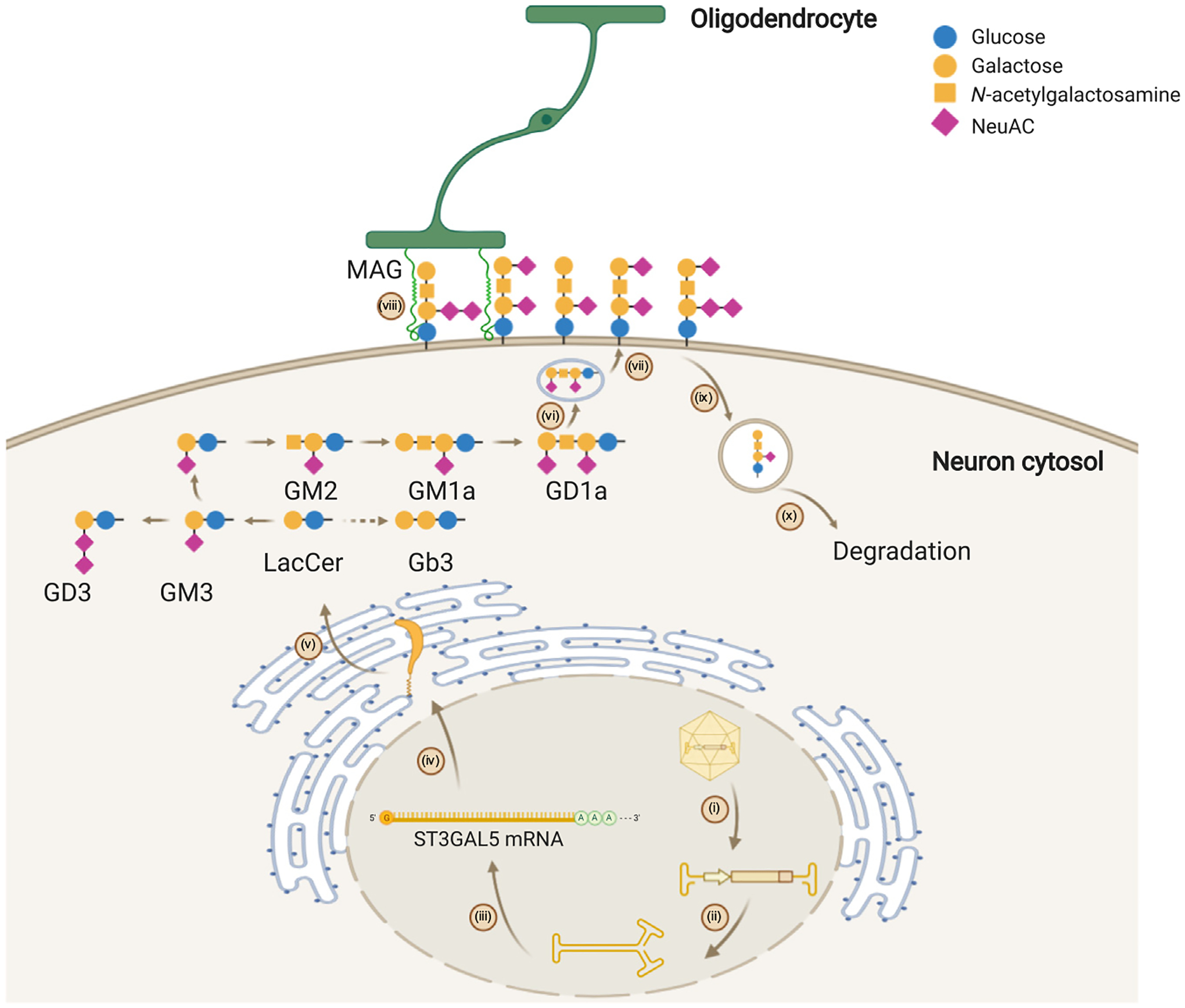
(i) AAV uncoating; (ii) second-strand synthesis; (iii) transcription and post-transcription modification; (iv) protein translation and transport; (v) ST3GAL5 protein restores the ganglioside biosynthesis network through the initiation of GM3 synthesis in the Golgi; (vi) ganglioside transportation via vesicles; (vii) ganglioside integration on the cell membrane; (viii) restored cellular communication mediated by gangliosides, such as the interaction with myelin-associated glycoprotein (MAG) of oligodendrocytes; (ix) ganglioside recycling from cell membrane; and (x) ganglioside degradation. Abbreviation: NeuAC, N-acetylneuraminic acid. Created with BioRender.com.
The aforementioned animal models can be used to evaluate therapeutic efficacy, based on both phenotypical and biochemical changes. However, substantial differences exist between murine models and humans, especially in brain size and anatomy. Large animal models, such as pig, can be developed to better understand disease pathology and serve as pre-clinical therapy assessment platforms.
Concluding Remarks
AAV vectors are capable of transducing a range of species and tissues in vivo with relatively low innate and adaptative immune responses and genotoxicity; therefore, they represent the leading gene delivery platform for the treatment of diverse human diseases. We expect that developing such gene therapies could ultimately fulfil the unmet medical needs of patients with GSL biosynthesis deficiencies.
Acknowledgments
G.G. is supported by grants from the University of Massachusetts Medical School (an internal grant) and by the National Institutes of Health (NIH) (R01NS076991-01, 4P01HL131471-02, UG3 HL147367-01, R01HL097088, and U19 AI149646-01).
Footnotes
Declaration of Interests
G.G. is a scientific cofounder of Voyager Therapeutics, Adrenas Therapeutics, and Aspa Therapeutics, and holds equity in these companies. G.G. is an inventor on patents with potential royalties licensed to Voyager Therapeutics, Aspa Therapeutics, and ten other biopharmaceutical companies. The remaining authors declare no competing interests.
References
- 1.Schengrund CL (2015) Gangliosides: glycosphingolipids essential for normal neural development and function. Trends Biochem. Sci 40, 397–406 [DOI] [PubMed] [Google Scholar]
- 2.Fridman V et al. (2015) Natural history and biomarkers in hereditary sensory neuropathy type 1. Muscle Nerve 51, 489–495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Eichler FS et al. (2009) Overexpression of the wild-type SPT1 subunit lowers desoxysphingolipid levels and rescues the phenotype of HSAN1. J. Neurosci 29, 14646–14651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Simpson MA et al. (2004) Infantile-onset symptomatic epilepsy syndrome caused by a homozygous loss-of-function mutation of GM3 synthase. Nat. Genet 36, 1225–1229 [DOI] [PubMed] [Google Scholar]
- 5.Bowser LE et al. (2019) Recessive GM3 synthase deficiency: Natural history, biochemistry, and therapeutic frontier. Mol. Genet. Metab 126, 475–488 [DOI] [PubMed] [Google Scholar]
- 6.Yamashita T et al. (2003) Enhanced insulin sensitivity in mice lacking ganglioside GM3. Proc. Natl. Acad. Sci. U. S. A 100, 3445–3449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yoshikawa M et al. (2009) Mice lacking ganglioside GM3 synthase exhibit complete hearing loss due to selective degeneration of the organ of Corti. Proc. Natl. Acad. Sci. U. S. A 106, 9483–9488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fishman PH et al. (1975) Deficient ganglioside biosynthesis: a novel human sphingolipidosis. Science 187, 68–70 [DOI] [PubMed] [Google Scholar]
- 9.Boukhris A et al. (2013) Alteration of ganglioside biosynthesis responsible for complex hereditary spastic paraplegia. Am. J. Hum. Genet 93, 118–123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sheikh KA et al. (1999) Mice lacking complex gangliosides develop Wallerian degeneration and myelination defects. Proc. Natl. Acad. Sci. U. S. A 96, 7532–7537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang D et al. (2019) Adeno-associated virus vector as a platform for gene therapy delivery. Nat. Rev. Drug Discov 18, 358–378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hudry E and Vandenberghe LH (2019) Therapeutic AAV gene transfer to the nervous system: a clinical reality. Neuron 101, 839–862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xie J et al. (2011) MicroRNA-regulated, systemically delivered rAAV9: a step closer to CNS-restricted transgene expression. Mol. Ther 19, 526–535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xie J et al. (2020) Effective and accurate gene silencing by a recombinant AAV-compatible microRNA scaffold. Mol. Ther 28, 422–430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lipina C and Hundal HS (2015) Ganglioside GM3 as a gatekeeper of obesity-associated insulin resistance: evidence and mechanisms. FEBS Lett. 589, 3221–3227 [DOI] [PubMed] [Google Scholar]