

Abstract
Studies have indicated that the abundance and community structure of gut microbiota are altered by diet. In this study, next‐generation sequencing of the 16S rRNA gene amplicon was performed to evaluate variations in the gut microbiota of wild and captive individuals of both sexes of Calotes versicolor. The results showed that there was a significant sex difference in microbial community structure for wild C. versicolor, Bacteroide was the dominant genus in wild females (WF), whereas Ochrobactrum was the dominant genus in wild males (WM). Acinetobacter and Hymenobacter were the dominant genera in WF, while Clostridium was the dominant genus in captive females (CF). The results indicated that differences in diet between wild and captive C. versicolor also resulted in variations in gut microbiota. Thus, it was not surprising that captivity and sex shape the gut microbiota in C. versicolor. In summary, the fundamental information presented about the gut microbiota of both sexes of wild (and captive females) C. versicolor, indicates that the artificial environments are not suitable for the wild C. versicolor.
Keywords: Calotes versicolor, captivity, diet, gut microbiota, sex
Relationships between bacterial and KEGG pathway by sex (a) and captive (b). Pairwise comparisons of bacterial were displayed with a color gradient denoting Spearman's correlation coefficient. Bacterial and KEGG community composition was related to each bacterium by Mantel test.

1. INTRODUCTION
Gut microbiota plays a critical role in host health and provides fundamental information about host physiology (Hale et al., 2018). Complex communities of gut microbiota are associated with host energy budget and nutrient metabolism (Cani, 2016; Rowland et al., 2018; Semova et al., 2012), foraging behavior (Heijtz et al., 2011), immune homeostasis (Dimitriu et al., 2013; Rastelli et al., 2019; Round & Mazmanian, 2009), and reproductive performance (Leftwich et al., 2017). Due to its size and complexity, the gut microbiota (microbiome) is known as the second genome (Weinstock, 2012). In recent years, the rapid development and decreasing cost of next‐generation sequencing has led to an increase in studies on the role of the gut microbiome in wildlife and human health (Debelius et al., 2016; Hird, 2017; Ingala et al., 2018; Zhu et al., 2011). In particular, studies have focused on explaining the coevolution of hosts and gut microbiota (Ingala et al., 2018; Montoya‐Ciriaco et al., 2020; Videvall et al., 2018), and diseases (Fan & Pedersen, 2021; Wang et al., 2021; Zhang et al., 2020).
Bacteroidetes, Firmicutes, and Proteobacteria are three of the most important components of the gut microbiota in vertebrate species (Kohl et al., 2017; Ren et al., 2016). Several studies have shown that gut microbiota may play a significant role in vertebrate evolution, as its diversity is correlated with the evolutionary history of these animals. However, vertebrate gut microbial communities are influenced by several other factors, such as host features (age, body size, and sex) and environment (diet and season) (Delsuc et al., 2014; Martin et al., 2010; Zhou, Nelson, et al., 2020). For example, in nonreproductive mice the relative abundance of Lactobacillus spp. was higher in males than in females, whereas the contrary was observed in reproductive mice (Maurice et al., 2015). Female Sceloporus virgatus display significantly lower microbial diversity and richness than males (Martin et al., 2010). Moreover, compared to males, Rhinella marina females display an increased relative abundance of Bacteroides, Comamonas, Flavobacterium, Microvirgula, Parabacteroides, and Pseudomonas species, with a decreased relative abundance of Cetobacterium, Clostridium, Epulopiscium, Plesiomonas, and Vibrio species (Zhou, Nelson, et al., 2020). However, no influences of sex have been observed in the microbial communities of Liolarmus and Phymaturus lizards (Kohl et al., 2017). Thus, the influence of sex on gut microbial communities in wildlife species is complex and largely unknown.
Recent studies have shown that captivity plays an important role in endangered species conservation by maintaining the breeding population, particularly for lizards; however, captivity has been shown to significantly alter the gut microbial community of lizards (Jiang et al., 2017; Kohl et al., 2017; Tang et al., 2020; Zhou, Zhao, et al., 2020), amphibians (Bataille et al., 2016; Tong et al., 2019), and other taxa (Chi et al., 2019; Hale et al., 2018; Martínez‐Mota et al., 2020; Oliveira et al., 2020). For instance, a study by Kohl et al. (2017) found that captive species exhibited less Firmicutes and Actinobacteria compared to wild species, and Bacteroidetes was only present in captive species. Furthermore, some studies have reported a significant difference between the alpha diversity of wild and captive animal gut microbiotas (Kohl et al., 2017; Ren et al., 2016), whereas other studies have indicated a general loss of microbial diversity as a result of captivity (Kohl & Dearing, 2014; Kohl et al., 2014).
Microbiome characterization and monitoring tools are being developed and are recommended for wild species conservation (Oliveira et al., 2020; Redford et al., 2012), particularly endangered species such as the giant panda (Ailuropoda melanoleuca) (Zhu et al., 2011), the Yangtze finless porpoise (Neophocaena asiaeorientalis asiaeorientalis) (Wan et al., 2016), and the Chinese crocodile lizard (Shinisaurus crocodilurus) (Jiang et al., 2017; Tang et al., 2020). Considering of the sex of individuals in captivity is important of conservation activities, particularly for health breeding programs. However, the important of gut microbiota to conservation efforts for lizards of both sexes in captive and wild environments is largely unknown.
Therefore, the main goal of this study was to characterize the gut microbiota of the lizard Calotes versicolor to address the following questions: (1) Do gut microbial communities vary with the sex of the lizard? (2) Does gut microbiota respond to captivity? (3) What functions are differentially determined by the bacteria? To answer these questions, we used 16S rRNA gene sequencing to characterize microbial communities sampled from: (1) wild lizards (nine individuals per sex), and (2) from lizards maintained in captivity and fed in a seminatural environment for 90 days. Calotes versicolor (Agamidae) is an oviparous, arboreal, multiple‐clutched, omnivorous lizard, widely distributed in Indo‐Malaya, that most feeds on Arthropoda, including Diptera, Coleoptera, Lepidoptera, and Orthoptera (Qiu et al., 2001). Adult lizards do not display sexually dimorphism in snout‐vent length (Qiu et al., 2001), but females have relatively narrow heads compared to males (Shanbhag & Parsad, 1993). Hindgut samples from C. versicolor tend to have more Firmicutes and Bacteroidetes, and less Proteobacteria than those of the small intestine (Zhang et al., 2021).
2. MATERIALS AND METHODS
2.1. Ethics
All experiments, including the sample collection, complied with the current laws of China for the care and use of experimental animals, were approved by Hainan Tropical Ocean University (September 2019), and followed the principles of the Ethical Committee for Experimental Animal Welfare of the Hangzhou Normal University (No. 2018135).
2.2. Sample collection
Eighteen healthy and nongravid adult lizards (9♀♀:9♂♂) from Hainan, China, were captured and numbered in June 2019. Hindgut samples were collected immediately after capture following Zhang et al. (2021) described, and labeled as wild females (WF) and wild males (WM). The collected lizards were maintained in captivity in a seminatural environment for 90 days, where they were fed Tenebrio molitor and Gryllulus chinensis. Hindgut samples were collected again at the end of this period. Samples from only four females were obtained after the 90‐days captivity period as most lizards had escaped, these samples were recorded as captive females (CF). All hindgut samples were collected separately and stored at −80°C prior to DNA extraction.
2.3. Gut microbiota analyses
DNA was extracted from all samples using the cetyltrimethylammonium bromide (CTAB)/sodium dodecyl sulfate (SDS) method. Universal primers were employed to amplify the V3–V4 regions of the bacterial 16S rRNA genes that contained Illumina sequences at the 5′‐end of forward primers harboring 7‒12 bp barcodes. Sequencing was performed using an Illumina MiSeq platform (San Diego, CA, USA) with Frasergen Bioinformatics (Wuhan, Hubei, China). All sequence analyses were performed using the QIIME software package (https://qiime.org/, Caporaso et al., 2010). Chloroplast and mitochondrial sequences were removed from the dataset. Operational taxonomic units (OTUs) with 97% similarity were defined using UCLUST (Edgar, 2010).
2.4. Statistical analyses
Variations in alpha diversity were analyzed using Tukey's HSD test. Nonmetric multidimensional scaling (NMDS) based on the Bray‐Curtis distance was constructed to determine the variations in beta‐diversity. To obtain the unique genus, features that occurred in ≥75% of the replicates in each group were retained. The linear discriminant analysis (LDA) effect size (LEfSe) method was employed to identify the variations in microbial communities based on LDA sources (Segata et al., 2011). To explore the functional profiles of gut microbiota between WM and WF, or between WF and CF, all OTUs were assigned to the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways by Tax4Fun (Aßhauer et al., 2015). The differences in gene function analyses were identified in STAMP (v2.1.3) (Parks et al., 2014), and Welch's t‐test was used for the comparisons between the two groups.
The relationships between various microbial communities were analyzed using Spearman's correlation coefficients. Partial Mantel tests were performed to evaluate relationships between the relative abundance of microbiota and gene functions. All analyses were conducted using the linKET package (Huang, 2021) in R version 4.0.4 (R Core Team, 2021).
3. RESULTS
In all lizards, Firmicutes, Proteobacteria, Bacteroidetes, and Verrucomicrobia, were the four dominant phyla identified (mean relative abundance >1%, Figure 1a). Actinobacteria only was a dominant phylum in WM, but not in WF (Figure 1a). Richness, Chao1, and ACE indices were lower in WF than in WM (all adj p < .01), but Shannon, Simpson, and Pielou's E diversity indices showed no differences (all adj p > .05) between WM and WF (Figure S1).
FIGURE 1.

Composition of the gut microbiota of each group at the phylum, family and genus levels
Fusobacteria and Deferribacteres were the two dominant phyla in CF, but Actinobacteria was a dominant phylum in WF. Captivity was correlated with the loss of the Firmicutes and Proteobacteria, and an introduction of the Bacteroidetes and Verrucomicrobia (Figure 1a). Nevertheless, the Shannon, Richness, Simpson, Pielou's E, Chao1, and ACE indices showed no differences (all adj p > .05) between WF and CF (Figure S2).
The NMDS plots showed significant differences in beta diversity between WF and WM (Figure 2a), and WF and CF (Figure 2b), respectively (Adonis test: WM‐WF, R 2 = .204, p = .03; WF‐CF, R 2 = .230, p < .01).
FIGURE 2.

The non‐metric multidimensional scaling (NMDS) of the gut microbiota composition. The variation explanation is indicated on each axis, respectively
We obtained 75, 107, 80, and 75 genera from WF and WM as well as WF and CF, respectively. WM and WF shared 61 genera (Figure 3a), but 46 and 14 genera were unique to WM and WF, respectively. In addition, WF and CF shared 63 genera (Figure 3b), but 17 and 12 genera were unique to WF and CF, respectively.
FIGURE 3.
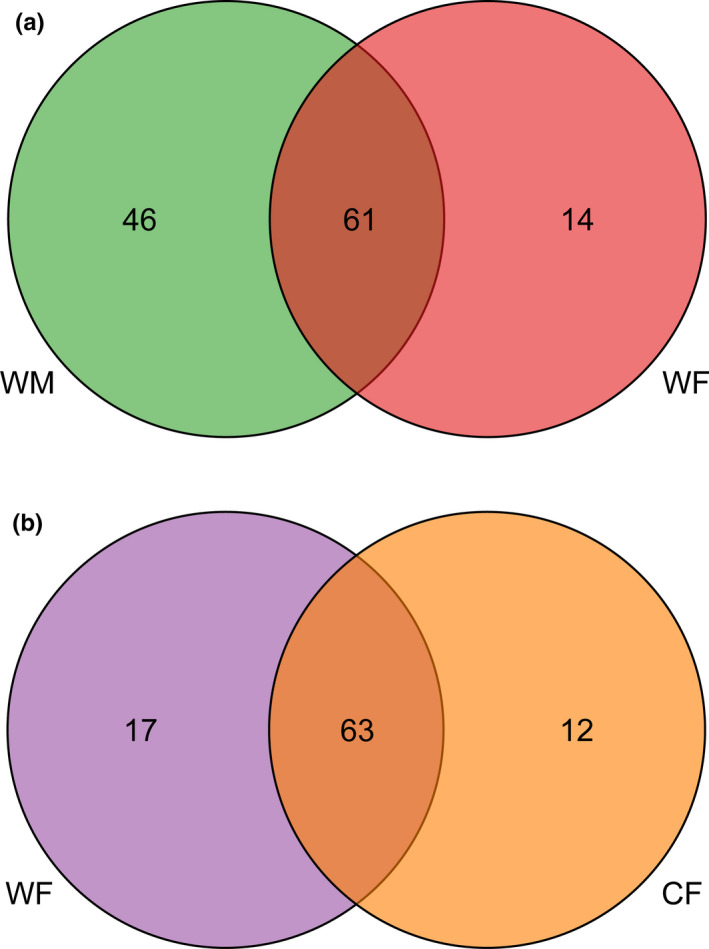
The Venn plot to show the unique and share genus between WM and WF (a) and WF and CF (b)
Linear discriminant analysis effect size was performed to identify the specific bacterial taxa in samples and to compare the gut microbiota of WF and WM (LDA > 3.0, p < .05, Figure 4a), and WF and CF (LDA > 2.0, p < .05, Figure 4b). In the first comparison, four discriminative features at the class level (Alphaproteobacteria, Bacilli, Betaproteobacteria, and Gammaproteobacteria), and three at the order level (Enterobacteriales, Lactobacillales, and Rhizobiales) were identified in the WM; at the family level, two discriminative features (Brucellaceae and Enterobacteriaceae) were identified in WM, and two discriminative features (Bacteroidaceae and Odoriobacteraceae) in WF, and at the genus level, one discriminative feature (Ochrobactrum) were identified in WM, and two discriminative features (Bacteroides and Odoribacter) in WF.
FIGURE 4.

Linear discriminative analysis of effect size (LEfSe) analysis of taxonomic biomarkers of gut microbiota. (a) Cladogram of significant changes at all taxonomic levels. The root of the cladogram represents the domain bacteria. The size of node represents the abundance of taxa. (b) Histogram of the LDA score computed for features differentially abundant taxon. LDA score >4 were shown
From the comparison made after the captivity period, at order level, one discriminative feature (Rhodospirillales) was found in the CF; at family level, two discriminative features (Moraxellaceae and Rhodospirillaceae) were identified in the CF, and two discriminative features (Bacillaceae and Peptostreptococcaceae) in the WF; and at genus level, two discriminative features (Acinetobacter and Hymenobacter) were identified in CF, and one discriminative feature (Clostridium) in WF.
There were including 172 (for sex) and 169 (for wild vs captivity) KEGG metabolic pathways were selected between WF and WM, and WF and CF, respectively, associated with metabolism (68.60% and 68.05%), genetic information processing (10.47% and 10.65%), cellular processes (3.49% and 5.33%), environmental information processing (4.07% and 3.55%), organismal systems (2.91% and 3.01%), and human diseases (9.30% and 8.28%). There was significant difference in organismal systems between WF and WM (adj p = .038), but no significant differences were detected between WF and CF (adj p > .20). Functional predictions identified 12 differentially present level 3 KEGG pathways between WF and WM (Figure 5a). The tetracycline biosynthesis and biosynthesis of type II polyketide product pathways were higher in WM, whereas other pathways were higher in WF. Six pathways differed between WF and CF (Figure 5b). The pentose phosphate pathway and epithelial cell signaling of the Helicobacter pylori infection were higher in WF, while other pathways were higher in CF.
FIGURE 5.

Functionally predicted KEGG pathways differing in (a) between wild males and wild females and (b) between wild females and captive females of Calotes versicolor. The bar plot shows mean proportions of differential level 3 of KEGG pathways predicted using Tax4Fun. The difference in proportions between the groups is shown with 95% confidence intervals. Only p value < .05 (Welch's t‐test, FDR adjusted) are shown and composition
Some discriminative features in WF and WM (Figure 6a) or WF and CF (Figure 6b) were negatively correlated. In the WM group, Bacilli and Gammaproteobacteria had a significant effect on the metabolism of other amino acids; Lactobacillales had a weak effect on tetracycline biosynthesis; and Betaproteobacteria, Rhizobiales, Brucellaceae, and Ochrobactrum had a significant effect on biosynthesis of the type II polyketide backbone. In the CF group, Moraxellaceae, Rhodospirillaceae, Acinetobacter, and Hymenobacter had a significant effect on the metabolism of cofactors and vitamins, transport and catabolism, pentose phosphate pathway, and epithelial cell signaling in the H. pylori infection.
FIGURE 6.

Relationships between bacterial and KEGG pathway by sex (a) and captive (b). Pairwise comparisons of bacterial were displayed with a color gradient denoting Spearman's correlation coefficient. Bacterial and KEGG community composition was related to each bacterium by Mantel test
4. DISCUSSION
Our results revealed that the core microbiota of C. versicolor consisted of Firmicutes, Proteobacteria, Bacteroidetes, and Verrucomicrobia at the phylum level, which is consistent with the results of previous studies (Jiang et al., 2017; Kohl et al., 2017; Tang et al., 2020; Zhou, Zhao, et al., 2020). Furthermore, our results showed that sex does influence gut microbial communities, with WF having significantly lower gut microbial diversity and richness compared to WM. Previous studies have detected sex‐related differences in the gut microbiota of S. virgatus (Martin et al., 2010), and R. marina (Zhou, Nelson, et al., 2020). Sexual dimorphism may be related to differences in the spatial and temporal niches (Butler, 2007), with male lizards having higher a perch than females for defending territories and remaining visible to potential mates (Logan et al., 2021). However, the snout‐vent length of C. versicolor is not sexually dimorphism (Qiu et al., 2001). A previous study showed that sex hormones intercept changes in gut microbiota via gonadectomy and testosterone hormone replacement in mice (Org et al., 2016). Hormonal changes and sex differences strongly affect bile acid profiles (Org et al., 2016), which respond to high‐fat/high‐sugar diets, which in turn affect gut microbiota (Islam et al., 2011; Li & Chiang, 2015). Moreover, the relatively narrow heads of female C. versicolor may be related to the selection of a small‐sized diet. The food niche overlap between sexes is 0.522 (Qiu et al., 2001). The main diet of adult females includes Orthoptera (Acrididae, 9.0%), Coleoptera (Chrysomelidae 14.7% and Scarabaeoidae 9.0%), and Diptera (Platypezidae 34.6%), whereas that of adult males includes Orthoptera (Acrididae 11.6%), Coleoptera (Chrysomelidae 24.5%), and Lepidoptera (Nymphalidae 11.1% and Papilionidae 17.6%) (Qiu et al., 2001). In the present study, we found that Bacteroides was the most dominant genus in WF, with Ochrobactrum being the dominant genus in WM. Bacteroides can employ dietary or host‐derived glycans and proteins according to the nutrient availability (Sonnenburg et al., 2005), improving the utilization rate of assimilated nutrients and incorporating external amino acids. The LEfSe showed that Bacilli, Gammaproteobacteria, Lactobacillales, Betaproteobacteria, Rhizobiales, Brucellaceae, and Ochrobactrum were discriminative features in the WM group. Based on the relationships between bacterial and KEGG pathways, we found that Bacilli and Gammaproteobacteria had a significant effect on the metabolism of other amino acids, Lactobacillales had a weak effect on tetracycline biosynthesis, and Betaproteobacteria, Rhizobiales, Brucellaceae, and Ochrobactrum had a significant effect on the biosynthesis of the type II polyketide backbone. Thus, the differences in diets may have directly driven variations in the gut microbial communities of C. versicolor males and females.
Previous studies have shown that captivity influences gut microbiota (Tang et al., 2020; Zhou, Zhao, et al., 2020). Our results showed that captivity was related to a loss of Firmicutes and Proteobacteria, and the introduction of Bacteroidetes and Verrucomicrobia to gut microbiota. Firmicutes play an important role in fiber and cellulose degradation by breaking down cellulose into volatile fatty acids, which can be used by the host. The higher abundance of Firmicutes in wild lizards probably leads to improved digestion and absorption of nutrients. Captive lizards were fed with an artificial fodder composed of Tenebrio molitor and Gryllulus chinensis, which had a relatively high‐fat content, but a relatively low fiber content. Moreover, two discriminative features (Acinetobacter and Hymenobacter) were identified in CF, while another (Clostridium) was identified in WF. Clostridium was positively correlated with the serum levels of total cholesterol, low‐density lipoprotein cholesterol, and triacylglycerols (Guo et al., 2018). The LEfSe showed that Moraxellaceae, Rhodospirillaceae, Acinetobacter, and Hymenobacter were discriminative features in the WF group. The relationships between bacterial and KEGG pathways indicated that the presence of Moraxellaceae, Rhodospirillaceae, Acinetobacter, and Hymenobacter had a significant effect on the metabolism of cofactors and vitamins, transport and catabolism, pentose phosphate pathway, and epithelial cell signaling in the H. pylori infection. Therefore, the results of this study indicated that a simple diet in captivity directly influences the gut microbial communities of C. versicolor.
5. CONCLUSIONS
In conclusion, the bacterial phyla Firmicutes, Proteobacteria, Bacteroidetes, and Verrucomicrobia dominated the core microbiota of C. versicolor. Our results led to the following conclusions: (1) WF having significantly lower microbial diversity and richness compared to WM; (2) captivity is related to a loss of Firmicutes and Proteobacteria, but also to the introduction of Bacteroidetes and Verrucomicrobia; (3) metabolic functions were differentially determined by the bacterial variations. The types and size of food items in the diet were significantly different for WM and WF, as well as CF and WF. It was not surprising we found that captivity and sex influence the gut microbiota in C. versicolor. The relationship between bacterial and KEGG pathways indicated that the artificial environments used here are not suitable for wild C. versicolor.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
AUTHOR CONTRIBUTIONS
Lin Zhang: Conceptualization (equal); Data curation (equal); Formal analysis (equal); Funding acquisition (equal); Methodology (equal); Project administration (equal); Supervision (equal); Visualization (equal); Writing – original draft (equal); Writing – review & editing (equal). Fang Yang: Data curation (equal); Formal analysis (equal); Writing – original draft (equal). Tangliang Li: Data curation (equal). Buddhi Dayananda: Writing – review & editing (equal). Longhui Lin: Resources (equal). Chixian Lin: Funding acquisition (equal).
Supporting information
Supplementary Material
ACKNOWLEDGMENTS
The authors wish to thank the members of Hainan Key Laboratory of Herpetological Research for assistance in the field and laboratory, and the anonymous reviewers and the editor of the journal for their valuable comments. The authors would like to thank Editage (www.editage.cn) for English language editing.
Zhang, L. , Yang, F. , Li, T. , Dayananda, B. , Lin, L. , & Lin, C. (2022). Lessons from the diet: Captivity and sex shape the gut microbiota in an oviparous lizard (Calotes versicolor). Ecology and Evolution, 12, e8586. 10.1002/ece3.8586
Lin Zhang and Fang Yang contributed equally to this work.
Funding information
This study was financially supported by the National Natural Science Foundation of China (31800337), State Key Laboratory of Microbial Technology Open Projects Fund (M2021‐18), Research Start‐up Fund of Hubei University of Chinese Medicine, and Hainan Key Program of Science and Technology (ZDXM20110008).
Contributor Information
Lin Zhang, Email: lzhangss@msn.com.
Chixian Lin, Email: chixianlin@hntou.edu.cn.
DATA AVAILABILITY STATEMENT
Sequencing data were exported as individual fastq files and have been deposited in Sequence Read Archive (SRA) NCBI (https://www.ncbi.nlm.nih.gov/) under the accession code: PRJNA673584 (https://www.ncbi.nlm.nih.gov/sra/PRJNA673584).
REFERENCES
- Aßhauer, K. P. , Wemheuer, B. , Daniel, R. , & Meinicke, P. (2015). Tax4Fun: Predicting functional profiles from metagenomic 16S rRNA data. Bioinformatics, 31, 2882–2884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bataille, A. , Lee‐Cruz, L. , Tripathi, B. , Kim, H. , & Waldman, B. (2016). Microbiome variation across amphibian skin regions: Implications for chytridiomycosis mitigation efforts. Microbial Ecology, 71, 221–232. 10.1007/s00248-015-0653-0 [DOI] [PubMed] [Google Scholar]
- Butler, M. A. (2007). Vive le difference! Sexual dimorphism and adaptive patterns in lizards of the genus Anolis . Integrative and Comparative Biology, 47, 272–284. [DOI] [PubMed] [Google Scholar]
- Cani, P. D. (2016). Gut microbiota: Changes in gut microbes and host metabolism: Squaring the circle? Nature Reviews Gastroenterology & Hepatology, 13, 563–564. [DOI] [PubMed] [Google Scholar]
- Caporaso, J. G. , Kuczynski, J. , Stombaugh, J. , Bittinger, K. , Bushman, F. D. , Costello, E. K. , Fierer, N. , Peña, A. G. , Goodrich, J. K. , Gordon, J. I. , Huttley, G. A. , Kelley, S. T. , Knights, D. , Koenig, J. K. , Ley, R. E. , Lozupone, C. A. , McDonald, D. , Muegge, B. D. , Pirrung, M. , … Knight, R. (2010). QIIME allows analysis of high‐throughput community sequencing data. Nature Methods, 7, 335–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi, X. , Gao, H. , Wu, G. , Qin, W. , Song, P. , Wang, L. , Chen, J. , Cai, Z. , & Zhang, T. (2019). Comparison of gut microbiota diversity between wild and captive bharals (Pseudois nayaur). BMC Veterinary Research, 15, 243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debelius, J. , Song, S. J. , Vazquez‐Baeza, Y. , Xu, Z. Z. , Gonzalez, A. , & Knight, R. (2016). Tiny microbes, enormous impacts: What matters in gut microbiome studies? Genome Biology, 17, 217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delsuc, F. , Metcalf, J. L. , Parfrey, L. W. , Song, S. J. , González, A. , & Knight, R. (2014). Convergence of gut microbiomes in myrmecophagous mammals. Molecular Ecology, 23, 1301–1317. [DOI] [PubMed] [Google Scholar]
- Dimitriu, P. A. , Boyce, G. , Samarakoon, A. , Hartmann, M. , Johnson, P. , & Mohn, W. W. (2013). Temporal stability of the mouse gut microbiota in relation to innate and adaptive immunity. Environmental Microbiology Report, 5, 200–210. [DOI] [PubMed] [Google Scholar]
- Edgar, R. C. (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics, 26, 2460–2461. [DOI] [PubMed] [Google Scholar]
- Fan, Y. , & Pedersen, O. (2021). Gut microbiota in human metabolic health and disease. Nature Reviews Microbiology, 19, 55–71. [DOI] [PubMed] [Google Scholar]
- Guo, W. L. , Pan, Y. Y. , Li, L. , Li, T. T. , Liu, B. , & Lv, X. C. (2018). Ethanol extract of Ganoderma lucidumameliorates lipid metabolic disorders and modulates the gut microbiota composition in high‐fat diet fed rats. Food Function, 9, 3419–3431. [DOI] [PubMed] [Google Scholar]
- Hale, V. L. , Tan, C. L. , Niu, K. , Yang, Y. , Knight, R. , Zhang, Q. , Cui, D. , & Amato, K. R. (2018). Diet Versus phylogeny: A comparison of gut microbiota in captive Colobine monkey species. Microbial Ecology, 75, 515–527. [DOI] [PubMed] [Google Scholar]
- Heijtz, R. D. , Wang, S. C. , Anuar, F. , Qain, Y. , Björkholm, B. , Samuelsson, A. , Hibberd, M. L. , Forssberg, H. , & Pettersson, S. (2011). Normal gut microbiota modulates brain development and behavior. Proceedings of the National Academy of Sciences of the United States of America, 108, 3047–3052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hird, S. M. (2017). Evolutionary biology needs wild microbiomes. Frontiers in Microbiology, 8, 725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, H. (2021). linkET: What the package does. R package version 0.0.1 (20200625). https://github.com/Hy4m/linkET [Google Scholar]
- Ingala, M. R. , Simmons, N. B. , Wultsch, C. , Krampis, K. , Speer, K. A. , & Perkins, S. L. (2018). Comparing microbiome sampling methods in a wild mammal: Fecal and intestinal samples record different signals of host ecology, evolution. Frontiers in Microbiology, 9, 803. 10.3389/fmicb.2018.00803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Islam, K. B. M. S. , Fukiya, S. , Hagio, M. , Fujii, N. , Ishizuka, S. , Ooka, T. , Ogura, Y. , Hayashi, T. , & Yokota, A. (2011). Bile acid is a host factor that regulates the composition of the cecal microbiota in rats. Gastroenterology, 141, 1773–1781. [DOI] [PubMed] [Google Scholar]
- Jiang, H.‐Y. , Ma, J.‐E. , Li, J. , Zhang, X.‐J. , Li, L.‐M. , He, N. , Liu, H.‐Y. , Luo, S.‐Y. , Wu, Z.‐J. , Han, R.‐C. , & Chen, J.‐P. (2017). Diets alter the gut microbiome of crocodile lizards. Frontiers in Microbiology, 8, 2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohl, K. D. , Brun, A. , Magallanes, M. , Brinkerhoff, J. , Laspiur, A. , Acosta, J. C. , Caviedes‐Vidal, E. , & Bordenstein, S. R. (2017). Gut microbial ecology of lizards: Insights into diversity in the wild, effects of captivity, variation across gut regions and transmission. Molecular Ecology, 26, 1175–1189. 10.1111/mec.13921 [DOI] [PubMed] [Google Scholar]
- Kohl, K. D. , & Dearing, M. D. (2014). Wild‐caught rodents retain a majority of their natural gut microbiota upon entrance into captivity. Environmental Microbiology Reports, 6, 191–195. [DOI] [PubMed] [Google Scholar]
- Kohl, K. D. , Skopec, M. M. , & Dearing, M. D. (2014). Captivity results in disparate loss of gut microbial diversity in closely related hosts. Conservation Physiology, 2, cou009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leftwich, P. T. , Clarke, N. V. E. , Hutchings, M. I. , & Chapman, T. (2017). Gut microbiomes and reproductive isolation in Drosophila . Proceedings of the National Academy of Sciences of the United States of America, 114, 12767–12772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, T. , & Chiang, J. Y. L. (2015). Bile acids metabolic regulators. Current Opinion in Gastroenterology, 31, 159–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logan, M. L. , Neel, L. K. , Nicholson, D. J. , Stokes, A. J. , Miller, C. L. , Chung, A. K. , Curlis, J. D. , Keegan, K. M. , Rosso, A. A. , Maayan, I. , Folfas, E. , Williams, C. E. , Casement, B. , Koyner, M. A. G. , Perez, D. J. P. , Falvey, C. H. , Alexander, S. M. , Charles, K. L. , Graham, Z. A. , … Cox, C. L. (2021). Sex‐specific microhabitat use is associated with sex‐biased thermal physiology in Anolis lizards. Journal of Experimental Biology, 224, jeb235697. [DOI] [PubMed] [Google Scholar]
- Martin, M. O. , Gilman, F. R. , & Weiss, S. L. (2010). Sex‐specific asymmetry within the cloacal microbiota of the striped plateau lizard, Sceloporus virgatus . Symbiosis, 51, 97–105. [Google Scholar]
- Martínez‐Mota, R. , Kohl, K. D. , Orr, T. J. , & Dearing, M. D. (2020). Natural diets promote retention of the native gut microbiota in captive rodents. ISME Journal, 14, 67–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurice, C. F. , Knowles, S. C. L. , Ladau, J. , Pollard, K. S. , Fenton, A. , Pedersen, A. B. , & Turnbaugh, P. J. (2015). Marked seasonal variation in the wild mouse gut microbiota. ISME Journal, 9, 2423–2434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montoya‐Ciriaco, N. , Gómez‐Acata, S. , Muñoz‐Arenas, L. C. , Dendooven, L. , Estrada‐Torres, A. , de la Vega‐Pérez, A. H. D. , & Navarro‐Noya, Y. E. (2020). Dietary effect on gut microbiota of the mesquite lizard Sceloporus grammicus (Wiegmann, 1828) across different altitudes. Microbiome, 8, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveira, B. C. M. , Murray, M. , Tseng, F. , & Widmer, G. (2020). The fecal microbiota of wild and captive raptors. Animal Microbiome, 2, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Org, E. , Mehrabian, M. , Parks, B. W. , Shipkova, R. , Liu, X. , Drake, T. A. , & Lusis, A. J. (2016). Sex differences and hormonal effects on gut microbiota composition in mice. Gut Microbes, 7, 313–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parks, D. H. , Tyson, G. W. , Hugenholtz, P. , & Beiko, R. G. (2014). STAMP: Statistical analysis of taxonomic and functional profiles. Bioinformatics, 30, 3123–3124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu, Q.‐B. , Ma, X.‐M. , & Ji, X. (2001). Ontogenetic shifts of morphology and food habits in the Oriental garden lizard, Calotes versicolor (Agamidae). Zoological Research, 22, 367–374. [Google Scholar]
- R Core Team (2021). R: A language and environment for statistical computing. R Foundation for Statistical Computing. https://www.R‐project.org/ [Google Scholar]
- Rastelli, M. , Cani, P. D. , & Knauf, C. (2019). The gut microbiome influences host endocrine functions. Endocrine Reviews, 40, 1271–1284. [DOI] [PubMed] [Google Scholar]
- Redford, K. H. , Segre, J. A. , Salafsky, N. , del Rio, C. M. , & McAloose, D. (2012). Conservation and the microbiome. Conservation Biology, 26, 195–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren, T. , Kahrl, A. F. , Wu, M. , & Cox, R. M. (2016). Does adaptive radiation of a host lineage promote ecological diversity of its bacterial communities? A test using gut microbiota of Anolis lizards. Molecular Ecology, 25, 4793–4804. [DOI] [PubMed] [Google Scholar]
- Round, J. L. , & Mazmanian, S. K. (2009). The gut microbiota shapes intestinal immune responses during health and disease. Nature Reviews Immunology, 9, 323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowland, I. , Gibson, G. , Heinken, A. , Scott, K. , Swann, J. , Thiele, I. , & Tuohy, K. (2018). Gut microbiota functions: Metabolism of nutrients and other food components. European Journal of Nutrition, 57, 1–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segata, N. , Izard, J. , Waldron, L. , Gevers, D. , Miropolsky, L. , Garrett, W. S. , & Huttenhower, C. (2011). Metagenomic biomarker discovery and explanation. Genome Biology, 12, R60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semova, I. , Carten, J. D. , Stombaugh, J. , Mackey, L. C. , Knight, R. , Farber, S. A. , & Rawls, J. F. (2012). Microbiota regulate intestinal absorption and metabolism of fatty acids in the zebrafish. Cell Host & Microbe, 13, 277–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shanbhag, B. A. , & Parsad, B. S. K. (1993). Follicular dynamics and germinal bed activity during the annual ovarian cycle of the lizard, Calotes versicolor . Journal of Morphology, 216, 1–7. [DOI] [PubMed] [Google Scholar]
- Sonnenburg, J. L. , Xu, J. , Leip, D. D. , Chen, C.‐H. , Westover, B. P. , Weatherford, J. , Buhler, J. D. , & Gordon, J. I. (2005). Glycan foraging in vivo by an intestine‐adapted bacterial symbiont. Science, 307, 1955–1959. [DOI] [PubMed] [Google Scholar]
- Tang, G. S. , Liang, X. X. , Yang, M. Y. , Wang, T. T. , Chen, J. P. , Du, W. G. , Li, H. , & Sun, B. J. (2020). Captivity influences gut microbiota in crocodile lizards (Shinisaurus crocodilurus). Frontiers in Microbiology, 11, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong, Q. , Liu, X.‐N. , Hu, Z.‐F. , Ding, J.‐F. , Bie, J. , Wang, H.‐B. , & Zhang, J.‐T. (2019). Effects of captivity and season on the gut microbiota of the brown frog (Rana dybowskii). Frontiers in Microbiology, 10, 1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Videvall, E. , Strandh, M. , Engelbrecht, A. , Cloete, S. , & Cornwallis, C. K. (2018). Measuring the gut microbiome in birds: Comparison of faecal and cloacal sampling. Molecular Ecology Resources, 18, 424–434. [DOI] [PubMed] [Google Scholar]
- Wan, X. , Ruan, R. , McLaughlin, R. W. , Hao, Y. , Zheng, J. , & Wang, D. (2016). Fecal bacterial composition of the endangered Yangtze finless porpoises living under captive and semi‐natural conditions. Current Microbiology, 72, 306–314. [DOI] [PubMed] [Google Scholar]
- Wang, R. , Tang, R. , Li, B. , Ma, X. , Schnabl, B. , & Tilg, H. (2021). Gut microbiome, liver immunology, and liver diseases. Cellular & Molecular Immunology, 18, 4–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinstock, G. M. (2012). Genomic approaches to studying the human microbiota. Nature, 489, 250–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, L. , Yang, F. , Li, N. , & Dayananda, B. (2021). Environment‐dependent variation in gut microbiota of an oviparous lizard (Calotes versicolor). Animals, 11, 2641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, X. , Chen, B.‐D. , Zhao, L.‐D. , & Li, H. (2020). The gut microbiota: Emerging evidence in autoimmune diseases. Trends in Molecular Medicine, 26, 862–873. [DOI] [PubMed] [Google Scholar]
- Zhou, J. , Nelson, T. M. , Lopez, C. R. , Sarma, R. R. , Zhou, S. J. , & Rollins, L. A. (2020). A comparison of nonlethal sampling methods for amphibian gut microbiome analyses. Molecular Ecology Resources, 20, 844–855. [DOI] [PubMed] [Google Scholar]
- Zhou, J. , Zhao, Y.‐T. , Jiang, Y.‐J. , Lin, L.‐H. , Li, H. , Qu, Y.‐F. , & Ji, X. (2020). Captivity affects diversity, abundance, and functional pathways of gut microbiota in the northern grass lizard Takydromus septentrionalis . MicrobiologyOpen, 9, e1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu, L. F. , Wu, Q. , Dai, J. Y. , Zhang, S. N. , & Wei, F. W. (2011). Evidence of cellulose metabolism by the giant panda gut microbiome. Proceedings of the National Academy of Sciences of the United States of America, 108, 17714–17719. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material
Data Availability Statement
Sequencing data were exported as individual fastq files and have been deposited in Sequence Read Archive (SRA) NCBI (https://www.ncbi.nlm.nih.gov/) under the accession code: PRJNA673584 (https://www.ncbi.nlm.nih.gov/sra/PRJNA673584).