

Abstract
Intracellular protein-protein interactions (PPIs) are challenging targets for traditional drug modalities. Macrocyclic peptides (MPs) prove highly effective PPI inhibitors in vitro and can be rapidly discovered against PPI targets by rational design or screening combinatorial libraries but are generally impermeable to the cell membrane. Recent advances in MP science and technology are allowing for the development of “drug-like” MPs that potently and specifically modulate intracellular PPI targets in cell culture and animal models. This article reviews the recent progress in generating cell-permeable MPs that enter the mammalian cell by passive diffusion, endocytosis followed by endosomal escape, or yet unknown mechanisms.
Keywords: Bicyclic peptide, Cyclic peptide, Drug discovery, Intracellular biologics, Permeability, Protein-protein interaction
MPs as PPI Inhibitors and Challenges
It is estimated that current drugs (i.e., small molecules and biologics) are effective against only ~20% of all disease relevant human proteins [1]. The other ~80%, which are predominantly proteins involved in intracellular PPIs (see Glossary), are “undruggable” because they do not contain well-defined hydrophobic pockets for small molecules to bind and are inaccessible to conventional biologics (e.g., antibodies), which are too large to enter the cell by passive diffusion. Over the past decade, MPs have emerged as an exciting modality for inhibition of PPIs. MPs in the molecular weight range of 700-2000 are 3-5 times larger than small-molecule drugs and capable of binding to flat protein surfaces with antibody-like affinity and specificity [2]. Moreover, powerful combinatorial library technologies are now available, allowing researchers to generate highly potent and specific MP ligands against essentially any protein target [2-5]. Proteolytic instability, previously considered as a major limitation of peptidyl drugs, is largely mitigated by cyclization and the incorporation of non-proteinogenic amino acids (e.g., Na-methylated and D-amino acids). In general, monocyclic peptides of ≤10 amino acids and bicyclic peptides of ≤20 amino acids are relatively stable against proteolytic degradation.
The “Achilles’ heel” of MPs is that they are generally impermeable to the cell membrane and, therefore, suffer from the same limitation as proteins with respect to intracellular targets. Although it has been known for decades that some peptides of both natural and synthetic origins, as well as proteins, can enter the cytosol of mammalian cells with varying efficiencies, their mechanism of cell entry was not well understood. This knowledge gap has historically hampered the rational design of cell-permeable MPs as PPI inhibitors. However, as described below, the recent discovery of highly active cell-penetrating peptides (CPPs) and elucidation of their cell entry mechanism have enabled researchers to develop cell-permeable, metabolically stable, potent, and specific MPs against intracellular PPIs. This review provides an overview of the key advancements in the science and technology of CPPs and MPs, the most notable examples of MP inhibitors against intracellular PPIs reported during the past four years (since 2017), and the outstanding questions/future directions of the MP field. For studies prior to 2017, the readers are referred to two previous reviews [6, 7].
Recent Advancements in CPP and MP Science/Technology
CPPs are small peptides (typically 5-30 aa) that enter the cell without causing significant damage to the cell membrane (Box 1). Initially, all CPPs were linear peptides, which undergo rapid proteolytic degradation in vivo and have low cytosolic entry efficiencies (e.g., ~2% for Tat [8]). A breakthrough was the discovery of cyclic CPPs, which are metabolically stable and have vastly improved cytosolic entry efficiencies (e.g., 120% for CPP12 [9]). CPPs enter the cell by endocytic mechanisms followed by endosomal escape, although at high concentrations some of them also directly translocate across the plasma membrane [10,11]. The greater cytosolic entry efficiency of cyclic CPPs stems primarily from their unique ability to efficiently escape the endosome [9, 12]. The latter in turn facilitated the mechanistic investigation of endosomal escape, which had been a long-standing mystery in the field of cell biology [13]. Pei and co-workers discovered that CPPs escape the endosome by inducing budding and collapse of small vesicles from the endosomal membrane [9,13]. Another key advancement is the development of robust assay methods that directly measure the relative and/or absolute cytosolic entry and endosomal escape efficiencies of CPPs, MPs, and other biomolecules [8, 9, 14-17].
Box 1. CPPs and Their Mechanism of Action.
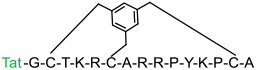
The CPP field emerged three decades ago, when transactivator of transcription (Tat) protein of human immunodeficiency virus (HIV) (in 1988) [89, 90] and Antennapedia homeodomain of Drosophila metanogaster (in 1991) [91] were found to enter the cell. Subsequent studies identified minimal peptide sequences responsible for cellular entry, which evolved into first CPPs – Tat (GRKKRRQRRRPQ) [92] and penetratin/Antp (RQIKIWFQNRRMKWKK) [93]. Since then, hundreds of CPPs have been discovered and applied to transport a variety of cargos, including small molecules, peptides, proteins, and nucleic acids, into the cell both in vitro and in vivo. However, therapeutic application of the linear CPPs is limited by low cytosolic delivery efficiency, susceptibility to proteolytic degradation, and poor PK properties [40]. The discovery of cyclic CPPs, such as CPP9 [cyclo(phe-Nal-Arg-arg-Arg-arg-Gln)] and CPP12 [cyclo(Phe-phe-Nal-Arg-arg-Arg-arg-Gln)] (Figure Ia), has greatly accelerated the therapeutic applications of CPPs, because they not only have vastly improved cytosolic delivery efficiencies (e.g., 120% for CPP12 compared to 2% for Tat) [9], but also are metabolically stable and exhibit good bioavailability and broad biodistribution.
Despite of intense research over three decades, the mechanism of action of CPPs has been a subject of contention since the inception of the CPP field. It is generally established that at lower concentrations, CPPs enter the cell primarily via energy-dependent endocytosis, followed by endosomal escape, while at high concentrations (≥10 μM), some of the CPPs can also directly translocate across the plasma membrane in an energy-independent manner [10-12]. Following endocytosis, CPPs must escape from the endosomal/lysosomal compartments to deliver their cargo into the cytosol (or nucleus). Pei and colleagues recently demonstrated that Tat, cyclic CPP12, and CPM3 (a non-peptidic cell-penetrating molecule) all exit the endosome by a novel vesicle budding-and-collapse (VBC) mechanism [9, 13]. In this mechanism, CPPs bind to the endosomal membrane and cluster into CPP-enriched lipid domains, which subsequently bud off as small vesicles (Figure Ib). The budded vesicles spontaneously and rapidly disintegrate into amorphous lipid/peptide aggregates, thereby releasing the vesicular content into the cytosol. This mechanism reconciles how CPPs can deliver macromolecular cargos (e.g., proteins and nucleic acids) into the cytosol without compromising the endosomal integrity. CPPs of high endosomal escape efficiencies are often amphipathic and conformationally constrained (e.g., by cyclization or α-helix formation). Conformational rigidity increases the binding affinity of CPPs for the endosomal membrane, while amphipathic structures allow CPPs to bind selectively to and stabilize the budding neck (and promote vesicle budding) [12]. It appears that highly efficient CPPs (e.g., cyclic CPPs) escape from the early endosome, whereas CPPs of poor endosomal escape/cytosolic entry efficiencies (e.g., Tat) exit from the later endosome. Early endosomal escape is advantageous for intracellular delivery of biomolecules, as it avoids acid denaturation and enzymatic degradation which may occur inside the late endosome and lysosome.
To achieve cell-permeability, MPs are often conjugated to or integrated with CPPs (see below). In addition to endocytosis/endosomal escape and direct translocation, MPs may also enter cells by passive diffusion or binding to membrane transporters [12]. Still other MPs enter the cell, but their entry mechanisms have not yet been well understood. Below we shall classify the recently developed cell-permeable MPs based on their mechanisms of cell entry.
MPs That Enter the Cell by Passive Diffusion
Most bioactive MPs violate Lipinski’s rules (Ro5), which are a set of criteria for achieving passive membrane permeability and oral bioavailability [18]. Despite violating those rules, some MPs (e.g., cyclosporine A) passively diffuse across cellular membranes and are orally bioavailable. Such MPs usually balance lipophilicity and polar surface area by various combinations of peptide backbone modification (e.g., Nα-methylation), nonpolar side chains, nonproteinogenic building blocks (e.g., D-amino acids), and formation of intramolecular hydrogen bonds [2, 12, 19]. Nα-Methylation and formation of intramolecular hydrogen bonds reduce the number of hydrogen bond donors (HBDs) and/or acceptors (HBAs) that usually impede membrane permeability. These strategies, unfortunately, also limit the structural diversity for target recognition and aqueous solubility [12]. As such, recently reported MPs with passive permeability continue to be dominated by model peptides devoid of any biological activity [20-24] but with a few notable exceptions. Hayashi et al. [25] designed cycloheptapeptidyl inhibitors of nicotinamide N-methyltransferase (NNMT). The most potent inhibitor (compound 1, Table 1) inhibited NNMT activity in HEK293 cells (IC50 = 0.77 μM). Compound 1 features four proline analogs and an Nα-methylation to minimize the number of backbone -NH’s. Although NNMT is not a PPI target, this work nevertheless demonstrates that relatively large MPs (MW > 1200) can be rendered cell permeable by passive diffusion and biologically active. Another class of cell-permeable macrocycles is cyclic peptoids and other peptidomimetics that have a reduced number of backbone - NH’s. Using in silico design, Schneider et al. [26] developed a cyclic peptoid (compound 2) targeting the β-catenin/TCF interaction, a critical modulator of Wnt signaling pathway. The peptoid inhibited Wnt pathway in a luciferase reporter assay (IC50 = 0.11 μM) as well as in a zebrafish model. Sulfono-γ-AApeptides, oligomers of N-sulfono-acylated N-aminoethyl amino acids, represent yet another class of cell-permeable and proteolytically stable Nα-substituted peptidomimetics. They fold into rigid and structurally defined helices with unique helical parameters that may be employed to disrupt α-helix-mediated PPIs [27]. Sulfono-γ-AApeptides demonstrate cell permeability owing to their pronounced helical conformation and a decrease in availability of amide bonds. Cai and co-workers rationally designed left- and right-handed sulfono-γ-AApeptidyl inhibitors of the p53/MDM2 (compound 3) [28, 29] and BCL9/β-catenin PPIs [30]. Given the demand of balancing multiple and often conflicting parameters, it is unclear whether passively permeable MPs will provide a general modality for intracellular targets.
Table 1.
Representative cell-permeable MPs reported since 2017. CPP motifs are highlighted in green color.
| Compd No. |
Compound structure | Target (KD
or IC50 value for target) |
Biological activity |
Cell entry mechanism |
Refs |
|---|---|---|---|---|---|
| 1 |
|
NNMT(IC50 = 1.9 nM for human NNMT) | In vitro (IC50 = 0.77 μM for NNMT inhibition in HEK293 cells) | Passive diffusion | [25] |
| 2 |
|
β-catenin (IC50 = 5.4 μM) | In vitro; in vivo (rescued eye development in zebrafish model) | Passive diffusion | [26] |
| 3 |
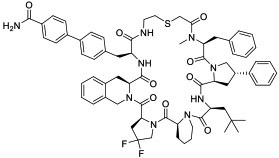
|
MDM2 (KD = 26 nM) | In vitro by stapled analog (activated p53 in U2OS cells at 30 μM) | Potentially passive diffusion | [28] |
| 4 |
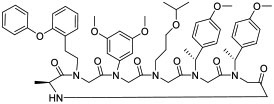
|
TEAD (KD = 0.7 pM for human TEAD) | In vitro (activated Hippo-associated genes in cardiomyocytes at 30 μM) | Endocytosis | [33] |
| 5 |
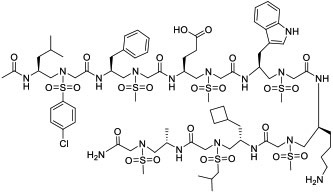
|
RbAp48 (KD = 8.6 nM) | In vitro (increased p53 level in U2OS cells) | Endocytosis | [34] |
| 6 |
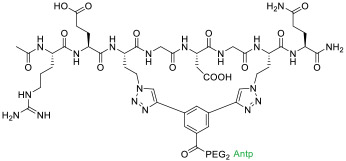
|
Tankyrase (KD = 440 nM) | In vitro (decreased Wnt reporter activity in HEK293T cells [IC50 ≈ 50 μM]) | Endocytosis | [36] |
| 7 |

|
Jun subunit of transcription factor activator protein-1 (KD =2 μM) | In vitro (antiproliferative activity against MCF-7 and ZR75-1 breast cancer cells) | Endocytosis | [37] |
| 8 |

|
HDAC (IC50 = 99-303 nM for different HDAC isoforms) | In vitro; in vivo (TGI @ 50 mg/kg in PA-1 and NTERA-2 mouse xenograft models) | Endocytosis | [38] |
| 9 |
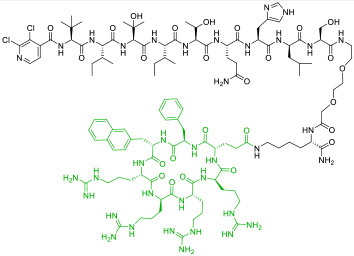
|
Calcineurin (KD = 16 nM) | In vitro; in vivo (prevented acute lung injury @ 3-5 mg/kg in a mouse model) | Endocytosis | [41] |
| 10 |
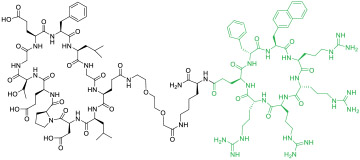
|
Keapl (IC50 = 153 nM) | In vitro (inhibited Keap1/Nrf2 PPI in ARE reporter-HepG2 cell line [EC50 ≈ 1.1 μM]) | Endocytosis | [43] |
| 11 |

|
Pin1 (IC50 = 220 nM) | In vitro (inhibited the proliferation of HeLa cervical cancer cells) | Endocytosis | [46] |
| 12 |
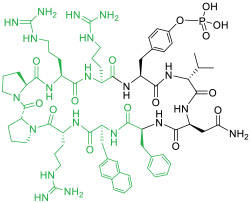
|
Grb2 SH2 domain (IC50 = 400 nM) | In vitro (inhibited Grb2/Ras signaling in MDA-MB-468 breast cancer cells [IC50 ≈ 15 MM]) | Endocytosis | [47] |
| 13 |
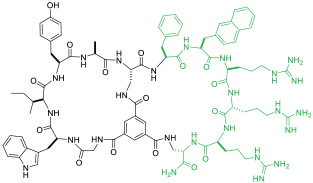
|
NEMO(IC50 = 1.0 μM) | In vitro (inhibition of TNFα-induced NF-κB activation [IC50 = 10 μM]) | Endocytosis | [49] |
| 14 |
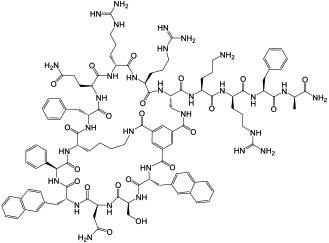
|
K-, H-, and N-Ras (KD = 21 nM for KRasG12V-GppNHp) | In vitro; in vivo (TGI @ 1-5 mg/kg in A549 and H358 mouse xenograft models) | Endocytosis | [50] |
| 15 |
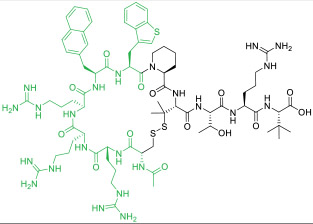
|
PDZ domain of CFTR-associated ligand (KD = 6 nM) | In vitro; ex vivo (improved ion channel activity of F508del-CFTR by 3-fold [EC50 ≈ 10 nM]) | Endocytosis | [51] |
| 16 |
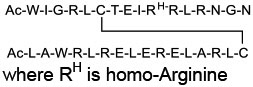
|
Ras (KD = 2 μM for HRas) | In vitro (antiproliferative activity in Ras mutant cancer cells) | Endocytosis | [53] |
| 17 |
|
Bcl2A1 (apparent Ki <0.1 nM) | In vitro (covalent modification of Bcl2A1 in U937 lymphoma cells) | Unknown | [68] |
| 18 |

|
β-catenin (KD = 123 nM) | In vitro (antiproliferative effect in Wnt-addicted colorectal cancer cells DLD-1 and SW480) | Unknown | [69] |
| 19 |
|
MDM2/X (KD = 0.87 and 3.9 nM for MDM2 and MDMX) | In vitro (antiproliferative effect in HCT116 cells [IC50 ≈ 25 μM]) | Unknown | [71] |
| 20 |
|
β-catenin (KD = 91 nM) | In vitro (inhibition of Wnt signaling in HEK293T reporter cell line [IC50 = 8 μM]) | Unknown | [74] |
| 21 |
|
Keap1 (KD = 35 nM) | In vitro (increased the expression of Nrf2-regulated genes in HEK293T cells) | Unknown (potentially endocytosis) | [79] |
| 22 |
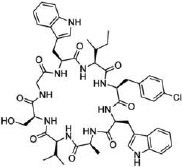
|
UEV domain of TSG101 protein (KD = 11.9 μM) | In vitro (viruslike particle budding assay in HEK293T cells [IC50 ≈ 2 μM]) | Unknown | [83] |
| 23 |
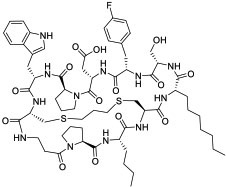
|
KRasG12D (EC50 = 22 nM) | In vitro; in vivo (TGI @40 mg/kg in PANC-1 mouse xenograft model) | Unknown | [84] |
| 24 |
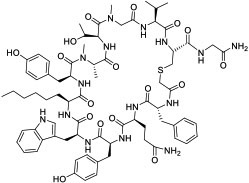
|
K48Ub tetramer (KD = 9 nM) | In vitro; in vivo (TGI @1 mg/kg in mouse model) | Unknown | [85] |
| 25 |
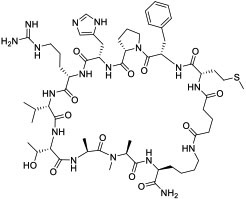
|
LC (KD = 120 and 192 nM for LC3A and LC3B, respectively) | In vitro (inhibited autophagy in HeLa cervical cancer cells) | Unknown | [87] |
| 26 |
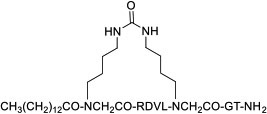
|
MyD88 | In vivo (reduced disease severity @ 10 mg/kg in sclerosis mouse models) | Unknown | [88] |
MPs That Enter the Cell by Endocytosis
To increase their cell-permeability, MPs have been covalently attached to CPPs to form CPP-MP conjugates or integrated with CPPs to produce CPP-MP hybrids (Figure 1) [2, 6, 7]. The advantage of CPP-MP conjugates is that they are straightforward to design, as each unit (CPP, MP, or linker) functions independently (usually) and can be individually optimized. Moreover, the same CPP may be used to deliver many different MPs. CPP-MP hybrids, on the other hand, allow the dual use of structural elements for both cell entry and target engagement. Compared to CPP-MP conjugates, CPP-MP hybrids are more challenging to design; but once successfully designed, they usually have simpler structures, making them easier and less expensive to produce. Both CPP-MP conjugates and hybrids are expected to enter cells by endocytosis followed by endosomal escape, at least at lower concentrations [12]. However, one should always bear in mind that the combination of a CPP with a MP may mutually interfere with each other’s function or result in off-target binding.
Figure 1.

Schematic representation of cargo delivery modes of CPPs. A cell-impermeable peptidyl cargo (blue) is conjugated to a CPP motif (green) to form cell-permeable CPP-MP conjugates or hybrids that can reach and modulate their intracellular PPI targets. (a) Generation of CPP-MP conjugates by covalent attachment of a linear or cyclic CPP to a cargo (linear, cyclic, or stapled peptide). (b) Formation of CPP-MP hybrids by insertion of a small peptidyl cargo into a cyclic CPP (endocyclic delivery). (c) Fusion of a cyclic CPP with an MP cargo results in a bicyclic hybrid (bicyclic delivery). (d) Reversible cyclization of a CPP-cargo fusion peptide into a monocyclic peptide via a disulfide bond. (e) Reversible bicyclization of a CPP-cargo fusion peptide into a bicyclic peptide via two disulfide bonds.
CPP-MP conjugates
Linear CPPs including Tat, penetratin, and oligoarginines remain popular choices for intracellular delivery of MPs including stapled peptides (SPs). SPs are stabilized α-helical peptides with their side chains on the same helical face (usually at positions i and i+4 or i+7) covalently crosslinked and represent a special type of MPs [31]. Because of their increased target binding affinity, proteolytic stability, and in some cases intrinsic cell permeability, SPs have become a popular modality for inhibiting intracellular PPIs mediated by α-helical motifs (see below). However, like MPs, most SPs are either impermeable to the cell membrane or have very limited cell permeability.
Depending on the specific structure of MPs/SPs, a CPP may be covalently attached to the N-terminus, C-terminus, a side chain of the MP/SP, or in the case of SPs, to the stapling unit. For example, Tat has been conjugated to linear and cyclic peptidyl inhibitors against the dimerization of Leishmania infantum trypanothione reductase (TryR) [32], a cyclic two-helix inhibitor against the interaction between transcription factor TEAD and its co-repressor VGL4 (compound 4, Table 1) [33], and a bicyclic peptidyl inhibitor targeting the interface between RbAp48 (KD = 8.6 nM) and scaffold protein MTA1 (compound 5) [34]. All three Tat-MP conjugates displayed biological activities in cell culture. Similarly, penetratin and nonaarginine were employed to improve the cellular entry of bicyclic peptidyl inhibitors of Grb7-SH2 domain [35] and MP inhibitors of tankyrase (compound 6) [36]. Baxter et al. [37] conjugated Tat and a nuclear localization signal (NLS) to the C-terminus of a lactam-stapled peptidyl inhibitor of Jun (KD = 2 μM) (20-NLS-Tat, compound 7, Table 1). The combination of a CPP with NLS promoted efficient cellular uptake and nuclear localization in breast cancer cells resulting in the decrease in cell proliferation. Conjugation of oligoarginines as short as R3 to the stapling unit of SPs against protein kinase CK2 [38] or the N-termini of SP inhibitors against histone deacetylase (HDAC) [39] greatly increased their cell permeability. One of the most active HDAC inhibitors, 16cyc-HxA (compound 8), resulted in cell-cycle arrest and apoptosis of ovarian teratocarcinoma PA-1 cells with minimal toxicity to normal cells. Intraperitoneal injection of the peptide (at 50 mg/kg) inhibited tumor growth by 80-90% in PA-1 and testicular embryonic carcinoma mouse xenograft models. However, in vivo application of linear CPPs is often complicated by their poor proteolytic stability and low cytosolic delivery efficiencies (typically <5%) [8, 40].
To overcome the limitations of linear CPPs, cyclic CPPs have recently been used to deliver linear peptides (LPs) [41,42], MPs [43], and SPs [44,45]. Dougherty et al. [41] discovered a nonapeptidyl inhibitor against the NFAT-binding site of calcineurin as a novel anti-inflammatory agent. Conjugation of cyclic CPP9 to its C-terminus generated CNI103 (compound 9, Table 1) as a metabolically stable (serum t1/2 >24 h), cell permeable, and potent inhibitor of the calcineurin/NFAT interaction in vitro (KD = 16 nM). Intranasal or intravenous administration of CNI103 (at 3-5 mg/kg) prevented lipopolysaccharide-induced acute lung injury in a mouse model. Similarly, conjugation of a previously reported MP inhibitor of the Kelch-like ECH-associated protein 1 (Keap1; KD = 18 nM) to CPP9 increased its cytosolic entry efficiency by 98-fold (compound 10) [43]. Finally, Dougherty et al. [44] conjugated CPP9 to a dozen different SPs, at their N-terminus, C-terminus, or stapling unit. The resulting CPP-SP conjugates were all highly cell permeable, irrespective of the intrinsic permeability of the SPs. These studies suggest that conjugation with a cyclic CPP represents a general solution to the intracellular delivery of LPs, MPs, and SPs.
CPP-MP hybrids
Small peptidyl cargos (≤5 aa) may be inserted into a cyclic CPP to form monocyclic CPP-MP hybrids (Figure 1b, endocyclic delivery). Bedewy et al. [46] designed a cycloheptapeptidyl inhibitor of peptidyl-prolyl isomerase Pin 1 as the smallest, biologically active CPP-MP hybrid known to date (compound 11, Table 1). MP 11, in which the naphthylalanine (Nal) residue performs the dual function of Pin1 binding and cell penetration. This method is, however, less effective for larger cargos, which greatly decrease the conformational rigidity and cell entry efficiency of cyclic CPPs. To accommodate larger peptidyl cargos, Wen et al. [47] introduced a D-Pro-L-Pro motif, which induces β-hairpin structures [48], into a CPP-MP hybrid. It was anticipated that a CPP containing the D-Pro-L-Pro motif would be conformationally constrained and retain high cell entry efficiency, even after the insertion of larger cargos. Through this approach, the investigators developed several cell-permeable MPs of large ring sizes, including a cyclodecapeptidyl inhibitor against the Grb2 SH2 domain (compound 12), although the generality of this approach remains to be demonstrated.
A more general approach to cell-permeable CPP-MP hybrids involves fusing cyclic CPPs and MPs to form bicyclic peptides, in which one ring is primarily responsible for cellular uptake while the other for target binding (Figure 1c, bicyclic delivery). Rhodes et al. [49] designed a bicyclic peptide library, in which one ring contained randomized peptide sequences for potential binding to targets of interest, while the other ring featured a family of CPP motifs for cell penetration. Screening of the library against NF-κB essential modulator (NEMO) identified a cell-permeable inhibitor (compound 13, Table 1) of the NEMO/IKKβ interaction (IC50 = 1.0 μM). Interestingly, alanine scanning revealed that three of the CPP residues contributed significantly to NEMO binding. Buyanova et al. [50] discovered a metabolically stable (serum t1/2 >24 h), cell-permeable, and potent bicyclic peptidyl pan-Ras inhibitor, B4-27 (compound 14; KD = 21 nM for KRasG12V-GppNHp), in which the cell-penetrating and Ras-binding sequences are fully integrated. SAR studies demonstrated that several residues of B4-27 (e.g., D-Arg, D-Phe) were critical for both KRas binding and cellular uptake. B4-27 binds selectively to the active (GTP-bound) form of K-, H-, and N-Ras isoforms, blocks their interaction with downstream effectors including Raf and PI3K, and induces apoptosis of Ras-mutant cancer cells. In A549 and H358 lung cancer mouse xenograft models, B4-27 effectively suppressed tumor growth at ≤5 mg/kg daily doses. Since the cargo does not affect the structure of the CPP ring, bicyclic delivery can in principle accommodate cargos of any size or sequence.
Some proteins require linear peptidyl ligands and cannot be targeted by the above CPP-MP hybrids. To this end, a reversible cyclization strategy has been developed to enhance the cell permeability and metabolic stability of peptidyl therapeutics (Figure 1d) [51, 52]. In brief, a CPP motif is conjugated to the N- or C-terminus of a linear peptidyl ligand and the fusion peptide is then cyclized via an intramolecular disulfide bond. Upon entering the cytosol of a target cell, the disulfide is reduced by intracellular glutathione to regenerate the linear peptide as the active drug. Dougherty et al. [51] designed a highly potent, selective, cell-permeable, and metabolically stable peptidyl inhibitor, PGD97 (compound 15), against the PDZ domain of CFTR-associated ligand (CAL; KD = 6 nM). PGD97 increased the cell surface expression of CFTR by blocking the CAL/CFTR interaction and CAL-mediated lysosomal degradation of CFTR. When tested against cystic fibrosis patient-derived primary lung epithelial cells, PGD97 improved the ion channel activity of F508del-CFTR by ~3-fold with an EC50 value of ~10 nM. It is notable that four of the five CPP residues in PGD97 contribute significantly to CAL binding. A reversible bicyclization strategy was also employed to produce a bicyclic peptidyl inhibitor of the NEMO/IKKβ interaction (Figure 1e) [52].
Proteomimetics
Conformationally stabilized peptides mimicking secondary and tertiary protein structures have been developed as PPI inhibitors. Arora and colleagues reported proteomimetic inhibitors of various PPIs termed crosslinked helix dimers (CHDs) by generating synthetic stabilized coiled coil mimics [53-56]. Some CHD examples include CHDSos-5 (compound 16), which mimics Ras effector protein Son-of-Sevenless (Sos) and exhibited selective toxicity towards Ras-mutant cancer cells [53], and CHD3NEMO, which mimics NEMO and blocks its interaction with vFLIP (Ki = 6.9 μM) [54], CHD3NEMO significantly reduced tumor growth in a PEL mouse xenograft model at 20 mg/kg via intraperitoneal injection. The authors proposed that CHDs are taken up by macropinocytosis and the uptake rate is dependent on the macropinocytic activity of the cells [55]. The arginine-rich nature of CHDs likely promotes both cellular uptake and endosomal escape. The same group also reported a proteolytically stable cell-permeable α3β chimeric Sos helix mimic that covalently targets RasG12C [56]. Scientists at FogPharma are also designing cell-penetrating miniproteins (CPMPs) to target intracellular PPIs such as β-catenin and Ras (https://fogpharma.com/#science).
MPs That Enter the Cell by Yet Unknown Mechanisms
Stapled peptides
Some SPs are intrinsically cell permeable, although their exact mechanism of cellular entry remains unknown. A hydrocarbon-stapled peptide against the MDM2/p53 interaction, ALRN-6924, is currently in phase II clinical trials [57]. This finding has inspired researchers to search for cell-permeable SPs against other intracellular PPI targets. These efforts have led to highly potent, biologically active (in cellular assays) hydrocarbon-stapled peptidyl inhibitors against a variety of intracellular PPI targets including Mcl-1 [58], the nuclear receptor coactivator 1 (NCOA1) [59], small GTPase Rab25 [60], cytokinesis regulating protein UNC119 [61], eukaryotic translation initiation factor 4E (eIF4E) [62], and cell-cycle regulator protein Id [63]. In general, hydrocarbon or other hydrophobic staples promote cellular uptake more effectively than hydrophilic ones [64, 65], although excessive hydrophobicity and positive charge may result in membrane leakage [66,67].
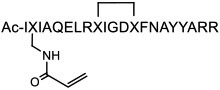
Researchers have explored innovative approaches to improving the potency, selectivity, and/or cell permeability of SPs. Fairlie and colleagues [68] appended an acrylamide warhead to an SP against oncogenic protein Bcl2A1 to increase its residence time on the target. The BimBH3 derivative (compound 17, Table 1) entered U937 lymphoma cells, became localized in the mitochondria (where Bcl2 proteins are mainly located), and covalently bound to Cys55 within the BH3-binding site of Bcl2A1. Grossman and colleagues [69] reported an integrative strategy to enhance the cellular uptake of poorly permeable SPs, including N-terminal modification with NLS and substitution of homoarginine for arginine. The optimized peptide against the β-catenin/TCF interaction, NLS-StAx-h (compound 18), bound to β-catenin with a KD of 123 nM, showed good cytosolic exposure, inhibited Wnt signaling in cell culture, and reduced the proliferation and migration of colorectal cancer cells. Partridge and colleagues [67] generated hydrocarbon-stapled D-peptides to simultaneously improve target binding, proteolytic resistance, and cell permeability. Pentelute and co-workers [70] developed a solution-phase library screening platform to rapidly discover peptidyl PPI inhibitors containing non-canonical amino acids. This platform was applied to identify a series of high-affinity, unnatural ligands that were further engineered into macrocyclic inhibitors of the p53/MDM2 interaction or the dimerization of a HIV capsid protein.
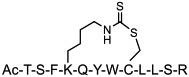
Efforts have also been made to develop alternative, simpler (and less costly) stapling methods. Li et al. [71] reported a dithiocarbamate (DTC)-based stapling method and applied it to generate peptidyl inhibitors of MDM2/X. One such inhibitor, DTCPMI (compound 19), showed a 50-fold tighter binding to MDM2 and MDMX (KD = 0.9 and 3.9 nM, respectively) than its linear counterpart. DTCPMI readily entered the mammalian cell and reduced the viability of cancer cells in a p53-dependent manner (IC50 = 25 μM). Zhang et al. [72] developed a solid-phase synthesis of bisthioether-stapled peptides of various architectures, including single-turn, double-turn, and double-stapled peptides. Pentelute and co-workers [73] demonstrated that perfluoroarene-based macrocyclic variant of Bim BH3 SP can cross the blood-brain barrier.
β-Sheet mimetics
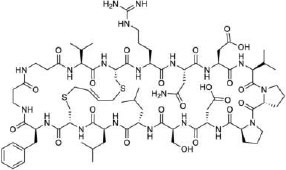
Compared to α-helical peptides, β-sheet structures remain relatively unexplored as PPI inhibitors. Most β-sheet mimetics are β-hairpin peptides consisting of two antiparallel β-strands connected by a turn. Grossman and co-workers [74] designed β-sheet mimicking bicyclic peptides to target the β-catenin/TCF interaction. The bicyclic β-sheet mimetic contained a D-Pro-L-Pro motif and was bicyclized using a butene bridge resulting in a pronounced β-sheet character, which may contribute to cell penetration. The most potent peptide A-b6 (compound 20, Table 1) bound β-catenin (KD = 91 nM) and inhibited Wnt signaling in a cell-based assay. The importance of a β-strand conformation was also demonstrated by Nadal-Bufi et al. [75], who grafted peptidyl inhibitors that block the oligomerization of lactate dehydrogenase 5 (LDH5) into a cyclic cell-penetrating β-hairpin scaffold to generate a cell-permeable LDH5 inhibitor of modest potency (IC50 = 2.5 μM). In addition to the β-strand conformation, the inhibitor contained five arginine residues and hydrophobic aromatic residues (e.g., L-Phe and L-Trp), which were important for its cell-penetrating properties. These structural features suggest that the LDH5 inhibitor likely enters the cell by endocytosis and the β-hairpin structure presumably constrains the CPP motif into a proper membrane-binding conformation(s), analogous to that of the Grb2 SH2 domain inhibitor (compound 12) [47].
Cyclotides
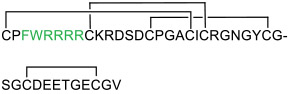
Cyclotides are plant-derived MPs of ~30 aa containing a head-to-tail cyclized backbone and multiple intramolecular disulfides [76]. Thanks to their exceptional stability and modest cellular permeability, cyclotides have been used as a scaffold for grafting of peptidyl inhibitors. Recent examples of this grafting strategy include a cell-permeable kalata B1-based cyclotide inhibitor of 14-3-3 proteins [77] and a cell-permeable MCoTI-II-based inhibitor of kallikrein-related peptidase 4 (KD = 0.1 nM) [78]. Another interesting strategy combined MCoTI-II grafting with the introduction of a CPP motif, FΦRRRR, resulting in a cell-permeable inhibitor of the Keap1/Nrf2 interaction, MCNr-2c (compound 21) [79]. Although the specific mechanism of cellular uptake of cyclotides remains unknown, there is some evidence that internalization of cyclotides occurs via multiple endocytic pathways and is dependent on the overall positive charge of the native sequences [78, 80-82].
Other cell-permeable MPs
Screening of combinatorial libraries has resulted in many highly potent MPs against PPI targets, some of which are cell permeable by yet unknown mechanisms. For example, Tavassoli and colleagues [83] reported a cell-permeable cyclooctapeptidyl inhibitor of the p6/UEV interaction (compound 22, IC50 = 5.4 μM) which reduced the release of virus-like particles in a cell-based assay. Through screening of a phage-display library and optimization by medicinal chemistry, Sakamoto et al. [84] developed a cell-permeable bicyclic peptide (KS-58, compound 23) which bound KRasG12D with an EC50 of 22 nM, inhibited Ras signaling and the proliferation of KRasG12D-mutant cancer cells in vitro, and suppressed tumor growth in a PANC-1 mouse xenograft model. By employing the RaPID system, Rogers et al. [85] discovered a cycloundecapeptide (compound 24) which bound to K48-linked ubiquitin (K48Ub) chains, caused the accumulation of Ub conjugates, induced apoptosis of cancer cells in vitro, and inhibited tumor growth in a mouse model. Similarly, Nawatha et al. [86] identified MPs that potently and specifically bind to K48Ub chains (e.g., KD = 6 nM for Ub4ix) but not K11Ub or K63Ub chains or mono-Ub. The MPs entered HeLa cells, protected Ub chains from recognition by deubiquitinases or the proteasome, and induced apoptosis of HeLa cells. Gray et al. [87] screened mRNA display libraries and discovered MP inhibitors of the autophagy protein LC3. The most promising candidate, compound 25, bound LC3A and LC3B with nanomolar affinity, entered the cytosol, and inhibited autophagosome formation in HeLa cells. Finally, Dishon et al. [88] demonstrated that N-terminal myristoylation of an MP inhibitor of myeloid differentiation primary response 88 (MyD88), c(myD4-4), rendered it cell permeable and orally bioavailable. Oral administration of Myr-c(MyD4-4) (compound 26) ameliorated disease severity in a mouse model for multiple sclerosis. These MPs violate most of Lipinski’s rules [18] and are not expected to passively diffuse across the cell membrane, suggesting that they may enter the cell by endocytosis and/or binding to membrane transporter proteins [12].
Concluding Remarks and Future Perspectives
Recent advances in MP science and technology have allowed the development of cell-permeable, metabolically stable, potent, and specific MPs against a variety of intracellular PPI targets. Some of these MPs have demonstrated in vivo efficacy, bioavailability, and suitable pharmacokinetics (PK) in animal models. We expect MPs to become a general modality for inhibition of intracellular PPIs and some of the MPs to reach the clinic/market during this decade. Among the different types of MPs, CPP-MP conjugates are most straightforward to design, since highly active CPPs are already available and specific MP ligands against PPI targets can be rapidly discovered by screening MP libraries. These molecules are, however, relatively complex and require lengthy chemical synthesis and usually purification by HPLC. Future innovations in their synthesis and non-chromatographic purification would facilitate their development as therapeutics. On the other hand, CPP-MP hybrids, MPs of passive permeability, and SPs have simpler structures and are easier to produce but are more challenging to design. Elucidation of the mechanism of cell entry (for SPs and other MPs) and improved understanding of the structure-activity relationship of cell penetration should help design the latter classes of MPs. Although cyclic CPPs are orally bioavailable on their own [9], oral bioavailability has rarely been demonstrated for biologically active MPs. Achieving therapeutically relevant levels of oral bioavailability for functional MPs would further unleash the power of this class of molecules. Finally, cell-permeable MPs (by endocytosis) appear to exhibit unique pharmacokinetics [41] that is inadequately described by current PK models, which were developed for small molecules and conventional biologics. New analytical methods and PK models need to be developed for this class of drugs. For MPs of passive permeability, the challenge continues to be engineering biological function into these hydrophobic molecules.
Figure I.

Schematic representation of the structures and mechanism of action of CPPs. (a) Structures of cyclic CPP9 and CPP12, two highly efficient CPPs. (b) CPPs (green stars) enter the cell primarily via endocytosis at low concentrations (<10 μM) but can also undergo direct translocation at high CPP concentrations (typically ≥10 μM). Inside the endosome, CPPs bind to the endosomal membrane and cluster into CPP-enriched lipid domains, which subsequently bud off as small vesicles. The budded vesicles disintegrate into lipid/peptide aggregates releasing the intraluminal contents into the cytosol. The molecular mechanism of direct translocation remains poorly defined.
Outstanding Questions.
Can MPs achieve tissue selectivity?
Can MPs be orally bioavailable?
How are MPs eliminated from the body?
Highlights.
Macrocyclic peptides are an emerging class of drug modality for PPI inhibition
Rational design and library screening are established to generate lead peptides
Membrane permeability can be attained by passive diffusion or endocytosis
Integration of membrane permeability and target binding is essential
Acknowledgements
The work in our laboratory was supported by the National Institutes of Health (GM122459 and CA234124). We thank Pei group members and collaborators for their contributions to the projects.
Glossary
- Alanine scanning
a mutagenesis method during which an amino acid is mutated to alanine to determine its contribution to a given function
- Apoptosis
a process of programmed cell death used by multicellular organisms
- Combinatorial Library
a large collection of structurally diverse compounds prepared from a small set of building blocks
- Cytosolic Entry Efficiency
the ratio of intracellular over extracellular concentration of a biomolecule (e.g., a CPP)
- Endocytosis
an energy-dependent process occurring at the cell surface and involving generation of small vesicles that transport cargo molecules into the cell
- Direct Translocation
an energy-independent process by which a molecule enters the cytosol by directly traversing the plasma membrane
- Endosomal Escape
a process by which a molecule exits the endosome after endocytic uptake
- mRNA display
a technology that enables in vitro selection and directed evolution of peptides/proteins that are covalently linked to their coding mRNAs
- Nuclear localization signal (NLS)
a short peptide sequence (e.g., PKKKRKV) that mediates direct import of proteins (or other cargo) into the nucleus
- Oral Bioavailability
fraction of an orally administered drug that reaches systemic circulation; usually positively correlated with membrane permeability
- Passive Diffusion
an energy-independent process by which a molecule can cross the plasma membrane
- Peptidomimetic
a compound that mimics the mode of action of a bioactive peptide but contains various modifications that improve the metabolic stability, bioavailability, and/or target binding affinity
- Peptoid
an oligomer of N-alkylated glycines
- Protein-Protein Interaction (PPI)
physical interaction between two or more proteins that execute and/or regulate various biological processes
- RaPID system
an in vitro system that combines mRNA display with flexible in vitro translation (FIT) to generate and screen large peptide libraries
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest
DP is a co-founder and holds equity of Entrada Therapeutics.
References
- 1.Verdine GL and Walensky LD (2007) The challenge of drugging undruggable targets in cancer: lessons learned from targeting BCL-2 family members. Clin. Cancer Res 13, 7264–7270. [DOI] [PubMed] [Google Scholar]
- 2.Dougherty PG et al. (2017) Macrocycles as protein-protein interaction inhibitors. Biochem. J 474, 1109–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Peacock H and Suga H (2021) Discovery of de novo macrocyclic peptides by messenger RNA display. Trends Pharmacol. Sci 42, 385–397. [DOI] [PubMed] [Google Scholar]
- 4.Deyle K et al. (2017) Phage selection of cyclic peptides for application in research and drug development. Acc. Chem. Res 50, 1866–1874. [DOI] [PubMed] [Google Scholar]
- 5.Tavassoli A (2017) SICLOPPS cyclic peptide libraries in drug discovery. Curr. Opin. Chem. Biol 38, 30–35. [DOI] [PubMed] [Google Scholar]
- 6.Qian Z et al. (2017) Targeting intracellular protein-protein interactions with cell-permeable cyclic peptides. Curr. Opin. Chem. Biol 38, 80–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.González-Muñiz R et al. (2021) Modulating protein-protein interactions by cyclic and macrocyclic peptides. Prominent strategies and examples. Molecules 26, 445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.LaRochelle JR et al. (2015) Fluorescence correlation spectroscopy reveals highly efficient cytosolic delivery of certain penta-arg proteins and stapled peptides. J. Am. Chem. Soc 137, 2536–2541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Qian Z et al. (2016) Discovery and mechanism of highly efficient cyclic cell-penetrating peptides. Biochemistry 55, 2601–2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Palm-Apergi C et al. (2012) Do cell-penetrating peptides actually “penetrate” cellular membranes? Mol. Ther 20, 695–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Takeuchi T and Futaki S (2016) Current understanding of direct translocation of arginine-rich cell-penetrating peptides and its internalization mechanisms. Chem. Pharm. Bull 64, 1431–1437. [DOI] [PubMed] [Google Scholar]
- 12.Dougherty PG et al. (2019) Understanding cell penetration of cyclic peptides. Chem. Rev 119, 10241–10287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sahni A et al. (2020) Cell-penetrating peptides escape the endosome by inducing vesicle budding and collapse. ACS Chem. Biol 15, 2485–2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Peraro L et al. (2018) Cell penetration profiling using the chloroalkane penetration assay. J. Am. Chem. Soc 140, 11360–11369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Peier A et al. (2021) NanoClick: a high throughput, target-agnostic peptide cell permeability assay. ACS Chem. Biol 16, 293–309. [DOI] [PubMed] [Google Scholar]
- 16.Teo SLY et al. (2021) Unravelling cytosolic delivery of cell penetrating peptides with a quantitative endosomal escape assay. Nat. Commun 12, 3721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lucchino M et al. (2021) Absolute quantification of drug vector delivery to the cytosol. Angew. Chem. Int. Ed 60, 14824–14830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lipinski CA et al. (2001) Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev 46, 3–26. [DOI] [PubMed] [Google Scholar]
- 19.Rezai T et al. (2006) Conformational flexibility, internal hydrogen bonding, and passive membrane permeability: Successful in silico prediction of the relative permeabilities of cyclic peptides. J. Am. Chem. Soc 128, 14073–14080. [DOI] [PubMed] [Google Scholar]
- 20.Furukawa A et al. (2020) Drug-like properties in macrocycles above MW 1000: Backbone rigidity versus side-chain lipophilicity. Angew. Chem. Int. Ed 59, 21571–21577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kelly CN et al. (2021) Geometrically diverse lariat peptide scaffolds reveal an untapped chemical space of high membrane permeability. J. Am. Chem. Soc 143, 705–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Golosov AA et al. (2021) Design of thioether cyclic peptide scaffolds with passive permeability and oral exposure. J. Med. Chem 64, 2622–2633. [DOI] [PubMed] [Google Scholar]
- 23.Hickey JL et al. (2016). Passive membrane permeability of macrocycles can be controlled by exocyclic amide bonds. J. Med. Chem 59, 5368–76. [DOI] [PubMed] [Google Scholar]
- 24.Hoang HN et al. (2021). Connecting hydrophobic surfaces in cyclic peptides increases membrane permeability. Angew. Chem. Int. Ed 60, 8385–8390. [DOI] [PubMed] [Google Scholar]
- 25.Hayashi K et al. (2021) Macrocyclic peptides as a novel class of NNMT inhibitors: A SAR study aimed at inhibitory activity in the cell. ACS Med. Chem. Lett 12, 1093–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schneider JA et al. (2018) Design of peptoid-peptide macrocycles to inhibit the β-catenin TCF interaction in prostate cancer. Nat. Commun 9, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sang P et al. (2020) Sulfono-γ-AApeptides as helical mimetics: Crystal structures and applications. Acc. Chem. Res 53, 2425–2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sang P et al. (2020) Rational design and synthesis of right-handed sulfono-γ-AApeptide helical foldamers as potent inhibitors of protein–protein interactions. J. Org. Chem 85, 10552–10560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sang P et al. (2020) α-Helix-mimicking sulfono-γ-AApeptide inhibitors for p53–MDM2/MDMX protein–protein interactions. J. Med. Chem 63, 975–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sang P et al. (2019) Inhibition of β-catenin/B cell lymphoma 9 protein–protein interaction using α-helix–mimicking sulfono-γ-AApeptide inhibitors. Proc. Natl. Acad. Sci. U. S. A 116, 10757–10762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sawyer TK et al. (2018) Macrocyclic α helical peptide therapeutic modality: A perspective of learnings and challenges. Bioorg. Med. Chem 26, 2807–2815. [DOI] [PubMed] [Google Scholar]
- 32.Ruiz-Santaquiteria M et al. (2017) First example of peptides targeting the dimer interface of Leishmania infantum trypanothione reductase with potent in vitro antileishmanial activity. Eur. J. Med. Chem 135, 49–59. [DOI] [PubMed] [Google Scholar]
- 33.Adihou H et al. (2020) A protein tertiary structure mimetic modulator of the Hippo signalling pathway. Nat. Commun 11, 5425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hart P et al. (2021) Structure based design of bicyclic peptide inhibitors of RbAp48. Angew. Chem. Int. Ed. Engl 60, 1813–1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Watson GM et al. (2017) Discovery, development, and cellular delivery of potent and selective bicyclic peptide inhibitors of Grb7 cancer target. J. Med. Chem 60, 9349–9359. [DOI] [PubMed] [Google Scholar]
- 36.Xu W et al. (2017) Macrocyclized extended peptides: inhibiting the substrate-recognition domain of tankyrase. J. Am. Chem. Soc 139, 2245–2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Baxter D et al. (2017) Downsizing proto-oncogene cFos to short helix-constrained peptides that bind Jun. ACS Chem. Biol 12, 2051–2061. [DOI] [PubMed] [Google Scholar]
- 38.Iegre J et al. (2019) Efficient development of stable and highly functionalised peptides targeting the CK2α/CK2β protein-protein interaction. Chem. Sci 10, 5056–5063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang D et al. (2019) Stabilized peptide HDAC inhibitors derived from HDAC1 substrate H3K56 for the treatment of cancer stem–like cells in vivo. Cancer Res. 79, 1769–1783. [DOI] [PubMed] [Google Scholar]
- 40.Guidotti G et al. (2017) Cell-penetrating peptides: From basic research to clinics. Trends Pharmacol. Sci 38, 406–424. [DOI] [PubMed] [Google Scholar]
- 41.Dougherty PG et al. (2020) A peptidyl inhibitor that blocks calcineurin–NFAT interaction and prevents acute lung injury. J. Med. Chem 63, 12853–12872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cerulli RA et al. (2020) Cytosolic delivery of peptidic STAT3 SH2 domain inhibitors. Bioorg. Med. Chem 28, 115542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Salim H. et al. (2020). Development of a cell-permeable cyclic peptidyl inhibitor against the Keap1-Nrf2 interaction. J. Org. Chem 85, 1416–1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dougherty PG et al. (2019) Enhancing the cell permeability of stapled peptides with a cyclic cell-penetrating peptide. J. Med. Chem 62, 10098–10107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Philippe GJ-B et al. (2021) Angler peptides: macrocyclic conjugates inhibit p53:MDM2/X interactions and activate apoptosis in cancer cells. ACS Chem. Biol 16, 414–428. [DOI] [PubMed] [Google Scholar]
- 46.Bedewy W et al. (2017) Generation of a cell-permeable cycloheptapeptidyl inhibitor against the peptidyl–prolyl isomerase Pin1. Org. Biomol. Chem 15, 4540–4543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wen J et al. (2020) Rational design of cell-permeable cyclic peptides containing a D-Pro-L-Pro motif. Bioorg. Med. Chem 28, 115711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Robinson JA (2008) Beta-hairpin peptidomimetics: design, structures and biological activities. Acc. Chem. Res 41, 1278–88. [DOI] [PubMed] [Google Scholar]
- 49.Rhodes CA et al. (2018) Cell-permeable bicyclic peptidyl inhibitors against NEMO-IκB kinase interaction directly from a combinatorial library. J. Am. Chem. Soc 140, 12102–12110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Buyanova M et al. (2021) Discovery of a bicyclic peptidyl pan-Ras inhibitor. J. Med. Chem 64, 13038–13053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dougherty PG et al. (2020) Cyclic peptidyl inhibitors against CAL/CFTR interaction for treatment of cystic fibrosis. J. Med. Chem 63, 15773–15784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Qian Z et al. (2017) Enhancing the cell permeability and metabolic stability of peptidyl drugs by reversible bicyclization. Angew. Chem. Int. Ed 56, 1525–1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hong SH et al. (2021) A Sos proteomimetic as a pan-Ras inhibitor. Proc. Natl. Acad. Sci. U. S. A 118, e2101027118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sadek J, et al. (2020) Modulation of virus-induced NF-κB signaling by NEMO coiled coil mimics. Nat. Commun 11, 1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yoo DY et al. (2020) Macropinocytosis as a key determinant of peptidomimetic uptake in cancer cells. J. Am. Chem. Soc 142, 14461–14471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yoo DY et al. (2020) Covalent targeting of Ras G12C by rationally designed peptidomimetics. ACS Chem. Biol 15, 1604–1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang S et al. (2021) Recent progress and clinical development of inhibitors that block MDM4/p53 protein-protein interactions. J. Med. Chem 64, 10621–10640. [DOI] [PubMed] [Google Scholar]
- 58.Araghi RR et al. (2018) Iterative optimization yields Mcl-1–targeting stapled peptides with selective cytotoxicity to Mcl-1-dependent cancer cells. Proc. Natl. Acad. Sci. U. S. A 115, E886–E895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lee Y et al. (2017) Targeted inhibition of the NCOA1/STAT6 protein-protein interaction. J. Am. Chem. Soc 139, 16056–16059. [DOI] [PubMed] [Google Scholar]
- 60.Mitra S et al. (2017) Stapled peptide inhibitors of RAB25 target context-specific phenotypes in cancer. Nat. Commun 8, 660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cromm PM et al. (2019) Lipidated stapled peptides targeting the acyl binding protein UNC119. ChemBioChem 20, 2987–2990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gallagher EE et al. (2019) Consideration of binding kinetics in the design of stapled peptide mimics of the disordered proteins eukaryotic translation initiation factor 4E-binding protein 1 and eukaryotic translation initiation factor 4G. J. Med. Chem 62, 4967–4978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Roschger C et al. (2017) Targeting of a helix-loop-helix transcriptional regulator by a short helical peptide. ChemMedChem 12, 1497–1503. [DOI] [PubMed] [Google Scholar]
- 64.Peraro L and Kritzer JA (2018) Emerging methods and design principles for cell-penetrant peptides. Angew. Chem. Int. Ed 57, 11868–11881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tian Y et al. (2017) Effect of stapling architecture on physiochemical properties and cell permeability of stapled α-helical peptides: a comparative study. ChemBioChem 18, 2087–2093. [DOI] [PubMed] [Google Scholar]
- 66.Partridge AW et al. (2019) Incorporation of putative helix-breaking amino acids in the design of novel stapled peptides: Exploring biophysical and cellular permeability properties. Molecules 24, 2292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kannan S et al. (2020) Macrocyclization of an all-D linear α-helical peptide imparts cellular permeability. Chem. Sci 11, 5577–5591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.De Araujo AD et al. (2017) Electrophilic helical peptides that bond covalently, irreversibly, and selectively in a protein–protein interaction site. ACS Med. Chem. Lett 8, 22–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Dietrich L et al. (2017) Cell permeable stapled peptide inhibitor of Wnt signaling that targets β-catenin protein-protein interactions. Cell Chem. Biol 24, 958–968. [DOI] [PubMed] [Google Scholar]
- 70.Touti F, et al. (2019) In-solution enrichment identifies peptide inhibitors of protein–protein interactions. Nat. Chem. Biol 15, 410–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Li X et al. (2019) Dithiocarbamate-inspired side chain stapling chemistry for peptide drug design. Chem. Sci 10, 1522–1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhang G et al. (2018) A Solid-phase approach to accessing bisthioether-stapled peptides resulting in a potent inhibitor of PRC2 catalytic activity. Angew. Chem. Int. Ed 57, 17073–17078. [DOI] [PubMed] [Google Scholar]
- 73.Fadzen CM et al. (2017) Perfluoroarene-based peptide macrocycles to enhance penetration across the blood-brain barrier. J. Am. Chem. Soc 139, 15628–15631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wendt M et al. (2021) Bicyclic β-sheet mimetics that target the transcriptional coactivator β-catenin and inhibit Wnt signaling. Angew. Chem. Int. Ed 60, 13937–13944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Nadal-Bufi F et al. (2021) Designed β-hairpins inhibit LDH5 oligomerization and enzymatic activity. J. Med. Chem 64, 3767–3779. [DOI] [PubMed] [Google Scholar]
- 76.De Veer SJ et al. (2019) Cyclotides: from structure to function. Chem. Rev 119, 12375–12421. [DOI] [PubMed] [Google Scholar]
- 77.Hellinger R et al. (2017) Chemical proteomics for target discovery of head-to-tail cyclized mini-proteins. Front. Chem 5, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Swedberg JE et al. (2018) Potent, selective, and cell-penetrating inhibitors of kallikrein-related peptidase 4 based on the cyclic peptide MCoTI-II. ACS Med. Chem. Lett 9, 1258–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yin H et al. (2021) An integrated molecular grafting approach for the design of Keap1-targeted peptide inhibitors. ACS Chem. Biol 16, 1276–1287. [DOI] [PubMed] [Google Scholar]
- 80.Henriques ST et al. (2015) The prototypic cyclotide kalata B1 has a unique mechanism of entering cells. Chem. Biol 22, 1087–1097. [DOI] [PubMed] [Google Scholar]
- 81.Contreras J (2011) Cellular uptake of cyclotide MCoTI-I follows multiple endocytic pathways. J. Control. Release 155, 134–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.D'Souza C et al. (2014) Structural parameters modulating the cellular uptake of disulfide-rich cyclic cell-penetrating peptides: MCoTI-II and SFTI-1. Eur. J. Med. Chem 88, 10–18. [DOI] [PubMed] [Google Scholar]
- 83.Lennard KR et al. (2019) Development of a cyclic peptide inhibitor of the p6/UEV protein-protein interaction. ACS Chem. Biol 14, 1874–1878. [DOI] [PubMed] [Google Scholar]
- 84.Sakamoto K et al. (2020). Generation of KS-58 as the first K-Ras(G12D)-inhibitory peptide presenting anti-cancer activity in vivo. Sci. Rep 10, 21671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Rogers JM et al. (2021) In vivo modulation of ubiquitin chains by N-methylated non-proteinogenic cyclic peptides. RSC Chem. Biol 2, 513–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Nawatha M et al. (2019) De novo macrocyclic peptides that specifically modulate Lys48-linked ubiquitin chains. Nat. Chem 11, 644–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Gray JP et al. (2021) Directed evolution of cyclic peptides for inhibition of autophagy. Chem. Sci 12, 3526–3543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Dishon S et al. (2019) Myristoylation confers oral bioavailability and improves the bioactivity of c(MyD 4-4), a cyclic peptide inhibitor of MyD88. Mol. Pharm 16, 1516–1522. [DOI] [PubMed] [Google Scholar]
- 89.Frankel AD and Pabo CO (1988) Cellular uptake of the tat protein from human immunodeficiency virus. Cell 55, 1189–1193. [DOI] [PubMed] [Google Scholar]
- 90.Green M and Loewenstein PM (1988) Autonomous functional domains of chemically synthesized human immunodeficiency virus Tat trans-activator protein. Cell 55, 1179–1188. [DOI] [PubMed] [Google Scholar]
- 91.Joliot A et al. (1991) Antennapedia homeobox peptide regulates neural morphogenesis. Proc. Natl. Acad. Sci. U. S. A 88, 1864–1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Vivès E et al. (1997) Truncated HIV-1 Tat protein basic domain rapidly translocates through the plasma membrane and accumulates in the cell nucleus. J. Biol. Chem 272, 16010–16017. [DOI] [PubMed] [Google Scholar]
- 93.Derossi D et al. (1994) The third helix of the Antennapedia homeodomain translocates through biological membranes. J. Biol. Chem 269, 10444–10450. [PubMed] [Google Scholar]