

Abstract
Metabolic alterations regulate cancer aggressiveness and immune responses. Given the poor response of pancreatic ductal adenocarcinoma (PDAC) to conventional immunotherapies, we investigated the link between metabolic alterations and immunosuppression. Our metabolic enzyme screen indicated that elevated expression of CD73, an ecto-5’–nucleotidase that generates adenosine, correlates with increased aggressiveness. Correspondingly, we observed increased interstitial adenosine levels in tumors from spontaneous PDAC mouse models. Diminishing CD73 by genetic manipulations ablated in vivo tumor growth, and decreased myeloid-derived suppressor cells (MDSC) in orthotopic mouse models of PDAC. A high-throughput cytokine profiling demonstrated decreased GM-CSF in mice implanted with CD73 knockdowns. Furthermore, we noted increased IFN-γ expression by intratumoral CD4+ and CD8+ T cells in pancreatic tumors with CD73 knockdowns. Depletion of CD4+ T cells, but not CD8+ T cells abrogated the beneficial effects of decreased CD73. We also observed that splenic MDSCs from Nt5e knockdown tumor-bearing mice were incompetent in suppressing T cell activation in the ex vivo assays. Replenishing GM-CSF restored tumor growth in Nt5e knockout tumors, which was reverted by MDSC depletion. Finally, anti-CD73 antibody treatment significantly improved gemcitabine efficacy in orthotopic models. Thus, targeting the adenosine axis presents a novel therapeutic opportunity for improving the anti-tumoral immune response against PDAC.
Keywords: CD73, NT5E, Adenosine, Anti-tumor Immune Response, Pancreatic Cancer
Introduction
Pancreatic cancer is the third leading cause of cancer-associated deaths in the United States, and exhibits a dismal 5-year survival rate of 9% [1]. Pancreatic cancer is predicted to become the second leading cause of cancer-associated deaths in the United States by 2030 [2]. New cases of pancreatic cancer are nearly equal to pancreatic cancer deaths each year, highlighting the urgent need to generate improved therapies [1].
Metabolic reprogramming and immune evasion are two widely recognized hallmarks of cancer [3]. Although a metabolic shift in cancer cells has long been observed [4], recent advances have shown how specific alterations in tumor metabolism can disrupt anti-tumor immunity [5–7]. Cancer cells can disable the effector function of immune cells in the microenvironment by virtue of their increased energy demand and concomitant glucose, glutamine, and oxygen consumption that deprives the immune cells of required nutrients. The tumor microenvironment can also increase expression of immunosuppressive enzymes, such as arginase, indoleamine 2,3-dioxygenase, tryptophan 2,3-dioxygenase, and CD73 [8].
CD73 is a cell surface ecto-5′-nucleotidase that helps clear extracellular adenosine triphosphate (ATP) by converting adenosine monophosphate (AMP) to adenosine [9–11]. ATP is released from dying and stressed cells in a variety of settings, including hypoxic and stressed environments, necrotic core of solid tumors, or as a result of chemotherapy treatment, and can be converted to adenosine. ATP serves as a potent proinflammatory signal that triggers recruitment of neutrophils, eosinophils, dendritic cells (DCs), natural killer cells, and macrophages [12]. Stimulation by ATP can further trigger cytokine release and promote immune clearance [12, 13]. Regulatory T cells (Tregs) are a chief mediator in controlling the inflammatory response, and one key mechanism which they oppose inflammation is through ATP degradation and the ultimate conversion to adenosine by CD73 [9, 14]. Immunosuppressive adenosine can impair multiple immune cells and their responses through several mechanisms [11]. In T cells, adenosine has the ability to counteract the T cell receptor signaling, which can promote T cell anergy and the conversion into inducible Tregs [15–17]. Thus, cancer cell expression of CD73 can promote a highly immunosuppressive microenvironment which may generate resistance to immunotherapy. Despite encouraging results in multiple cancers to popular checkpoint inhibitors, discouraging results were observed in pancreatic cancer [18, 19]. PDAC frequently exists in highly hypoxic microenvironments [20], which promotes CD73 expression [21]. We therefore postulated that CD73-mediated immune suppression directly contributes to aggressiveness in PDAC [18, 19].
Given the wide array of metabolic gene expression perturbations across cancers, we screened The Cancer Genome Atlas (TCGA) data for enzymatic pathways and genes that had the most apparent influence on PDAC patient survival. This screen revealed the importance of nucleotide metabolism, for which CD73 was a top hit. We observed increased tumoral expression of CD73, adenosine levels in PDAC tissues, and confirmed the role of tumoral CD73 expression in patient survival with an independent database. Implantations in syngeneic immune-competent mice revealed a dramatic reduction in mouse tumor burden and improved survival upon Nt5e knockdown in PDAC cells. In contrast, athymic nude mice lacking T cells showed only a marginal alteration. Tumor cytokine profile revealed a decrease in GM-CSF in orthotopically implanted Nt5e knockdown tumors, which correlated with decreased intratumoral MDSCs and increased IFN-γ production by CD4+ and CD8+ T cells in the tumors. Restoring GM-CSF levels increased CD73 knockout tumor burden similar to control, and this was dependent on MDSCs. We furthermore observed that the tumors with stable Nt5e knockdowns were highly sensitive to CD4 depletion. Combination therapy with anti-CD73 antibody and gemcitabine also showed an improvement in tumor volume and survival in orthotopic models of PDAC. Thus, we propose CD73 as a critical immune checkpoint inhibitor in pancreatic cancer that promotes GM-CSF-mediated MDSC recruitment to inhibit CD4+ T cells, promoting tumor growth.
Results
NT5E is one of the strongest metabolic enzyme contributors to overall PDAC patient survival.
To investigate the potential roles metabolic enzymes play in regulating tumor aggressiveness, we sought to correlate the mRNA expression with overall survival through an unbiased screen of TCGA dataset. We chose to examine the patients through two strategies. In the first approach, we separated patients through the interquartile range (IQR) that had either the highest or lowest 25% expression (Supplementary Table 1). The second approach examined if the mRNA expression was statistically increased or decreased compared to normal adjacent tissue with a Z-score ≥2 or ≤−2, respectively (Supplemental Table 2). Significant hits (p<0.05) from both approaches were examined through pathway enrichment to examine if a particular enzyme function is associated with an impact on survival (Fig. 1A). Both approaches revealed pyrimidine metabolism as the top hit and purine metabolism ranked second when combining both approaches. We further investigated the genes that impacted patient survival within these two nucleotide metabolism pathways. Interestingly, enzyme commission (EC) number 3.1.3.5 showed the greatest survival impact in both pyrimidine and purine metabolic pathways when examining Z-score (Supplementary Fig. 1A, B). IQR-based survival separation showed EC 3.1.3.5 had the largest significant survival impact on pyrimidine metabolism and the second best hit in purine metabolism (Supplementary Fig. 1C, D). EC 3.1.3.5 activity is performed by a few enzymes that catalyze a 5’-nucleotidase function.
Figure 1. NT5E is overexpressed and impacts survival in pancreatic cancer.
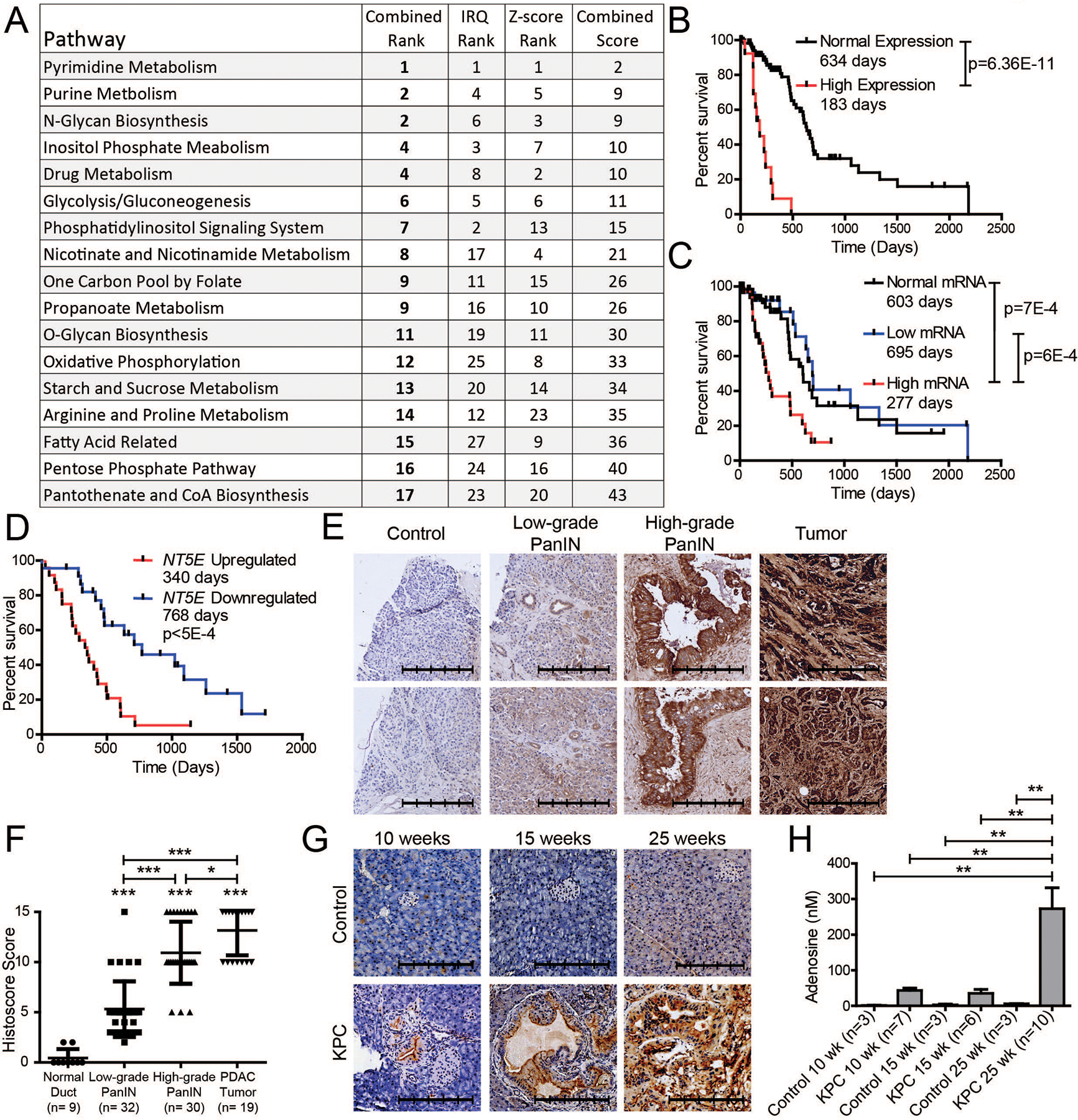
A Pathway enrichment of enzymes contributing to a significant (p<0.05) overall survival (OS) in The Cancer Genome Atlas (TCGA) when the patient’s mRNA expression was separated by Z-score or through the interquartile range (IQR). Pathways were ordinally ranked for each approach and added together to generate a combined score, which was subsequently ranked. B TCGA pancreatic cancer OS when comparing patients two standard deviations above adjacent normal mRNA and those within two standard deviations. C IQR range OS comparing TCGA patients with low (0%−25%), normal (25%−75%), and high (75–100%) NT5E mRNA expression. D International Cancer Genome Consortium (ICGC) OS when comparing patients with high and low IQR NT5E mRNA. E,F Representative immunohistochemistry (IHC) images of CD73 in control, low-grade and high-grade PanINs (E), and tumor tissue used in histological scoring of sections (F). One-way ANOVA with Tukey’s multiple comparison test was performed for significance. G Representative IHC images of Cre control (top) and KPC (bottom) mice pancreas at 10 (control n=3; KPC n=7), 15 (control n=3; KPC n=6), and 25 (control n=3; KPC n=9) weeks after birth, as well as the interstitial fluid levels of adenosine in these mice (H). One-way ANOVA with Tukey’s multiple comparison test was performed for significance. The scale bar denotes 250 μm. Error bars depict the standard error of the mean. *P < 0.05, **P < 0.01, ***P < 0.001.
When ranking the metabolic enzymes’ survival correlation with mRNA utilizing Z-score or IQR approach, NT5E was the only gene that consistently showed a robust correlation across both approaches, as exemplified by being the only gene shared between both methods when comparing the top 10 log-rank p-values of all cohorts together (Supplementary Fig. 1E, F, and Supplementary Tables 1, 2). Interestingly, NT5E is a 5’-nucleotidase, which is the strongest contributor to the survival correlations of EC 3.1.3.5 in Supplementary Fig. 1A–D. It is interesting to note the top ranked enzymes in IQR are GMPS, NT5E, TYMS (Supplementary Fig. 1F), which highlight the significance of nucleotide monophosphate enzymes and nucleotide metabolism pathways on survival (Fig. 1A).
We sought to verify the role of NT5E in survival and observed significantly decreased survival in patients with mRNA levels two standard deviations higher than the normal tissues (Fig. 1B). We also observed a similar significant change for elevated NT5E by the IQR method (Fig. 1C). No difference in survival was seen when comparing patients with normal and low expression of NT5E mRNA. To validate these findings, we examined the ICGC data and noted the same significant trend (Fig. 1D). We therefore hypothesized that elevated NT5E drives tumor progression and correlates with overall patient survival in PDAC.
CD73 expression increases throughout tumor progression.
We sought to determine if CD73, the protein encoded by NT5E, and its metabolite product, adenosine, correlate with tumor progression. We examined pancreatic tumors obtained through the University of Nebraska Medical Center’s Rapid Autopsy Program and observed significantly increased CD73 expression with human PDAC progression from low-grade PanIN to high-grade PanIN to malignant tumor (Fig. 1E, F). Furthermore, increased CD73 expression through tumor progression was observed in our LSL-KrasG12D/+;LSL-Trp53R172H/+;Pdx-1-Cre (KPC) autochthonous mouse model (Fig. 1G). Lastly, we observed that the functional product of CD73, adenosine, was significantly increased in the interstitial fluid of the KPC tumors at a relatively advanced timepoint (Fig. 1H). These data suggest that CD73 becomes progressively elevated in pancreatic cancer.
Knocking down CD73 expression decreases tumor growth and increases survival.
To assess the impact of CD73 on tumor growth and survival, we used KPC1245 cells that were engineered to express luciferase (KPC1245Lum) and knocked down Nt5e by stable short hairpin RNA (shRNA) expression. Knockdowns achieved >95% reduction in mRNA expression compared to the control (Fig. 2A). We performed orthotopic implantations of these cells in syngeneic B6(Cg)-Tyrc−2J/JC57BL/6J mice and observed a significant increase in mice survival and a decrease in tumor volume for both knockdowns (Fig. 2B–D). After re-validating the knockdowns (Fig. 2E), we implanted fewer cells (5×103) to extend survival to evaluate differences at early and late timepoints. Differences were much larger at a later timepoint (Fig. 2F–H). An examination of the tumors through hematoxylin and eosin (H&E) revealed increased levels of intra-tumoral immune cells and a drastic increase in the immune cell penetration of the tissue edge in Nt5e knockdowns (Fig. 2I). Control and Nt5e-b implanted mice with 5×103 cells were examined for long-term survival. Control mice met euthanasia criteria; however, a durable progression-free survival was observed for more than a year in shNt5e-b, in which no tumor was palpable after a year, except for two mice, one of which relapsed around day 43 and another expired 175 days later (Supplementary Fig. 2).
Figure 2. Decreased CD73 improves survival and decreases tumor burden.
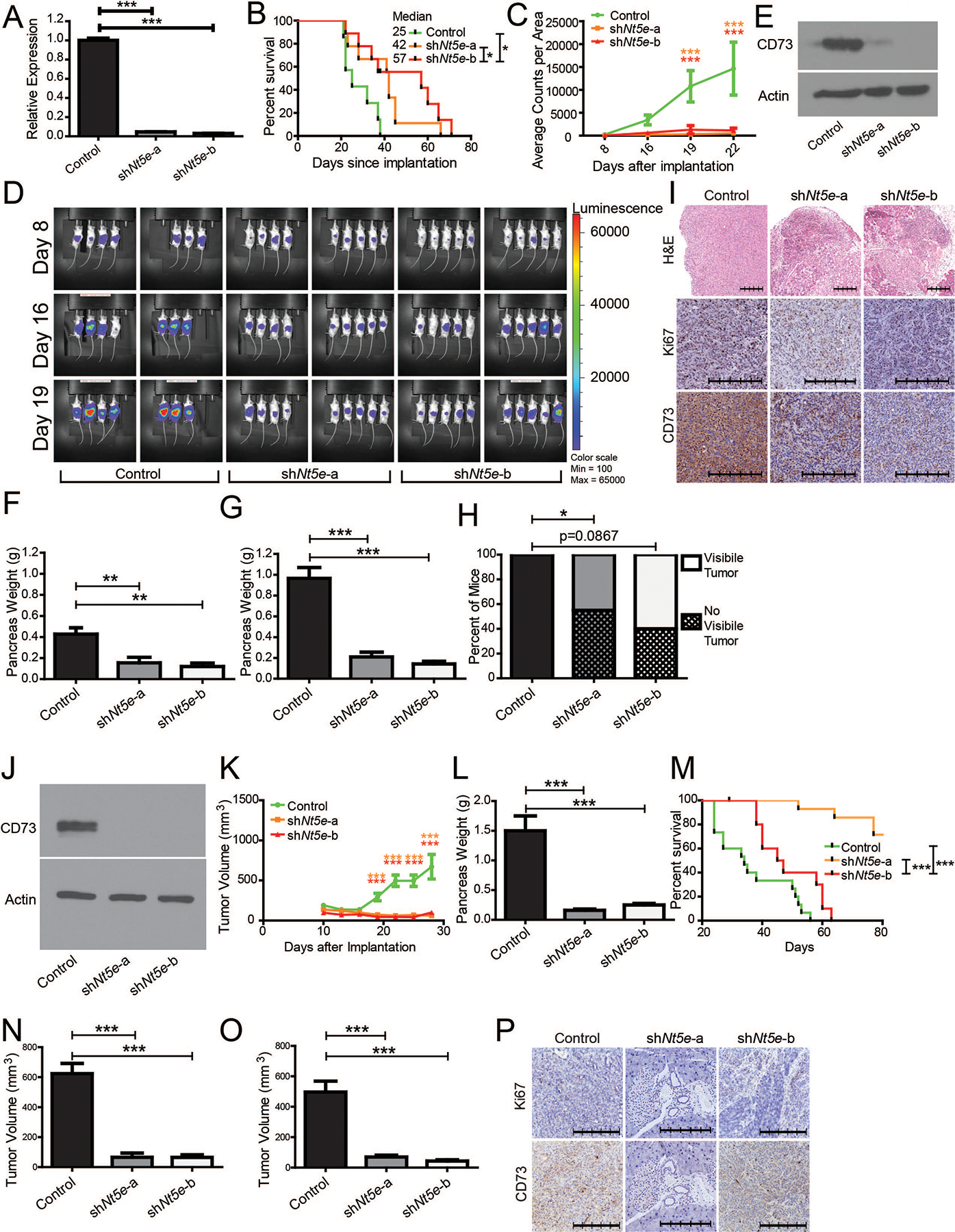
A Nt5e mRNA measured by qPCR in control and Nt5e knockdown KPC1245 cells with four technical replicates each. B Overall survival of B6(Cg)-Tyrc-2J/J mice orthotopically implanted with 5 × 104 syngeneic KPC1245 cells C and tumor burden as measured by luminescence imaging. D Images with luminescence signal indicating tumor burden. E Immunoblot showing CD73 levels in control and Nt5e knockdown KPC1245 cells. F B6(Cg)-Tyrc-2J/J mice were orthotopically implanted with 5 × 103 syngeneic KPC1245 cells and pancreas weight was recorded upon necropsy at day 20 (G) and day 30 (H), with macroscopic evaluation of the excised pancreas at day 30. I Representative pancreas tissue H&E and IHC images from mice necropsied at day 30. J Immunoblot showing CD73 levels in control and Nt5e knockdown KPC1199 cells. K Tumor growth rates in mice orthotopically implanted with 5 × 103 syngeneic KPC1199 cells, as measured by calipers, along with pancreas weight upon necropsy at day 30 (L), and overall survival to day 80 (M). N-O Caliper measurements in male (N) and female (O) B6(Cg)-Tyrc-2J/J mice orthotopically implanted with control and Nt5e knockdown KPC1199 cells at day 25. P Representative IHC images of pancreas tissue. The scale bar denotes 250 μm. Bar charts were compared with one-way ANOVA with Tukey’s multiple comparison test, while longitudinal measurements were compared with two-way ANOVA with Bonferroni’s multiple comparison test for significance. Percent of mice with tumors in panel H were compared with Fisher’s exact test. Error bars depict the standard error of the mean. *P < 0.05, **P < 0.01, ***P < 0.001.
To confirm these results, we utilized another independent cell line, KPC1199, which is derived from a KPC tumor. After confirming Nt5e knockdown (Fig. 2J), we performed orthotopic implantations and observed a very significant decrease in tumor growth, as well as decreased tumor weight at necropsy (Fig. 2K, L). Mice implanted with Nt5e knockdown KPC1199 cells showed improved survival, compared to the controls (Fig. 2M) and the results were consistent between male and female mice (Fig. 2N, O). We also confirmed Nt5e knockdown by evaluating CD73 protein expression in tumors by IHC staining (Fig. 2P). Collectively, these data establish that CD73 expression supports tumor growth and that knocking down Nt5e diminishes tumor burden and increases survival of tumor-bearing mice, possibly through an immune-related function.
Nt5e knockdown increases the activation of CD4+ and CD8+ T cells.
To investigate the mechanistic basis of decreased growth of Nt5e knockdown tumor cells in mice, we investigated the tumoral and circulating immune cells using flow cytometry using the gating strategy described in supplementary figure 3. We observed a significant decrease in the proportion of intratumoral monocytic MDSCs, and a trend toward a decrease in the proportion of granulocytic MDSCs (Fig. 3A, Supplementary Fig. 4) in Nt5e knockdown tumor-bearing mice. We also noted a significant increase in IFN-γ expression in CD8+ and CD4+ T cells from Nt5e knockdown tumor-bearing mice compared to control counterparts (Fig. 3B–C). In parallel, we noted a significant decrease in circulating MDSCs and increased CD8+ and CD4+ T cells in Nt5e knockdown tumor-bearing mice as compared to control mice (Fig. 3D–G). Next, we noted an increased frequency of M1 but not M2 macrophages and activated CD4/CD8 T cells (CD69+ CD4+/CD8+) in Nt5e knockdown tumors compared to tumors from control cohorts (Supplementary Fig. 4). Because the half-life of adenosine in circulation is only a few seconds [22], it was unclear how the immune cell composition of peripheral blood could be altered by Nt5e knockdown. We therefore examined a panel of 34 cytokines in the tumor of KPC1199 implanted mice and observed that the largest significant changes were associated with GM-CSF and TGFβ−1 (Fig. 3H, I). These data suggest that decreased tumoral secretion of GM-CSF and/or TGFβ−1 could be responsible for the decrease in MDSCs and the increased activation of CD4+ and CD8+ T cells (IFN-γ expression) in Nt5e knockdown tumor-bearing mice. This observation is supported by a previous report showing that pancreatic cancer cell expression of GM-CSF is seen to increase MDSC infiltration [23]. Furthermore, MDSCs are known to secrete TGFβ−1, which can promote CD4+ transformation into Tregs that can secrete immunosuppressive TGFβ−1 [24]. Hence, we evaluated Treg (CD4+FoxP3+) population in the circulation and tumors from control and Nt5e knockdown tumor-bearing mice. Intriguingly, the proportion of Tregs (FoxP3+) remained unaltered in the tumors and circulation from different mice cohorts (Supplementary Fig. 5).
Figure 3. Tumoral Nt5e expression regulates GM-CSF, T cells, and MDSCs.
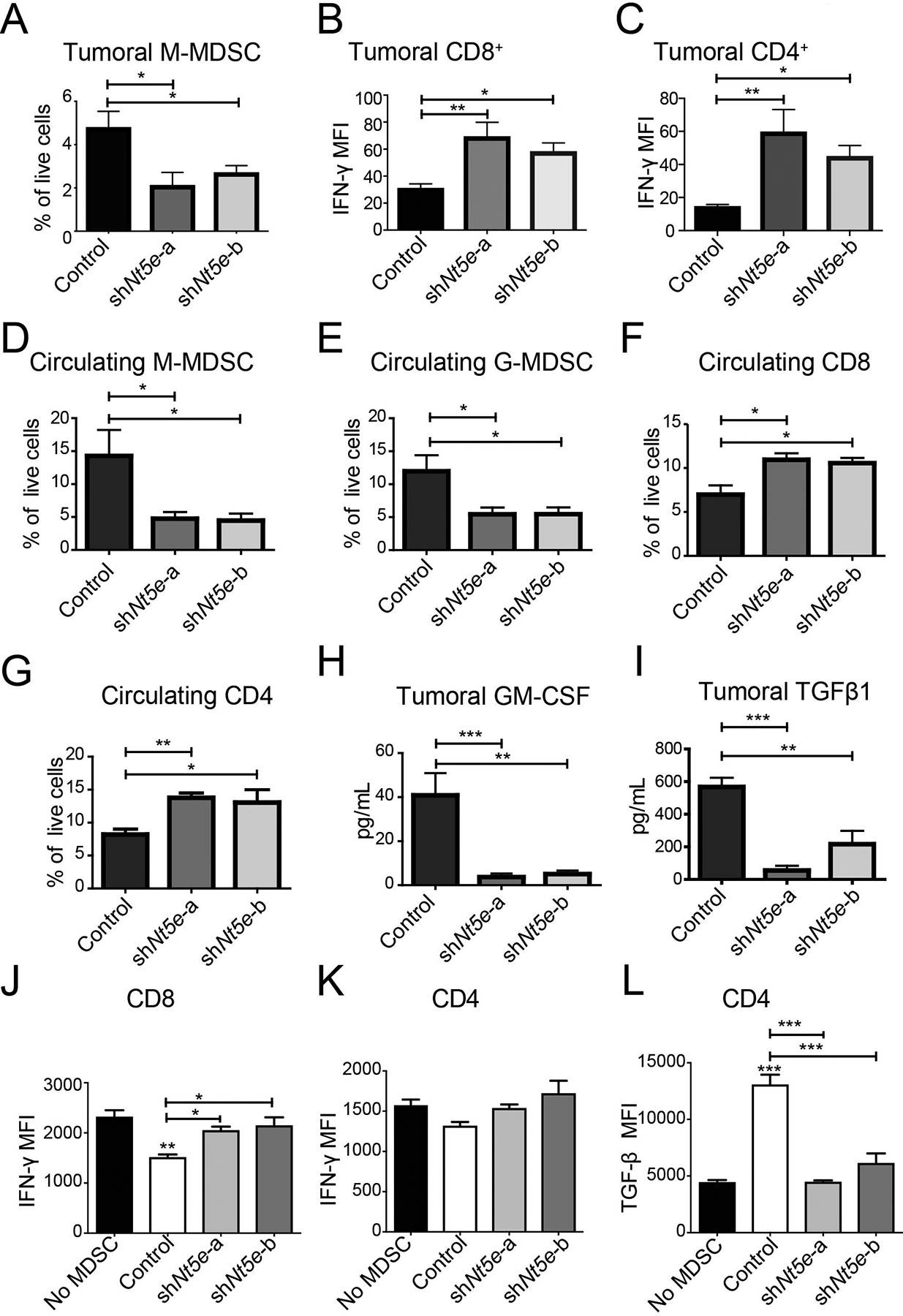
A-C Flow cytometry quantification for intratumoral M-MDSCs (Control, n=7; shNt5e-a, n=6; shNt5e-b, n=9) (A), and the median fluorescence intensity (MFI) for IFN-γ on CD8+ T cells (B) and CD4+ T cells (C). D-G Flow cytometry-based quantification of the percentage of circulating M-MDSC (D), G-MDSC (E), CD8+ T cells (F), and CD4+ T cells (G). H-I GM-CSF (H) and TGFβ−1 (I) levels in the tumors. J-L Spleen derived MDSCs from mice containing scrambled control or Nt5e knockdown were co-cultured with isolated CD8+ or CD4+ T cells from healthy mice stimulated with Dynabeads for 48 hr. MFI for IFN-γ on CD8+ (J) and CD4+ (K) cells post stimulation with CD3/C28 (Dynabeads) in the presence or absence of MDSCs derived from different mice. MFI for TGFβ−1 in CD4+ post stimulation with CD3/C28 (Dynabeads) in the presence or absence of MDSCs (L). Bar charts were compared with one-way ANOVA with Dunn’s multiple comparison test and Kruskal-Wallis test with Dunn’s multiple comparison test examining MFI. The co-culture utilized a one-way ANOVA with Tukey’s multiple comparison test. All mice used in this figure were B6(Cg)-Tyrc-2J/J. Error bars depict the standard error of the mean. *P < 0.05, **P < 0.01, ***P < 0.001.
To investigate if Nt5e knockdown can also alter the function of MDSCs, we assessed the immunosuppressive activities of MDSCs in ex vivo co-culture assays. Given that Nt5e knockdown tumors exhibit decreased numbers of circulating MDSCs, we utilized splenic MDSCs from tumor bearing mice and splenic CD4+ and CD8+ T cells from the healthy mice in the ex vivo assays. We noted that splenic MDSCs from Nt5e knockdown tumor-bearing mice have attenuated suppressive activities on the T cell function but not on the proliferation capabilities (Fig. 3J, K, Supplementary Fig. 6). Additionally, we noted that CD4+ T cells significantly decreased TGFβ−1 expression upon co-culture with splenic MDSCs derived from Nt5e knockdowns as compared to scrambled control mice (Fig. 3L). This perhaps explains, in part, why there is significantly increased expression of IFN-γ in intratumoral CD4+ and CD8+ T from Nt5e knockdown cell lines, but not from scrambled control tumors-bearing mice. Together our data suggest that tumoral Nt5e expression not only regulates the activity of MDSCs, but also modulates the systemic levels of MDSCs. In parallel, it also increases the IFN-γ expression of CD8+ and CD4+ T cells in the tumor and the abundance of circulating cells within Nt5e knockdown tumor-bearing mice.
Nt5e knockdown leads to enhanced anti-tumor immune response.
Increased circulatory CD4+ and CD8+ T cells in Nt5e knockdown tumor-bearing mice encouraged us to examine the T cell-mediated immune response. To achieve this, we implanted athymic nude mice, which lack T cells, with KPC1199 Nt5e knockdown or control cells. To our surprise, all tumors were palpable (Fig. 4A). This is in contrast to our results showing very small tumors or non-palpable tumors in immunocompetent mice implanted with Nt5e knockdown KPC1199 cells, as well as KPC1245 cells (Fig. 2 and Supplementary Fig. 2). Furthermore, the tumor volume and weight upon necropsy were not significantly different for shNt5e-a (Fig. 4B–D). While shNt5e-b showed significant differences in tumor volume and weight as compared to control counterparts, the tumor sizes in athymic nude mice were significantly greater than the immunocompetent mice and all had palpable tumors. Furthermore, we did not notice a significantly reduced metastatic foci in one of the Nt5e knockdown tumor set (shNt5e-a) compared to the other and the control tumor-bearing athymic nude mice (Supplementary Fig. 7). We posit that the observed discrepancy in the metastatic incidences between the two knockdown sets results from different tumor growth rates in mice (Fig. 4A–C, Supplementary Fig. 7B). Thus, the effect of Nt5e knockdown was much less pronounced in athymic nude mice than in their immunocompetent counterparts. Overall, these data suggest a possible implication of T cell-dependent antitumor immunity in Nt5e knockdown tumors.
Figure 4. Nt5e knockdown facilitates CD4+ T cell-mediated anti-tumor immune response.
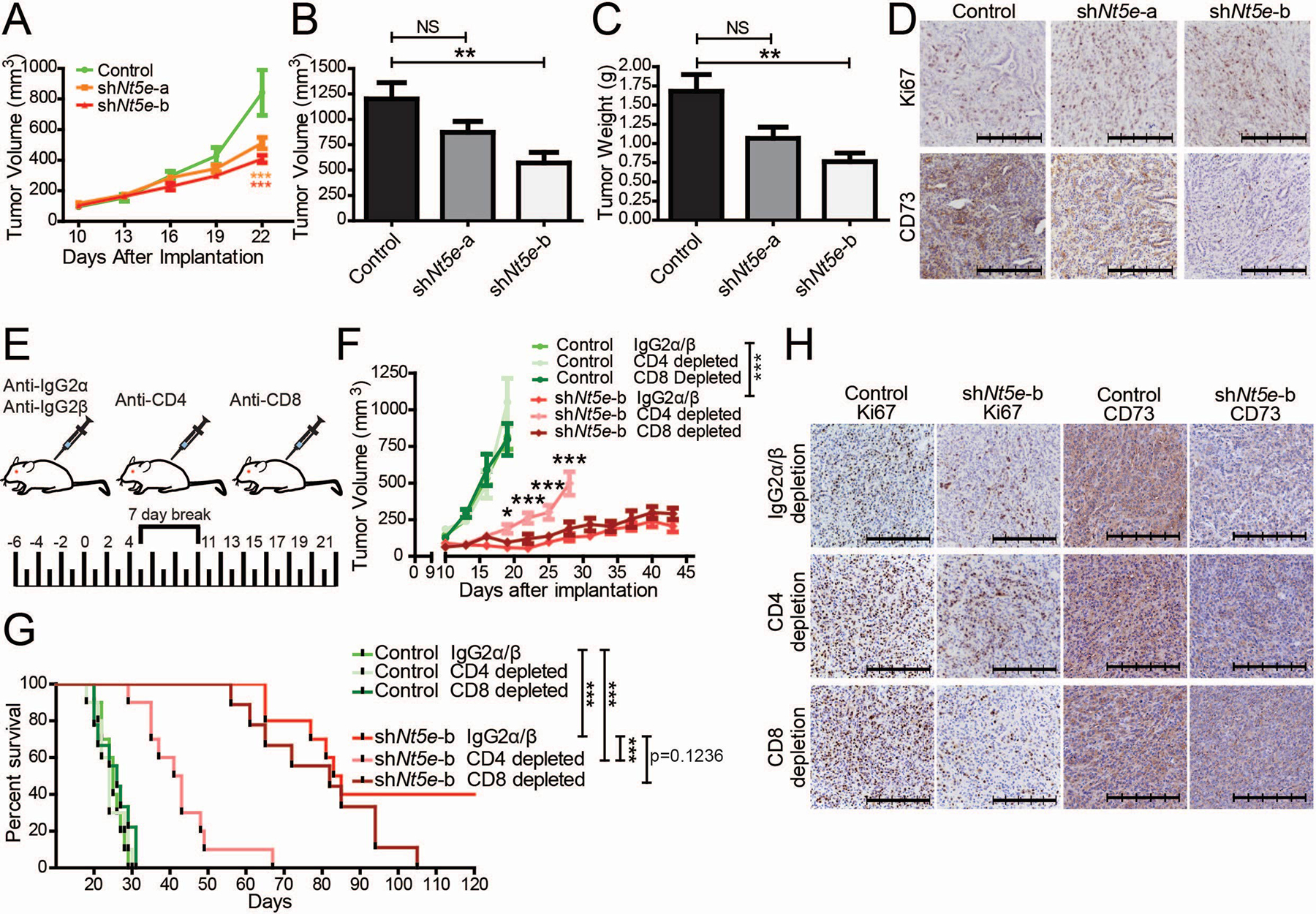
A Tumor growth rates in athymic nude mice orthotopically implanted with KPC1199 cells. Data were were compared using a two-way ANOVA with Bonferroni’s multiple comparison test. B-D Tumor volume (B) and tumor weight (C) at necropsy, and representative images of tumor sections stained with Ki67 or CD73 antibodies (D) from athymic nude mice euthanized at 23 days after implantation. E Schematic illustration of immune depletion regimen in immunocompetent C57BL/6J mice orthotopically implanted with KPC1245 cells. F Tumor growth rates in mice orthotopically implanted with KPC1245 cells. Statistical significance was calculated by using a generalized linear mixed model for longitudinal log-normal data incorporating compound symmetry correlation among longitudinal data from the same subjects. * above the CD4-depleted cohort indicates CD4-depletion in comparison to IgG2α/β-treatment in knockdown tumors. *** in the legend denotes the significance between the IgG2α/β treatments. Control cell-implanted mice with IgG2α/β-treatment, CD4-depletion, and CD8-depletion did not show significant differences compared to each other. G Kaplan-Meier survival plot indicating overall survival with pairwise Mantel-Cox log-rank comparisons. H Representative IHC tumor images of CD4- or CD8-depleted mice orthotopically implanted with control and shNt5e-b KPC1245 cells. The scale bar denotes 250 μm. Error bars depict the standard error of the mean. *P < 0.05, **P < 0.01, ***P < 0.001.
To understand the possible involvement of T cells in regulating the growth of Nt5e knockdown tumors, we utilized our least aggressive Nt5e knockdown, KPC1245 shNt5e-b, to investigate if T cell depletion could restore tumor growth in the knockdown cell-implanted mice. First, we confirmed the efficacy of CD8 and CD4 depletion in the circulation and tumors in KPC1245 orthotopic tumor-bearing mice (Supplementary Fig. 8, 9). We observed that CD4 depletion, but not CD8 depletion, drastically decreased the survival and increased the tumor burden in Nt5e knockdown tumor-bearing mice (Fig. 4E–G). Collectively, these results suggest that CD4+ T cells underlie the anti-tumor immune response in Nt5e knockdown tumor-bearing mice.
GM-CSF and MDSCs are critical to the functional role of CD73.
Nt5e knockdown tumor-bearing mice exhibit decreased intratumoral GM-CSF and circulating MDSCs (Fig. 3A, D, E, H). GM-CSF promotes the recruitment of MDSCs in early lesions in PDAC [25]. Increased levels of GM-CSF have been recorded in the intra-pancreatic lesion as well as peripheral circulation. Similar to PDAC, other cancer models highlight the importance of GM-CSF in early influx of MDSCs in premalignant lesions [26]. At these pre-malignant lesions, MDSCs suppress anti-tumor immune response and create a permissive environment for tumor growth. Relevant to our study, MDSCs can also regulate CD4+ T cell activity. Since we have observed attenuated levels of GM-CSF in Nt5e knockdown tumors, we investigated if increasing GM-CSF would restore growth of Nt5e inhibited cancer cells. To achieve this, we confirmed the CD73 knockout in KPC1245 (Fig. 5A) and administered GM-CSF via IP injections in mice implanted with control and Nt5e knockout KPC1245 cancer cells (Fig. 5B–G). In our study, the Nt5e knockout cancer cells recapitulated the decrease in tumor weight and volume observed in the Nt5e knockdown cells, along with a similar fraction appearing tumor-free to palpitation and under visual inspection (Fig. 5B, E). However, upon GM-CSF treatment, there was no significant difference in the tumor weight and volume between the control and Nt5e knockout tumors (Fig. 5C, F). To establish whether the restored growth of knockout tumors supplemented with GM-CSF depended on MDSC influx, we performed MDSC depletion by utilizing anti-Gr1 antibody. Nt5e knockout tumor-bearing mice, compared to the control, showed significantly decreased tumor weight and volume upon MDSC depletion despite GM-CSF treatment (Fig. 5D, G). Of note, we also examined the effect of MDSC depletion alone on the growth of control and Nt5e knockdown tumors in immunocompetent mice (Supplementary Fig. 10). As GM-CSF also regulates the function and differentiation of other myeloid cells such as macrophages, we examined the impact of macrophage depletion on tumor growth in our model. Our results demonstrated that MDSCs but not macrophage depletion significantly reduced tumor burden in KPC 1245-Sg NT control tumor-bearing mice compared to the isotype antibody-treated control counterparts (Supplementary Fig. 10). Given the lower frequency MDSCs in Nt5e knockdown tumors, we did not observe any differences in the tumor volume between control and Nt5e knockdown tumors upon MDSC depletion (Supplementary Fig. 10). Thus, it appears that the growth inhibitory effects of CD73 attenuation are mediated by GM-CSF and this mechanism, at least in part, requires MDSCs.
Figure 5. Decreased tumor burden by CD73 knockouts is rescued by GM-CSF supplementation, and the effect is abolished by MDSC depletion.
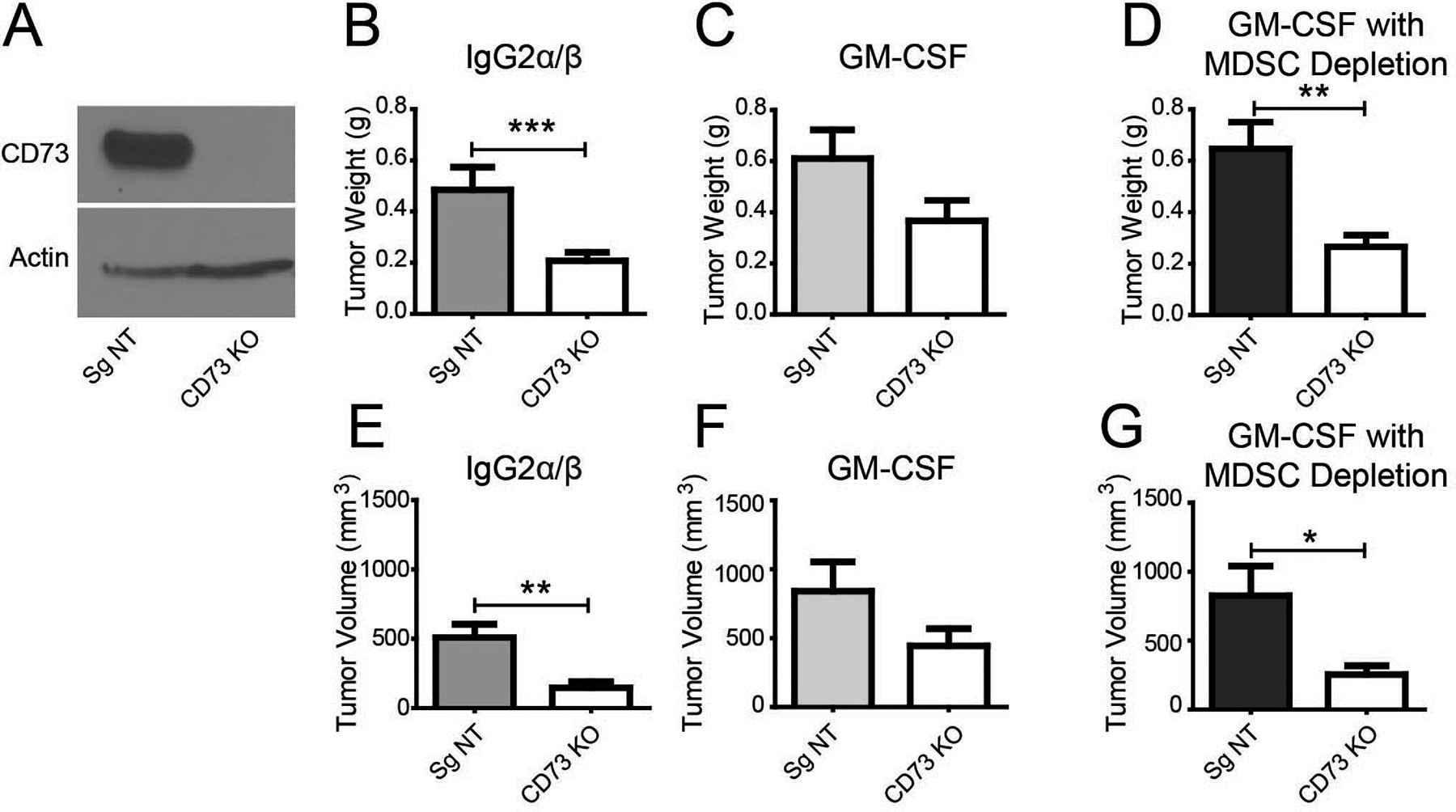
A Western blot analysis for CD73 expression in KPC1245 cells stably transduced with guide RNA targeting Nt5e (CD73 KO) or a non-targeting guide RNA (Sg NT). B-D Tumor weight (top) and tumor volume (bottom) of B6(Cg)-Tyrc-2J/J mice orthotopically implanted with Nt5e knockout or control cells treated with IgG2β (B), GM-CSF (C), and GM-CSF with anti-Gr-1 antibody to deplete MDSCs (D). Bar charts were compared with a Student’s t-test. Error bars depict the standard error of the mean. *P < 0.05, **P < 0.01, ***P < 0.01.
Combination therapy with anti-CD73 antibody and gemcitabine.
With such drastic changes in growth kinetics upon decreased CD73, we next investigated if these effects could potentially be exploited for therapeutic purposes. We examined the effect of combining CD73 inhibitory antibody with gemcitabine, a chemotherapy agent commonly utilized against PDAC. We noted a significant improvement in the survival and significantly decreased tumor growth rates in mice orthotopically-implanted with KPC1245 cells upon treatment with a combination of gemcitabine and anti-CD73 antibody, as compared to either treatment alone or the solvent control (Fig. 6). Of note we also assessed the combination of anti-CD73 and gemcitabine in the presence and absence of a checkpoint inhibitor using anti-PD-L1 antibodies. Our study demonstrates moderate efficacy of anti-CD73 and checkpoint inhibitor combination compared to anti-CD73 and gemcitabine combination in KPC1245 orthotopic tumor-bearing mice (Supplementary Fig. 11). Further, we did not observe any additional benefit of combining all three therapies together on the tumor burden. Nonetheless our data suggest that targeting CD73 with a chemotherapy can decrease tumor burden and promote survival in pancreatic cancer.
Figure 6. Gemcitabine and anti-CD73 combination therapy diminish tumor burden and improves survival.
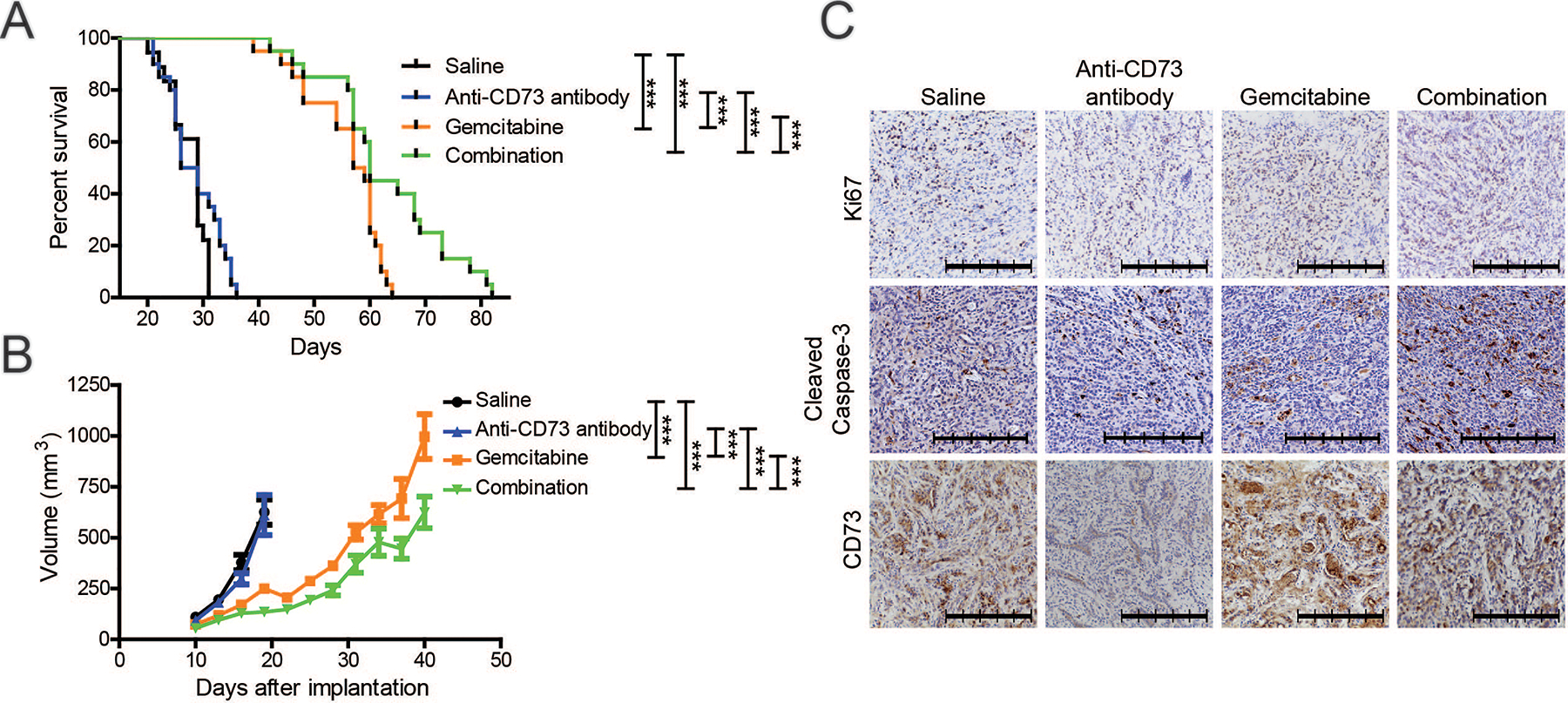
A-C Overall survival (A), tumor volume (B), and representative IHC images of tumor sections (C) from C57BL/6J mice orthotopically implanted with KPC1245 cells and treated with saline control, gemcitabine, anti-CD73 antibody, or the combination of gemcitabine and anti-CD73 antibody. Survival analysis was examined with Mantel-Cox log-rank comparison. Statistical significance for tumor growth was calculated by using a generalized linear mixed model for longitudinal log-normal data incorporating compound symmetry correlation among longitudinal data from the same subjects. Error bars depict the standard error of the mean. ***P < 0.001.
Discussion
This study sought to identify metabolic enzymes that may be targeted to improve patient survival in PDAC. Our unbiased screen identified enzymes in the nucleotide biosynthesis pathway that significantly correlate with patient survival. Our results highlight how altered expression of metabolic enzymes, in this case CD73, can allow tumor cells to escape antitumor immunity.
Our pathway enrichment analysis on enzymes that impacted survival demonstrates that pyrimidine and purine metabolic pathways significantly impact PDAC patient prognosis (Fig. 1A). The skew towards nucleotide metabolism could, at least in part, reflect the tumor response to nucleotide analog-based chemotherapies, which can increase nucleotide metabolism as a mechanism of drug resistance [27]. Chemotherapy can promote tumor-associated antigenicity, which can be further impacted by DNA damage that, in turn, can be regulated by nucleotide biosynthesis [28–30]. Therefore, targeting these pathways may also enhance the efficacy of chemotherapy, as observed in this study (Fig. 6). Because nucleotide metabolism pathways were enriched in enzymatic genes contributing to survival differences, it is likely that other metabolic enzymes in these pathways, beyond CD73, may present actionable targets for PDAC. Two genes flanking NT5E in the IQR enzyme screen were TYMS and GMPS, the latter of which ranked the highest in the IQR enzyme screen (Supplementary Fig. 1F) and was the fourth most significant when examining high vs. low expression with all of the genes (Supplementary Table 3). When examining all genes with high vs. low IQR expression, IL17D and ARID5A are the first and third top hits (Supplementary Table 3), which carry multiple immune functions [31] and can promote CD4+ T cell differentiation into pro-inflammatory Th17 cells [32]. However, this could also be related to the frequent upregulation of RORC in pancreatic cancer stem cells [33]. The association of increased NT5E and survival has been seen before in other cancers [34, 35], but our studies are the first to show that NT5E is one of the top metabolic enzymes whose expressions have significant impact on patient survival in PDAC in an immune-dependent mechanism.
CD73 expression was evident in early stage PanINs and became drastically elevated in tumors, suggesting that cancer evolution of immune suppression begins at a very early stage. The increased CD73 could be generated as a byproduct of increased hypoxia-inducible factor (HIF)-1α accumulation [21] or due to the selection of neoplastic cells with high CD73 expression due to pro-tumorigenic roles of extracellular adenosine [11, 36]. Accordingly, previous studies have observed increasing CD73 expression with increasing stages of progression in multiple cancers [34, 35]. Our studies are the first to report increased CD73 levels through PanIN development and increased activity as indicated by increased adenosine levels in KPC mice.
One of the major physiological responses of the increased CD73 expression is an alteration in the tumor immune microenvironment [9, 37]. Using multiple immunocompetent mouse models, we investigated the effect of targeting and altering CD73 expression in pancreatic cancer. We consistently observed drastically increased survival and decreased tumor burden when knocking down CD73, knocking out CD73, or targeting CD73 in combination with gemcitabine.
We show here a potential feed-forward mechanism whereby cancer-associated CD73 regulates GM-CSF-induced MDSCs levels, which inversely correlate with T cells. Decreased levels of MDSCs inducing cytokine, GM-CSF [38], were observed in Nt5e knockdowns, which correlated with the reduced M-MDSCs and increased CD8+ and CD4+ T cells in the peripheral blood. It was observed that tumoral CD4+ and CD8+ T cells are indeed affected by CD73 knockdowns, in which activated T cells produce more IFN-γ (Fig. 3B, C). To investigate if the diminished tumor burden in CD73 low tumors was mediated by a GM-CSF–MDSC axis, we implanted CD73 knockout tumor cell line and treated mice with GM-CSF or with GM-CSF and MDSC depletion (Fig. 5). GM-CSF supplementation abrogated the significant difference in tumor burden between CD73 knockout and control tumors. However, depleting MDSCs greatly reduced the GM-CSF-mediated rescue of tumor growth in CD73 knockouts. Thus, CD73-mediated regulation of tumor growth is, at least in part, mediated by GM-CSF that in turn regulates MDSCs and T cells. We furthermore observed decreased TGFβ−1 in Nt5e knockdowns tumors and that MDSCs derived from mice with Nt5e knockdowns had reduced capacity of inducing TGFβ−1 in CD4+ T cells. TGFβ−1 is one of the major immunosuppressive cytokines MDSCs utilize for inhibiting T cells in the tumor microenvironment [24]. TGFβ−1 is also capable of promoting immunosuppressive Tregs and MDSCs [39–41]. We observed that MDSCs from hosts implanted with control or knockdown cells had different abilities in generating TGFβ−1 positive CD4+ T cells which will likely impact the immunosuppressive environment of the tumor, and might be why activated T cells were observed to have more IFN-γ production (Fig. 3B, C, J–L). Notably, we observed no significant alterations in the Treg population between control or knockdown tumors (Supplementary Figure 5). Besides regulating immune response, TGF-β actively participates in tumor growth, invasion, and metastasis. We have seen drastic reduction in tumor growth in Nt5e knockdown tumors. This suggests that while attenuated TGF-β levels did not impact the Treg levels, it mitigated tumor growth and expansion upon CD73 knockdown in tumor cells. Furthermore, studies have shown that Tregs can be further characterized into Th1-like Tregs (T-bet+IFNγ+Foxp3+), Th2-like Tregs (Gata3+IRF4+IL4+Foxp3+) and Th17-like Tregs (IL-17+RORγt+Foxp3+), which we have not investigated in the present study [42]. Perhaps the effect of CD73 knockdown and absence of TGFβ−1 alters a particular subset of Treg, which is not mirrored in the total FOXP3+ population. Hence future investigations, such as scRNA-seq on Nt5e knockdown and control tumors will solve this conundrum.
We further investigated the role of T cells in the orthotopic models. In contrast to stagnating tumors and tumor-free phenotype in immunocompetent mice, we observed rapidly growing tumors without any incidence of the tumor-free phenotype in Nt5e knockdowns implanted athymic nude mice. Interestingly, the tumor-free phenotype in pancreatic cancer has been observed before when altering GM-CSF and MDSCs [23]. Finally, the direct role of CD4+ T cells was seen to significantly impact the ability of Nt5e knockdown to improve tumor burden. Surprisingly, CD8-depletion at day 120 had a marginal effect, suggesting a CD8-independent mechanism; however, the remainder of the knockdown IgG2α/β cohort lived for more than a year, which ultimately generated significance in comparison to CD8 depletion. These data suggest decreased CD73 largely acts through a beneficial CD4+ subtype, which could be through mechanisms such as CD4-dependent senescence surveillance or IFN-γ mediated anti-tumor responses [43, 44]. These data also show that MDSCs from CD73 tumors can promote TGFβ−1 expression in CD4 T cells, suggesting that tumoral CD73 impacts CD4 T cells through more than one mechanism. Of note, previous studies have also shown an important role of adenosine on the proposed mechanism. It has been previously seen that targeting the adenosine receptor in CD4+ T cells could block autoimmune destruction of the host [17] and that adenosine promotes MDSC expansion [45]. Hence, pancreatic tumors facilitate adenosine accumulation to generate a multipronged immunosuppressive microenvironment.
Recent advances in immune modulators have shown tremendous clinical advantages, while offering little-to-no benefit in the majority of PDAC where it is resistant due to its immunosuppressive and quiescent immune microenvironment [46–48]. Gemcitabine is known to increase tumor antigenicity [28, 29] and facilitate a beneficial immune response in pancreatic cancer [49, 50]. Targeting CD73 in vivo in combination with gemcitabine resulted in decreased tumor burden and enhanced survival compared to gemcitabine alone (Fig. 6). These data suggest a therapeutic avenue for targeting CD73 in the immunosuppressive microenvironment of pancreatic cancer [20].
Previous studies have shown that depleting CD73 is an effective way to decrease metastasis [51–54]. This study also observed a difference with CD73 expression and gross examination of metastasis to the mesentery, liver, and spleen in athymic nude mice (Supplementary Fig. 7A). However, we speculate that the differences in the incidence of metastasis are primarily due to differences in the tumor size (Fig. 4A–C and Supplementary Fig. 7B). Similarly, we do not believe metastasis to be the reason for the anti-CD73 antibody response. Other studies have reported that anti-CD73 antibodies can reduce metastasis and may even help target non-cancer cell CD73 to further reduce tumor burden [51–54]. It might be that a higher concentration of CD73 antibody is needed or that a more robust immune stimulator is needed in conjunction with the removal of immunosuppressive adenosine, such as cell death through gemcitabine. However, due to the lack of anti-CD73 response to the antibody alone, it stands strong to rationalize that altered cancer cell growth kinetics alone did not cause the enhanced response when combined with gemcitabine.
Overall, our study shows that CD73 expression is upregulated in human and mouse tumors and that genetic or pharmacological targeting of CD73 expression in tumors leads to reduced tumor burden and prolonged survival. Unlike other studies, we observed a novel GM-CSF – MDSCs-regulated mechanism that utilized CD4+ immune cells largely independent of CD8+ T cells. Thus, targeting CD73 may reverse the innate and/or adaptive resistance to immune checkpoints in pancreatic cancer.
Materials and Methods
Cell Culture and Reagents
C57BL/6J-congenic LSL-KrasG12D/+;LSL-Trp53R172H/+;Pdx-1-Cre (KPC) tumor-derived pancreatic cancer cell lines KPC1245 and KPC1199 were obtained from Dr. David Tuveson’s lab. Cell lines were validated by PCR to detect mutant Kras and p53. Cells were also tested by STR profiling by the Genetics Core at University of Arizona. Cells were routinely tested for mycoplasma contaminations by PCR-based methods. Cells were cultured in Dulbecco’s modified Eagle medium (Sigma-Aldrich, St Louis, MO, USA) supplemented with 50 IU/mL penicillin, 50 μg/mL streptomycin, and incubated at 37°C in a humidified incubator with 5% CO2. Cells were maintained at 5% fetal bovine serum (FBS). Upon reaching 70–80% confluency, cells were passaged by washing with phosphate-buffered saline (PBS) before adding 0.25% trypsin (Caisson Labs, Smithfield, UT, USA) and plating at 25% confluency.
MDSC and CD4 T cell co-culture was conducted by isolating each cell type using the respective kits from Miltenyi Biotec (Bergisch Gladbach, Germany) and following the manufacturer’s protocol. MDSCs were isolated from spleens from tumor bearing mice, with the cancer containing scrambled shRNA or shNt5e knockdown in KPC1245, while T cells were isolated from the spleens of healthy tumor-free mice. MDSCs were co-cultured with T cells in a 2:1 ratio in Gibco RPMI 1640 Media (Thermo Fisher Scientific, Waltham, MA) with 10% FBS and anti-CD3/CD28 dynabeads (Thermo Fisher Scientific) for 48 hr.
Lentivirus Transfection
Nt5e-targeting short hairpin RNA (shRNA) constructs were obtained from Sigma-Aldrich and sgRNA all-in-one constructs containing Cre were purchased from Cellecta (Mountain View, CA, USA). Lentiviral knockdowns were produced by utilizing Nt5e-targeting constructs (TRCN0000080813 and TRCN0000080814, denoted as shNt5e-a and shNt5e-b respectively). Cells were infected two times with replication-incompetent lentivirus with polybrene in 48 hr. After transduction, cells were selected using puromycin for two to three weeks. For knockout and knockout non-targeting (Sg NT) control cells, a low amount of cells were seeded in a 100mm2 dish and single colonies were isolated and grown in a 96-well before transitioning to 24-,12-, and 6-well plates. All genetic manipulations were confirmed with western blotting and/or qPCR.
Animal Studies
All animal experiments were approved by the University of Nebraska Medical Center IACUC. Unless otherwise noted, in-house bred 6- to 8-week-old same-sex B6(Cg)-Tyrc−2J/J mice were used for orthotopic implantations. Further details can be found in the supplementary methods section. Given that the investigator responsible for overall project coordination was also responsible for most of the animal studies, the investigator was not blinded to the mice cohorts. Mice cages, with up to 5 mice, were randomized upon weaning and implantation took cages with similar sex and age range. Furthermore, unless stated otherwise, 5×103 cells in 50μL PBS were orthotopically injected with a 27.5-gauge needle to the pancreas. Tumor sizes were measured by utilizing a Spectrum In Vivo Imaging System (PerkinElmer, Waltham, MA, USA) for luciferase imaging or by caliper measurements.
The sample size calculation was conducted to optimize the power of detecting the differences in tumor size or survival between cohorts. Therapy survival studies were performed by orthotopically implanting 5×103 cells in male C57BL/6NTac mice and beginning treatments on day 7 through intraperitoneal (IP) injections. Treatments consisted of either 100μL saline, 1.32mg of gemcitabine (Sagent, Schaumburg, IL, USA), 100μg of anti-CD73 antibody (BE0209, BioXCell, Lebanon, NH, USA), or the combination of gemcitabine and anti-CD73 antibody every three days. For immunodeficient mice studies, 6–8-week-old athymic nude mice were utilized.
Immune depletions in C57BL/6NTac male mice were performed by administering 100μg of anti-CD4 or anti-CD8α antibodies (BE0003–1 and BE0004–1 respectively, BioXCell) in 100μL saline via IP injections. IgG2α and IgG2β (BE0089 and BE0090 respectively, BioXCell) were injected together as a control. Antibodies for immune depletions were injected six times, once every two days, starting 6 days prior to implantation. After 6 injections of immuno-depleting antibodies, mice were given a week of rest before resuming the cycle. Each cohort contained 10 mice, except for CD8 depletion, which consisted of 9 mice per group.
Studies with recombinant GM-CSF and MDSC depletion utilized B6(Cg)-Tyrc−2J/J male mice between 9–11 weeks. The IgG2β control was the same as above, but with 200μg per mouse. Based on a previous publication [55], 100ng recombinant GM-CSF (Sino Biological, Beijing, China) was dissolved in saline and administered daily beginning 7 days post implantation. Similar to previous studies [56, 57], 200μg of anti-Gr-1 (BE0075, BioXCell) was injected IP every two days starting two days before implantation.
TCGA and ICGC Analysis
TCGA survival analysis was performed as previously described [58]. In brief, clinical and primary mRNA data was downloaded through the TCGA Data Matrix. The donor information for the International Cancer Genome Consortium (ICGC) PDAC dataset collected by the Australian consortium was downloaded from the ICGC, while the normalized mRNA expression was downloaded from Supplemental Table 2 in a previous publication [59].
TCGA survival was analyzed in R 3.3.2 through the function “survdiff” in the R package “survival” using the Mantel-Haenszel log-rank test. ICGC and mouse survival was examined in Graphpad Prism 5 using Mantel-Cox log-rank test for significance. Pathway analysis was conducted through Database for Annotation, Visualization and Integrated Discovery v6.8 [60, 61]. Kyoto Encyclopedia of Genes and Genomes was used for pathway visualization and enzyme gene list [62, 63]. ActiveState Perl v5.24.1 generated the tables and controlled R as the top-level script.
Cytokine Quantification
Cytokine quantification was performed by Eve Technologies (Calgary, Canada) on a 31-plex (MD31) and a 3-plex (TGFB1–3) mouse cytokine array/chemokine array. For values exceeding standard curve, values were replaced with the lowest or highest obtained value as recommended by Eve Technologies.
Experimental Results and Statistical Analysis
Description of the experiments, sample size, and statistics can be found in the figure legends. Statistical tests were two-sided when applicable and the assumptions were checked. Results show the center as the arithmetic mean with the standard error of the mean, unless noted otherwise. Animal studies were generally performed once. Exceptions include observing the knockdown effect in KPC1245 and KPC1199 orthotopically implanted mice that were performed more than three times. Western blots to confirm knockdowns were performed before implanting, with exception to figure 1B–D, which was confirmed by qPCR.
More detailed information is described in Supplementary Methods.
Supplementary Material
Acknowledgements:
We would like to thank the University of Nebraska Medical Center Rapid Autopsy Pancreatic Program and the patients who generously donated their samples. This work was supported in part by funding from the National Institutes of Health grant (R01CA163649, R01CA210439, and R01CA216853, NCI) to PKS; the Specialized Programs of Research Excellence (SPORE, 2P50 CA127297, NCI) to PKS and MAH; SPORE Career Development Award (2P50 CA127297, NCI) to KM; the PCDC U01CA210240 to MAH; NCI Research Specialist award (5R50CA211462) to PMG. We would also like to acknowledge the Fred & Pamela Buffett Cancer Center Support Grant (P30CA036727, NCI) for supporting shared resources.
Funding:
This work was supported in part by funding from the National Institutes of Health grant (R01CA163649, R01CA210439, and R01CA216853, NCI) to PKS; the Specialized Programs of Research Excellence (SPORE, 2P50 CA127297, NCI) to PKS and MAH; Specialized Programs of Research Excellence (SPORE, 2P50 CA127297, NCI) Career Enhancement Award to KM; the PCDC U01CA210240 to MAH; NCI Research Specialist award (5R50CA211462) to PMG. We would also like to acknowledge the Fred & Pamela Buffett Cancer Center Support Grant (P30CA036727, NCI) for supporting shared resources.
Footnotes
Conflicts of interest: The authors declare no potential conflicts of interest.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin 2020; 70: 7–30. [DOI] [PubMed] [Google Scholar]
- 2.Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, Matrisian LM. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res 2014; 74: 2913–2921. [DOI] [PubMed] [Google Scholar]
- 3.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011; 144: 646–674. [DOI] [PubMed] [Google Scholar]
- 4.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 2009; 324: 1029–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sugiura A, Rathmell JC. Metabolic Barriers to T Cell Function in Tumors. J Immunol 2018; 200: 400–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rivadeneira DB, Delgoffe GM. Antitumor T-cell Reconditioning: Improving Metabolic Fitness for Optimal Cancer Immunotherapy. Clin Cancer Res 2018; 24: 2473–2481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gupta S, Roy A, Dwarakanath BS. Metabolic Cooperation and Competition in the Tumor Microenvironment: Implications for Therapy. Front Oncol 2017; 7: 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Molinier-Frenkel V, Castellano F. Immunosuppressive enzymes in the tumor microenvironment. FEBS Lett 2017; 591: 3135–3157. [DOI] [PubMed] [Google Scholar]
- 9.Allard B, Longhi MS, Robson SC, Stagg J. The ectonucleotidases CD39 and CD73: Novel checkpoint inhibitor targets. Immunol Rev 2017; 276: 121–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vigano S, Alatzoglou D, Irving M, Menetrier-Caux C, Caux C, Romero P et al. Targeting Adenosine in Cancer Immunotherapy to Enhance T-Cell Function. Front Immunol 2019; 10: 925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vijayan D, Young A, Teng MWL, Smyth MJ. Targeting immunosuppressive adenosine in cancer. Nat Rev Cancer 2017; 17: 709–724. [DOI] [PubMed] [Google Scholar]
- 12.Junger WG. Immune cell regulation by autocrine purinergic signalling. Nat Rev Immunol 2011; 11: 201–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dosch M, Gerber J, Jebbawi F, Beldi G. Mechanisms of ATP Release by Inflammatory Cells. Int J Mol Sci 2018; 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhao H, Liao X, Kang Y. Tregs: Where We Are and What Comes Next? Front Immunol 2017; 8: 1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Linden J, Cekic C. Regulation of lymphocyte function by adenosine. Arterioscler Thromb Vasc Biol 2012; 32: 2097–2103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zarek PE, Powell JD. Adenosine and anergy. Autoimmunity 2007; 40: 425–432. [DOI] [PubMed] [Google Scholar]
- 17.Zarek PE, Huang CT, Lutz ER, Kowalski J, Horton MR, Linden J et al. A2A receptor signaling promotes peripheral tolerance by inducing T-cell anergy and the generation of adaptive regulatory T cells. Blood 2008; 111: 251–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.O’Reilly EM, Oh DY, Dhani N, Renouf DJ, Lee MA, Sun W et al. Durvalumab With or Without Tremelimumab for Patients With Metastatic Pancreatic Ductal Adenocarcinoma: A Phase 2 Randomized Clinical Trial. JAMA Oncol 2019; 5: 1431–1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med 2012; 366: 2455–2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Daniel SK, Sullivan KM, Labadie KP, Pillarisetty VG. Hypoxia as a barrier to immunotherapy in pancreatic adenocarcinoma. Clin Transl Med 2019; 8: 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Synnestvedt K, Furuta GT, Comerford KM, Louis N, Karhausen J, Eltzschig HK et al. Ecto-5’-nucleotidase (CD73) regulation by hypoxia-inducible factor-1 mediates permeability changes in intestinal epithelia. J Clin Invest 2002; 110: 993–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Moser GH, Schrader J, Deussen A. Turnover of adenosine in plasma of human and dog blood. Am J Physiol 1989; 256: C799–806. [DOI] [PubMed] [Google Scholar]
- 23.Bayne LJ, Beatty GL, Jhala N, Clark CE, Rhim AD, Stanger BZ et al. Tumor-derived granulocyte-macrophage colony-stimulating factor regulates myeloid inflammation and T cell immunity in pancreatic cancer. Cancer Cell 2012; 21: 822–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Salminen A, Kaarniranta K, Kauppinen A. Immunosenescence: the potential role of myeloid-derived suppressor cells (MDSC) in age-related immune deficiency. Cell Mol Life Sci 2019; 76: 1901–1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pylayeva-Gupta Y, Lee KE, Hajdu CH, Miller G, Bar-Sagi D. Oncogenic Kras-induced GM-CSF production promotes the development of pancreatic neoplasia. Cancer Cell 2012; 21: 836–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ostrand-Rosenberg S, Fenselau C. Myeloid-Derived Suppressor Cells: Immune-Suppressive Cells That Impair Antitumor Immunity and Are Sculpted by Their Environment. J Immunol 2018; 200: 422–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shukla SK, Purohit V, Mehla K, Gunda V, Chaika NV, Vernucci E et al. MUC1 and HIF-1alpha Signaling Crosstalk Induces Anabolic Glucose Metabolism to Impart Gemcitabine Resistance to Pancreatic Cancer. Cancer Cell 2017; 32: 71–87 e77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gravett AM, Trautwein N, Stevanovic S, Dalgleish AG, Copier J. Gemcitabine alters the proteasome composition and immunopeptidome of tumour cells. Oncoimmunology 2018; 7: e1438107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jackaman C, Majewski D, Fox SA, Nowak AK, Nelson DJ. Chemotherapy broadens the range of tumor antigens seen by cytotoxic CD8(+) T cells in vivo. Cancer Immunol Immunother 2012; 61: 2343–2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gunda V, Souchek J, Abrego J, Shukla SK, Goode GD, Vernucci E et al. MUC1-Mediated Metabolic Alterations Regulate Response to Radiotherapy in Pancreatic Cancer. Clin Cancer Res 2017; 23: 5881–5891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.O’Sullivan T, Saddawi-Konefka R, Gross E, Tran M, Mayfield Stephen P, Ikeda H et al. Interleukin-17D Mediates Tumor Rejection through Recruitment of Natural Killer Cells. Cell Reports 2014; 7: 989–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Masuda K, Ripley B, Nyati KK, Dubey PK, Zaman MM, Hanieh H et al. Arid5a regulates naive CD4+ T cell fate through selective stabilization of Stat3 mRNA. J Exp Med 2016; 213: 605–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lytle NK, Ferguson LP, Rajbhandari N, Gilroy K, Fox RG, Deshpande A et al. A Multiscale Map of the Stem Cell State in Pancreatic Adenocarcinoma. Cell 2019; 177: 572–586 e522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang R, Zhang Y, Lin X, Gao Y, Zhu Y. Prognositic value of CD73-adenosinergic pathway in solid tumor: A meta-analysis and systematic review. Oncotarget 2017; 8: 57327–57336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jiang T, Xu X, Qiao M, Li X, Zhao C, Zhou F et al. Comprehensive evaluation of NT5E/CD73 expression and its prognostic significance in distinct types of cancers. BMC Cancer 2018; 18: 267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhou L, Jia S, Chen Y, Wang W, Wu Z, Yu W et al. The distinct role of CD73 in the progression of pancreatic cancer. J Mol Med (Berl) 2019; 97: 803–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Deaglio S, Dwyer KM, Gao W, Friedman D, Usheva A, Erat A et al. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J Exp Med 2007; 204: 1257–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ma N, Liu Q, Hou L, Wang Y, Liu Z. MDSCs are involved in the protumorigenic potentials of GM-CSF in colitis-associated cancer. Int J Immunopathol Pharmacol 2017; 30: 152–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee CR, Lee W, Cho SK, Park SG. Characterization of Multiple Cytokine Combinations and TGF-beta on Differentiation and Functions of Myeloid-Derived Suppressor Cells. Int J Mol Sci 2018; 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tran DQ. TGF-beta: the sword, the wand, and the shield of FOXP3(+) regulatory T cells. J Mol Cell Biol 2012; 4: 29–37. [DOI] [PubMed] [Google Scholar]
- 41.Andersson J, Tran DQ, Pesu M, Davidson TS, Ramsey H, O’Shea JJ et al. CD4+ FoxP3+ regulatory T cells confer infectious tolerance in a TGF-beta-dependent manner. J Exp Med 2008; 205: 1975–1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li C, Jiang P, Wei S, Xu X, Wang J. Regulatory T cells in tumor microenvironment: new mechanisms, potential therapeutic strategies and future prospects. Mol Cancer 2020; 19: 116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kang TW, Yevsa T, Woller N, Hoenicke L, Wuestefeld T, Dauch D et al. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature 2011; 479: 547–551. [DOI] [PubMed] [Google Scholar]
- 44.Bhat P, Leggatt G, Waterhouse N, Frazer IH. Interferon-gamma derived from cytotoxic lymphocytes directly enhances their motility and cytotoxicity. Cell Death Dis 2017; 8: e2836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Condamine T, Mastio J, Gabrilovich DI. Transcriptional regulation of myeloid-derived suppressor cells. J Leukoc Biol 2015; 98: 913–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yarchoan M, Hopkins A, Jaffee EM. Tumor Mutational Burden and Response Rate to PD-1 Inhibition. N Engl J Med 2017; 377: 2500–2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Looi CK, Chung FF, Leong CO, Wong SF, Rosli R, Mai CW. Therapeutic challenges and current immunomodulatory strategies in targeting the immunosuppressive pancreatic tumor microenvironment. J Exp Clin Cancer Res 2019; 38: 162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Foley K, Kim V, Jaffee E, Zheng L. Current progress in immunotherapy for pancreatic cancer. Cancer Lett 2016; 381: 244–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Johnson BA 3rd, Yarchoan M, Lee V, Laheru DA, Jaffee EM. Strategies for Increasing Pancreatic Tumor Immunogenicity. Clin Cancer Res 2017; 23: 1656–1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Plate JM, Plate AE, Shott S, Bograd S, Harris JE. Effect of gemcitabine on immune cells in subjects with adenocarcinoma of the pancreas. Cancer Immunol Immunother 2005; 54: 915–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang B CD73 promotes tumor growth and metastasis. Oncoimmunology 2012; 1: 67–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang L, Fan J, Thompson LF, Zhang Y, Shin T, Curiel TJ et al. CD73 has distinct roles in nonhematopoietic and hematopoietic cells to promote tumor growth in mice. J Clin Invest 2011; 121: 2371–2382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stagg J, Divisekera U, Duret H, Sparwasser T, Teng MW, Darcy PK et al. CD73-deficient mice have increased antitumor immunity and are resistant to experimental metastasis. Cancer Res 2011; 71: 2892–2900. [DOI] [PubMed] [Google Scholar]
- 54.Stagg J, Divisekera U, McLaughlin N, Sharkey J, Pommey S, Denoyer D et al. Anti-CD73 antibody therapy inhibits breast tumor growth and metastasis. Proc Natl Acad Sci U S A 2010; 107: 1547–1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Szomolay B, Eubank TD, Roberts RD, Marsh CB, Friedman A. Modeling the inhibition of breast cancer growth by GM-CSF. J Theor Biol 2012; 303: 141–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Srivastava MK, Zhu L, Harris-White M, Kar UK, Huang M, Johnson MF et al. Myeloid suppressor cell depletion augments antitumor activity in lung cancer. PLoS One 2012; 7: e40677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mauti LA, Le Bitoux MA, Baumer K, Stehle JC, Golshayan D, Provero P et al. Myeloid-derived suppressor cells are implicated in regulating permissiveness for tumor metastasis during mouse gestation. J Clin Invest 2011; 121: 2794–2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.King RJ, Yu F, Singh PK. Genomic alterations in mucins across cancers. Oncotarget 2017; 8: 67152–67168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bailey P, Chang DK, Nones K, Johns AL, Patch AM, Gingras MC et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016; 531: 47–52. [DOI] [PubMed] [Google Scholar]
- 60.Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 2009; 4: 44–57. [DOI] [PubMed] [Google Scholar]
- 61.Huang da W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 2009; 37: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kanehisa M, Sato Y, Furumichi M, Morishima K, Tanabe M. New approach for understanding genome variations in KEGG. Nucleic Acids Res 2019; 47: D590–D595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res 2000; 28: 27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.