

ABSTRACT
Background
Biallelic mutations in the GBA1 gene encoding glucocerebrosidase cause Gaucher's disease, whereas heterozygous carriers are at risk for Parkinson's disease (PD). Glucosylsphingosine is a clinically meaningful biomarker of Gaucher's disease but could not be assayed previously in heterozygous GBA1 carriers.
Objective
The aim of this study was to assess plasma glucosylsphingosine levels in GBA1 N370S carriers with and without PD.
Methods
Glucosylsphingosine, glucosylceramide, and four other lipids were quantified in plasma from N370S heterozygotes with (n = 20) or without (n = 20) PD, healthy controls (n = 20), idiopathic PD (n = 20), and four N370S homozygotes (positive controls; Gaucher's/PD) using quantitative ultra‐performance liquid chromatography tandem mass spectrometry.
Results
Plasma glucosylsphingosine was significantly higher in N370S heterozygotes compared with noncarriers, independent of disease status. As expected, Gaucher's/PD cases showed increases in both glucocerebrosidase substrates, glucosylsphingosine and glucosylceramide.
Conclusions
Plasma glucosylsphingosine accumulation in N370S heterozygotes shown in this study opens up its future assessment as a clinically meaningful biomarker of GBA1‐PD. © 2021 The Authors. Movement Disorders published by Wiley Periodicals LLC on behalf of International Parkinson Movement Disorder Society.
Keywords: Parkinson's, Gaucher's, glucocerebrosidase, lipidomics
The GBA1 gene encodes the lysosomal enzyme, acid β‐glucocerebrosidase (GCase; EC 3.2.1.45). Biallelic mutations in GBA1 cause the lysosomal lipid storage disorder, Gaucher's disease (GD). 1 A common coding variant of GBA1, N370S, is pathogenic for GD and represents one of the most common genetic risk factors for Parkinson's disease (PD) in the heterozygous carriers,2, 3, 4 but the mechanism underlying this increased risk for PD remains unknown.
Pathogenic variants in GBA1, including N370S, decrease GCase activity. GCase metabolizes glycosphingolipids, a class of lipids essential for membrane structure and a range of cellular functions. The prevailing hypothesis for PD associated with GBA1 mutations (ie, GBA‐PD) is that a decrease in GCase activity alters the lipid composition of membranes and results in increased α‐synuclein aggregation and accumulation in the lysosomes. 5 The two key substrates of GCase are glucosylceramide (GluCer) and glucosylsphingosine (GluSph). Recent studies suggest that GluCer and GluSph accumulation may promote α‐synuclein aggregation,6, 7, 8 further indicating the importance of studying these lipids as biomarkers of GBA‐PD. However, accumulation of GluCer or GluSph in GBA‐PD brain or cerebrospinal fluid (CSF) has not been unequivocally demonstrated,9, 10, 11 posing a major challenge to the loss‐of‐function hypothesis for GBA‐PD.
GluCer is by far more abundant than GluSph and is shown to accumulate in macrophages and plasma of people with GD. However, it is not a clinically useful biomarker for GD. 12 By contrast, plasma GluSph levels correlate with disease burden in GD, 13 leading to its routine use as a biomarker for GD severity, as well as response to therapy. 14 GluSph is also a key regulator of the immune system. Thus, it would be important to develop sensitive assays to monitor GluSph levels and determine its utility as a biomarker of GBA‐PD.
In this study, we measured GCase substrates, GluSph and GluCer, as well as the product, ceramide, and additional lipids (galactosyl ceramide, galactosyl sphingosine, and glucosyl cholesterol) as negative controls in a cohort of GBA1 N370S mutation carriers and noncarriers with and without PD. We aimed to test whether GluSph levels were elevated in GBA1 N370S mutation carriers and whether levels were associated with PD status. We hypothesized, based on the GD literature, that plasma GluSph would be a superior marker of reduced GCase activity in comparison with plasma GluCer levels.
Materials and Methods
Participants in this study were participants of the Spot study, which was previously described. 15 The Spot study is an ongoing biomarker recruitment effort at Columbia University Irving Medical Center and the Icahn School of Medicine at Mount Sinai (ISMMS), through which we have identified (and reported on) 218 carriers of various GBA1 pathogenic variants, including 135 carriers with PD and 83 carriers without PD.15, 16 In brief, GBA1 carriers with and without PD were recruited among Columbia University Irving Medical Center patients with PD and spouse controls, and GBA1 carriers without PD were recruited among family members of patients with GD (ie, parents or children, who are by definition obligate carriers) at the Lysosomal Storage Diseases Program at ISMMS. Demographics, as well as the Unified Parkinson's Disease Rating Scale (UPDRS) score in the on state and the Montreal Cognitive Assessment score, were collected from all participants. To reduce heterogeneity, we included only GBA1 N370S heterozygotes in this study (and not other GBA1 pathogenic variants). We further excluded LRRK2 G2019S mutation carriers. In addition, we included four samples of N370S homozygotes with PD, who also by definition have GD (ie, GD/PD). Two of the four GD/PD participants were treated with enzyme replacement therapy at the time of recruitment. A total of 10 mL of whole blood per subject was collected in Ethylenediaminetetraacetic acid (EDTA) tubes, which were centrifuged, and from which 1 mL plasma aliquots per subject were extracted within 60 minutes of collection. Samples were stored in a −80°C freezer until processing. All clinical study procedures were approved by the Columbia University Institutional Review Board (and ISMMS Institutional Review Board, if collected at Mount Sinai, NY, USA), and all participants signed informed consents. All samples were shipped to Nextcea (Woburn, MA, USA) on dry ice in one batch.
Quantitative Ultraperformance Liquid Chromatography Tandem Mass Spectrometry Analyses
The bioanalytics were conducted by Nextcea, where scientists remained blinded to participants' PD status and genotype before analysis.
A multiplexed quantitative ultraperformance liquid chromatography tandem mass spectrometry method was used to simultaneously quantitate plasma ceramides (Cer d18:1/16:0, d18:1/18:0, d18:1/20:0, d18:1/22:0, d18:1/24:0, d18:1/24:1), GluCers (GluCer d18:1/16:0, d18:1/18:0, d18:1/22:0, d18:1/24:0, d18:1/24:1), galactosylceramides (GalCer d18:1/16:0, d18:1/18:0, d18:1/22:0, d18:1/24:0, d18:1/24:1), GluSph (GluSph 18:1), galactosylsphingosine (GalSph 18:1), and glucosylcholesterol. The GluSph (GluSph 18:1) was well separated from galactosylsphingosine (GalSph 18:1). Standard curves were prepared from related standards using a class‐based approach. Internal standards were used for each analyte reported. A SCIEX Triple Quad 7500 mass spectrometer was used in positive electrospray ionization mode for detection (SCIEX, Framingham, MA, USA). Injections were made using a Shimadzu Nexera XR UPLC (ultraperformance liquid chromatography) system (Shimadzu Scientific Instruments, Kyoto, Japan). The instruments were controlled by SCIEX OS 2.0 software.
The parameters for assay validation included 3‐day intra‐assay and interassay accuracy and precision, sensitivity (lower limit of quantitation [LLOQ]), specificity, carryover, recovery, matrix effect, dilution integrity, and stability in human plasma. The LLOQ for all lipids measured was 5 pg/mL. Interassay and intra‐assay coefficients of variation for all dilutions (including LLOQ) were ≤20%. The specificity of the GluSph versus GluCer assay was evaluated at the LLOQ in six different lots of human plasma with stable isotope‐labeled GluCer and GluSph, where we observed no interference.
Calibration and Data Processing
The intensities of the analytes and internal standards were determined by integration of extracted ion peak areas using SCIEX OS 2.0 software. Calibration curves were prepared by plotting the peak area ratios for each analyte to internal standard versus concentration. The model for the calibration curves was linear with (1/x 2) weighting. The concentrations of all lipids were quantitated in nanograms per milliliter (ng/mL) of plasma.
Statistical Analysis
Demographics and disease characteristics were compared across the four groups based on PD status and genotype (GBA − /PD − , GBA +/PD − , GBA − /PD+, GBA + /PD+). Groups were a priori matched by sex and age (PD − versus PD+). We then compared the lipid species concentrations across the four groups. We computed total concentrations of ceramides, GluCer and GalCer, by summing all fatty acid analogs for each species. Although primary analyses compared the four groups, we included the four GD/PD subjects (positive controls) in Figure 1 for illustrative purposes. We further constructed regression models to predict GBA1 mutation status (outcome), including sex, age, PD status, and lipid concentrations as predictors. Analyses were computed in SPSS. Figure 1 (and post hoc Tukey analyses) was computed using GraphPad Prism version 9.1.1 for macOS, GraphPad Software (GraphPad, San Diego, CA, USA; https://www.graphpad.com).
Results
Demographic Comparisons
Demographic and disease characteristics of the four groups (GBA − /PD−, GBA +/PD−, GBA − /PD+, GBA + /PD+) are presented in Table 1. The groups were matched by sex and age (PD versus non‐PD), and the PD groups were also similar in age at onset, disease duration, education, Montreal Cognitive Assessment performance, levodopa‐equivalent daily dose, and Unified Parkinson's Disease Rating Scale, Part III scores. Figure 1 demonstrates the plasma concentration of total ceramides, total GluCer, and GluSph d18:1 in GBA1 N370S heterozygotes and noncarriers with and without PD and in four patients with GD/PD. The plasma lipid concentrations across the four groups are presented in Table 1 and Supporting Information Table S1. Next, we performed logistic regression models to test the association between GBA1 status and plasma lipid concentrations, including age, sex, PD status, and the six lipid groups tested. GluSph was the only variable (and the only lipid) associated with GBA mutation status (P < 0.001).
TABLE 1.
Demographics, disease characteristics, and lipid concentration data
| GBA −/PD− | GBA +/PD− | GBA −/PD+ | GBA +/PD+ | P value a | |
|---|---|---|---|---|---|
| n | 20 | 20 | 20 | 20 | |
| Sex (male/female) | 10/10 | 10/10 | 10/10 | 10/10 | |
| Age, y | 71.1 (9.1) | 59.8 (9.2) | 65.9 (12.8) | 66.2 (9.5) | 0.010 |
| AAO | N/A | N/A | 59.4 (14.2) | 60.6 (9.6) | 0.756 |
| Education, y | 16.6 (3.1) | 18 (3.5) | 16.4 (3.5) | 16.4 (2.7) | 0.329 |
| MoCA score | 26.6 (3.1) | 26.5 (1.8) | 24.9 (3.2) | 25.1(2.9) | 0.108 |
| UPDRS III score | 16.0 (8.3) | 15.8 (7.7) | 0.944 | ||
| LEDD | 593 (560) | 447 (359) | 0.332 | ||
| Glucosylsphingosine d18:1 | 0.48 (0.14) | 0.73 (0.20) | 0.50 (0.14) | 0.82 (0.24) | <0.001 |
| Total glucosylceramides | 2443.75 (706.45) | 3217.22 (814.86) | 2910.08 (1030.44) | 2701.03 (1139.59) | 0.071 |
| Total ceramides | 3038.51 (1554.18) | 2338.91 (876.27) |
2719.79 (1260.67) |
2350.19 (1148.74) |
0.228 |
| Glucosylcholesterol | 165.92 (178.04) | 99.33 (63.37) | 116.45 (80.43) | 130.16 (148.91) | 0.401 |
| Total galactosylceramides | 323.08 (110.55) | 387.35 (154.74) | 341.73 (144.46) | 324.25 (81.96) | 0.341 |
| Galactosylsphingosine d18:1 | 0.11 (0.05) | 0.13 (0.07) | 0.13 (0.06) | 0.14 (0.08) | 0.417 |
Values are presented as mean (standard deviation). Data were analyzed by one‐way ANOVA. Participants were grouped by the presence or absence of either Parkinson's disease (PD− or PD+) or the GBA1 N370S mutation (GBA − or GBA +).
P value represents overall effect by ANOVA.
AAO, age at motor onset; MoCA, Montreal Cognitive Assessment; UPDRS III, Unified Parkinson's Disease Rating Scale, Part III; LEDD, levodopa‐equivalent daily dose; N/A, not applicable.
FIG. 1.
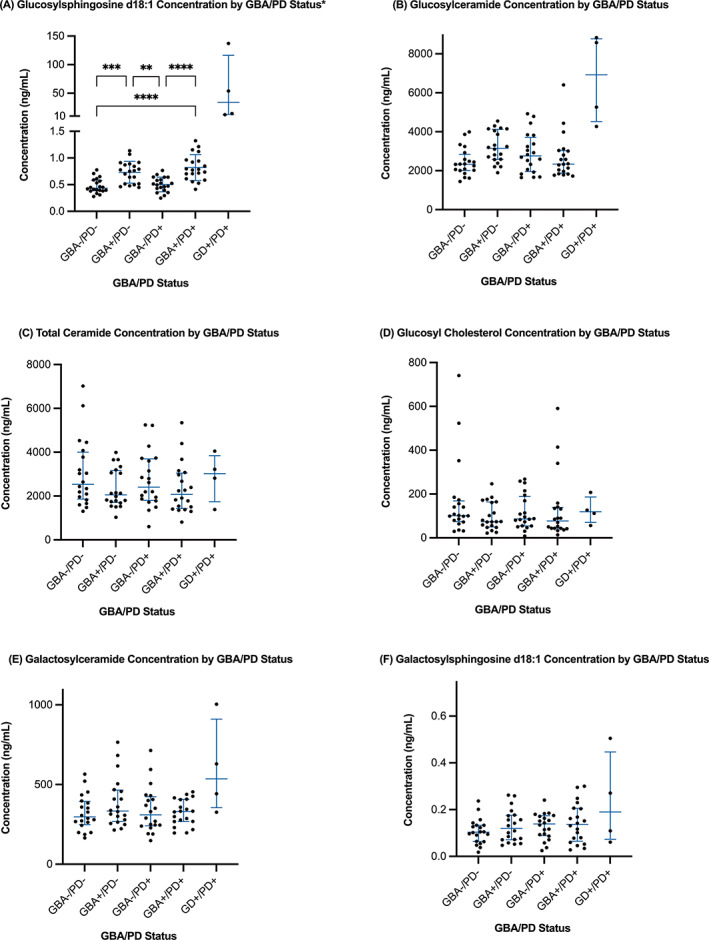
Plasma lipid concentration for glucolipids in GBA −/PD−, GBA +/PD−, GBA −/PD+, GBA +/PD+, and Gaucher's disease (GD)+/PD+, including: (A) glucosylsphingosine d18:1 (GluSph), (B) glucosylceramide (GluCer), (C) total ceramides, (D) glucosylcholesterol (GluChol), (E) galactosylceramide (GalCer), and (F) galactosylsphingosine d18:1 (GalSph). Patients with GD/PD had significantly higher plasma levels of GluSph and GluCer than all other groups (A and B). GBA +/PD− and GBA +/PD+ had significantly higher GluSph levels than GBA −/PD− and GBA −/PD+ (A: GBA −/PD− vs. GBA + /PD−, P = 0.0002; GBA −/PD− vs. GBA +/PD+, P < 0.0001; GBA −/PD+ vs. GBA + /PD−, P = 0.0012; GBA −/PD+ vs. GBA +/PD+, P < 0.0001). **P < 0.005; ***P < 0.0005; ****P < 0.0001.
Discussion
The link between GBA1 pathogenic variants and PD has opened a window of opportunity for multiple therapeutics targeting the GCase pathway. The mechanisms of interventions range from augmentation of GCase activity via chaperones or allosteric activators, inhibition of GluCer synthase, and GBA gene therapy.17, 18, 19 The therapeutic development trials require reliable biomarkers to establish target engagement/modulation and inform effective dose range. Furthermore, the penetrance of PD among carriers of pathogenic variants in GBA1 is estimated at 10–30%, indicating that the majority of mutation carriers will never develop PD.20, 21, 22, 23 Thus, biomarkers that can prognosticate which GBA1 pathogenic variant carriers are more likely to develop PD are required to conduct patient‐enriched trials.
To the best of our knowledge, this is the first study to successfully detect GluSph in the plasma of non‐GD GBA1 mutation carriers. Importantly, our data demonstrate that a single allele of GBA1 N370S is sufficient to result in a significant elevation in plasma GluSph levels. This effect was specific for GluSph among the panel of six lipids tested, including GluCer (Table 1, Fig. 1). The validity of our assays is also confirmed by the elevation in GluSph and GluCer in the four GD/PD cases. Thus, plasma GluSph offers a promising biomarker of target engagement/modulation in GCase‐targeted therapies for PD. However, it is possible (given our sample size) that we were underpowered to detect differences of smaller effect size, for example, in GluCer levels between carriers and noncarriers, or in GluSph levels between carriers with and without PD. Previous studies support the hypothesis that we were underpowered to identify small differences in GlucCer levels between carriers and noncarriers. Specifically, in the Parkinson's Progression Markers Initiative study, noncarriers had lower GluCer levels in CSF than carriers, but similar analysis in plasma (as done in this study) was not reported. 24
It is important to note that GluSph, rather than GluCer, is emerging as a more disease‐relevant biomarker. A recent study reported higher levels of GluCer in the CSF in conjunction with a decrease in blood GCase activity in GBA‐PD compared with idiopathic PD, but the differences were observed primarily in those with severe GBA mutations (eg, L444P), but not the mild GBA mutation, N370S 11 (GluSph was not assayed in this study). When GCase activity is deficient, the enzyme acid ceramidase metabolizes GluCer into GluSph. 25 GluSph has been shown to cause oligomerization of α‐synuclein in vitro more efficiently than GluCer. 7 More importantly, GluSph, and not GluCer, induced pathogenic templating of α‐synuclein in cells, including in induced pluripotent stem cell–derived neurons. 7 Furthermore, a role of GluSph in immune cell activation is well documented, 26 and GluSph levels in the brain correlated with neurological manifestations in GD. 27 Because aberrant immune activation in PD is thought to contribute to pathogenesis, the elevation in plasma GluSph in N370S mutation carriers suggests that this lipid has the potential to be a disease pathophysiology marker even for prodromal GBA‐PD. However, the exact mechanism by which GBA mutations cause PD remains to be determined. For example, it is possible that reduced activity of GCase leads to lower intralysosomal levels of ceramide, which negatively affect the maturation and activation of cathepsin‐D.28, 29
We have not identified an association between plasma GluSph levels and PD status in the current samples of N370S carriers or with idiopathic PD. However, because GluSph levels were numerically higher in carriers with PD compared with carriers without PD (0.82 ± 0.24 versus 0.73 ± 0.20), and in noncarriers with PD compared with controls (0.50 ± 0.14 versus 0.48 ± 0.14), we posit that a larger cohort of GBA mutation carriers and noncarriers will be needed to investigate whether GluSph levels are associated PD status. Furthermore, future studies should include a wide range of GBA1 pathogenic variants (eg, E326K) to test potential correlation between variant severity and GluSph levels. Studies that will examine GluSph levels in the brain and CSF in addition to blood plasma may firmly test its relevance as a disease marker, as is the case for GD.
In summary, these results indicate that plasma GluSph levels may be a useful target engagement/modulation biomarker for interventions aiming to increase GCase activity. Studies on larger cohorts would be required to test whether elevated GluSph levels are associated with PD risk or progression.
Author Roles
Study concept and design: R.N.A., F.H., K.S.M., K.M.M., S.P.
Subject recruitment, assessment, genotyping, and blood collection: M.S., M.B., Z.G.‐O.
Biomarker assays: F.H., T.H., L.S.
Statistical analysis of data: R.N.A.
Initial drafting of manuscript: R.N.A., K.M.M., M.S., A.H.
Editing and approval of submitted manuscript: all authors.
Financial Disclosures
Disclosures unrelated to the study: Manisha Balwani: research support (NIH/NIDDK, Genzyme, Alnylam, Mitsubishi‐Tanabe), honoraria (Genzyme, Takeda, Alnylam, Prevail, Freeline, Alexion). Cheryl Waters: research support (Biogen, Roche, Sanofi); consulting fees (Kyowa, Alexza, Sunovion); speaker's honoraria (Acadia, Acorda, Adamas, Amneal, Kyowa, Neurocrine, US WorldMeds). Ziv Gan‐Or: research support (The Michael J. Fox Foundation, Fonds de recherche du Québec–Santé, Canadian Consortium for Neurodegeneration in Aging, Canada First Research Excellence Fund through the Healthy Brain for Healthy Lives project), consulting fees (Denali, Inception Sciences [now Ventus], Idorsia, Lysosomal Therapeutics Inc., Prevail Therapeutics, Deerfield, Lighthouse, Neuron23, Ono Therapeutics, Handl Therapeutics, and Bial Biotech Inc.). Kalpana M. Merchant: research support (The Michael J. Fox Foundation), advisor/paid consultant (The Michael J. Fox Foundation, Caraway Therapeutics, Sinopia Biosciences, Nitrome, NuraBio, Retromer Therapeutics, Vanqua Biosciences). Roy N. Alcalay: research support (NIH, DoD, the Parkinson's Foundation, and The Michael. J. Fox Foundation), consultation fees (Avrobio, Caraway, GSK, Merck, Ono Therapeutics, and Sanofi).
Supporting information
Table S1 Concentrations of each lipid analyzed in carriers and non‐carriers with and without PD
Values are presented as mean (standard deviation). Data were analyzed by one‐way ANOVA. Participants were grouped by the presence or absence of either Parkinson's disease (PD− or PD+) or the GBA1 N370S mutation (GBA− or GBA+). a P value represents overall effect by ANOVA.
Acknowledgments
This research was supported by the Parkinson's Foundation (Stanley Fahn Junior Faculty Award), The Michael J. Fox Foundation, and the National Institutes of Health (Grants K02NS080915 and UL1 TR000040).
Relevant conflicts of interest/financial disclosures: The authors have no financial disclosures or conflicts of interest to report. Disclosures unrelated to the study: Matthew Surface: Mr. Surface received compensation for employment at Ipsos. Manisha Balwani: Research support (NIH/NIDDK, Genzyme, Alnylam, Mitsubishi‐Tanabe), Honoraria (Genzyme, Takeda, Alnylam, Prevail, Freeline, Alexion). Cheryl Waters: Research support (Neuraly,Biogen, Roche, Sanofi); consulting fees (Kyowa, Sunovion); speaker's honoraria (Adamas, Amneal, Kyowa, Neurocrine). Ziv Gan Or: Research support (Michael J. Fox Foundation, Fonds de recherche du Québec – Santé, Canadian Consortium for Neurodegeneration in Aging, Canada First Research Excellence Fund through the Healthy Brain for Healthy Lives project); Consulting fees (Denali, Inception Sciences (now Ventus), Idorsia, Lysosomal Therapeutics Inc., Prevail Therapeutics, Deerfield, Lighthouse, Neuron23, Ono Therapeutics, Handl Therapeutics and Bial Biotech Inc).; Kalpana Merchant: research support from the Michael J Fox Foundation, advisor/paid consultant to Michael J Fox Foundation, Caraway Therapeutics, Sinopia Biosciences, Nitrome, NuraBio, Retromer Therapeutics. Roy N. Alcalay is funded by the NIH, DoD, the Parkinson's Foundation and the Michael. J. Fox Foundation. He received consultation fees from Avrobio, Caraway, GSK, Merck, Ono Therapeutics and Sanofi.
Funding agencies: The research was funded by the Parkinson's Foundation (Stanley Fahn Junior Faculty Award), the Michael J. Fox Foundation, the and the National Institutes of Health (K02NS080915, and UL1 TR000040).
Contributor Information
Kalpana M. Merchant, Email: kalpana.merchant@northwestern.edu.
Roy N. Alcalay, Email: rna2104@columbia.edu.
Data Availability Statement
The data that support the findings of this study are available on request from the corresponding author.
References
- 1. Hruska KS, LaMarca ME, Scott CR, Sidransky E. Gaucher disease: mutation and polymorphism spectrum in the glucocerebrosidase gene (GBA). Hum Mutat 2008;29:567–583. [DOI] [PubMed] [Google Scholar]
- 2. Neudorfer O, Giladi N, Elstein D, et al. Occurrence of Parkinson's syndrome in type I Gaucher disease. QJM 1996;89:691–694. [DOI] [PubMed] [Google Scholar]
- 3. Sidransky E, Nalls MA, Aasly JO, et al. Multicenter analysis of glucocerebrosidase mutations in Parkinson's disease. N Engl J Med 2009;361:1651–1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gan‐Or Z, Giladi N, Rozovski U, et al. Genotype‐phenotype correlations between GBA mutations and Parkinson disease risk and onset. Neurology 2008;70:2277–2283. [DOI] [PubMed] [Google Scholar]
- 5. Mazzulli JR, Xu YH, Sun Y, et al. Gaucher disease glucocerebrosidase and alpha‐synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell 2011;146:37–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Suzuki M, Sango K, Wada K, Nagai Y. Pathological role of lipid interaction with alpha‐synuclein in Parkinson's disease. Neurochem Int 2018;119:97–106. [DOI] [PubMed] [Google Scholar]
- 7. Taguchi YV, Liu J, Ruan J, et al. Glucosylsphingosine promotes alpha‐synuclein pathology in mutant GBA‐associated Parkinson's disease. J Neurosci 2017;37:9617–9631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zunke F, Moise AC, Belur NR, et al. Reversible conformational conversion of alpha‐synuclein into toxic assemblies by glucosylceramide. Neuron 2018;97:92–107. e110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Huebecker M, Moloney EB, van der Spoel AC, et al. Reduced sphingolipid hydrolase activities, substrate accumulation and ganglioside decline in Parkinson's disease. Mol Neurodegener 2019;14:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hallett PJ, Huebecker M, Brekk OR, et al. Glycosphingolipid levels and glucocerebrosidase activity are altered in normal aging of the mouse brain. Neurobiol Aging 2018;67:189–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lerche S, Sjodin S, Brinkmalm A, et al. CSF protein level of neurotransmitter secretion, synaptic plasticity, and autophagy in PD and DLB. Mov Disord 2021. doi: 10.1002/mds.28704 [DOI] [PubMed] [Google Scholar]
- 12. Barton NW, Brady RO, Dambrosia JM, et al. Replacement therapy for inherited enzyme deficiency‐macrophage‐targeted glucocerebrosidase for Gaucher's disease. N Engl J Med 1991;324:1464–1470. [DOI] [PubMed] [Google Scholar]
- 13. Murugesan V, Chuang WL, Liu J, et al. Glucosylsphingosine is a key biomarker of Gaucher disease. Am J Hematol 2016;91:1082–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dekker N, van Dussen L, Hollak CE, et al. Elevated plasma glucosylsphingosine in Gaucher disease: relation to phenotype, storage cell markers, and therapeutic response. Blood 2011;118:e118–e127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Alcalay RN, Levy OA, Waters CC, et al. Glucocerebrosidase activity in Parkinson's disease with and without GBA mutations. Brain 2015;138:2648–2658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Galper J, Balwani M, Fahn S, et al. Cytokines and Gaucher biomarkers in glucocerebrosidase carriers with and without Parkinson disease. Mov Disord 2021;36:1451–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Schneider SA, Alcalay RN. Precision medicine in Parkinson's disease: emerging treatments for genetic Parkinson's disease. J Neurol 2020;267:860–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sardi SP, Cedarbaum JM, Brundin P. Targeted therapies for Parkinson's disease: from genetics to the clinic. Mov Disord 2018;33:684–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Menozzi E, Schapira AHV. Enhancing the activity of glucocerebrosidase as a treatment for Parkinson disease. CNS Drugs 2020;34:915–923. [DOI] [PubMed] [Google Scholar]
- 20. Anheim M, Elbaz A, Lesage S, et al. Penetrance of Parkinson disease in glucocerebrosidase gene mutation carriers. Neurology 2012;78:417–420. [DOI] [PubMed] [Google Scholar]
- 21. Alcalay RN, Dinur T, Quinn T, et al. Comparison of Parkinson risk in Ashkenazi Jewish patients with Gaucher disease and GBA heterozygotes. JAMA Neurol 2014;71:752–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rana HQ, Balwani M, Bier L, Alcalay RN. Age‐specific Parkinson disease risk in GBA mutation carriers: information for genetic counseling. Genet Med 2013;15:146–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. McNeill A, Duran R, Hughes DA, Mehta A, Schapira AH. A clinical and family history study of Parkinson's disease in heterozygous glucocerebrosidase mutation carriers. J Neurol Neurosurg Psychiatry 2012;83:853–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lerche S, Schulte C, Wurster I, et al. The mutation matters: CSF profiles of GCase, sphingolipids, alpha‐synuclein in PDGBA. Mov Disord 2021;36:1216–1228. [DOI] [PubMed] [Google Scholar]
- 25. Ferraz MJ, Marques AR, Appelman MD, et al. Lysosomal glycosphingolipid catabolism by acid ceramidase: formation of glycosphingoid bases during deficiency of glycosidases. FEBS Lett 2016;590:716–725. [DOI] [PubMed] [Google Scholar]
- 26. van Eijk M, Ferraz MJ, Boot RG, Aerts J. Lyso‐glycosphingolipids: presence and consequences. Essays Biochem 2020;64:565–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Orvisky E, Park JK, LaMarca ME, et al. Glucosylsphingosine accumulation in tissues from patients with Gaucher disease: correlation with phenotype and genotype. Mol Genet Metab 2002;76:262–270. [DOI] [PubMed] [Google Scholar]
- 28. Bellomo G, Paciotti S, Gatticchi L, Parnetti L. The vicious cycle between alpha‐synuclein aggregation and autophagic‐lysosomal dysfunction. Mov Disord 2020;35:34–44. [DOI] [PubMed] [Google Scholar]
- 29. Murphy KE, Gysbers AM, Abbott SK, et al. Reduced glucocerebrosidase is associated with increased alpha‐synuclein in sporadic Parkinson's disease. Brain 2014;137:834–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 Concentrations of each lipid analyzed in carriers and non‐carriers with and without PD
Values are presented as mean (standard deviation). Data were analyzed by one‐way ANOVA. Participants were grouped by the presence or absence of either Parkinson's disease (PD− or PD+) or the GBA1 N370S mutation (GBA− or GBA+). a P value represents overall effect by ANOVA.
Data Availability Statement
The data that support the findings of this study are available on request from the corresponding author.