

Abstract
Modern drug delivery technology began in 1952 with the advent of the Spansule® sustained-release capsule technology, which can deliver a drug for 12 hours after oral administration through an initial immediate dose followed by the remaining released gradually. Until the 1980s, oral and transdermal formulations providing therapeutic durations up to 24 hours for small molecules dominated the drug delivery field and the market. The introduction of Lupron Depot® in 1989 opened the door for long-acting injectables and implantables, extending the drug delivery duration from days to months and occasionally years. Notably, the new technologies allowed long-term delivery of peptide and protein drugs, although limited to parenteral administration. The introduction of the first PEGylated protein, Adagen®, in 1990 marked the new era of PEGylation, resulting in Doxil® (doxorubicin in PEGylated liposome) in 1995, Movantik (PEGylated naloxone - naloxegol) in 2014, and Onpattro® (Patisiran - siRNA in PEGylated lipid nanoparticle) in 2018. Drug-polymer complexes were introduced, e.g., InFed® (iron-dextran complex injection) in 1974 and Abraxane® (paclitaxel-albumin complex) in 2005. In 2000, both Mylotarg™ (antibody-drug conjugate – gemtuzumab ozogamicin) and Rapamune® (sirolimus nanocrystal formulation) were introduced. The year 2000 also marked the launching of the National Nanotechnology Initiative by the U.S. government, which was soon followed by the rest of the world. Extensive work on nanomedicine, particularly formulations designed to escape from endosomes after being taken by tumor cells, along with PEGylation technology, ultimately resulted in the timely development of lipid nanoparticle formulations for COVID-19 vaccine delivery in 2020.
While the advances in drug delivery technologies for the last seven decades are breathtaking, they are only the tip of an iceberg of technologies that have yet to be utilized in an approved formulation or even to be discovered. As life expectancy continues to increase, more people require long-term care for various diseases. Filling the current and future unmet needs requires innovative drug delivery technologies to overcome age-old familiar hurdles, e.g., improving water-solubility of poorly soluble drugs, overcoming biological barriers, and developing more efficient long-acting depot formulations. The lessons learned from the past are essential assets for developing future drug delivery technologies implemented into products. As the development of COVID-19 vaccines demonstrated, meeting the unforeseen crisis of the uncertain future requires continuous cumulation of failures, knowledge, and technologies. Conscious efforts of supporting diversified research topics in the drug delivery field are urgently needed more than ever.
Keywords: drug delivery, history, evolution, drug targeting, biological barriers, long-term treatment
1. Evolution of Drug Delivery Systems
The term “drug delivery system” means a drug formulation, e.g., tablet, capsule, ointment, and solutions. The term “controlled release drug delivery system” or “controlled drug delivery system” means that a formulation has a built-in technology to control the drug release kinetics over time. The controlled release drug delivery systems are distinguished from conventional formulations that release most or all loaded drug(s) immediately without any control. Thus, conventional formulations are usually called “immediate release” or IR formulations. The term “controlled” had an additional meaning of maintaining relatively constant (i.e., the same) drug concentration in the blood over time. However, maintaining a constant drug concentration was difficult, especially for oral controlled release formulations.
The evolution of drug delivery technologies can be described in different ways, e.g., classes of therapeutics and delivery paradigms [1]. Here, the evolution is described by introducing new technologies using the products approved by the U.S. Food and Drug Administration (FDA). Figure 1 describes landmark developments in drug delivery systems that have shaped the history of controlled drug delivery systems. While drug delivery technologies improve constantly, the ultimate measure of the success of a formulation is demonstrated safety and efficacy through the approval by the FDA, enabling patients to benefit from the new technologies. As shown on the left of Figure 1, the initial developments were based on oral and transdermal formulations, all of which were based on four drug release mechanisms: diffusion-, dissolution-, osmosis-, and ion exchange-controlled drug release. Out of the four, dissolution- and diffusion-controlled mechanisms have been used most widely. Modulated drug delivery, also called self-regulated drug delivery, was mainly designed to control insulin delivery, but it has not been achieved yet.
Figure 1.
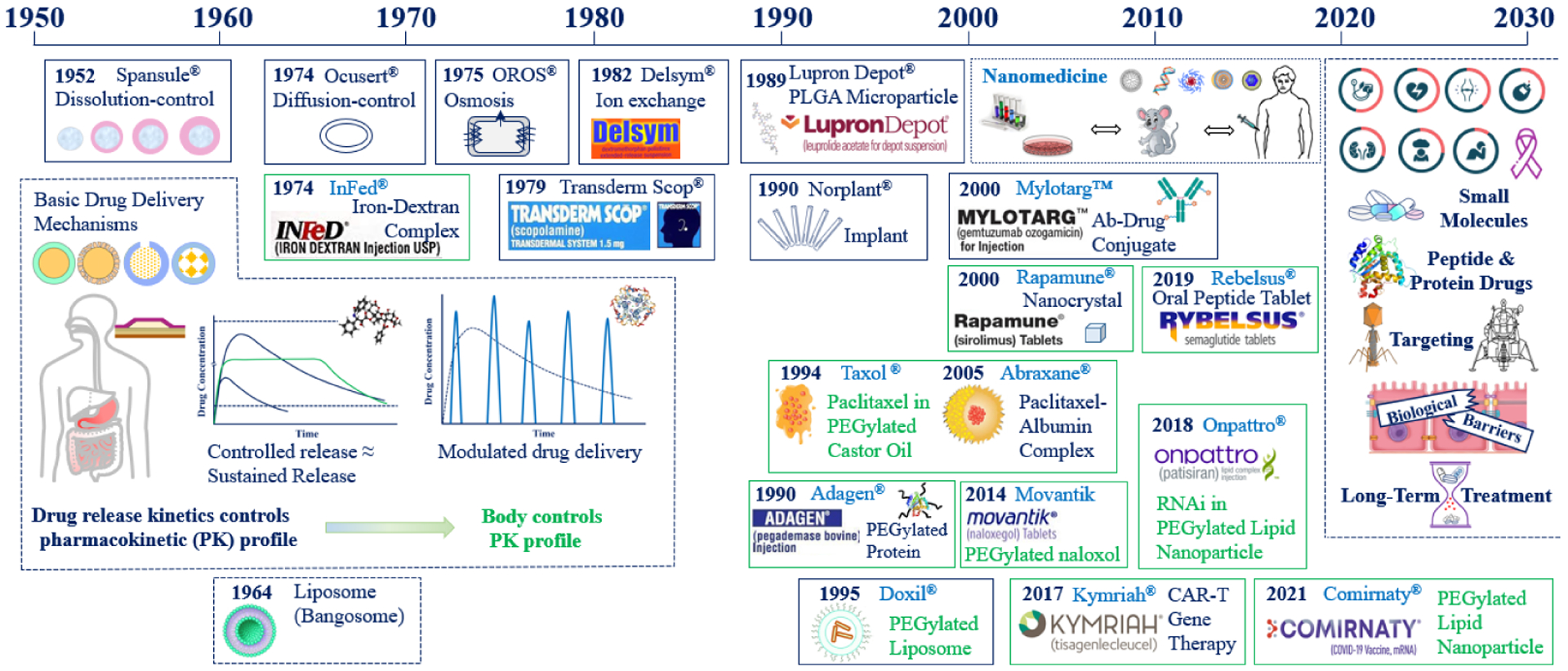
Evolution of drug delivery systems through the introduction of FDA-approved drug products from the Spansule® oral controlled release formulation to the lipid nanoparticle formulations for COVID-19 vaccine delivery. 24-hour release oral controlled release formulations in the 1950–1970s were followed by 24-week injectable depot formulations in the 1990s and nanomedicine in the 2000s. Technological advances made in the last seven decades provide a foundation for further developing new technologies for the next few decades.
A formulation providing longer durations of release is theoretically as equally effective as an IR formulation, as long as the drug concentration in the blood remains below the maximum safe concentration (Cmax) and above the minimum effective concentration (Cmin). The ratio Cmax/Cmin is known as a therapeutic index. Since most drugs have a therapeutic index large enough to be safe even if excess drugs are consumed, certain fluctuations of the drug concentration in the blood typically result in the same efficacy. Furthermore, controlled release drug delivery systems can minimize the number of peaks and troughs in plasma concentrations, thereby minimizing potential side effects and periods of non-efficacy. Since the drug concentration in the blood does not have to be constant to be safe and effective, various formulations were developed, called “sustained” or “extended” release systems. Over time, different terms have been used interchangeably. Other terms, such as “therapeutic system” or “modified system,” were also used. Still, they all mean controlled release formulations – whereby controlled release is referred to as the ideal release of the drug, i.e., at a zero- or first-order rate constant. According to the FDA, modified-release solid oral dosage forms refer to extended-release and delayed/enteric-coated formulations [2]. The terms “extended-release” and “modified-release” are used by the US Pharmacopeia (USP), and their requirements for the respective drug release are specified in their individual monographs (USP <724> and <1088>) [3]. Nowadays, almost all formulations are made for sustained drug delivery, and thus, the term “drug delivery system” is used to represent all types of formulations. Conventional tablet and capsule formulations are distinguished by calling them immediate-release formulations. Since the late 1980s, sustained-release has also meant drug delivery for months. Thus, the meaning of sustained-release needs to be understood in the context of drug delivery technologies and their representative administration routes.
1.1. The Beginning
1.1.1. The Spansule Technology and Beyond
The dawning of the controlled release drug delivery systems began with the introduction of the Spansule® 12-hour release technology by Smith, Kline & French Laboratories, which was first applied to develop Dexedrine® (delivering dextroamphetamine sulfate), followed by Contac® 600 (delivering phenylpropanolamine hydrochloride and chlorpheniramine maleate). [4–6]. The Spansule technology is based on controlling the dissolution of the drug core through a coating barrier that limits access to GI fluids, and thus, the dissolution-controlled mechanism. It spurred the development of other oral formulations utilizing different release mechanisms, such as diffusion-controlled (e.g., Ocusert® delivering pilocarpine), osmotic pressure-controlled (Oral Osmotic (OROS®) formulation, such as Acutrim® delivering phenylpropanolamine, and Concerta delivering methylphenidate), and ion-exchange-controlled (e.g., Delsym® delivering dextromethorphan). Out of these, dissolution- and diffusion-controlled systems dominate the number of formulations approved by the FDA. There are only about 20 products based on osmosis, while there is only one product, Delsym, based on an ion-exchange mechanism. Since the ion-exchange mechanism is not suited for sustained release in the high salt environment, the resin particles are also coated with diffusion-limiting polymers. The diffusion-controlled release mechanisms were also used to develop various transdermal drug delivery systems, e.g., Transderm Scop®. Other controlled-release mechanisms were developed and explored during the 1950s-1980s, but most commercial products were based on the dissolution- and diffusion-controlled mechanisms and the mixture of the two. Norplant, a cylinder-shaped tube the size of a match stick, contains a contraceptive agent that is released through the silicone rubber tube for 5 years. It received FDA approval in 1990, much later than other formulations utilizing the same reservoir technology simply because it had to have a long clinical testing period. Although, it was removed from the US market in 2002, mainly due to insertion and removal issues and lack of experience in administering the implants. During the first three decades, mathematical modeling for drug release was developed to further mature the drug delivery field [7].
1.1.2. Zero-Order Drug Release and Modulated Drug Release
The ability of drug release at a zero-order rate attracted many scientists to develop oral formulations that can maintain a constant drug concentration in the blood. Although, making such oral formulations was difficult due to a few physiological limitations. First, as an oral formulation transits from the stomach to the intestine, the drug absorption usually decreases because of the reduced absorption abilities of the lower segments of the intestine. This problem was usually compounded by the reduced drug amount released from the formulation over time. The only example of maintaining the constant blood concentration for about 16 hours was phenylpropanolamine HCl release from Acutrim® (an OROS formulation) [8, 9]. Compensating decreased drug absorption as a formulation transits through the intestine requires more drug released over time, which is much more complicated than making zero-order release formulations. Furthermore, maintaining a constant drug concentration in the blood is not necessary, as most drugs have a therapeutic index (The maximum safe drug concentration/the minimum effective drug concentration, or Cmax/Cmin) that is large enough to be safe and effective over the order of magnitude different blood concentrations.
Another extensive effort was focused on developing modulated, also known as self-regulated, delivery formulations. This type of delivery system is essential for insulin delivery because insulin needs to be delivered in an exact amount and at the right time as the glucose concentration in the blood fluctuates [10]. But the controlled technology developed to date has been limited to continuous release at a specific rate, and modulated release is still out of reach [11]. For insulin delivery, e.g., a formulation needs to contain a glucose sensor, actuator, and feedback system controlling the insulin release. The modulated drug delivery system remains the most complex technical issue. Hopefully, fully implantable modulated delivery systems can be developed and commercialized soon.
1.1.3. Boundaries of the Early Drug Delivery Technologies
The critical point of the technological advances in the beginning was that the controlled release formulations were almost exclusively dedicated to oral formulations for 12- or 24-hour delivery, and to some extent, to transdermal formulations. For oral formulations, in vitro drug release profiles are usually correlated to the in vivo pharmacokinetic (PK) profiles. Thus, the PK profiles could be adjusted by controlling the formulation parameters. The controlled release formulations started to deliver more prolonged periods, ranging from weeks to months. However, the in vitro drug release kinetics no longer dictated the PK profiles. Developing formulations that deliver drugs for 24 hours is significantly different from developing formulations designed for 24 weeks.
1.2. Long-Acting Injectable Formulations
1.2.1. PLGA-based systems
The first long-acting injectable formulation approved by the United States Food and Drug Administration (FDA) was Lupron Depot® delivering leuprolide acetate for 1 month. Since the first approval in 1989, the PLGA microparticle formulation has expanded to deliver the drug up to 6 months by adjusting the lactide:glycolide (L:G) ratios and molecular weight(s) of the polymer(s). This new long-acting injectable formulation was followed by other systems that delivered not only small molecules but also peptides and proteins [12, 13]. All polymer-based, biodegradable long-acting injectable formulations approved by the FDA are based on PLGA polymers due to their long history of safety. While other biodegradable polymers may provide better functionality, such as better properties of controlling drug release and higher drug loading, the absence of any use in FDA-approved products requires additional regulatory hurdles such as toxicology studies to demonstrate the safety and fate of the polymer after injection/implantation.
Controlling the drug release kinetics for weeks and months is significantly different from drug release for a day. The long-acting, ranging up to 6 months, formulations require large(r) doses, and the drug release needs to be controlled throughout their lifetimes. Currently, three different formulation types can deliver drugs up to 6 months: microparticles, in situ forming implants, and solid implants. Microparticle formulations have been most common due to their ability to load large amounts of drugs (up to ~35% of the total solids content), control the drug release, and administer by either intramuscular or subcutaneous injection.
While two dozen products have been developed using PLGA polymers, surprisingly, our understanding of PLGA polymers has been minimal. Despite more than 3 decades of PLGA formulation development, as shown in Figure 2, there are currently no FDA-approved generic products of long-acting injectable formulations available on the market. One of the reasons for this is the absence of thorough characterization of PLGA polymers. To approve a generic product, FDA needs to consider whether a proposed product is qualitatively (Q1) and quantitatively (Q2) the same as the reference listed drug (RLD) with respect to inactive ingredients [14]. To determine whether PLGA polymers used are the same component (Q1) and the same components in the same concentration (Q2), they must be analyzed for their L:G ratio, molecular weight, and end-group. The issue becomes complicated when an RLD utilizes more than one PLGA type or unconventional PLGA polymer, e.g., branched PLGA instead of linear PLGA [13, 15, 16]. Thorough characterization of PLGAs presents additional benefits for optimizing drug loading and release by using the optimum PLGA.
Figure 2.

Injectable long-acting PLGA formulations approved by the U.S. FDA. All products are based on microparticle, in situ forming implant, and solid implant formulations.
1.2.2. Nanocrystal suspensions
Nanocrystal suspensions are another category of long-acting injectable formulations, composed mainly of hydrophobic drugs with minimal amounts of excipients or surfactants, resulting in very high drug loadings. High energy milling is widely used as a preparation method for this type of formulation. Furthermore, the active molecule can be synthesized into a prodrug, often with a long-chain fatty acid, to further decrease the aqueous solubility and/or dissolution rate, and requires enzymatic or hydrolytic cleavage to become active [17]. Paliperidone palmitate (Invega Sustenna®) is a nanocrystal suspension of the palmitate ester of paliperidone, of which active metabolite of risperidone. An alternative solid-state mechanism is formulating the drug as a salt or co-crystal with a hydrophobic counterion/conformer. Olanzapine pamoate (Zyprexa Relprevv®) is a nanocrystal salt formulation of olanzapine with pamoic acid acting as the counterion. While these formulations are advantageous due to the extraordinarily high drug loading, issues that may arise include changes in the crystal polymorph, aggregation within the suspension, and tissue irritation [18, 19].
1.3. PEGylation
1.3.1. PEGylated Proteins and PEGylated Liposomes
PEGylation is a process of attaching poly(ethylene glycol) (PEG) to protein molecules so that proteins can circulate in the blood longer than the control with substantially reduced immunogenic responses [20–23]. However, the subsequent studies on PEGylated proteins and drug delivery systems showed that the body produced antibodies against PEG, causing accelerated blood clearance (ABC) [24, 25]. The ABC phenomenon is a concern if PEGylated formulations are administered repeatedly, and thus, more effective use of PEGylation requires better understanding. The hydrophilic PEG molecules are known to wobble on the protein surface to reduce uptake by the reticuloendothelial cells, proteolytic degradation, and immune responses, resulting in enhanced therapeutic efficacy. PEGylation also reduces protein drug bioactivity by reducing the binding to the target site, but the more prolonged circulation overall results in enhanced therapeutic efficacy. The concept of PEGylation was born in the late 1970s by Professor Frank Davis of Rutgers University [22, 26, 27]. The main advantage of PEGylation is that it increases blood circulation time, and the idea was used to improve the residence time of liposomes. The first successful PEGylated liposomal formulation was Doxil approved in 1995, demonstrating increased uptake by tumors and decreased toxicity, specifically the cardiotoxicity of doxorubicin [28–30].
The first injectable formulation of PEGylated protein was approved (Adagen®, pegademase bovine injection) in 1990, shortly after the first long-acting injectable PLGA formulation, Lupron Depot®, was approved. Figure 3 shows the introduction of PEGylated protein drugs approved by the FDA. Over the last 30 years, about 20 PEGylated protein formulations have been developed. PEGylated interferon-α2b is currently used for treating COVID-19.
Figure 3.
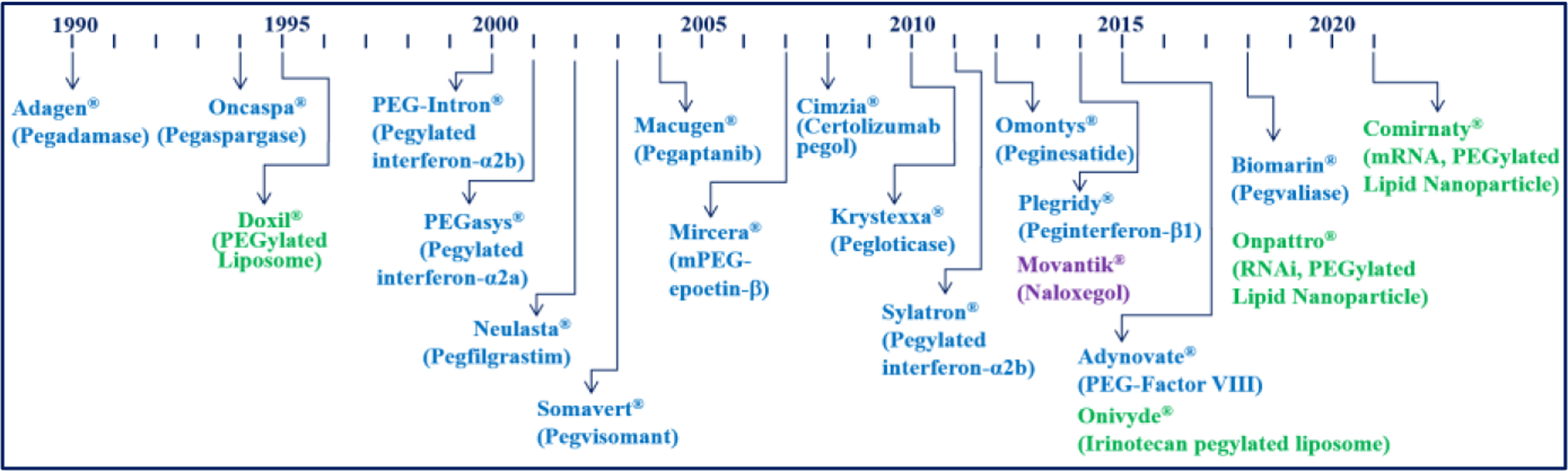
Development of PEGylated drugs for the last two decades. PEGylation was also critical in the development of liposome and lipid nanoparticle formulations.
The significance of the PEGylation technology goes beyond increasing the circulation times of modified proteins, and its application to liposomal formulations, starting from Doxil® (PEGylated liposome delivery doxorubicin) in 1995, was the beginning to other vital formulations. Liposomes were initially referred to as Bangosomes or smectic mesophase [31] after Alec Bangham first observed multilamellar lipid structures in 1964 [32]. The name was changed later to liposomes as the lipid structures were formulated using phospholipids [33]. The liposome’s potential as a drug delivery system was first suggested by Gregory Gregoriadis [34, 35]. Later, liposomes were also used to deliver DNAs to cells [36]. Since the first approval of a PEGylated liposome formulation, Doxil®, more than a dozen lipid formulations have been in clinical use [29]. In addition to liposomes, lipid nanoparticles containing PEGylated lipid have shown successes in delivering oligonucleotides, e.g., Onpattro® (siRNA in PEGylated lipid nanoparticle) in 2018. One of the essential uses of PEGylation technology was the rapid development of mRNA-based COVID-19 vaccines in PEGylated lipid nanoparticles [37–41]. Comiranty® (COVID-19 vaccine, mRNA by Pfizer/BioNTech) received full approval by the FDA in August 2021. The Covid-19 pandemic has dramatically reinvigorated research in lipid nanoparticles, where new and old lipid-based nanoparticles could be crucial for delivering next-generation Covid-19 vaccines, new mRNA-based vaccines, and gene-editing therapies such as CRISPR-Cas9.
PEGylation has also been used to modify small molecules. Movantik®, approved in 2014, is a PEGylated α-naloxol derivative (naloxegol) that can reduce transport across the blood-brain barrier (BBB) to prevent the side effects in the central nervous system [42, 43]. Most drugs need to be delivered to overcome the BBB, but preventing transport across the BBB found a successful application here.
2. Nanomedicine
In the Year 2000, the United States government introduced a new initiative called the National Nanotechnology Initiative (NNI). Its application to the drug discovery and delivery field has been known as nanomedicine. Since its beginning, the nanomedicine field has almost exclusively focused on tumor-targeted drug delivery [44]. Still, the outcome has been less than anticipated, and the number of new anticancer formulations approved has been few [45–47]. Mylotarg®, Doxil®, and Abraxane® (albumin-paclitaxel complex approved in 2005) have become the face of nanomedicine. The approvals of Doxil® and Abraxane® were based primarily on reduced side effects rather than superior efficacy of the drugs. The reduction in side effects is an essential contribution to making better products. However, the results are not entirely based on the prevailing theory of “enhanced permeability and retention,” commonly known as the EPR effect, which nanomedicines were supposed to possess for tumor-targeted drug delivery, theoretically resulting in more efficacious products with reduced side effects.
While tumor-targeting of nanomedicine has yet to be achieved, the research on various forms of nanoformulations has produced a few critical side achievements. First, by being “nano in size,” nanoformulations end up increasing the water-solubility of poorly soluble drugs. Nanosized drug crystals, or nanocrystals, increase the dissolution kinetics of poorly soluble drugs fast enough to continuously release drug molecules as the dissolved drugs are absorbed by the body. Thus, the overall result appears to be increased water solubility. The first nanocrystal formulation approved by the FDA was Rapamune® in 2000.
Another critical improvement by nanomedicine research is the manipulation of lipid molecular structures to escape from endosomes more efficiently. Realizing that once nanoparticles are entrapped by endosomes, limiting access to other subcellular components of cells, engineering escape from the endosome is the first step toward efficacy, and formulations were designed with this in mind [48–50]. One of the results has been the lipid formulations used in Onpattro® for the delivery of siRNA, approved by the FDA in 2018 [51]. Onpattro contains an ionizable cationic lipid that was optimized for RNA encapsulation and intracellular delivery and the PEG-containing lipid that regulates the nanoparticle size. The lipid molecular and assembled structures have been the key to developing COVD-19 mRNA-based vaccines delivered via lipid nanoparticles (see below).
2.1. Drug-Polymer Complexes and Conjugates
A complex is a molecular assembly of individual molecules held together by non-covalent bonds, such as electrostatic and/or hydrophobic interactions. Thus, the associated molecules can be dissociated upon changes in the environmental conditions. Drug complexes are usually made using water-soluble polymers, such as low molecular weight dextran used in InFed®, approved in 1974 [52, 53], and Dexferrum® in 1992 [54, 55], and albumin in Abraxane®, approved in 2005. InFed is an iron complex with low molecular weight (5,000–7,000 Da) dextran that has been used to correct iron deficiency [54, 56]. Iron complexed with low molecular weight dextran has been clinically demonstrated to be safer than the complex with high molecular weight dextran (96,000 Da) [55, 57–59]. Another widely known drug-polymer complex is Abraxane®, nanoparticles of 100 mg paclitaxel coated with 900 mg human albumin for cancer treatment [60]. The main advantage of making drug-polymer complexes is to increase the water-solubility of the poorly soluble drugs.
A drug-polymer conjugate is a polymeric carrier having covalently bound drug molecules [61, 62]. The carrier polymer, due to its macromolecular structure with multiple functional groups, can carry a large number of covalently bound drug molecules. The idea is attractive, as this allows incorporation of a targeting moiety and an imaging agent, and other useful agents, in addition to many drug molecules to either the polymer backbone or functional side groups depending on the linker chemistry. Since then, numerous drug-polymer conjugates have been tested in clinical studies [63, 64], but with disappointing results. Adding a targeting moiety, e.g., ligand or antibody, does not increase the targeting property because the distribution is mainly controlled by blood circulation. Ligand-receptor and antigen-antibody interactions occur only after they successfully reach the target site(s). Here, drug-polymer conjugates exclude PEGylated drugs, as the PEG chains used in PEGylation do not have the functions of polymeric carriers of the proposed drug-polymer conjugates. Monoclonal antibodies themselves have played a vital role in drug development. Subsequently, antibodies were conjugated with drugs (Ab-drug conjugates, or ADC). The first antibody-drug conjugate approved for clinical use was Mylotarg® (gemtuzumab ozogamicin) in 2009 [65]. An N-acetyl-γ-calicheamicin molecule is covalently linked to the CD33-directed monoclonal antibody in Mylotarg [66]. Binding with the CD33 antigen results in internalization and hydrolytic release of the calicheamicin moiety, resulting in DNA damage and cell death. Eight additional antibody-drug conjugates were approved by the FDA up to 2020 [67].
2.2. Lessons Learned from Nanomedicine
Although the progress made by the nanomedicine field has been slow in developing tumor-targeted drug delivery systems, the technology developed through the period has been a source of ultrafast development of mRNA vaccines against COVID-19. It took only 2 months from the announcement of the genetic sequence of SARS-CoV-2 to initiating the clinical trials of mRNA vaccines [68].
As of December 2021, nine vaccines have been approved for full use [70], and the first two vaccines approved by the FDA, Pfizer/BioNTech and Moderna vaccines, use mRNA. mRNAs are very unstable and thus, require proper protection. Protection must be followed by efficient delivery into the cells and escape from the endosome to be effective as a vaccine. The PEGylated lipid nanoparticles turned out to be working just as they were designed to. This is, by any measure, a fabulous achievement by formulation scientists. This speedy development of functional mRNA delivery lipid nanoparticles was possible only because of the cumulation of the decades-long progress in the sophisticated delivery systems for delivering genetic drugs, such as siRNA, mRNA, and plasmid DNA [71]. The fruit of the decades-long research activities was collected in the area that the initial nanomedicine researchers did not consider as their primary objective.
While lipid nanoparticles worked well for RNA delivery, further advances are necessary, including “endosomal escape” to reach the cytosol [72]. Ionizable lipids form destabilizing non-bilayer structures at the acidic pH present in endosomes, ultimately resulting in endosomal escape. Along with the optimal extent of PEGylation, the concept of ionizable lipids [73] was introduced to form ionizable lipid nanoparticles for the efficient delivery of nucleic acids [72]. Ionizable lipids, forming cations in the acidic environment of the endosome, interact with anionic lipids to form an inverted cone shape, resulting in the membrane fusion, endosomal disruption, and release of the entrapped nucleic acid into the cytosol [74–77].
Figure 5 summarizes the contribution that decades-long nanomedicine research has made to the COVID-19 vaccines. The majority of nanomedicine research was centered around tumor-targeting. Two factors were critical in targeted delivery to tumors: PEGylation for more extended circulation and improved stability of nanomedicine, and effective endosomal escape for drug release in the cytosol. For the delivery of unstable RNAs, lipid nanoparticles consisting of PEGylated lipid and ionizable cationic lipid were developed to maximize the stability and endosomal escape of the delivered RNAs. The lipid nanoparticle formulation was successfully used to develop Onpattro®, the first formulation approved by the FDA for delivery of siRNA targeting transthyretin [51, 78]. Lipid nanoparticle formulations with different ionizable cationic lipids were optimized for use in mRNA vaccines.
Figure 5.

The essence of the contribution of nanomedicine in the fast development of COVID-19 vaccines. The technologies developed for tumor-targeted drug delivery were combined with siRNA delivery using ionizable cationic lipids, leading to the fast development of mRNA COVID-19 vaccines.
2.3. Significance of the Lesson
2.3.1. Unexpected results in clinical trials
Since the identification of HIV as the cause of AIDS in 1984, the development of AIDS vaccines has been ongoing. Since 1987, a dozen HIV vaccine clinical trials have been completed, but none has been successful. Some trials were halted due to safety concerns, while others have not demonstrated protection against HIV as of 2021 [79, 80]. Understandably, a test vaccine cannot provide protection, as HIV is not a singular type of virus but composed of different types, groups, subtypes, and strains. Some clinical trials, however, have even shown that vaccines resulted in an increased risk of HIV infection [81–83]. The increased risk is suspected to be due to the dampened vaccine-induced responses by the pre-existing immunity against a recombinant adenovirus type-5 vector used in the studies [84]. Only in 2021, a promising result was obtained using a new HIV immunogen (delivered using Alhydrogel® alum) that elicits broadly neutralizing antibodies binding to conserved regions of HIV [85].
Initially, DNA vaccines were developed. They are simple in design and development, quick and straightforward in manufacturing with better quality control, and heat-stable with simpler transportation in lyophilized form relative mRNA-based vaccines. But the practical issues have been a low cellular uptake, the requirement of entering into the nucleus, and the lack of efficiency in responses [86, 87]. While RNA vaccines are effective, they are inherently unstable compared to DNA with poor thermal stability, requiring sub-freezing temperatures [88, 89]. In 2021, Moderna started a Phase 1 study to evaluate the safety and immunogenicity of its mRNA vaccine for HIV-1[90]. Even with the issues in manufacturing and transport, the efficacy may outweigh the other variables, providing opportunities for novel delivery vehicles, logistics, and other opportunities to make these formulations more viable to the developing world.
It seems that 2021 is the dawning of mRNA vaccines for the future [69, 91]. The success of mRNA vaccines and RNA therapeutics, however, is a result of using the suitable delivery systems, lipid nanoparticles, which were developed over more than a decade [92]. The compositions of the Pfizer-BioNTech and Moderna lipid nanoparticles are very similar, and the story behind it can shed some light on how the science of lipid nanoparticle formulations mixed with the business aspect to create and deliver (administratively and logistically) the vaccines [93, 94].
Since the first clinical trial of the HIV vaccine in 1986, more than three decades have passed. The fact that new approaches based on broadly neutralizing antibodies and mRNA have been highly successful indicates that paradigms change and a prevailing opinion at a given period may be wrong, coupled with different vaccine concepts that need to be tested in parallel [95]. It is essential to accept that the development of an HIV vaccine, and any new formulations for that matter, is complicated; the temptation of just following the fashion of the time should be avoided, where long-term commitment is necessary by both funding agencies and scientists [95]. At present, mRNA is bound to be the fashion for the coming decade, but only time will tell if the current stunning success of the mRNA-based COVD-19 vaccine will translate into additional medical breakthroughs [96]. Considering significant differences in development between vaccines and disease treatment that requires steady delivery of therapeutic agents, one can expect only several clinical products in the next decade or two.
While the COVID-19 pandemic has been shockingly disastrous, it also brought the best of our ability to develop vaccines and novel anti-viral molecules at an unprecedented rate. Such speed can be applied to other health priorities, such as cancer, antibiotic-resistant bacteria, and chronic diseases [68].
3. Future
3.1. Chronic Diseases
As the average lifespan increases, more years will be spent by patients living with chronic diseases requiring further long-term treatment. The 10 most common chronic conditions include hypertension (high blood pressure), high cholesterol, arthritis, ischemic heart disease (or coronary heart disease), diabetes, chronic kidney disease, heart failure, depression, Alzheimer’s disease and dementia, and chronic obstructive pulmonary disease [97]. Taking drugs once a day may not be convenient enough, not to mention a few times a day. For many elderly people who have to take medicines for the rest of their lives, once-a-day, or even once-a-month, may not be convenient enough.
3.2. Small Molecule Drugs & Biologics
Table 1 shows the top 20 best-selling drugs in 2010 in the United States [98] and worldwide in 2015 [99] and 2020 [100]. Most drugs are used long-term for treating chronic diseases. The list of drugs in Table 1 shows that the number of biologics has increased significantly over the last decade. According to the report by EvaluatePharma, biologics occupied only 34% of the top 100 products in 2010. Its share gradually increased to 53% in 2018 and remained around 50% for the next several years [101]. This clearly indicates that future drugs will be an equal mixture of small molecules and biologics. Currently, most biologics are administered by injection for short-term delivery. It is necessary to develop long-acting injectable formulations that can deliver the drug(s) for weeks and months to provide enhanced convenience and compliance to patients.
Table 1.
Examples of Top 20 drugs sold in selected years.
| $7.2 B (Atorvastatin) | $14.0 B (Adalimumab) | $20.4 B (Adalimumab) |
| $6.3 B (Esomeprazole) | $13.9 B (Ledipasvir, sofosbuvir) | $14.4 B (Pembrolizumab) |
| $6.1 B (Clopidogrel) | $12.2 B (Lenalidomide) | |
| $4.7 B (Salmeterol, fluticasone) | $8.7 B (Etanercept) | $9.2 B (Apixaban) |
| $8.4 B (Infliximab) | $8.4 B (Ibrutinib) | |
| $4.6 B (Aripiprazole) | $8.5 B (Rituximab) | $8.4 B (Aflibercept) |
| $4.3 B (Quetiapine) | $7.0 B (Insulin Glargine) | $7.9 B (Ustekinumab) |
| $4.1 B (Montelukast) | $6.8 B (Bevacizuma) | $7.9 B (Nivolumab) |
| $3.8 B (Rosuvastatin) | $6.6 B (Trastuzumab) | $7.3 B (Emtricitabine, bictegravir, tenofovir) |
| $3.5 B (Pioglitazone) | $5.8 B (Lenalidomide) | |
| $3.3 B (Epoetin Alfa) | $5.3 B (Sofosbuvir) | $6.9 B (Rivaroxaban) |
| $3.3 B (Infliximab) | $5.2 B (Salmeterol) | $6.4 B (Rivaroxaban) |
| $3.3 B (Rivaroxaban) | $5.0 B (Rosuvastatin Calcium) | $6.0 B (Diphtheria protein) |
| $3.2 B (Duloxetine) | $4.8 B (Pregabalin) | $5.4 B (Palbociclib) |
| $3.1 B (Bevacizuma) | $4.7 B (Pegfilgrastim) | $5.3 B (Bevacizumab) |
| $3.1 B (Oxycodone) | $4.7 B (Imatinib) | $5.1 B (Dulaglutide) |
| $3.0 B (Pegfilgrastim) | $4.3 B (Rivaroxaban) | $4.6 B (Ocrelizumab) |
| $3.0 B (Olanzapine) | $4.0 B (Glatiramer) | $4.5 B (Rituximab) |
| $2.9 B (Adalimumab) | $3.9 B (Sitagliptin) | $4.4 B (Enzalutamide) |
| $2.8 B (Escitalopram) | $3.8 B (Aripiprazole) | $4.3 B (Osimertinib) |
| $2.8 B (Rituximab) | $3.6 B (Dimethyl Fumarate) | $4.2 B (Infliximab) |
3.3. Targeted Drug Delivery
The concept of targeted drug delivery needs to be clarified because it has different meanings to scientists depending on their disciplines. The original concept of the magic bullet (targeted drug) by Paul Ehrlich is that antitoxins, or antibodies, have selective targeting to a bacterium without affecting other organisms. Here, selective targeting simply means “selective killing” of a bacterium. The antibodies still distribute throughout the body but selectively interact with the targets. Most of the drugs we consume mainly interact with the targets, but they also interact with normal cells, causing side effects. Thus, the magic bullet concept did not mean that antibodies, or drug molecules, only go to targets [102–104].
The current concept of targeted drug delivery using nanomedicine has evolved into something quite different from the original magic bullet concept. Nowadays, the term “targeted drug delivery” is used if a nanomedicine delivers slightly more drugs than the control, which is usually a drug solution, to target tumors [45, 47]. All anticancer drugs are still harmful to normal cells, and thus, the idea of using “targeted drug delivery” without affecting normal cells does not apply. Furthermore, delivering a drug or nanoparticles to the solid tumor is not enough, as the drug has to diffuse through the tumor microenvironment and get into tumor cells to be effective [105]. Thus, delivery of a drug to a target site is not necessarily the same as the efficacy. Nevertheless, the concept of targeted drug delivery using nanomedicine has permeated the field deeply, and this needs to be changed to make genuinely targeted drug delivery systems in the future.
Targeted drug delivery is essential in gene therapy. If the genes of interest are entering non-target cells, the side effect will surely follow. In the early days of gene therapy and even current clinical gene therapy trials, some volunteers died due to the unexpected delivery to the wrong places by inactivated adenovirus vectors which still can stimulate an immune response [106–108]. The same issue persists in 2021: patients died during the gene therapy trials [109] or developed cancers [110]. While gene therapies and cell therapies will continue to improve their safety and efficacy, they will have serious competition by the small molecule therapeutics, which are always easier to produce and administer. It is important to remember Ehrlich’s adage that success in science requires the four big G’s: “Geduld, Geschick, Glück, und Geld” (patience, cleverness, luck, and money) [111]. While the mRNA vaccine development was performed at “Warp Speed,” we should not necessarily conclude that all therapies will follow this same speed and continue to remember and practice the four G’s.
3.4. Overcoming Biological Barriers
Biological barriers to drug delivery scientists are considered to be a nuisance. Without biological barriers, drug delivery will be much easier. At the same time, we all know that biological barriers are essential for protecting us from harmful materials. Biological barriers can be divided into epithelial barriers and endothelial barriers. Epithelial barriers include mucosal tissues (e.g., cornea, nose, gastrointestinal tract, and lung) and the epidermis [112, 113]. The endothelial barriers, e.g., blood-brain barrier, consist of the endothelial cell membrane, tight junctions, apical surface glycocalyx, and basement membrane [114].
Over the years, various approaches have been proposed to overcome biological barriers [115–122], and many have shown promises in increasing drug absorption. The higher drug bioavailability, however, comes with a significant price. Once biological barriers are compromised, not only beneficial drugs but also all pathogens enter the system together. To date, there has been no “safe” approach to overcoming biological barriers. It is necessary to distinguish the potential of overcoming biological barriers in small animal experiments from clinical applications, which requires a clear demonstration of safety and efficacy.
3.4.1. Oral Drug Delivery: From Small Molecules to Peptide Drugs
Oral drug delivery is the most convenient and most widely used means of drug delivery. Oral drug delivery has formulation barriers, such as low water solubility and low stability, and biological barriers, particularly short GI transit time, low permeability/absorption, and presystemic clearance. The biopharmaceutics classification system was developed to account for two crucial drug properties critical to drug bioavailability, water-solubility and permeability [123, 124]. To overcome the short GI transit time, formulation scientists have developed various gastro-retentive formulations, including floating, mucoadhesion, swellable, high-density systems, superporous hydrogel, magnetic systems, and unfoldable/extendible designs [125]. Although, gastric physiology differences, including gastric pH and motility, can largely influence the absorption and residence time and thus the resultant pharmacokinetic profile [126]. Still, genuine gastro-retentive systems are yet to be developed.
The gastrointestinal (GI) tract is designed to absorb small molecules, such as carbohydrates, amino acids, and fat-soluble molecules. All orally administered drugs are low molecular weight drugs. It has been a dream of drug delivery scientists to deliver peptide and protein drugs by oral administration, as its benefit is unmeasurable. Various approaches and devices have been tested to overcome poor absorption of peptide and protein drugs [127–130], but again they are all only in the research and development stage.
The reality of oral drug delivery is that the bioavailability of peptide and protein drugs is so low that it is not practical regardless of the delivery system used [131, 132]. The difficulty becomes insurmountable when it comes to the oral delivery of insulin. Many studies indicate that insulin can be absorbed from the GI tract and control the in vivo glucose level [133]. This may be true in mice. But even then, one needs to carefully dissect the details of the experimental conditions. The mice were kept in the fasted condition before administering insulin, at orders of magnitude larger than that used in humans. Microneedle transdermal delivery systems were also used to deliver insulin to observe the decrease in the blood glucose level for several hours [134]. Remember, insulin is not a drug that can be delivered at an arbitrary concentration range anytime. It has to be delivered at the exact amount at the exact time to be effective, and it has to be responsive to the fluctuations of the glucose level in the blood. Furthermore, the insulin release has to be stopped after the glucose level is lowered. The timely delivery of an accurate dose at the right time is critical for insulin delivery, and currently, there are no drug delivery systems that can fulfill such requirements reproducibly. The development of self-regulated insulin delivery systems is one of the holy grails in drug delivery.
Oral drug delivery has been the most preferred administration route. However, further mechanistic understanding of processes critical to oral absorption needs to be obtained, such as interindividual variations, differences of drug absorption between different genders and ages, gastric retention, and formulation limitations (of poorly water-soluble drugs and poorly permeable drugs) [135].
3.4.2. The blood-brain barrier
The blood-brain barrier (BBB) has been the main hurdle in treating central nervous system diseases. To overcome the BBB, various drug delivery systems have been developed, e.g., adeno-associated virus vectors [136], virus-mimetic nanoparticles [137], erythrocyte-mimicking nanovehicles (nanovehicles coated with erythrocyte membranes) [138], cell-based delivery nanocarriers [139], cell-penetrating peptide-linked nanovehicles [140], extracellular vesicles [141], injectable hydrogels [142], tight junction modulators [143], and immunomodulators [144]. While most have shown promising efficacy in small animal models, translations to clinical use are still far away. It is encouraging to observe successful Phase II clinical trials of paclitaxel trevatide (ANG1005, a conjugate of paclitaxel with Angiopep-2) that effectively crosses the BBB via a low-density lipoprotein receptor-related protein [145]. To demonstrate clinical safety and efficacy, precision dosing, off-target effects, and scale-up manufacturing issues must be resolved [144].
The lack of suitable BBB models is a compounding factor in slow progress in developing formulations for diseases like Alzheimer’s disease, Parkinson’s disease, multiple sclerosis, epilepsy, and ischemic stroke [146]. The lack of suitable models is not limited to drug delivery to the brain, and other disease models, particularly cancer treatment [147, 148], also need to be improved. Drugs have also been delivered directly to the brain by intranasal deposition and absorption on the olfactory and respiratory epithelia, reducing systemic exposure and providing greater bioavailability [149, 150]. Such administration routes, however, require more work to understand the underlying mechanisms to be practical. The drug distribution pattern in the brain is unknown, making it difficult to use the drugs that become effective only after binding to specific receptors in specific brain regions [151].
3.5. Long-Term Treatment for Chronic Diseases
A chronic illness is a long-term health condition that may not have a cure. Examples of chronic illnesses are addiction, Alzheimer disease and dementia, arthritis, asthma, cancer, chronic obstructive pulmonary disease, Crohn’s disease, cystic fibrosis, diabetes, depression, epilepsy, heart disease, human immunodeficiency virus/acquired immunodeficiency syndrome (HIV/AIDS), hypertension, multiple sclerosis, Parkinson disease, and tuberculosis. 60% of adults have a chronic disease in the U.S., and 40% of adults have two or more [152]. Treating chronic diseases requires ongoing medical attention and intervention for years. Adherence to long-term therapy, however, is averaging 50% even in developed countries [153]. Adherence to long-term therapy requires more than taking prescribed pharmaceuticals. It also encompasses numerous therapeutic behaviors, including seeking medical attention, filling prescriptions, taking medication appropriately, and attending follow-up appointments [153]. Here, long-acting formulations are undoubtedly helpful for increasing patients’ compliance and convenience, particularly long-acting injectable formulations that deliver daily doses for weeks and months.
Since 1989 when the first long-acting injectable formulation was introduced, only about 20 drugs have been made into long-acting injectable formulations with a duration ranging up to 6 months. The duration of each formulation will increase beyond the 6-month mark currently set. Still, the longer-term delivery depends on the daily dose of each drug, drug stability, and the therapeutic index.
Long-acting formulations also include other formulations [154], e.g., devices that require surgery to implant in the body and another one for removal, unless the formulation is designed to biodegrade. The surgically implantable devices may be necessary if a drug is released for years, but Norplant®, a contraceptive device delivering a drug for 5 years, had to be removed from the market due to incidences of being unable to remove them. Thus, using biodegradable polymers is preferred over using non-degradable formulations that have to be removed after their use.
4. The Future of Drug Delivery Systems
4.1. Hindsight Analysis of Drug Delivery History
The beauty of fundamental research is that there may be no immediate application of the findings, yet they may provide the solid foundation for a quantum leap in another field [47]. Developing new formulations, even if it is solely for acquiring a research grant rather than the medical need, may turn out to be life-saving formulations. For this process to be more productive, however, it needs to be changed. For the last few decades, the current research and funding system has been in place to encourage so-called innovation. Innovation, however, is difficult to define. It is common to prepare a proposal that appears to be highly innovative to convince the review panel, even if the real innovation is only minutely incremental at best. Such proposals are usually accompanied by articles published in high-impact journals (or even too many articles to count based on the ever-expanding open access journal numbers), resulting in the vicious cycle of high-impact publication and research funding. In the process, the desire to solve existing medical and pharmaceutical problems has been minimized. Developing formulations that can treat the existing diseases requires an iterative process to optimize the formulation for safety and efficacy. This, however, has been treated as an issue that should be dealt with by biomedical device and pharmaceutical companies, not by the national funding agencies. Furthermore, obtaining this funding mentioned above is even more difficult as in vivo data is essentially “required” to be competitive; further hindering newly formed labs, universities that may not have access to facilities or groups that do not have access to pilot funding even to perform these types preliminary studies.
As the brief review of the evolution of drug delivery technologies in Figure 1 highlights, only a dozen breakthrough drug delivery formulations have been developed during the last 7 decades. This illustrates how difficult it is to develop breakthrough technologies for clinical use. Whenever a new technology is introduced, the field tends to focus on that particular idea, and the majority follows down that path. This is where diversity in research ideas becomes very narrow, and the field does not evolve. We need to build a new system that encourages diversity and different points of view in research, coupled with respective funding allocations. One example that illustrates the current problem well is the field of “organ-on-a-chip.” Growing cells in multiple layers on chips by no means make them an “organ,” yet it dominates the field to the point that research funding and publications must use such words to be competitive. There came human-on-a-chip to surpass organ-on-a-chip. Such utterly inadequate terminology still attracts considerable research funding, and thus, others adapt to the same nonsense. This occurs because so many scientists, particularly new, follow this “hot” trend not to be left behind when the future is uncertain. The human organs-on-chips research will be obsolete soon, as it is like practicing golf on a driving range or simulator instead of an actual golf course. The human is just so different from human organs-on-chips. The differences in drug absorption, distribution, metabolism, and elimination between humans and animals must be appreciated before presenting new in vitro “gadgets”. Advances in treating diseases occur not because of a handful of fancy terms and by a handful of famous scientists. Individual new drug formulations and biomedical technologies result from the collective progress of various scientists, across different teams and disciplines, and now across many companies. To continue accelerating the progress, we need to examine whether the current system allows the influx of new ideas or encourages me-too research. Since lipid nanoparticles seem to be today’s research fashion, most research will focus on a similar subject. This may hinder trials of new ideas. Remember that no single tool is suitable for everything; even a Swiss army knife has its limitations.
4.2. Foresight Analysis of Drug Delivery of the Future
The future events are impossible to predict, as they occur unpredictably. Who could have predicted COVID-19 would have paralyzed the world and lipid nanoparticles would have become the savior of Homo sapiens? Despite the deadly COVID-19 pandemic, we can find a remedy and continue our lives. The future is uncertain, and we need to prepare for an uncertain future by adapting to the new environment by expanding our drug delivery technologies. Individual scientists need to think and evaluate problems independently and often “outside the box.” Diversified drug delivery technologies will make it easier and faster to adapt to new problems. Since no single formulation or technology will suffice for all diseases, investing in many different technologies will allow an improvised adaptation repeatedly.
We need to be honest in accepting an uncomfortable number of mistakes the field has made by focusing on specific topics in fashion at a given time. When the decade-long investment has not worked out, it has been a tendency to cover them up or emphasize many publications to construe the investment as a success. The best we can do is to learn from our mistakes. Often, mistakes, unsuccessful designs, or experiments that do not align with the prevailing theory produce the most significant impact in advancing science/technology.
As Figure 1 shows, each technological advance provides a partial solution to a complex set of problems. PEGylation technology has been suitable for developing products that are “good enough for better efficacy and safety,” and continuous, sequential advances will lead to clinical products that help patients to live longer and better. The evolutionary process has no foresight [155], and we need to have diverse technologies ready to challenge any future, where unexpected diseases may appear from anywhere. Scientific progress will train us to know what is possible or impossible with the technology available now. We need to focus on what we did and, more importantly, what we can do differently in the future.
The progress of drug delivery research is slow. The extent of the slowness can be easily recognized by comparing the progress made in the computer industry (Figure 6). Liposomes were discovered in 1964, and in the same year, UNIVAC 1108 computer based on integrated circuits with 1 MB memory was introduced at the price of $566,460 [156, 157]. Fast forward to 2021, and the computer memory increased 30,000 times with 200 times lower price and equally smaller size. On the other hand, it took more than 30 years to translate liposomes into Doxil®, a PEGylated liposome, and more than 50 years to develop PEGylated lipid nanoparticles for siRNA and mRNA delivery. The introduction of new drug delivery formulations is bound to be slow and expensive due to the clinical studies requiring time to demonstrate safety and efficacy. On the other hand, electronic systems can be introduced as soon as improvements are made and able to be made at scale. The perceived slowness of the development of new drug delivery systems is mainly based on the safety mentioned above and efficacy studies, which are not linearly dependent on technological improvements. One way to accelerate the formulation development is to focus on clinical applications more than minor technological improvements.
Figure 6.

Comparison of evolutions of computer and liposome drug delivery systems from 1964 to 2021.
4.3. The Ultimate Goal
The ultimate goal of drug delivery research is to develop formulations that can deliver drugs to target sites with predefined release kinetics and duration. Basic research is essential, but not all basic research results in products used by patients. There are fundamental differences between showing the potential of a particular drug delivery technology and developing clinical products. Almost always, it takes decades to translate new technologies into clinical products. Ensuring their safety and efficacy in humans and scale-up production requires years of research and development. Identification of antibacterial activity of penicillin would not have been enough to save us from infection without figuring out scale-up production. This is why the Nobel Prize in Physiology or Medicine was awarded jointly to Ernst B. Chain and Sir Howard W. Florey, in addition to Sir Alexander Fleming.
Research on lipid nanoparticles for oligonucleotide delivery, especially mRNA delivery, is expected to surge for a while. Such intensive efforts will add a flood of new information to the current [71, 72, 76–78, 92, 158, 159]. It, in turn, will provide critical information essential for developing clinically valuable formulations. When mRNA was successful as a vaccine for COVID-19, mRNA research skyrocketed. The research funding follows the most popular research topic of the day.
The current funding situation is such that it does not favor the research on product development, and this may need to change. Without timely translation of the basic research to clinical products, appreciation of the basic research is bound to be slow, causing the public to lose the patience of playing a long game. To be fair, the nature of basic research is different from that of introducing products to the market. The former is focused on finding new information, while the latter is mainly focused on solving practical problems that may not appear to be grandiose, even though highly important. COVID-19 vaccine development is a good example. If COVID-19 is not a life-or-death disease spread throughout the world in such a short time, the development of its vaccines likely would have taken much longer. mRNA vaccines for Zika virus and cancer were already in human studies when COVID-19 started [160, 161], and the robustness and versatility of mRNA-based vaccine technology allowed the fast production of COVID-19 vaccines. While most diseases do not seem as urgent as COVID-19, each disease is equally devastating to those affected by it. It is time for drug delivery scientists to deal with each disease as the most dangerous one we face. This small change in the mindset will make a big difference in research focus and move us from generating paper-knowledge to life-knowledge [162].
Figure 4.
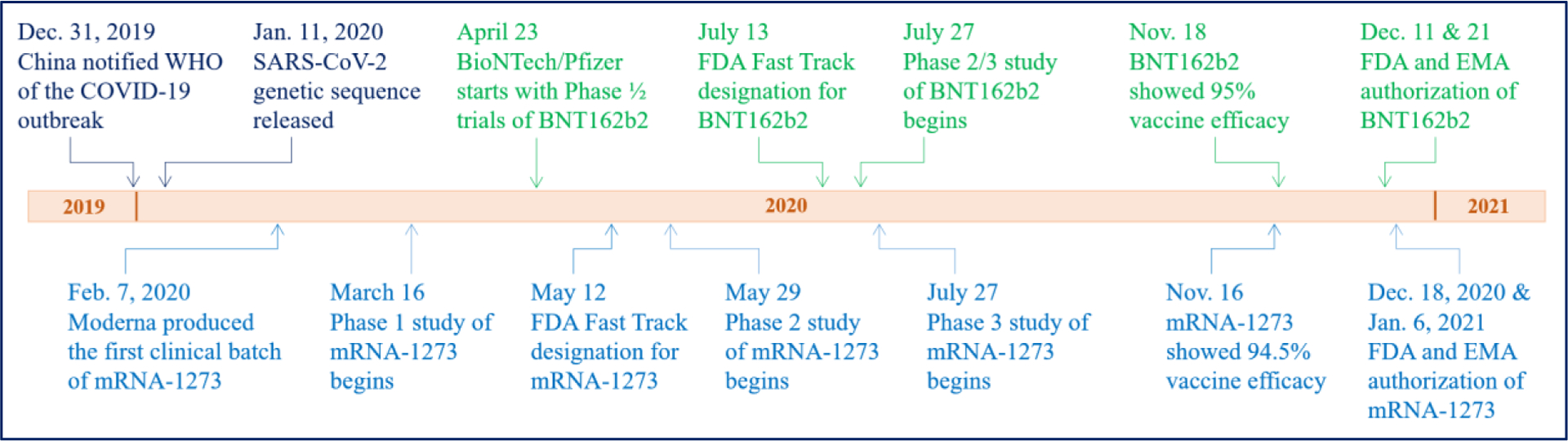
Timeline of the development of mRNA vaccines against COVID-19 by BioNTech/Pfizer (BNT162b2, Green) and Moderna (mRNA-1273, Blue). (Modified from Reference [69]).
Acknowledgments
This study was supported by the Showalter Research Trust Fund and in part by UG3 DA048774 from the National Institute on Drug Abuse (NIDA).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Vargason AM, Anselmo AC, Mitragotri S. The evolution of commercial drug delivery technologies. Nature Biomedical Engineering, (2021). [DOI] [PubMed] [Google Scholar]
- [2].FDA/CDER. SUPAC-MR: Modified release solid oral dosage forms scale-up and post approval changes: Chemistry, manufacturing, and controls; in vitro dissolution testing and in vivo bioequivalence documentation. (1997).
- [3].USP. USP-NF 2021 (Issue 1). (2021). [Google Scholar]
- [4].Blythe RH, Sympathomimetic preparation (1956)
- [5].Ullyot GE, Ullyot BH, Slater LG. The metamorpohsis of Smith-Kline & French Laboratories to Smith Kline Beecham: 1925–1998. Bull. Hist. Chem, 25 (2000) 16–20. [Google Scholar]
- [6].Hillery AM, Hoffman AS. Historical introduction to the field of controlled drug delivery, in: Hillery AM, Park K (Eds.) Drug Delivery: Fundamentals & Applications. Second Edition, CRC Press, Boca Raton, FL. 2016, pp. 1–21. [Google Scholar]
- [7].Peppas NA. Historical perspective on advanced drug delivery: How engineering design and mathematical modeling helped the field mature. Adv. Drug Del. Rev, 65 (2013) 5–9. [DOI] [PubMed] [Google Scholar]
- [8].Liu J-C, Farber M, Chien YW. Comparative release of phenylpropanolamine HCl from long-acting appetite suppressant products: Acutrim vs. Dexatrim. Drug Development and Industrial Pharmacy, 10 (1984) 1639–1661. [Google Scholar]
- [9].Chien YW. Novel Drug Delivery Systems, Second Edition. Second Edition. Revised and Expanded ed., Marcel Dekker, New York. 1992. 797 pages. [Google Scholar]
- [10].Scharp DW, Marchetti P. Encapsulated islets for diabetes therapy: History, current progress, and critical issues requiring solution. Adv. Drug Deliv. Rev, 67–68 (2014) 35–73. [DOI] [PubMed] [Google Scholar]
- [11].Park K. Controlled drug delivery systems: Past forward and future back. J. Control. Release, 190 (2014) 3–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Yun YH, Lee BK, Park K. Controlled drug delivery: Historical perspective for the next generation. J. Control. Release, 219 (2015) 2–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Park K, Skidmore S, Hadar J, Garner J, Park H, Otte A, Soh BK, Yoon G, Yu D, Yun Y, Lee BK, Jiang XJ, Wang Y. Injectable, long-acting PLGA formulations: Analyzing PLGA and understanding microparticle formation. J. Control. Release, 304 (2019) 125–134. [DOI] [PubMed] [Google Scholar]
- [14].FDA/CDER. Controlled correspondence related to generic drug development. file:///C:/Users/kp/AppData/Local/Temp/GUI_Final_ControlledCorrespondenceDecember2020.pdf (2020).
- [15].Skidmore S, Hadar J, Garner J, Park H, Park K, Wang Y, Jiang XJ. Complex sameness: Separation of mixed poly(lactide-co-glycolide)s based on the lactide:glycolide ratio. J. Control. Release, 300 (2019) 174–184. [DOI] [PubMed] [Google Scholar]
- [16].Park K, Otte A, Sharifi F, Garner J, Skidmore S, Park H, Jhon YK, Qin B, Wang Y. Formulation composition, manufacturing process, and characterization of poly(lactide-co-glycolide) microparticles. J. Control. Release, 329 (2021) 1150–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Gautam N, McMillan JM, Kumar D, Bade AN, Pan Q, Kulkarni TA, Li W, Sillman B, Smith NA, Shetty BLD, Szlachetka A, Edagwa BJ, Gendelman HE, Alnouti Y. Lipophilic nanocrystal prodrug-release defines the extended pharmacokinetic profiles of a year-long cabotegravir. Nat. Commun, 12 (2021) 3453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Jarvis M, Krishnan V, Mitragotri S. Nanocrystals: A perspective on translational research and clinical studies. Bioeng. Transl. Med, 4 (2018) 5–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Lu Y, Qi J, Dong X, Zhao W, Wu W. The in vivo fate of nanocrystals. Drug Discov, 22 (2017) 744–750. [DOI] [PubMed] [Google Scholar]
- [20].Abuchowski A, McCoy JR, Palczuk NC, van Es T, Davis FF. Effect of covalent attachment of polyethylene glycol on immunogenicity and circulating life of bovine liver catalase. J. Biol. Chem, 252 (1977) 3582–3586. [PubMed] [Google Scholar]
- [21].Davis FF, Van Es T, Palczuk NC, Non-immunogenic polypeptides (1979) 4,179,337.
- [22].Davis FF. The origin of pegnology. Adv. Drug Del. Rev, 54 (2002) 457–458. [DOI] [PubMed] [Google Scholar]
- [23].Halford B. Frank F Davis, pegylation pioneer, dies at 100. https://cen.acs.org/people/obituaries/Frank-F-Davis-pegylation-pioneer-dies-at-100/99/i20 (2021).
- [24].Park K. To PEGylate or not to PEGylate, that is not the question., J. Control. Release, 142 (2010) 147–148. [DOI] [PubMed] [Google Scholar]
- [25].Park K. Impact of anti-PEG antibodies on PEGylated nanoparticles fate in vivo. J. Control. Release, 287 (2018) 257. [DOI] [PubMed] [Google Scholar]
- [26].Veronese FM, Harris JM. Introduction and overview of peptide and protein pegylation. Advanced Drug Delivery Reviews, 54 (2002) 453–456. [DOI] [PubMed] [Google Scholar]
- [27].Hoffman AS, Lai JJ. Three significant highlights of controlled drug delivery over the past 55 years: PEGylation, ADCs, and EPR. Adv, Drug Del. Rev, 158 (2020) 2–3. [DOI] [PubMed] [Google Scholar]
- [28].Barenholz Y. Doxil®- The first FDA-approved nano-drug: lessons learned. J. Control. Release, 160 (2012) 117–134. [DOI] [PubMed] [Google Scholar]
- [29].Bulbake U, Doppalapudi S, Kommineni N, Khan W. Liposomal formulations in clinical use: An updated review. Pharmaceutics, 9 (2017) 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Mohamed M, Abu Lila AS, Shimizu T, Alaaeldin E, Hussein A, Sarhan HA, Szebeni J, Ishida T. PEGylated liposomes: immunological responses. Sci Technol Adv Mater, 20 (2019) 710–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Ellory C. AD Bangham (1922–2010). Physiology News, 79 (2010) 55–56. [Google Scholar]
- [32].Bangham AD, Horne RW. Negative staining of phospholipids and their structural modification by surface-active agents as observed in the electron microscope. J. Mol. Biol, 8 (1964) 660–668. [DOI] [PubMed] [Google Scholar]
- [33].Makwana V, Karanjia J, Haselhorst T, Anoopkumar-Dukie S, Rudrawar S. Liposomal doxorubicin as targeted delivery platform: Current trends in surface functionalization. Int. J. Pharm, 593 (2021) 120117 (120114 pages). [DOI] [PubMed] [Google Scholar]
- [34].Gregoriadis G, Leathwood PD, Ryman BE. Enzyme entrapment in liposomes. FEBS Letters, 14 (1971) 95–99. [DOI] [PubMed] [Google Scholar]
- [35].Gregoriadis G. Liposomes in drug delivery: How it all happened. Pharmaceutics, 8 (2016) 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Smith JG, Walzem RL, Bruce German J. Liposomes as agents of DNA transfer. Biochimica et Biophysica Acta (BBA) - Reviews on Biomembranes, 1154 (1993) 327–340. [DOI] [PubMed] [Google Scholar]
- [37].Elia U, Ramishetti S, Rosenfeld R, Dammes N, Bar-Haim E, Naidu GS, Makdasi E, Yahalom-Ronen Y, Tamir H, Paran N, Cohen O, Peer D. Design of SARS-CoV-2 hFc-conjugated receptor-binding domain mRNA vaccine delivered via lipid nanoparticles. ACS Nano, DOI: 10.1021/acsnano.0c10180 (2021). [DOI] [PubMed] [Google Scholar]
- [38].Khurana A, Allawadhi P, Khurana I, Allwadhi S, Weiskirchen R, Banothu AK, Chhabra D, Joshi K, Bharani KK. Role of nanotechnology behind the success of mRNA vaccines for COVID-19. Nano Today, 38 (2021) 101142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Park KS, Sun X, Aikins ME, Moon JJ. Non-viral COVID-19 vaccine delivery systems. Adv. Drug Deliv. Rev, 169 (2021) 137–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Schoenmaker L, Witzigmann D, Kulkarni JA, Verbeke R, Kersten G, Jiskoot W, Crommelin DJA. mRNA-lipid nanoparticle COVID-19 vaccines: Structure and stability. Int. J. Pharm, 601 (2021) 120586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Brader ML, Williams SJ, Banks JM, Hui WH, Zhou ZH, Jin L. Encapsulation state of messenger RNA inside lipid nanoparticles. Biophys. J, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].BroadPharm. PEGylated small molecule drugs. https://broadpharm.com/web/blog.php?post=60 (2018).
- [43].Wu T, Chen K, He S, Liu X, Zheng X, Jiang Z-X. Drug Development through Modification of Small Molecular Drugs with Monodisperse Poly(ethylene glycol)s. Org. Process Res. Dev, 24 (2020) 1364–1372. [Google Scholar]
- [44].Wang J, Li Y, Nie G. Multifunctional biomolecule nanostructures for cancer therapy. Nature Reviews Materials, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Bae YH, Park K. Targeted drug delivery to tumors: Myths, reality, and possibility. J. Control. Release, 153 (2011) 198–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Park K. The drug delivery field at the inflection point: Time to fight its way out of the egg. J. Control. Release, 267 (2017) 2–14. [DOI] [PubMed] [Google Scholar]
- [47].Bae YH, Park K. Advanced drug delivery 2020 and beyond: Perspectives on the future. Adv. Drug Del. Rev, 158 (2020) 4–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Xu E, Saltzman WM, Piotrowski-Daspit AS. Escaping the endosome: assessing cellular trafficking mechanisms of non-viral vehicles. J. Control. Release, 335 (2021) 465–480. [DOI] [PubMed] [Google Scholar]
- [49].Wei P, Sun M, Yang B, Xiao J, Du J. Ultrasound-responsive polymersomes capable of endosomal escape for efficient cancer therapy. J. Control. Release, 322 (2020) 81–94. [DOI] [PubMed] [Google Scholar]
- [50].Tang M, Svirskis D, Leung E, Kanamala M, Wang H, Wu Z. Can intracellular drug delivery using hyaluronic acid functionalised pH-sensitive liposomes overcome gemcitabine resistance in pancreatic cancer? J. Control. Release, 305 (2019) 89–100. [DOI] [PubMed] [Google Scholar]
- [51].Akinc A, Maier MA, Manoharan M, Fitzgerald K, Jayaraman M, Barros S, Ansell S, Du X, Hope MJ, Madden TD, Mui BL, Semple SC, Tam YK, Ciufolini M, Witzigmann D, Kulkarni JA, van der Meel R, Cullis PR. The Onpattro story and the clinical translation of nanomedicines containing nucleic acid-based drugs. Nat. Nanotechnol, 14 (2019) 1084–1087. [DOI] [PubMed] [Google Scholar]
- [52].Li Y. Advances in iron colloid products: Quality considerations when conducting comparability studies, in Complex generics: Complex injectables, opthalmic and otic products Part 1. https://www.youtube.com/watch?v=1w35BOB2HmE(25:40/1:25:58), https://www.fda.gov/drugs/news-events-human-drugs/advancing-generic-drug-development-translating-science-approval-09212021-09222021?utm_medium=email&utm_source=govdelivery (2021). [Google Scholar]
- [53].FDA/CDER. Workshop: Advancing generic drug development: Translating science to approval https://www.fda.gov/drugs/news-events-human-drugs/advancing-generic-drug-development-translating-science-approval-09212021-09222021?utm_medium=email&utm_source=govdelivery (2021).
- [54].Cançado RD, Muñoz M. Intravenous iron therapy: how far have we come? Rev. Bras. Hematol. Hemoter, 33 (2011) 461–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Wysowski DK, Swartz L, Vicky Borders-Hemphill B, Goulding MR, Dormitzer C. Use of parenteral iron products and serious anaphylactic-type reactions. Am. J. Hematol, 85 (2010) 650–654. [DOI] [PubMed] [Google Scholar]
- [56].Lawrence R. Development and comparison of iron dextran products. PDA J. Pharm. Sci. Technol, 52 (1998) 190–197. [PubMed] [Google Scholar]
- [57].Rodgers GM, Auerbach M, Cella D, Chertow GM, Coyne DW, Glaspy JA, Henry DH. High-molecular weight iron dextran: A wolf in sheep’s clothing? J. Am. Soc. Nephrol, 19 (2008) 833–834. [DOI] [PubMed] [Google Scholar]
- [58].Shander A, Spence RK, Auerbach M. Can intravenous iron therapy meet the unmet needs created by the new restrictions on erythropoietic stimulating agents? Transfusion, 50 (2010) 719–732. [DOI] [PubMed] [Google Scholar]
- [59].Auerbach M, Macdougall IC. Safety of intravenous iron formulations: facts and folklore. Blood Transfus, 12 (2014) 296–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].CDER/FDA. Chemistry review(s)- Abraxane (paclitaxel protein bound particles for injectable suspension) for injectable suspension, 100 mg. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2005/21660_AbraxaneTOC.cfm (2004).
- [61].Ringsdorf H. Structure and properties of pharmacologically active polymers. J. Polym. Sci.: Polym. Symp, 51 (1975) 135–153. [Google Scholar]
- [62].Greco F, Vicent MJ. Combination therapy: Opportunities and challenges for polymer-drug conjugates as anticancer nanomedicines. Adv. Drug Deliv. Rev, 61 (2009) 1203–1213. [DOI] [PubMed] [Google Scholar]
- [63].Duncan R, Kopečková-Rejmanová P, Strohalm J, Hume I, Cable HC, Pohl J, Lloyd JB, Kopeček J. Anticancer agents coupled to N-(2-hydroxypropyl)methacrylamide copolymers. I. Evaluation of daunomycin and puromycin conjugates in vitro. Brit. J. Cancer, 55 (1987) 165–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Ekladious I, Colson YL, Grinstaff MW. Polymer–drug conjugate therapeutics: Advances, insights and prospects. Nat. Rev. Drug Discov, 18 (2019) 273–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Lambert JM. Drug-conjugated monoclonal antibodies for the treatment of cancer. Curr. Opin. Pharmacol, 5 (2005) 543–549. [DOI] [PubMed] [Google Scholar]
- [66].RxList. Mylotarg https://www.rxlist.com/mylotarg-drug.htm#description (2020).
- [67].Joubert N, Beck A, Dumontet C, Denevault-Sabourin C. Antibody-drug conjugates: The last decade. Pharmaceuticals (Basel), 13 (2020) 31 pages. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Rappuoli R, De Gregorio E, Del Giudice G, Phogat S, Pecetta S, Pizza M, Hanon E. Vaccinology in the post–COVID-19 era. Proc. Natl. Acad. Sci, 118 (2021) e2020368118 (2020368117 pages). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Verbeke R, Lentacker I, De Smedt SC, Dewitte H. The dawn of mRNA vaccines: The COVID-19 case. J. Controlled Rel, 333 (2021) 511–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Zimmer C, Corum J, Wee S-L. Coronavirus vaccine tracker. https://www.nytimes.com/interactive/2020/science/coronavirus-vaccine-tracker.html (2021).
- [71].Cullis PR, Hope MJ. Lipid nanoparticle systems for enabling gene therapies. Molecular Therapy, 25 (2017) 1467–1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Schlich M, Palomba R, Costabile G, Mizrahy S, Pannuzzo M, Peer D, Decuzzi P. Cytosolic delivery of nucleic acids: The case of ionizable lipid nanoparticles. Bioeng. Transl. Med, 6 (2021) e10213 (10216 pages). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Bailey AL, Cullis PR. Modulation of membrane fusion by asymmetric transbilayer distributions of amino lipids. Biochemistry, 33 (1994) 12573–12580. [DOI] [PubMed] [Google Scholar]
- [74].Cullis PR, Hope MJ. Effects of fusogenic agent on membrane structure of erythrocyte ghosts and the mechanism of membrane fusion. Nature, 271 (1978) 672–674. [DOI] [PubMed] [Google Scholar]
- [75].Hafez IM, Cullis PR. Roles of lipid polymorphism in intracellular delivery. Adv. Drug Deliv. Rev, 47 (2001) 139–148. [DOI] [PubMed] [Google Scholar]
- [76].Veiga N, Diesendruck Y, Peer D. Targeted lipid nanoparticles for RNA therapeutics and immunomodulation in leukocytes. Adv. Drug Del. Rev, 159 (2020) 364–376. [DOI] [PubMed] [Google Scholar]
- [77].Dammes N, Goldsmith M, Ramishetti S, Dearling JLJ, Veiga N, Packard AB, Peer D. Conformation-sensitive targeting of lipid nanoparticles for RNA therapeutics. Nature Nanotechnology, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Witzigmann D, Kulkarni JA, Leung J, Chen S, Cullis PR, van der Meel R. Lipid nanoparticle technology for therapeutic gene regulation in the liver. Adv. Drug Del. Rev, 159 (2020) 344–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].NIAID. History of HIV vaccine research. https://www.niaid.nih.gov/diseases-conditions/hiv-vaccine-research-history (2018).
- [80].Ng’uni T, Chasara C, Ndhlovu ZM. Major scientific hurdles in HIV vaccine development: Historical perspective and future directions. Front. Immunol, 11 (2020) Article 590780 (590717 pages). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Morrow MP, Weiner DB. DNA drugs come of age. Sci. Am, July (2010) 49–53. [DOI] [PubMed] [Google Scholar]
- [82].Hammer SM, Sobieszczyk ME, Janes H, Karuna ST, Mulligan MJ, Grove D, Koblin BA, Buchbinder SP, Keefer MC, Tomaras GD, Frahm N, Hural J, Anude C, Graham BS, Enama ME, Adams E, DeJesus E, Novak RM, Frank I, Bentley C, Ramirez S, Fu R, Koup RA, Mascola JR, Nabel GJ, Montefiori DC, Kublin J, McElrath MJ, Corey L, Gilbert PB. Efficacy trial of a DNA/rAd5 HIV-1 preventive vaccine. New Eng. J. Med, 369 (2013) 2083–2092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Sohn E. The four-decade quest for an HIV vaccine yields new hope. https://www.nationalgeographic.com/science/article/the-four-decade-quest-for-an-hivvaccine-yields-new-hope (2021).
- [84].Buchbinder SP, McElrath MJ, Dieffenbach C, Corey L. Use of adenovirus type-5 vectored vaccines: a cautionary tale. The Lancet, 396 (2020) e68–e69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Lee JH, Hu JK, Georgeson E, Nakao C, Groschel B, Dileepan T, Jenkins MK, Seumois G, Vijayanand P, Schief WR, Crotty S. Modulating the quantity of HIV Env-specific CD4 T cell help promotes rare B cell responses in germinal centers. J. Exp. Med, 218 (2020) e20201254 (20201216 pages). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Giri M, Ugen KE, Weiner DB. DNA vaccines against human immunodeficiency virus type 1 in the past decade. Clinical Microbiology Reviews, 17 (2004) 370–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].IAVI. First-in-human clinical trial confirms novel HIV vaccine approach developed by IAVI and Scripps Research. https://www.iavi.org/news-resources/press-releases/2021/first-in-human-clinical-trial-confirms-novel-hiv-vaccine-approach-developed-by-iavi-and-scripps-research (2021).
- [88].Brown DM, Magrath DI, Neilson AH, Todd AR. Hydrolysis of esters of monoribonucleotides. Nature, 177 (1956) 1124–1125. [Google Scholar]
- [89].Mikkola S, Lönnberg T, Lönnberg H. Phosphodiester models for cleavage of nucleic acids. Beilstein J. Org. Chem, 14 (2018) 803–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].ClinicalTrials.gov. A Phase 1 study to evaluate the safety and immunogenicity of eOD-GT8 60mer mRNA vaccine (mRNA-1644) and Core-g28v2 60mer mRNA vaccine (mRNA-1644v2-Core). https://clinicaltrials.gov/ct2/show/NCT05001373?term=mRNA&cond=HIV&draw=2&rank=4 (2021).
- [91].Dammes N, Peer D. Paving the road for RNA therapeutics. Trends Pharmacol. Sci, 41 (2020) 755–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Kulkarni JA, Witzigmann D, Thomson SB, Chen S, Leavitt BR, Cullis PR, van der Meel R. The current landscape of nucleic acid therapeutics. Nature Nanotechnology, 16 (2021) 630–643. [DOI] [PubMed] [Google Scholar]
- [93].Vardi N. Covid’s forgotten hero: The untold story of the scientist whose breakthrough made the vaccines possible. https://www.forbes.com/sites/nathanvardi/2021/08/17/covids-forgotten-hero-the-untold-story-of-the-scientist-whose-breakthrough-made-the-vaccines-possible/?sh=6f7bca3a354f (2021).
- [94].Dolgin E. The tangled history of mRNA vaccines. Nature, 597 (2021) 318–324. [DOI] [PubMed] [Google Scholar]
- [95].Esparza J. What has 30 Years of HIV vaccine research taught us? Vaccines (Basel), 1 (2013) 513–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Dunn A. Biotech companies pivot to RNA research after COVID-19 vaccines. https://www.businessinsider.com/biotech-pivots-to-rna-research-after-covid-19-vaccines-2021-9 (2021).
- [97].NCO. The top 10 most common chronic conditions in older adults https://www.ncoa.org/article/the-top-10-most-common-chronic-conditions-in-older-adults (2021).
- [98].Lindsley CW. The top prescription drugs of 2010 in the United States: antipsychotics show strong growth. ACS Chem Neurosci, 2 (2011) 276–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Pharmacompass. Top drugs by sales revenue in 2015: Who sold the biggest blockbuster drugs? https://www.pharmacompass.com/radio-compass-blog/top-drugs-by-sales-revenue-in-2015-who-sold-the-biggest-blockbuster-drugs (2016).
- [100].Sagonowsky E. The top 20 drugs by worldwide sales in 2020. https://www.fiercepharma.com/special-report/top-20-drugs-by-2020-sales (2021).
- [101].EvaluatePharma®. World preview 2019, outlook to 2024. https://info.evaluate.com/rs/607-YGS-364/images/EvaluatePharma_World_Preview_2019.pdf (2019).
- [102].Strebhardt K, Ullrich A. Paul Ehrlich’s magic bullet concept: 100 years of progress. Nat. Rev. Cancer, 8 (2008) 473–480. [DOI] [PubMed] [Google Scholar]
- [103].Valent P, Groner B, Schumacher U, Superti-Furga G, Busslinger M, Kralovics R, Zielinski C, Penninger JM, Kerjaschki D, Stingl G, Smolen JS, Valenta R, Lassmann H, Kovar H, Jäger U, Kornek G, Müller M, Sörgel F. Paul Ehrlich (1854–1915) and his contributions to the foundation and birth of translational medicine. Journal of Innate Immunity, 8 (2016) 111–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Science-History-Institute. Paul Ehrlich. https://www.sciencehistory.org/historical-profile/paul-ehrlich (2017).
- [105].Park K, Han B, Korc M. Targeting the tumor microenvironment, in: Hartshorn CM (Ed.) Cancer Nanotechnology Plan 2015, National Cancer Insititute, National Institute of Health. 2015, pp. 25–28. [Google Scholar]
- [106].Lehrman S. Virus treatment questioned after gene therapy death. Nature, 401 (1999) 517–518. [DOI] [PubMed] [Google Scholar]
- [107].Frank KM, Hogarth DK, Miller JL, Mandal S, Mease PJ, Samulski RJ, Weisgerber GA, Hart J. Investigation of the cause of death in a gene-therapy trial. N. Eng. J. Med, 361 (2009) 161–169. [DOI] [PubMed] [Google Scholar]
- [108].Philippidis A. Fourth boy dies in trial of Astellas gene therapy candidate. https://www.genengnews.com/news/fourth-boy-dies-in-trial-of-astellas-gene-therapy-candidate/ (2021). [DOI] [PubMed]
- [109].Paulk N. Gene therapy: It’s time to talk about high-dose AAV. https://www.genengnews.com/commentary/gene-therapy-its-time-to-talk-about-high-dose-aav/ (2020).
- [110].Liu A. Bluebird Bio hits pause on rollout for ill-fated gene therapy Zynteglo as trial flags 2 cancer cases. https://www.fiercepharma.com/pharma/bluebird-bio-halts-selling-ill-fated-gene-therapy-zynteglo-as-trial-flags-2-cancer-cases (2021).
- [111].Israeli A. Paul Ehrlich’s ingredients for success. Lancet, 352 (1998) 1712. [DOI] [PubMed] [Google Scholar]
- [112].Wels M, Roels D, Raemdonck K, De Smedt SC, Sauvage F. Challenges and strategies for the delivery of biologics to the cornea. J. Control. Release, 333 (2021) 560–578. [DOI] [PubMed] [Google Scholar]
- [113].Liu X, Anissimov YG, Grice JE, Cheruvu HS, Ghosh P, Raney SG, Maibach HI, Roberts MS. Relating transdermal delivery plasma pharmacokinetics with in vitro permeation test (IVPT) findings using diffusion and compartment-in-series models. J. Control. Release, 334 (2021) 37–51. [DOI] [PubMed] [Google Scholar]
- [114].Benoit DSW, Overby CT, Sims KR Jr, Ackun-Farmmer MA. Drug delivery systems, in: Wagner WR, Sakiyama-Elbert SE, Zhang G, Yaszemski MJ (Eds.) Biomaterials Science (Fourth Edition), Academic Press. 2020, pp. 1237–1266 (Ch. 1232.1235.1212). [Google Scholar]
- [115].Bally MB, Harvie P, Wong FM, Kong S, Wasan EK, Reimer DL. Biological barriers to cellular delivery of lipid-based DNA carriers. Adv. Drug Deliv. Rev, 38 (1999) 291–315. [DOI] [PubMed] [Google Scholar]
- [116].Kuno N, Fujii S. Recent advances in ocular drug delivery systems. Polymers, 3 (2011) 193–221. [Google Scholar]
- [117].Ruge CA, Kirch J, Lehr CM. Pulmonary drug delivery: from generating aerosols to overcoming biological barriers-therapeutic possibilities and technological challenges. Lancet Respir. Med, 1 (2013) 402–413. [DOI] [PubMed] [Google Scholar]
- [118].Kim SM, Faix PH, Schnitzer JE. Overcoming key biological barriers to cancer drug delivery and efficacy. J. Control. Release, 267 (2017) 15–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].von Roemeling C, Jiang W, Chan CK, Weissman IL, Kim BYS. Breaking down the barriers to precision cancer nanomedicine. Trends Biotechnol, 35 (2017) 159–171. [DOI] [PubMed] [Google Scholar]
- [120].Yang R, Wei T, Goldberg H, Wang W, Cullion K, Kohane DS. Getting drugs across biological barriers. Adv. Mater, 29 (2017) 1606596 (1606525 pages). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Henrich-Noack P, Nikitovic D, Neagu M, Docea AO, Engin AB, Gelperina S, Shtilman M, Mitsias P, Tzanakakis G, Gozes I, Tsatsakis A. The blood–brain barrier and beyond: Nano-based neuropharmacology and the role of extracellular matrix. Nanomedicine: Nanotechnology, Biology and Medicine, 17 (2019) 359–379. [DOI] [PubMed] [Google Scholar]
- [122].Deprez J, Lajoinie G, Engelen Y, De Smedt SC, Lentacker I. Opening doors with ultrasound and microbubbles: Beating biological barriers to promote drug delivery. Adv. Drug Del. Rev, 172 (2021) 9–36. [DOI] [PubMed] [Google Scholar]
- [123].Amidon GL, Lennernäs H, Shah VP, Crison JR. A theoretical basis for a biopharmaceutic drug classification: the correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm. Res, 12 (1995) 413–420. [DOI] [PubMed] [Google Scholar]
- [124].FDA/CDER. Guidance for Industry: Waiver of in vivo bioavailability and bioequivalence studies for immediate release solid oral dosage forms based on a Biopharmaceutics Classification System. (2000).
- [125].Hwang SJ, Park H, Park K. Gastric retentive drug delivery systems. Critical Reviews in Therapeutic Drug Carrier Systems, 15 (1998) 243–284. [PubMed] [Google Scholar]
- [126].Vinarov Z, Abdallah M, Agundez JAG, Allegaert K, Basit AW, Braeckmans M, Ceulemans J, Corsetti M, Griffin BT, Grimm M, Keszthelyi D, Koziolek M, Madla CM, Matthys C, McCoubrey LE, Mitra A, Reppas C, Stappaerts J, Steenackers N, Trevaskis NL, Vanuytsel T, Vertzoni M, Weitschies W, Wilson C, Augustijns P. Impact of gastrointestinal tract variability on oral drug absorption and pharmacokinetics: An UNGAP review. Eur. J. Phar,m. Sci, 162 (2021) 105812 (105833 pages). [DOI] [PubMed] [Google Scholar]
- [127].Ahadian S, Finbloom JA, Mofidfar M, Diltemiz SE, Nasrollahi F, Davoodi E, Hosseini V, Mylonaki I, Sangabathuni S, Montazerian H, Fetah K, Nasiri R, Dokmeci MR, Stevens MM, Desai TA, Khademhosseini A. Micro and nanoscale technologies in oral drug delivery. Adv. Drug Del. Rev, 157 (2020) 37–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Brayden DJ, Hill TA, Fairlie DP, Maher S, Mrsny RJ. Systemic delivery of peptides by the oral route: Formulation and medicinal chemistry approaches. Adv Drug Deliv Rev, 157 (2020) 2–36. [DOI] [PubMed] [Google Scholar]
- [129].Brown TD, Whitehead KA, Mitragotri S. Materials for oral delivery of proteins and peptides. Nat. Rev. Mater, 5 (2020) 127–148. [Google Scholar]
- [130].Byrne J, Huang H-W, McRae JC, Babaee S, Soltani A, Becker SL, Traverso G. Devices for drug delivery in the gastrointestinal tract: A review of systems physically interacting with the mucosa for enhanced delivery. Adv. Drug Del. Rev, 177 (2021) 113926. [DOI] [PubMed] [Google Scholar]
- [131].Chaplin S. Rybelsus: an oral formulation of the GLP-1 agonist semaglutide. Prescriber, 31 (2020) 32–33. [Google Scholar]
- [132].Fattah S, Ismaiel M, Murphy B, Rulikowska A, Frias JM, Winter DC, Brayden DJ. Salcaprozate sodium (SNAC) enhances permeability of octreotide across isolated rat and human intestinal epithelial mucosae in Ussing chambers. Eup. J. Pharm. Sci, 154 (2020) 105509. [DOI] [PubMed] [Google Scholar]
- [133].Ito S, Torii Y, Chikamatsu S, Harada T, Yamaguchi S, Ogata S, Sonoda K, Wakayama T, Masuda T, Ohtsuki S. Oral coadministration of Zn-insulin with D-form small intestine-permeable cyclic peptide enhances its blood glucose-lowering effect in mice. Mol. Pharm, 18 (2021) 1593–1603. [DOI] [PubMed] [Google Scholar]
- [134].Li J, Zhou Y, Yang J, Ye R, Gao J, Ren L, Liu B, Liang L, Jiang L. Fabrication of gradient porous microneedle array by modified hot embossing for transdermal drug delivery. Mater. Sci. Eng. C, 96 (2019) 576–582. [DOI] [PubMed] [Google Scholar]
- [135].Vinarov Z, Abrahamsson B, Artursson P, Batchelor H, Berben P, Bernkop-Schnürch A, Butler J, Ceulemans J, Davies N, Dupont D, Flaten GE, Fotaki N, Griffin BT, Jannin V, Keemink J, Kesisoglou F, Koziolek M, Kuentz M, Mackie A, Meléndez-Martínez AJ, McAllister M, Müllertz A, O’Driscoll CM, Parrott N, Paszkowska J, Pavek P, Porter CJH, Reppas C, Stillhart C, Sugano K, Toader E, Valentová K, Vertzoni M, De Wildt SN, Wilson CG, Augustijns P. Current challenges and future perspectives in oral absorption research: An opinion of the UNGAP network. Adv. Drug Del. Rev, 171 (2021) 289–331. [DOI] [PubMed] [Google Scholar]
- [136].Chen W, Yao S, Wan J, Tian Y, Huang L, Wang S, Akter F, Wu Y, Yao Y, Zhang X. BBB-crossing adeno-associated virus vector: An excellent gene delivery tool for CNS disease treatment. J. Control. Release, 333 (2021) 129–138. [DOI] [PubMed] [Google Scholar]
- [137].Figueroa SM, Fleischmann D, Goepferich A. Biomedical nanoparticle design: What we can learn from viruses. J. Control. Release, 329 (2021) 552–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Castro F, Martins C, Silveira MJ, Moura RP, Pereira CL, Sarmento B. Advances on erythrocyte-mimicking nanovehicles to overcome barriers in biological microenvironments. Adv. Drug Del. Rev, 170 (2021) 312–339. [DOI] [PubMed] [Google Scholar]
- [139].Mendanha D, Vieira de Castro J, Ferreira H, Neves NM. Biomimetic and cell-based nanocarriers – New strategies for brain tumor targeting. J. Control. Release, 337 (2021) 482–493. [DOI] [PubMed] [Google Scholar]
- [140].Khan MM, Filipczak N, Torchilin VP. Cell penetrating peptides: A versatile vector for co-delivery of drug and genes in cancer. J. Control. Release, 330 (2021) 1220–1228. [DOI] [PubMed] [Google Scholar]
- [141].de Castilla PEM, Tong L, Huang C, Sofias AM, Pastorin G, Chen X, Storm G, Schiffelers RM, Wang J-W. Extracellular vesicles as a drug delivery system: A systematic review of preclinical studies. Adv. Drug Del. Rev, 175 (2021) 113801. [DOI] [PubMed] [Google Scholar]
- [142].Akhtar A, Andleeb A, Waris TS, Bazzar M, Moradi A-R, Awan NR, Yar M. Neurodegenerative diseases and effective drug delivery: A review of challenges and novel therapeutics. J. Control. Release, 330 (2021) 1152–1167. [DOI] [PubMed] [Google Scholar]
- [143].Brunner J, Ragupathy S, Borchard G. Target specific tight junction modulators. Adv. Drug Del. Rev, 171 (2021) 266–288. [DOI] [PubMed] [Google Scholar]
- [144].Nash A, Aghlara-Fotovat S, Hernandez A, Scull C, Veiseh O. Clinical translation of immunomodulatory therapeutics. Adv. Drug Del. Rev, 176 (2021) 113896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Kumthekar P, Tang S-C, Brenner AJ, Kesari S, Piccioni DE, Anders C, Carrillo J, Chalasani P, Kabos P, Puhalla S, Tkaczuk K, Garcia AA, Ahluwalia MS, Wefel JS, Lakhani N, Ibrahim N. ANG1005, a brain-penetrating peptide–drug conjugate, shows activity in patients with breast cancer with leptomeningeal carcinomatosis and recurrent brain metastases. Clin. Cancer Res, 26 (2020) 2789. [DOI] [PubMed] [Google Scholar]
- [146].Hanafy AS, Dietrich D, Fricker G, Lamprecht A. Blood-brain barrier models: Rationale for selection. Adv. Drug Del. Rev, 176 (2021) 113859. [DOI] [PubMed] [Google Scholar]
- [147].Pozzi S, Scomparin A, Israeli Dangoor S, Rodriguez Ajamil D, Ofek P, Neufeld L, Krivitsky A, Vaskovich-Koubi D, Kleiner R, Dey P, Koshrovski-Michael S, Reisman N, Satchi-Fainaro R. Meet me halfway: Are in vitro 3D cancer models on the way to replace in vivo models for nanomedicine development? Adv. Drug Del. Rev, 175 (2021) 113760. [DOI] [PubMed] [Google Scholar]
- [148].Boix-Montesinos P, Soriano-Teruel PM, Armiñán A, Orzáez M, Vicent MJ. The past, present, and future of breast cancer models for nanomedicine development. Adv. Drug Del. Rev, 173 (2021) 306–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Quintana DS, Guastella AJ, Westlye LT, Andreassen OA. The promise and pitfalls of intranasally administering psychopharmacological agents for the treatment of psychiatric disorders. Mol. Psychiatry, 21 (2016) 29–38. [DOI] [PubMed] [Google Scholar]
- [150].Erdő F, Bors LA, Farkas D, Bajza Á, Gizurarson S. Evaluation of intranasal delivery route of drug administration for brain targeting. Brain Res. Bull, 143 (2018) 155–170. [DOI] [PubMed] [Google Scholar]
- [151].Crowe TP, Greenlee MHW, Kanthasamy AG, Hsu WH. Mechanism of intranasal drug delivery directly to the brain. Life Sci, 195 (2018) 44–52. [DOI] [PubMed] [Google Scholar]
- [152].CDC. About chronic diseases. https://www.cdc.gov/chronicdisease/about/index.htm (2021).
- [153].WHO. Adherence to long-term therapies. Evidence for action. file:///C:/Users/kp/AppData/Local/Temp/adherence_full_report.pdf (2003). [PubMed] [Google Scholar]
- [154].Abdelkader H, Fathalla Z, Seyfoddin A, Farahani M, Thrimawithana T, Allahham A, Alani AWG, Al-Kinani AA, Alany RG. Polymeric long-acting drug delivery systems (LADDS) for treatment of chronic diseases: Inserts, patches, wafers, and implants. Adv. Drug Del. Rev, 177 (2021) 113957. [DOI] [PubMed] [Google Scholar]
- [155].Bazerman MH. The Power of Noticing: What the Best Leaders See. Simon & Schuster. 2015. 240 pages. [Google Scholar]
- [156].SolarWinds. Retro delight: Gallery of early computers (1940s - 1960s). https://www.pingdom.com/blog/retro-delight-gallery-of-early-computers-1940s-1960s/ (2019).
- [157].Walker J. Typical UNIVAC® 1108 Prices: 1968. https://www.fourmilab.ch/documents/univac/config1108.html (1996).
- [158].Leung AK, Tam YY, Cullis PR. Lipid nanoparticles for short interfering RNA delivery. Adv. Genet, 88 (2014) 71–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [159].Cheng X, Lee RJ. The role of helper lipids in lipid nanoparticles (LNPs) designed for oligonucleotide delivery. Adv. Drug Deliv. Rev, 99 (2016) 129–137. [DOI] [PubMed] [Google Scholar]
- [160].Richner J. Modified mRNA vaccines protect against Zika virus infection. Cell, 168 (2017) pp. 1114–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].Pardi N, Hogan MJ, Porter FW, Weissman D. mRNA vaccines — a new era in vaccinology. Nat. Rev. Drug Discov, 17 (2018) 261–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].Park K, Mrsny R. Are controlled release scientists doing enough for our environment? J. Controlled Rel, 332 (2021) 620–622. [DOI] [PubMed] [Google Scholar]