

Two recent papers have linked narcolepsy to dysfunction of the newly discovered hypocretin (Hcrt) (orexin) peptide system. The paper by Lin et al. (1999 [previous issue of Cell]) examined the genetic correlate of canine narcolepsy, using the well-characterized Doberman pinscher and Labrador retriever models. They found a deletion in the transcripts of the hypocretin receptor 2 (Hcrtr2) gene in the narcoleptic Doberman and a different deletion in transcripts of the same receptor gene in the narcoleptic Labrador. Lin et al. speculate that these changes disrupt the proper membrane localization or transduction functions of this receptor. The paper by Chemelli et al. (1999 [this issue of Cell]) used a different approach to arrive at a similar conclusion. They created a knockout of the Hcrt gene in mice. Mice lacking Hcrt had abnormalities of sleep control resembling aspects of narcolepsy. Together these two studies implicate dysfunction of the Hcrt system or systems closely linked to it in the pathophysiology of narcolepsy. The implications of these findings can best be understood by reviewing the nature of narcolepsy and of the Hcrt system.
What Is Narcolepsy?
Narcolepsy is a disease affecting approximately 1 in 2,000 individuals (about 125,000 in the U.S.) and usually develops in the second or third decade of life with symptoms progressing over a period of 1 or more years and then stabilizing. Narcolepsy is characterized by sleepiness and cataplexy, which is a loss of muscle tone triggered by sudden strong emotions such as laughter and anger. Attacks of cataplexy are usually over within seconds but in some narcoleptic patients can last for minutes and be disabling. In contrast to sleep attacks, consciousness is maintained during cataplexy. In most narcoleptic patients, sleepiness rather than cataplexy is the more troublesome symptom. Narcoleptics go through life feeling the way most of us would feel if we had been awake for 24 hr. They awake refreshed from naps, but soon are sleepy again. Their nighttime sleep is fragmented with less of the deeper stages of sleep.
Narcolepsy also has been reported in horses, cattle, and dogs. Some cases of canine narcolepsy are sporadic and these dogs cannot be bred to produce narcoleptic offspring. Occasionally, entire litters are born that develop the symptoms of narcolepsy at 1–4 months of age and show both sleepiness and cataplexy. In the early 1970s, Dement, Mitler, and their colleagues found that it was possible to breed these familial narcoleptic Doberman pinschers or Labrador retrievers. The disorder is transmitted in the Dobermans and Labradors as a single-gene autosomal recessive trait with full penetrance.
The sleepiness of narcolepsy can be treated by a number of agents, such as amphetamine-like drugs and Modafinil, which increase arousal. Cataplexy is commonly treated with tricyclic antidepressants and by selective serotonin reuptake inhibitors. Nishino and colleagues have shown that the selective serotonin reuptake inhibitors and the antidepressants inhibit cataplexy in proportion to their activation of norepinephrine receptors. In contrast, prazosin, which blocks α1 noradrenergic receptors, greatly exacerbates cataplexy in both dogs and humans. Cholinergic agonists and the cholinesterase blocker physostigmine also exacerbate cataplexy, consistent with a role of cholinoceptive neurons in triggering muscle tone suppression. Thus, the balance between the noradrenergic and cholinergic systems is a major factor in the control of cataplexy.
In normal individuals, postural muscles maintain some level of tone throughout waking and non-REM sleep. Only in REM sleep is muscle tone completely abolished. This suppression of tone prevents the motor programs generated in REM sleep from causing dangerous and sleep-disrupting movements. The similarity between the atonia in REM sleep and the atonia in cataplexy, and the abnormally short latency to REM sleep onset shown by narcoleptics, have led to the hypothesis that cataplexy may represent a triggering, in waking, of the mechanism that normally functions to suppress muscle tone in REM sleep. Studies in the narcoleptic dog have supported this hypothesis and provided insights into the nature of the mechanisms controlling posture in waking. Recordings of brainstem neuronal activity in narcoleptic dogs have shown that cataplexy is linked to the activation of a population of cells in the medulla and pons that in normal animals is active only during REM sleep (Siegel et al., 1991; Siegel, 1994). During REM sleep and presumably during cataplexy, this pontomedullary system causes the release of glycine onto motoneurons, producing hyperpolarization (Figure 1).
Figure 1.
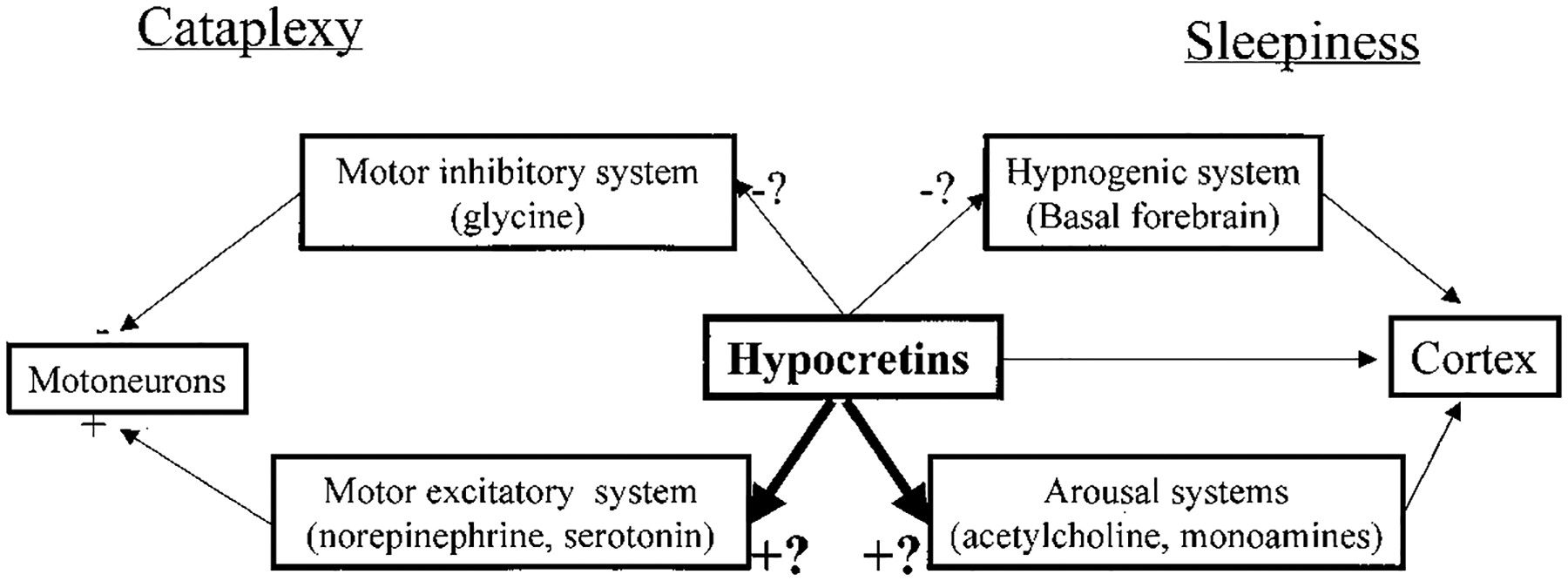
Possible Role of the Hypocretins (Orexins) in the Pathology of Narcolepsy
The hypocretin (orexin)–containing neurons are known to project to brainstem regions linked to motor inhibition as well as to locus coeruleus (norepinephrine), raphe (serotonin), and laterodorsal tegmental nuclei (acetylcholine) and ventral tegmental (dopamine) neurons. They also project to forebrain regions including posterior hypothalamus (histamine), septal nucleus, and diagonal band (acetylcholine) neurons and have major projections to the amygdala and basal forebrain hypnogenic regions. Loss of function of the hypocretin system could cause cataplexy by disfacilitating the brainstem’s motor excitatory systems or by disinhibiting the brainstem’s motor inhibitory system. Loss of function of the hypocretins could increase sleepiness by disfacilitating the cholinergic and aminergic arousal systems or by disinhibiting the forebrain’s hypnogenic systems. The few physiological studies done to date suggest that the hypocretins are usually excitatory; therefore, these two possibilities are highlighted in the figure.
At cataplexy onset, at the same time as glycine begins to be released, the norepinephrine-containing neurons of the locus coeruleus, which are normally continuously active in waking, abruptly and completely cease discharge (Wu et al., 1999). They also cease discharge in REM sleep. Norepinephrine released from these neurons is known to facilitate motoneurons (Figure 1). Thus, a combination of active inhibition and disfacilitation (reduced excitatory input) appears to underlie both cataplexy and the muscle atonia of REM sleep.
What Are the Hypocretins (Orexins)?
In 1998, de Lecea et al., using directional tag PCR subtraction, described a hypothalamus-specific mRNA that encoded “preprohypocretin,” which was thought to be the precursor of two peptides, Hcrt1 and −2. They named these peptides hypocretins (Hcrts) to indicate their hypothalamic localization and similarity to the gut hormone secretin. At about the same time, Sakurai et al. (1998) were searching for ligands for certain cDNA sequences that resembled G protein–coupled receptors but had no known ligands, i.e., orphan receptors. They identified peptides that bind to and activate two related receptors. Because they found that these peptide ligands stimulated food intake, they named them “orexins” after the Greek word for appetite. The peptides described by de Lecea et al. and Sakurai et al. are identical.
Anatomical studies determined that the somas of the Hcrt-producing cells were restricted to the hypothalamus and concentrated in the perifornical nucleus and in the dorsal, lateral, and posterior hypothalamus. The mRNA for the Hcrts is expressed by embryonic day 18 in rats and increases dramatically after postnatal day 21 (de Lecea et al., 1998). Although the cell bodies of Hcrt-producing neurons are entirely restricted to the hypothalamus, they have widespread axonal projections. In addition to dense hypothalamic projections, the limbic system, thalamus, subthalamic nucleus, substantia nigra, raphe, locus coeruleus, ventral tegmental area, medullary reticular formation, nucleus of the solitary tract, and other brainstem regions are innervated by these cells (Peyron et al., 1998). The neocortex also receives projections. Hcrt receptors are distributed throughout the innervated regions with a striking segregation of the two receptor types within rat brain (Trivedi et al., 1998). Hcrtr2 mRNA is mainly expressed in cerebral cortex, nucleus accumbens, subthalamic and paraventricular thalamic nuclei, and anterior pretectal nucleus. Lin et al. (1999) implicate the Hcrtr2 in canine narcolepsy.
Physiological studies showed that Hcrt release raises cytoplasmic calcium levels by opening plasma membrane channels and increases the release of both gamma amino butyric acid (GABA) and glutamate from axon terminals (van den Pol et al., 1998). Hcrt neurons have close and reciprocal connections with hypothalamic neurons containing neuropeptide Y (NPY) (Horvath et al., 1999), which strongly stimulates appetite, suggesting that Hcrt neurons have a role in feeding and metabolic control. The entire lateral hypothalamic region in which the Hcrt neurons are embedded has long been thought of as an appetite control “center.”
Pharmacological and physiological studies indicate that Hcrt neurons have a role in appetite (Sakurai et al., 1998). Hcrt-producing cells express leptin receptor immunoreactivity (Horvath et al., 1999). Insulin-induced hypoglycemia greatly increases the level of the Hcrt precursor protein, whereas leptin treatment decreases Hcrt levels. Genetically obese mice have downregulated Hcrt gene expression. Microinjection of Hcrt and in some cases intracerebroventricular administration triggers feeding (Sweet et al., 1999) and increases metabolic rate.
The Link between Narcolepsy and the Hypocretins
The two apparently unrelated topics of a putative lateral hypothalamic feeding hormone and a disease characterized by episodes of sleepiness and sudden losses of muscle tone have now been forever united by the work of Lin et al. (1999) and Chemelli et al. (1999). The narcoleptic dogs studied by Lin et al. have clear episodes of cataplexy, disturbed sleep, and evidence of sleepiness resembling that shown by human narcoleptics. They are also similar to human narcoleptics in their response to drugs that exacerbate and ameliorate cataplexy and sleepiness.
It remains to be determined if the Hcrt gene knockout mice created by Chemelli et al. have the features of human and canine narcolepsy. These mice lack the Hcrt peptides, in contrast to the dogs, which have mutations in the Hcrt2 receptor. Although they have periods of “behavioral arrest,” it is not yet clear if these include periods of cataplexy or if they are all direct transitions from waking into REM sleep. The triggers for these episodes differ from those in the dogs, with emotional excitement associated with food intake not being a common instigator of attacks in the mice. It may well be that an identical knockout in a dog would produce symptoms similar to those seen in the narcoleptic Doberman and Labrador, but that the brain of the mouse does not contain the machinery required for full expression of the syndrome seen in humans and dogs. Conversely, an Hcrtr2 knockout mouse might mimic more closely the symptoms seen in dogs and humans. Sleep is disrupted in the Hcrt knockout mice although it is not clear that they are “sleepy.” Finally the pharmacologic response of these symptoms is not reported in the Chemelli et al. paper. Clearly much work remains to be done to fully characterize this new model of narcolepsy. Nevertheless, the presence of sleep onset REM sleep periods and disrupted sleep in both dog and knockout mouse provides strong support for the proposed involvement of Hcrt in narcolepsy.
How Does the Finding of Hcrt Linkage Explain the Symptoms of Narcolepsy?
A remarkable link appears to exist between the anatomy of the Hcrt system and the triggering of cataplexy. Peyron et al. (1998) report that the densest extrahypo-thalamic projection of the Hcrt system is directed at the locus coeruleus. As outlined above, cessation of activity in locus coeruleus cells precedes and accompanies cataplexy, and narcoleptic dogs have a relatively low rate of locus coeruleus discharge (Wu et al., 1999). In normal animals, locus coeruleus cells never cease discharge during waking. Hcrt administration has been shown to greatly increase c-Fos labeling in locus coeruleus neurons (Date et al., 1999), an indication that Hcrts increase discharge in these cells. Thus, it can be argued that a malfunctioning Hcrt system, by removing a source of excitation from the locus coeruleus, would increase the propensity for cataplexy. However, the Hcrt2 receptor that is mutated in narcoleptic dogs is not present at high levels in the locus coeruleus of rats. If this is also true in the dog, one may hypothesize that the Hcrt2 receptor mutation leads to a more widespread loss of Hcrt function than would be caused by Hcrt2 receptor malfunction alone. This may occur through disturbed feedback regulation or degeneration of Hcrt neurons resulting in a direct or indirect disfacilitation of locus coeruleus cells.
Although we now understand key elements of the system responsible for cataplexy, the neurons responsible for the sleepiness of narcolepsy have not been identified. Several populations of cells known to increase arousal or increase sleepiness exist. Reduced excitation of the noradrenergic cells of the locus coeruleus, serotonergic cells of the raphe, dopaminergic cells of the hypothalamus and mesopontine region, or histaminergic cells of the posterior hypothalamus could reduce arousal levels. Reduced excitation of cholinergic neurons of the basal forebrain and brainstem would also impair behavioral and EEG arousal.
Conversely, the Hcrts could directly or indirectly inhibit sleep active neurons in the basal forebrain, amygdala, or the hypnogenic regions of the nucleus solitarius. If this were the case, dysfunction of these projections would increase sleepiness. If the Hcrts have a role in monoamine and acetylcholine release similar to the role they have been shown to have in GABA and glutamate release, Hcrt hypofunction could cause the accumulation of dopamine and the upregulation of several receptor types that is observed in the narcoleptic dogs. It is significant that the hypocretin neurons have direct projections to all of the arousal and sleepiness modulating cell groups mentioned above.
Recent work has shown that there is neuronal degeneration in limbic forebrain regions in the narcoleptic dogs. This degeneration peaks before symptom onset (Siegel et al., 1999). Degeneration is maximal in the amygdala and septal nuclei and is also elevated in the diagonal band, thalamus, and hypothalamic regions. These areas receive a moderate to heavy Hcrt innervation. Tafti et al. (1996) have reported increased expression of major histocompatibility class II molecules in brain microglia of narcoleptic dogs, with a time course similar to that of the degeneration, consistent with direct or indirect immune activation. Could the Hcrt2 gene mutation be linked to the degenerative changes? The human Hcrt precursor gene is located at chromosome 17q21–22, a gene locus that has been implicated in a series of neurodegenerative disorders (Basun et al., 1997), suggesting a possible role for Hcrts in other neurodegenerative diseases. An intriguing clue to the possible role of Hcrts in degeneration comes from the recent report of Ichinose et al. (1998). They found that Hcrt2 induced an outward current in peritoneal macrophages, indicating that the Hcrts can modulate macrophage functions through the activation of Ca2+-dependent K+ channels. It remains to be determined if the Hcrts have similar action on brain microglia. However, such a role could unite the loss of Hcrt function with the postulated but unproven immune link to narcolepsy.
Malfunction of the Hcrt system might be expected to affect several systems not implicated in sleep and arousal control. van den Pol has shown that Hcrt neurons strongly innervate the spinal cord. The innervation is targeted to dorsal regions involved in autonomic and pain control and may explain the profound analgesia produced by lateral hypothalamic stimulation. Hypofunction of this system might be involved in pain syndromes, but there has so far been no report of any linkage of narcolepsy to altered pain perception.
A simplistic prediction based on the role of Hcrts in appetite stimulation would be that narcoleptics should have a gross abnormality of food intake resulting in anorexia. However, there is no evidence for anorexia in unmedicated narcoleptics. Conversely, although there may be some modulation of narcoleptic symptomatology as a function of food intake, there is no strong evidence for the major change in symptoms that would be predicted by the effects of hypoglycemia on Hcrt neurons. Similarly, hypofunction of the nucleus accumbens, a site for reward integration, might suggest a syndrome of ahedonia in narcoleptics, but none has been reported. However, there is evidence for higher rates of depression in narcolepsy. This might be explained by reduced Hcrt excitation in the nucleus accumbens.
In general the deficits seen in animals with gene knockouts must be thought of as due to a combination of loss of gene function combined with the body’s compensatory response to the loss. Receptor upregulation, synaptic sprouting, biochemical changes within the affected cells, loss of trophic factors, immune interactions, and other changes may combine to produce the resulting phenotype. NPY neurons are adjacent to and have close synaptic interactions with Hcrt cells. NPY administration produces a more potent activation of food intake than does Hcrt administration. However, the NPY knockout mouse has normal weight regulation and response to food deprivation. On the other hand, these mice are prone to seizures and have elevated responses to leptin (Erickson et al., 1996). It may well be a similar reorganization of brain systems interacting with Hcrt neurons that causes the symptoms of narcolepsy, rather than the immediate effects of the Hcrt receptor malfunction or Hcrt disruption.
Relevance to Human Disease
Mutations of the Hcrt system may be responsible for some proportion of human narcolepsy cases. However, it is unlikely that most human narcoleptics have a mutation in their Hcrt or Hcrtr genes. Most narcoleptics have no narcoleptic relatives, ruling out the autosomal recessive mode of inheritance seen in the dogs. The typical onset of symptoms in the second decade of life or later suggests that damage has occurred to a normally functioning sleep and motor control system. Approximately 75% of the pairs of identical twins examined are discordant for the disease, suggesting that environmental factors are critical in the triggering of human narcolepsy.
More than 85% of all narcoleptic patients with cataplexy share a specific HLA allele, HLA DQB1*0602, compared with 12% to 38% of the general population (Mig-not, 1998). Because of the role of HLA gene products in immune regulation, because most HLA-linked diseases are autoimmune in nature, and because of the evidence that environmental triggers might be involved, it has been speculated that narcolepsy might be an autoimmune disorder. An obvious question is whether there is evidence for an immune attack on the Hcrt2 receptor in human narcolepsy. Since receptors are continuously regenerated, any immune attack that affected only the receptors would have to continue for the duration of the disease. No such autoimmune process has so far been detected. Alternatively, irreversible damage to axon terminals or to the hypocretin neurons themselves, creating the equivalent of an Hcrt knockout, might cause the disorder. The deficit in human narcolepsy may also be downstream of the Hcrt system. For example, immune-mediated damage to neurons or receptors in the locus coeruleus, amygdala, or basal forebrain sleep related regions might produce the syndrome even with normal function of the Hcrt neurons and the Hcrt2 receptor.
Therapeutic Implications
Current treatments for narcolepsy provide some symptomatic relief at the expense of substantial side effects. The nature of the altered function caused by the Hcrt2 receptor mutation in the dogs is at present unclear. If the mutations cause a moderately diminished functional response to their ligand, administration of Hcrts might improve symptoms. Similarly, in the knockout mouse, the effects of the loss of Hcrt might be reversed by Hcrt administration. If similar pathologies exist in humans, the same treatments could be effective. However, since Hcrt has been reported to increase eating, metabolic rate, and gastric acid secretion, and Hcrts may have a role in the control of anterior pituitary hormones and pain regulation, a large number of brain systems would likely be affected by Hcrt administration. The possibility of effects on the immune system should also be considered. Moreover, Hcrts produce “wet dog shakes” in rats, an indication of stress or anxiety, and high doses produce seizures in rats (Ida et al., 1999). Since the Hcrt system is strongly connected with the noradrenergic and serotonergic systems, themselves diffusely projecting, both therapeutic and deleterious effects could be mediated by secondary alteration in the activity of these aminergic systems. The development of specific Hcrt agonists for Hcrtr2, the receptor mutated in narcoleptic dogs, might reduce these problems. Since some of the most successful neuroactive therapeutic agents manipulate the widely projecting aminergic systems, there is reason to hope that manipulation of the Hcrt system will also lead to useful drug therapies for narcolepsy.
Selected Reading
- Basun H, Almkvist O, Axelman K, Brun A, Campbell TA, Col-linge J, Forsell C, Froelich S, Wahlund L, Wetterberg L, and Lannfelt L (1997). Arch. Neurol 54, 539–544. [DOI] [PubMed] [Google Scholar]
- Chemelli RM, Willie JT, Sinton CM, Elmquist JK, Scammell T, Lee C, Richardson JA, et al. (1999). Cell 98, this issue, 437–451. [DOI] [PubMed] [Google Scholar]
- Date Y, Ueta Y, Yamashita H, Yamaguchi H, Matsukura S, Kangawa K, Sakurai T, Yanagisawa M, and Nakazato M (1999). Proc. Natl. Acad. Sci. USA 96, 748–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lecea L, Kilduff TS, Peyron C, Gao X, Foye PE, Danielson PE, Fukahara C, Battenberg EL, Gautvik VT, Bartlett FS II, et al. (1998). Proc. Natl. Acad. Sci. USA 95, 322–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erickson JC, Clegg KE, and Palmiter RD (1996). Nature 381, 415–421. [DOI] [PubMed] [Google Scholar]
- Horvath TL, Diano S, and van den Pol AN (1999). J. Neurosci 19, 1072–1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichinose M, Asai M, Sawada M, Sasaki K, and Oomura Y (1998). FEBS Lett. 440, 51–54. [DOI] [PubMed] [Google Scholar]
- Ida T, Nakahara K, Katayama T, Murakami N, and Nakazato M (1999). Brain Res. 821, 526–529. [DOI] [PubMed] [Google Scholar]
- Lin L, Faraco J, Li R, Kadotani H, Rogers W, Lin X, Qiu X, de Jong PJ, Nishino S, and Mignot E (1999). Cell 98, 365–376. [DOI] [PubMed] [Google Scholar]
- Mignot E (1998). Neurology 50(2), S16–S22. [DOI] [PubMed] [Google Scholar]
- Peyron C, Tighe DK, van den Pol AN, de Lecea L, Heller HC, Sutcliffe JG, and Kilduff TS (1998). J. Neurosci 18, 9996–10015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakurai T, Amemiya A, Ishii M, Matsuzaki I, Chemelli RM, Tanaka H, Williams SC, Richardson JA, Kozlowski GP, Wilson S, et al. (1998). Cell 92, 573–585. [DOI] [PubMed] [Google Scholar]
- Siegel JM (1994). In Principles and Practice of Sleep Medicine, Kryger MH, Roth T, and Dement WC, eds. (Philadelphia: W.B. Saunders Company; ), pp. 125–144. [Google Scholar]
- Siegel JM, Nienhuis R, Fahringer H, Paul R, Shiromani P, Dement WC, Mignot E, and Chiu C (1991). Science 262, 1315–1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel JM, Nienhuis R, Gulyani S, Ouyang S, Wu MF, Mignot E, Switzer RC, and Cornford M (1999). J. Neurosci 19, 248–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweet DC, Levine AS, Billington CJ, and Kotz CM (1999). Brain Res. 821, 535–538. [DOI] [PubMed] [Google Scholar]
- Tafti M, Nishino S, Aldrich MS, Liao W, Dement WC, and Mignot E (1996). J. Neurosci 16, 4588–4595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trivedi P, Yu H, MacNeil DJ, Van der Ploeg LH, and Guan XM (1998). FEBS Lett. 438, 71–75. [DOI] [PubMed] [Google Scholar]
- van den Pol AN, Gao XB, Obrietan K, Kilduff TS, and Belousov AB (1998). J. Neurosci 18, 7962–7971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu MF, Gulyani S, Yao E, Mignot E, Phan B, and Siegel JM (1999). Neuroscience 91, 1389–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]