

Abstract
Bruton’s tyrosine kinase (BTK), expressed in B cells and cells of innate immunity, including microglia, is an essential signaling element downstream of the B‐cell receptor and Fc‐receptors. Tolebrutinib (PRN2246, SAR442168) is a potent BTK inhibitor that covalently binds the kinase, resulting in durable inhibition with the potential to target inflammation in the periphery and central nervous system (CNS). Tolebrutinib crosses the blood‐brain barrier and potently inhibits BTK in microglial cells isolated from the CNS. A first‐in‐human randomized, double‐blind, placebo‐controlled study of tolebrutinib was conducted. The trial design consisted of five single ascending dose arms with oral administration of a single dose of 5, 15, 30, 60, and 120 mg (n = 6 per arm, n = 2 placebo), five multiple ascending dose arms with oral administration of 7.5, 15, 30, 60, and 90 mg (n = 8 per arm, n = 2 placebo) over 10 days, and one arm (n = 4) in which cerebral spinal fluid (CSF) exposure was measured 2 h after a single 120 mg dose. Tolebrutinib was well‐tolerated in the study and all treatment‐related treatment emergent adverse events were mild. Tolebrutinib was rapidly absorbed following oral administration with a rapid half‐life of ~ 2 h. Peripheral BTK occupancy was assessed at various timepoints by an enzyme‐linked immunosorbent assay‐based readout using an irreversible probe. Assessments demonstrated extensive and prolonged peripheral BTK occupancy at steady‐state with once daily doses as low as 7.5 mg. Further, CSF exposure was demonstrated 2 h after administration at 120 mg.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
Bruton’s tyrosine kinase (BTK) plays an important role in B‐cell driven antibody production as well as cells involved in innate immunity, such as brain resident microglia pointing to potential importance in treatment of multiple sclerosis (MS). Clinical stage BTK inhibitors designed for immune‐mediated inflammatory disorders have not previously shown the ability to cross the blood‐brain barrier.
WHAT QUESTION DID THIS STUDY ADDRESS?
The covalent BTK inhibitor, tolebrutinib, was designed to cross the blood‐brain barrier. Tolebrutinib was administered in the experimental autoimmune encephalomyelitis model to demonstrate efficacy in a neuroinflammatory animal model and confirm that BTK protein in the brain was effectively inhibited. A phase I study in healthy human volunteers explored safety, levels of BTK inhibition, and pharmacokinetics, including exposure within the cerebral spinal fluid (CSF).
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
Tolebrutinib was well‐tolerated in single doses up to 120 mg and 90 mg with 10 days q.d. dosing. High levels of systemic BTK occupancy were achieved with doses at 15 mg and above and CSF exposure at levels above the cellular 90% inhibitory concentration was confirmed after administration of a single 120 mg dose.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
Results of the phase I study confirmed CSF exposure of tolebrutinib and enabled the design of a phase II dose range finding study to explore clinical outcomes in patients with MS.
INTRODUCTION
Multiple sclerosis (MS) is an inflammatory, demyelinating, and degenerative disease of the central nervous system (CNS) that affects ~ 2.5 million people worldwide. MS represents the leading cause of nontraumatic neurologic disability in young and middle‐aged adults. The clinical course of MS typically manifests as relapsing forms of MS (RMS), characterized by initial episodes of transient neurological compromise (synonymously called relapses, clinical exacerbations, or attacks) with variable recovery (remissions), eventually leading to cumulative deficits that may increase acutely with each new relapse. In progressive MS subtypes, a category that includes primary and secondary progressive MS (PPMS and SPMS, respectively) cumulative deficits may increase gradually independently of, or in the absence of, relapses, although recent findings support the view that such progression independent of relapse activity (PIRA) is also observed in people living with RMS. RMS includes relapsing‐remitting multiple sclerosis (RRMS) and SPMS “with activity”.
Immunomodulatory drugs targeting adaptive immunity in the periphery have been the mainstay of MS therapy. Recent results from clinical studies have focused attention on agents that target B lymphocytes, especially B cell depleting agents like ocrelizumab (anti‐CD20). 1 , 2 The contributions of immune cells residing in the CNS (microglia as an example) to MS pathology are also well‐known and are thought to represent the chief unmet medical need for people living with MS. 3 Thus, there is a need for therapies that directly target neuroinflammation in the CNS with a goal of slowing disability accumulation and neurodegeneration across the full spectrum of MS (RMS, PPMS, and SPMS). 4
The Bruton’s tyrosine kinase (BTK) pathway is critical to signaling in B lymphocytes and resident immune cells of the CNS known as microglia. Each of these cell types has been implicated in the pathophysiology of MS. BTK is critical for B‐cell activation, differentiation, and maturation. BTK also regulates the activation of cells from other hematopoietic lineages, such as basophils, mast cells, macrophages, and neutrophils, with the notable exception of T lymphocytes. BTK inhibitors have been approved for hematological malignancies, including ibrutinib, acalabrutinib, and zanubrutinib. 5 , 6 , 7 However, there is only limited data in CNS infiltrating oncology indications, demonstrating the ability of these inhibitors to cross the blood‐brain barrier. Accordingly, a CNS penetrant BTK inhibitor has the potential to act via multiple mechanisms of action in MS and targets both adaptive and innate immune cells simultaneously in the periphery and CNS. Data from a phase IIb trial in patients with RMS treated with the BTK inhibitor evobrutinib have demonstrated reductions in the number of new contrast‐enhancing T1 lesions and new or enlarging T2 lesions. 8 However, it is uncertain whether evobrutinib can engage the target signaling mechanisms in the CNS, and currently no data are available as to whether a CNS‐penetrant BTK inhibitor might have direct effects. Tolebrutinib was designed to effectively cross the blood‐brain barrier and to target BTK‐expressing cells within the CNS. Tolebrutinib is a potent inhibitor of BTK, with selectivity driven by covalent binding to a specific conserved cysteine present in only 11 kinases. 9 In vitro, tolebrutinib bound tightly to BTK in Ramos B cells, blocking the binding of an irreversible probe molecule with a 90% inhibitory concentration (IC90) of 2 nM and potently inhibited BCR signaling and B cell activation, reducing cell surface expression of the activation marker CD69 with a 50% inhibitory concentration IC50 of 10 nM in stimulated human whole blood. It also potently bound to BTK in microglia, blocking probe binding with an IC50 of 0.7 nM. High levels of BTK occupancy in peripheral blood mononuclear cells (PBMCs) and microglia persisted 18 h after washout. In vivo, tolebrutinib demonstrated dose‐dependent protection from disease induction in a neuroinflammatory mouse model of experimental autoimmune encephalomyelitis (EAE; unpublished data). In addition, pharmacodynamics of BTK inhibition were assessed by use of an irreversible probe to determine free BTK levels at end of study in the periphery (spleen) and within the brain. This highlights the potential for the covalent binding of tolebrutinib to impart a prolonged impact on BTK inhibition even systemic clearance of free drug. It also allowed the potential for translation of occupancy observed in human clinical studies in healthy volunteers to estimate doses required to inhibit BTK signaling expected in clinical studies.
This first‐in‐human randomized, double‐blind, placebo‐controlled trial was comprised of five single ascending dose (SAD) arms, five multiple ascending dose (MAD) arms of 10 days of daily administration, and one arm in which cerebrospinal fluid (CSF) exposure was measured following lumbar puncture 2 h after administration of a single dose. In one cohort of the SAD (60 mg), participants returned following a washout of 1 week and were subsequently administered a second single 60 mg dose of tolebrutinib after a moderately fat meal. Frequent measurement of pharmacokinetics, BTK receptor occupancy, and safety were conducted with the goal of enabling selection of doses for administration in therapeutic studies. Results of a phase IIb study of tolebrutinib in patients with relapsing MS have recently been reported 10 and phase III studies in patients with relapsing as well as non‐relapsing secondary progressive MS (SPMS) and progressive MS are ongoing.
METHODS
Study design
This first‐in‐human healthy volunteer study was comprised of three parts: part A (SAD), a part B (MAD), and part C, an open label single dose arm with lumbar puncture to measure tolebrutinib CSF concentrations. Parts A and B followed a randomized, double blind, placebo‐controlled design, whereas part C was conducted open label. Part A included five cohorts (n = 8; 6 active, 2 placebo per cohort) of doses 5, 15, 30, 60, and 120 mg, with two sentinel participants treated (1 active:1 placebo) 24 h prior to the rest of the cohort. One cohort (60 mg dose) in part A returned to the site after a 6‐day washout to receive a second 60 mg administration with a moderate fat meal. Part B included 5 cohorts (n = 10; 8 active and 2 placebo), of doses 7.5, 15, 30, 60, and 90 mg administered once daily for 10 days. Part C was a single open label dose of 120 mg (n = 4). Tolebrutinib was administered as an oral liquid formulation or matching placebo. Unless otherwise indicated, all doses were administered under fasting conditions.
The study protocol was approved by the local ethics committee, and all participants provided written informed consent prior to participating in any study procedures. Safety and pharmacokinetics were reviewed by a Safety Review Committee prior to each dose escalation.
Safety assessments
Safety assessments included regular monitoring of the frequency and type of adverse events, physical examination, clinical laboratory testing, and 12‐lead electrocardiograms (ECGs) and vital signs. Laboratory investigations included hematology, coagulation, clinical chemistry, and urinalysis. Participants in part A remained in the clinical study unit from day −1 until 48 h postdosing, and returned for a 7‐day safety follow‐up visit. Participants in part B were admitted to the study unit on day −1, remained until day 12 (2 days after the last dose of study drug), and returned for a follow‐up safety visit on day 17. Participants in part C remained in the clinical study unit for 24 h following dosing and lumbar puncture, with a 7‐day safety follow‐up visit.
Pharmacokinetics
Plasma samples to determine concentrations of tolebrutinib were obtained at prespecified times, per protocol. In part A, sampling was conducted at 0 (predose), 0.25, 0.5, 1, 1.5, 2, 2.5, 3, 4, 6, 8, 10, 12, and 24 h after dosing. Urine was collected from the 60 mg cohort in part A over 12 h postdosing to determine the urinary excretion of tolebrutinib. In part B, plasma samples were obtained at 0 (predose), 0.25, 0.5, 1, 1.5, 2, 2.5, 3, 4, 6, 8, 10, 12, and 24 h after dosing on days 1 and 10, with predose samples collected on days 3, 5, and 7. In part C, plasma sampling followed part A. In addition, CSF concentrations were determined following lumbar puncture 2 h post‐administration of the single 120 mg dose.
Concentrations of tolebrutinib in human K2EDTA plasma, urine, CSF, and plasma protein binding were determined using four validated liquid‐chromatography tandem mass spectrometry (LC‐MS/MS) methods by Alturas (Moscow, ID, USA). Tolebrutinib and its deuterated internal standard PRN2737 were extracted from biological matrices by acetonitrile precipitation before analysis by liquid chromatography with C18 column and tandem mass spectrometry (Sciex API). The methods were validated with standard curves range 0.1–100 ng/ml for plasma, 0.5–500 ng/ml for urine, and 0.01–2 ng/ml for CSF, use quadratic 1/x2 regression. Each analytical run included two calibration curves, appropriate blanks, and duplicate quality control samples at three concentrations within the calibration range. The inter run accuracy and precision for plasma was 101–102% and 1.3–8.1%, respectively; for urine was 93.5–117% and 2.5–5.6%, respectively; for CSF was 92.7–104% and 3.1–7.4%, respectively. For the protein binding experiment, a 96‐well Micro‐Equilibrium Dialysis Device from HTDialysis was utilized to determine the protein binding of tolebrutinib in human plasma. Samples from the protein binding experiment were analyzed together with plasma samples.
Plasma concentration data were analyzed by standard noncompartmental methods (Phoenix WinNonlin Version 8, Certara USA). Results were summarized using descriptive statistics and reported by cohort. Dose proportionality following single dose administration in part A was assessed with a power model. No formal hypothesis testing or sample size calculations were conducted.
Flow cytometry analysis of immune cell subsets
Measurement of lymphocyte subpopulations was assessed in whole blood samples drawn predose on days 1, 4, and 10 in part B only and performed at Sonic Clinical Laboratories, New South Wales, Australia. T/B/NK/monocyte counts were measured by flow cytometry using antibodies for CD3, CD4, CD8, CD14, CD19, and CD16/56. Additionally, naïve (CD19+ CD27‐) and memory (CD19+ CD27+) B cell subpopulations were also analyzed by flow cytometry.
RESULTS
Participant disposition and demographics
Ninety‐four healthy volunteers were screened, enrolled, and received at least one dose of tolebrutinib or placebo (74 active and 20 placebo). Of these, there were 40 participants in part A (30 active and 10 placebo), 50 participants in part B (40 active and 10 placebo), and four participants in part C (all active). In part A, all participants were men, with a median (range) age of 26.0 years (18–52 years) and mean (SD) body weight of 78.5 kg (11.2 kg). The majority of participants were White (77.5%) and not Hispanic or Latino (95%). The demographics for parts B and C participants were very similar to part A. Full demographic details are available in Table S1.
Safety and tolerability
Tolebrutinib was well‐tolerated by all participants in part A of the study with no serious treatment‐emergent adverse events (TEAEs; Table S2). In part B of the study, all treatment‐related TEAE’s for all combined dose groups were mild in nature with only diarrhea and headache occurring in greater than 10% of all participants (Table 1). There were no treatment‐related clinically significant or drug‐related changes in vital signs, ECG parameters, or laboratory values, except for one mild treatment‐related event of decreased platelet count in part B for one participant who presented with low platelet values at baseline trending downward from day 7. Study drug was withheld following dosing on day 9, and the adverse event was considered resolved 15 days after onset. The frequency of tolebrutinib‐related events of diarrhea was greater in the highest‐level dose group. There were no other clinically significant patterns of note in treatment‐related TEAEs, regardless of assigned treatment and single or repeat dose administration, and all treatment related TEAEs were mild. BTK inhibitors are not B cell depleters, and CD19 expressing B cells increased were seen to modestly increase after 4 days of administration of tolebrutinib in part B of the study (vide infra).
TABLE 1.
Summary of adverse events occurring in greater than 10% of participants in part B (MAD)
| 7.5 mg | 15 mg | 30 mg | 60 mg | 90 mg | All tolebrutinib | Placebo | |
|---|---|---|---|---|---|---|---|
| Number of participants | 8 | 8 | 8 | 8 | 8 | 40 | 10 |
| Treatment‐related TEAE, N | 4 | 2 | 3 | 2 | 5 | 16 | 2 |
| Diarrhea | 1 | 1 | 2 | 0 | 5 | 9 | 1 |
| Headache | 2 | 1 | 1 | 0 | 0 | 4 | 0 |
Abbreviations: MAD, multiple ascending dose; TEAE, treatment‐emergent adverse event.
Pharmacokinetics
After oral administration under fasting conditions, tolebrutinib displayed rapid absorption with a median time to maximum concentration (Tmax) of ~ 1 h. The mean half‐life was ~ 1.5 to 2 h. Assessment of dose proportionality from 5 to 120 mg using the power model revealed that PRN2246 pharmacokinetic increases in a more than dose proportional manner for maximum plasma concentration (Cmax) and area under the curve (AUC). However, the more than dose proportional increase in exposure is primarily driven by the 120 mg dose. Dose levels between 15 and 60 mg were approximately dose proportional in exposure. Plasma protein binding was determined to be ~ 93.5%. A summary of tolebrutinib plasma pharmacokinetics from part A are provided in Table 2. Mean concentration‐time profiles are illustrated in Figure 1. The primary metabolic clearance pathways are oxidative in nature. Further details regarding the metabolites from the drug disposition study will be reported in due course.
TABLE 2.
Mean pharmacokinetic parameters of tolebrutinib part A (SAD)
| Dose, mg | Cmax, ng/ml | Tmax, h a | AUClast, ng.h/ml | AUC0‐inf, ng.h/ml | t 1/2, h |
|---|---|---|---|---|---|
| 5 (n = 6) | 0.369 (62%) | 1.0 (0.5–1.5) | 0.570 (84%) | 1.39 (n = 1) | 1.92 (87%) |
| 15 (n = 6) | 1.97 (51%) | 1.0 (1.0–1.0) | 3.67 (43%) | 4.06 (37%) | 1.39 (11%) |
| 30 (n = 6) | 3.12 (38%) | 1.0 (0.5–1.0) | 6.04 (29%) | 6.46 (27%) | 1.47 (25%) |
| 60 (n = 6) | 6.89 (57%) | 1.5 (1.0–1.5) | 15.6 (54%) | 15.9 (53%) | 1.67 (32%) |
| 120 (n = 6) | 32.3 (35%) | 1.0 (0.5–1.0) | 61.6 (54%) | 62.6 (54%) | 2.18 (32%) |
Abbreviations: AUC, area under the time‐concentration curve; Cmax, peak serum concentration after a single dose; SAD, single ascending dose; t 1/2, terminal half‐life; Tmax, time at which Cmax is obtained.
Median value (min – max).
FIGURE 1.
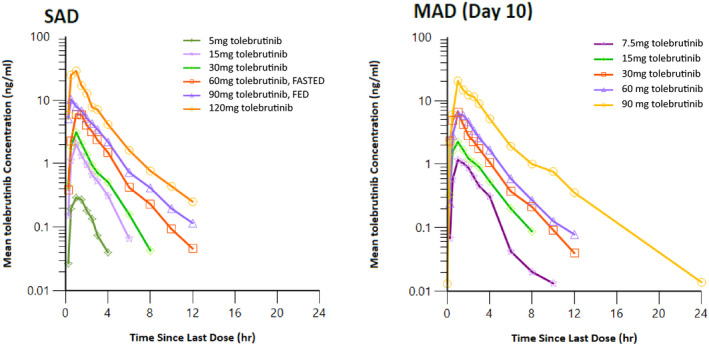
Mean SAD and steady‐state (MAD day 10) concentration‐time profiles. MAD, multiple ascending dose; SAD, single ascending dose
Multiple dose pharmacokinetic parameters were similar and consistent with the results from part A (Table 2). Following once daily (q.d.) dosing for 10 consecutive days, low accumulation was observed on day 10 compared to day 1, with mean accumulation ratios of 1.57, 1.36, and 1.07 for AUC0‐24, Cmax, and concentration at 24 h postdose (C24), respectively. The reason for the increased exposure seen at day 10 of the MAD portion of the study is unclear, but significant accumulation is unlikely given the rapid clearance. A summary of tolebrutinib plasma pharmacokinetics from part B are provided in Table 3.
TABLE 3.
Mean pharmacokinetic parameters of tolebrutinib part B (MAD)
| Dose, mg | Day 1 | Day 10 | ||||||
|---|---|---|---|---|---|---|---|---|
| Cmax, ng/ml | Tmax, h a | AUC0‐24, ng.h/ml | t 1/2, h | Cmax, ng/ml | Tmax, h a | AUC0‐24, ng.h/ml | t 1/2, h | |
| 7.5 (n = 8) | 1.17 (57%) | 1.0 (0.5–1.5) | 2.12 (51%) | 1.45 (22%) | 1.32 (31%) | 1.0 (0.5–2.0) | 3.07 (51%) | 1.57 (50%) |
| 15 (n = 8) | 1.96 (41%) | 1.0 (0.5–1.5) | 3.82 (43%) | 1.40 (16%) | 2.51 (60%) | 1.0 (1.0–1.5) | 5.85 (33%) | 1.68 (21%) |
| 30 (n = 8) | 6.25 (75%) | 0.75 (0.5–1.0) | 9.67 (68%) | 1.6 (24%) | 7.46 (74%) | 1.0 (0.5–2.0) | 14.9 (75%) | 2.02 (34%) |
| 60 (n = 8) | 8.47 (80%) | 1.25 (1.0 −3.0) | 24.3 (61%) | 1.98 (45%) | 6.69 (66%) | 1.5 (1.0–2.0) | 18.2 (55%) | 2.37 (44%) |
| 90 (n = 8) | 19.3 (48%) | 1.0 (0.5–1.0) | 35.5 (46%) | 2.1 (30%) | 21.7 (60%) | 1.5 (1.0–2.5) | 56.6 (54%) | 2.83 (30%) |
Abbreviations: AUC, area under the time‐concentration curve; Cmax, peak serum concentration after a single dose; MAD, multiple ascending dose; t 1/2, terminal half‐life; Tmax, time at which Cmax is obtained.
Median value (min – max).
Urine pharmacokinetics was assessed following a single oral 60 mg dose (fasted) of tolebrutinib. The total mean amount of tolebrutinib recovered unchanged in urine after 12 h was 4.2 µg, and the mean renal clearance was estimated to be ~ 0.3 L/h (total clearance 5.2 L/h). Based on these results, renal excretion appears to be a minor route of elimination for the parent drug.
The impact of a moderately fat meal on the pharmacokinetics of tolebrutinib was assessed in four participants at 60 mg (2 participants did not return to receive the fed dose) and demonstrated a modest food effect for tolebrutinib, with the geometric least squares mean ratios for Cmax and AUCinf being ~ 137% and 155%, respectively. Given the small sample size (n = 4), these results should be interpreted with caution.
Assessment of CNS penetration
Penetration of tolebrutinib into CSF was measured 2 h after a single dose of 120 mg tolebrutinib in four participants. The mean CSF concentration was 1.87 ng/ml (4.1 nM). The CSF:total plasma concentration ratio at 2 h post‐administration was 0.14, whereas the CSF:free plasma concentration ratio was 2.25. With a plasma Tmax of tolebrutinib of ~ 1 h and half‐life of 90 to 120 min, CSF concentrations measured at 2 h postdose may be an under‐representation of maximal CSF concentrations following a single dose of tolebrutinib. Additional CSF timepoints would be necessary to fully characterize the pharmacokinetic profile of tolebrutinib in CSF. Concentration‐time profiles for total, free, and CSF concentrations are provided in Figure 2.
FIGURE 2.
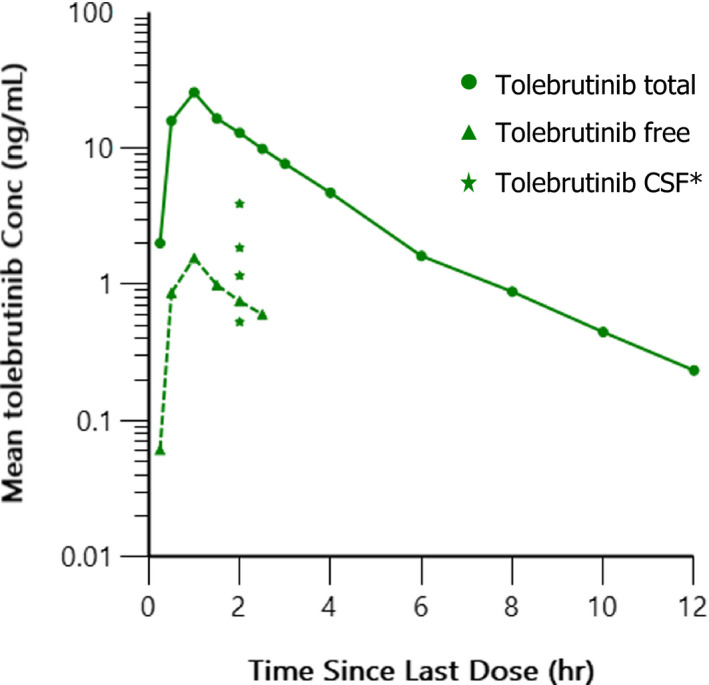
Mean total, free, and individual (N = 4) CSF concentration‐time profiles after 120 mg single dose administration of tolebrutinib. Individual concentrations (Conc) of tolebrutinib measure in CSF at 2 h are shown. CSF, cerebrospinal fluid
Pharmacodynamics
BTK occupancy was measured at 4, 12, and 24 h after dosing in the SAD portion of the study and day 1 of the MAD portion. Additionally, in the MAD study, occupancy was measured predose on days 3, 7, and 10, and at multiple timepoints out to 168 h (7 days) after administration of the final dose. All occupancy measurements were normalized to predose levels from day 1. Single doses of tolebrutinib at 5 mg to 120 mg in the SAD portion of the study resulted in a dose dependent increase in occupancy of BTK in human PBMC clinical pharmacodynamic samples. Maximal mean BTK occupancy of 96% ±2% and 97% ±0% was achieved after administration of single tolebrutinib doses of 60 mg and 120 mg, respectively (~ 2–3 h after Tmax). BTK occupancy was not changed by food intake. Daily doses of tolebrutinib for 10 consecutive days at 7.5 mg to 90 mg in the MAD portion of the study resulted in a dose dependent increase in BTK occupancy (Figure 3, Table 4). Within each cohort, occupancy levels increased with sequential doses. Mean BTK occupancy of greater than 75% was achieved and maintained in all dose cohorts by day 10 (7.5 mg to 90 mg daily dosing) and had returned toward baseline by 7 days after dosing in all cohorts. In part C (120 mg), maximal BTK occupancies (97%) were achieved with a single 120 mg dose, consistent with the results from part A.
FIGURE 3.

BTK occupancy observed during MAD (part B). Levels of BTK occupancy were measured at 4, 12, and 24 h after dosing on day 1, predose on days 3, 7, and 10, and at multiple timepoints out to 168 h (7 days) after administration of the final dose. All occupancy measurements were normalized to predose levels from day 1. BTK, Bruton’s tyrosine kinase; MAD, multiple ascending dose
TABLE 4.
Tolebrutinib BTK occupancy part B (MAD)
| Dose (h postdose) | Placebo a | 7.5 mg | 15 mg | 30 mg | 60 mg | 90 mg |
|---|---|---|---|---|---|---|
| D1 (4) | 4 ± 5 | 21 ± 16 | 56 ± 13 | 71 ± 22 | 94 ± 1 | 94 ± 1 |
| D1 (24) | 4 ± 7 | 21 ± 18 | 51 ± 13 | 64 ± 22 | 93 ± 2 | 91 ± 2 |
| D10 (4) | 7 ± 8 | 87 ± 5 | 95 ± 1 | 96 ± 1 | 96 ± 1 | 97 ± 1 |
| D10 (24) | 4 ± 8 | 78 ± 6 | 88 ± 3 | 82 ± 2 | 93 ± 2 | 93 ± 2 |
| D10 (48) | 4 ± 6 | 62 ± 10 | 74 ± 3 | 74 ± 3 | 85 ± 3 | 82 ± 4 |
| D10 (168) | 1 ± 1 | 7 ± 8 | 13 ± 13 | 12 ± 10 | 6 ± 9 | 26 ± 7 |
Abbreviations: BTK, Bruton’s tyrosine kinase; MAD, multiple ascending dose.
Placebo from all MAD cohorts at respective nominal time points.
Flow cytometry analysis of immune cell subsets was performed for part B (MAD) and evaluated as exploratory pharmacodynamic markers in addition to the safety assessment. Treatment with tolebrutinib did not result in B cell depletion. Elevated circulating total B cell levels (both naïve and memory B cells) were observed at day 4 and day 10 in all part B cohorts (Figure 4, Figure S1). Similar to the BTK occupancy results, elevated B cells were observed at all dose levels but were less pronounced for the lowest dose cohort (7.5 mg). No changes were observed in the other measured immune cell populations, including T cells (total CD3, CD4, and CD8), monocytes and NK (CD16+ and CD56+).
FIGURE 4.
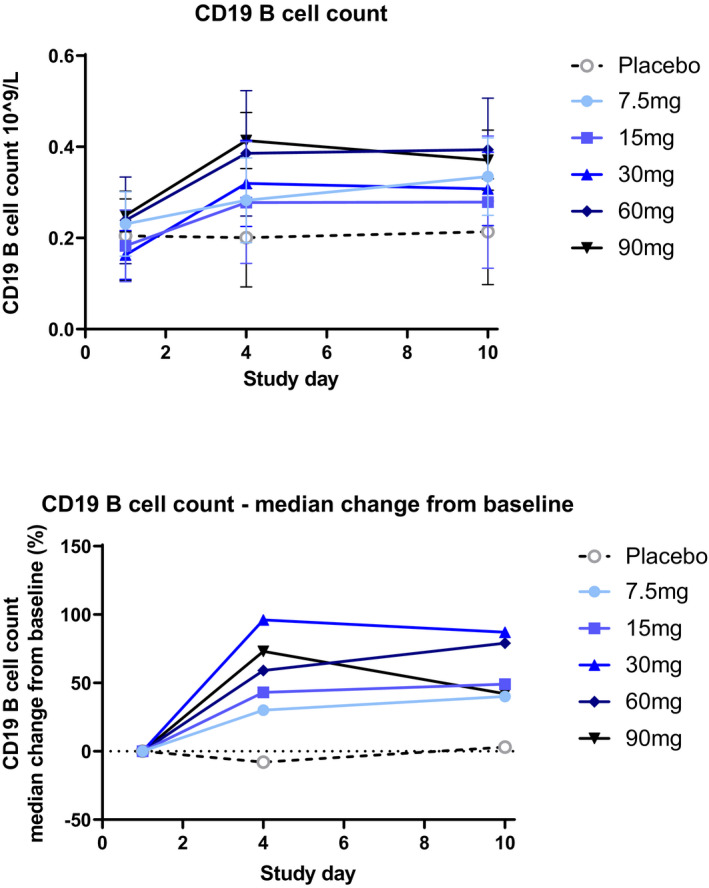
Changes in CD19 B cell counts by dose during MAD (part B). Numbers of CD19+ B cells were measured predose on days 1, 4, and 10 by flow cytometry in part B only. Notes: Error bars represent the SD. MAD, multiple ascending dose
DISCUSSION
Tolebrutinib was found to be well‐tolerated in healthy volunteers with single doses up to 120 mg and once daily multiple doses of up to 90 mg. In the MAD portion of the study, all treatment‐related TEAE’s for all combined dose groups were mild in nature with only diarrhea and headache occurring in greater than 10% of all participants. In both SAD and MAD portions of the study, tolebrutinib pharmacokinetics were characterized by rapid absorption and clearance from systemic circulation. Exposure was generally dose proportional and consistent with rapid clearance of drug and limited accumulation was seen after 10 days of administration. Although limited to data from four participants in the 60 mg cohort, exposures increased somewhat when tolebrutinib was administered after a moderately fat meal. CSF exposure was assessed via lumbar puncture at 2 h in four participants administered a single 120 mg dose. The mean CSF concentration of tolebrutinib was 1.87 ng/ml (~ 4.1 nM) which exceeds mean Cmax plasma (total) exposure at 15 mg dose and is likely sufficient to achieve significant CNS target engagement. Exposure also exceeds the IC90 of 2.0 nM value obtained in a competitive binding assay in Ramos cells. The free fraction was assessed and tolebrutinib was ~ 93.5% bound to plasma proteins resulting in a CSF:plasma ratio of free tolebrutinib of 2.25.
The use of covalent BTK inhibitors allows the assessment of target binding by evaluating the levels of free BTK in ex vivo peripheral blood samples at specific time points. 11 BTK occupancy approaching saturation was seen in PBMC’s after single doses greater than 60 mg and after 10 days, complete inhibition was seen in the MAD portion with doses as low as 15 mg. Free BTK levels returned toward normal over ~ 7 days as new protein was synthesized as reported previously with BTK inhibitors. 12 Importantly, whereas exposures varied between individual participants, levels of BTK occupancy were consistently high. Although occupancy was not directly measured within the brain, exposures of unbound drug in excess of the cellular IC90 (2.0 nM in Ramos cells) combined with the irreversible mode of action point to the potential for direct effects on BTK expressing cells in the CNS with repeat doses of tolebrutinib.
CONCLUSIONS
In this phase I study of tolebrutinib, no safety or tolerability concerns were identified in healthy volunteers receiving single oral doses up to 120 mg and repeated doses up to 90 mg for 10 days. Complete peripheral target BTK occupancy (>95% at 4 h post‐administration) was achieved with single doses of 60 mg and 120 mg. Binding of BTK increased with repeat dosing, and full occupancy could be achieved with doses as low as 15 mg after 10 days. Exposure of tolebrutinib within the CNS was also confirmed, with mean exposure greater than IC90 observed 2 h after a 120 mg dose. Although the 2 h timepoint was selected to align shortly after the observed peripheral Tmax, this may not correlate with maximal concentrations and exposure over time was not evaluated. In this phase I study, tolebrutinib was well‐tolerated and demonstrated high levels of peripheral BTK coverage. These features combined with the potential to directly affect cells expressing BTK within the CNS, support further development in relapsing and progressive forms of MS and other autoimmune and inflammatory indications.
CONFLICT OF INTEREST
T.D.O., Y.X., J.S., D.E.K., M.R.F., C.L.L., P.A.N. and S.G.G. were, or are, employees of Principia Biopharma, a Sanofi Company. P.F.S. and S.H. were, or are employees of Certara, who received financial support from the Sponsor, Principia Biopharma. A.R. declared no competing interests for this work.
AUTHOR CONTRIBUTIONS
T.D.O. and P.F.S. wrote the manuscript. T.D.O., P.F.S., D.E.K., S.G.G., P.A.N., and C.L.L. designed the research.; A.R., M.R.F., Y.X., and J.S. performed the research. All authors analyzed the data.
Supporting information
Figure S1
Table S1
Table S2
Table S3
ACKNOWLEDGEMENTS
The authors acknowledge Timothy Turner for careful review and helpful comments during the preparation of this manuscript.
Owens TD, Smith PF, Redfern A, et al. Phase 1 clinical trial evaluating safety, exposure and pharmacodynamics of BTK inhibitor tolebrutinib (PRN2246, SAR442168). Clin Transl Sci. 2022;15:442–450. doi: 10.1111/cts.13162
Funding information
Principia Biopharma, a Sanofi Company provided funding to Certara and Linear Clinical Research for the conduct of this study.
REFERENCES
- 1. Hauser SL, Bar‐Or A, Comi G, et al. Ocrelizumab versus interferon beta‐1a in relapsing multiple sclerosis. N Engl J Med. 2017;376:221‐234. [DOI] [PubMed] [Google Scholar]
- 2. Montalban X, Hauser SL, Kappos L, et al. Ocrelizumab versus placebo in primary progressive multiple sclerosis. N Engl J Med. 2016;376:209‐220. [DOI] [PubMed] [Google Scholar]
- 3. Hemmer B, Nessler S, Zhou D, Kieseier B, Hartung HP. Immunopathogenesis and immunotherapy of multiple sclerosis. Nat Clin Pract Neurol. 2006;2:201‐211. [DOI] [PubMed] [Google Scholar]
- 4. Stys PK, Zamponi GW, van Minnen J, Geurts JJ. Will the real multiple sclerosis please stand up? Nat Rev Neurosci. 2012;13:507‐514. [DOI] [PubMed] [Google Scholar]
- 5. Sharman JP, Egyed M, Jurczak W, et al. Acalabrutinib with or without obinutuzumab versus chlorambucil and obinutuzumab for treatment‐naive chronic lymphocytic leukaemia (ELEVATE TN): a randomised, controlled, phase 3 trial. Lancet. 2020;395:1278‐1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Burger JA, Barr PM, Robak T, et al. Long‐term efficacy and safety of first‐line ibrutinib treatment for patients with CLL/SLL: 5 years of follow‐up from the phase 3 RESONATE‐2 study. Leukemia. 2020;34:787‐798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Tam CS, Opat S, D’Sa S, et al. A randomized phase 3 trial of zanubrutinib vs ibrutinib in symptomatic Waldenström macroglobulinemia: the ASPEN study. Blood. 2020;136:2038‐2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Montalban X, Arnold DL, Weber MS, et al. Placebo‐controlled trial of an oral BTK inhibitor in multiple sclerosis. N Engl J Med. 2019;380:2406‐2417. [DOI] [PubMed] [Google Scholar]
- 9. Leproult E, Barluenga S, Moras D, Wurtz J‐M, Winssinger N. Cysteine mapping in conformationally distinct kinase nucleotide binding sites: application to the design of selective covalent inhibitors. J Med Chem. 2011;54:1347‐1355. [DOI] [PubMed] [Google Scholar]
- 10. Reich DS, Arnold DL, Vermersch P, et al. Safety and efficacy of tolebrutinib, an oral, central nervous system‐penetrant Bruton’s tyrosine kinase inhibitor, in relapsing multiple sclerosis: a phase 2, randomised, placebo‐controlled trial. Lancet Neurol. 2021;20:729‐738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Smith PF, Krishnarajah J, Nunn PA, et al. A phase I trial of PRN1008, a novel reversible covalent inhibitor of Bruton’s tyrosine kinase, in healthy volunteers. Br J Clin Pharmacol. 2017;83:2367‐2376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Catlett IM, Nowak M, Kundu S, et al. Safety, pharmacokinetics and pharmacodynamics of branebrutinib (BMS‐986195), a covalent, irreversible inhibitor of Bruton’s tyrosine kinase: Randomised phase I, placebo‐controlled trial in healthy participants. Br J Clin Pharmacol. 2020;86:1849‐1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1
Table S1
Table S2
Table S3