

Abstract
Milvexian (BMS‐986177/JNJ‐70033093) is a small molecule, active‐site inhibitor of factor XIa (FXIa) being developed to prevent and treat thrombotic events. The safety, tolerability, pharmacokinetics (PKs), and pharmacodynamics (PDs) of milvexian were assessed in a two‐part, double‐blind, placebo‐controlled, sequential single ascending dose (SAD) and multiple ascending dose (MAD) study in healthy adults. Participants in SAD panels (6 panels of 8 participants; n = 48) were randomized (3:1) to receive milvexian (4, 20, 60, 200, 300, or 500 mg) or placebo. The 200‐ and 500‐mg panels investigated the pharmacokinetic impact of a high‐fat meal. Participants in MAD panels (7 panels of 8 participants; n = 56) were randomized (3:1) to receive milvexian (once‐ or twice‐daily) or placebo for 14 days. All milvexian dosing regimens were safe and well‐tolerated, with only mild treatment‐emergent adverse events and no clinically significant bleeding events. In SAD panels, maximum milvexian plasma concentration occurred 3 h postdose in all fasted panels. The terminal half‐life (T1/2) ranged from 8.3 to 13.8 h. In fasted panels from 20 to 200 mg, absorption was dose‐proportional; results at higher doses (300 and 500 mg) were consistent with saturable absorption. Food increased milvexian bioavailability in a dose‐dependent fashion. In MAD panels, steady‐state milvexian plasma concentration was reached within 3 and 6 dosing days with once‐ and twice‐daily dosing, respectively. Renal excretion was less than 20% in all panels. Prolongation of activated partial thromboplastin time was observed and was directly related to drug exposure. These results suggest that the safety, tolerability, PK, and PD properties of milvexian are suitable for further clinical development.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
Factor XI (FXI) amplifies thrombin generation and has a limited role in hemostasis. Targeted FXI inhibition may reduce the burden of vascular and thromboembolic diseases while preserving hemostasis.
WHAT QUESTION DID THIS STUDY ADDRESS?
This study evaluated the safety/tolerability, pharmacokinetics (PKs), and pharmacodynamics (PDs) of the selective, direct, small molecule FXIa inhibitor milvexian.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
Single and multiple ascending doses of milvexian up to 500 mg were generally safe and well‐tolerated, with no clinically significant bleeding events. Milvexian plasma concentration was dose proportional at doses up to 200 mg q.d. The milvexian half‐life is suitable for q.d. or b.i.d. dosing. Milvexian exhibited low renal excretion and low overall variability in PK and PD parameters.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
These results can inform the future clinical development of milvexian.
INTRODUCTION
Anticoagulant and/or antiplatelet therapies are cornerstones of treatment to reduce the risk of serious adverse thromboembolic events in patients with a broad range of conditions, including nonvalvular atrial fibrillation, or a history of ischemic stroke, acute coronary syndrome, coronary artery disease (CAD), and peripheral artery disease. 1 , 2 , 3 , 4 Although approved therapies are effective for the prevention of thrombosis, an increased risk of major bleeding is a significant concern. 5 Thus, drug discovery efforts have focused on identification of novel therapies with better risk‐benefit profiles compared to current standards of care.
The coagulation cascade is a complex but well‐controlled process that regulates clot formation, preserves tissue perfusion, and stimulates reparative processes. 6 Hemostasis involves the formation of a fibrin‐ and platelet‐rich clot to seal a damaged blood vessel and prevent blood loss. Thrombin plays a key role in the formation of a clot, catalyzing the conversion of fibrinogen into fibrin and activating platelets. 6 Coagulation factor XI (FXI), a zymogen, is activated to FXIa, a protease, by thrombin and factor XIIa. Activation of FXI leads to enhanced thrombin formation, thus forming a positive feedback loop for thrombin formation and consolidation of coagulation. 7 , 8 Although hemostasis is essential to life, pathological blood clotting is the basis of thromboembolic cardiovascular diseases. FXIa plays a role in pathological thrombosis but has a limited role in normal hemostasis. 9
In patients with FXI deficiency, spontaneous bleeding is rare and bleeding risk is poorly correlated with plasma levels of FXI. 10 In most cases, patients with FXI deficiency do not exhibit symptoms or require treatment, and the only clinical manifestation is an increased tendency for mild bleeding after serious injury or surgery. 11 , 12 , 13 Importantly, clinical, preclinical, and epidemiologic studies have shown that FXI deficiency is associated with reduced risk of adverse cardiovascular events and venous thromboembolism. 14 , 15 , 16 , 17 , 18 In contrast, a large, population‐based, case‐control study (the Leiden Thrombophilia Study) found that patients who had suffered a deep vein thrombosis had significantly higher FXI levels compared with healthy controls, with the risk of thromboembolism doubled for individuals with FXI levels above the 90th population percentile compared with those who had lower values. 19 Furthermore, studies in patients with a history of or high risk for cardiovascular disease have shown that high levels of FXI are associated with an increased risk of stroke, transient ischemic attack, myocardial infarction, and severe CAD, suggesting that high levels of FXI contribute to an increased risk of thrombosis. 20 , 21 , 22 Preclinical models and clinical studies suggest that inhibitors of FXIa reduce thrombus formation and that the FXI amplification pathway is not critical for normal hemostasis. 23 , 24 , 25 , 26 Taken together, these results support the hypothesis that targeted inhibition of FXIa represents a novel mechanism for systemic anticoagulation with a potentially improved benefit‐risk profile that may be achieved by reducing the burden of vascular and thromboembolic diseases while preserving hemostasis. 17 , 18 , 20 , 27
Milvexian (formerly referred to as BMS‐986177/JNJ‐70033093) is a small molecule, active‐site inhibitor of FXIa with high affinity and selectivity for FXIa and oral bioavailability in preclinical species. 28 In preclinical models of arterial and venous thrombosis, milvexian has demonstrated antithrombotic activity while preserving homeostasis. 29 , 30 These findings supported the initiation of an extensive clinical development program that included this first‐in‐human study of milvexian in healthy participants. Currently, phase II studies of milvexian are underway for the secondary prevention of major cardiovascular events in patients with acute ischemic stroke and for the prevention of total venous thromboembolism events in patients undergoing total knee replacement surgery. 31 , 32
Investigations of the safety, tolerability, pharmacokinetic (PK), and pharmacodynamic (PD) properties of milvexian are essential components to support further clinical development efforts. The primary objective of the current study was to assess the safety and tolerability of single ascending doses (SADs) and multiple ascending doses (MADs) of milvexian in healthy participants. Secondary objectives included assessment of PK and dose proportionality of escalating doses of milvexian, and assessment of PD effects, including activated partial thromboplastin time (aPTT) and FXI clotting activity.
METHODS
This study was conducted in accordance with the ethical principles that have their origin in the Declaration of Helsinki and the International Council for Harmonization of Technical Requirements for Pharmaceuticals for Human Use Note for Guidance on Good Clinical Practice (CPMP/ICH/135/95) and with the ethical principles underlying European Union Directive 2001/20/EC and the US Code of Federal Regulations, Title 21, Part 50 (21CFR50). The study was registered with ClinicalTrials.gov (NCT02608970). The study protocol and all amendments were reviewed and approved by an independent ethics committee and/or institutional review board according to specifications outlined in applicable regulations. Prior to beginning the study, all participants provided written informed consent. The study was conducted at one clinical center (SGS Life Sciences, Antwerp, Belgium) from November 25, 2015, to July 23, 2017.
Study design
This was a two‐part, randomized, double‐blind, placebo‐controlled, sequential, SAD and MAD study of milvexian in healthy volunteers (Figure 1). Both parts of the study included a screening period of up to 21 days.
FIGURE 1.
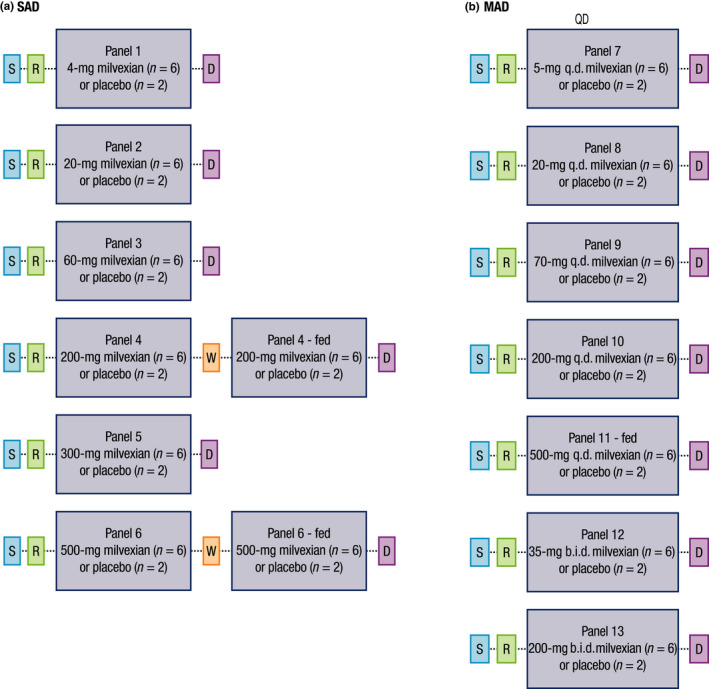
Study design. SAD, single ascending dose; S, screening; R, randomization; W, washout; D, discharge; MAD, multiple ascending dose
SAD
The SAD analyses included six sequential ascending dose panels that evaluated milvexian doses of 4, 20, 60, 200, 300, and 500 mg (panels 1, 2, 3, 4, 5, and 6, respectively). Each panel included eight participants who were randomized according to a computer‐generated randomization scheme prepared by a randomization coordinator in a 3:1 ratio to receive milvexian or matched placebo. Participants were dosed by investigators on day 1 after fasting for ~ 10 h and remained fasted on day 1 for 4 h postdose.
Dose escalation occurred only after a review of PK analyses from the prior dose panels and a review of the safety and tolerability data up to 48 h postdose for at least six participants from the preceding dose panel were performed and the preceding dose was deemed safe by the investigator and the sponsor’s medical monitor. Dose escalation to panels 5 and 6 occurred after the PK data from at least the first two panels had been assessed and deemed sufficient to model exposure for panels 5 and 6.
Participants in panels 4 and 6 participated in a food effect arm. Following SAD dosing on day 1 and at least 2 (panel 4) or 3 (panel 6) days of washout (period 1), participants entered period 2. In period 2, participants completed a standard high‐fat breakfast and then received the same dose of milvexian or matching placebo that they received in period 1 (30 min after the start of the high‐fat breakfast in panel 6).
MAD
The MAD analyses included five sequential ascending dose panels that evaluated milvexian doses of 5, 20, 70, 200, and 500 mg (panels 7, 8, 9, 10, and 11, respectively) given q.d. The MAD analyses included two additional sequential ascending dose panels that evaluated milvexian doses of 35 and 200 mg given b.i.d. (panels 12 and 13, respectively). Each panel included eight participants who were randomized in a 3:1 ratio to receive milvexian or placebo for 14 days. Participants began daily dosing on day 1 and fasted overnight for ~ 10 h every night prior to dosing, except in panel 11. In panel 11, breakfast was provided 30 min prior to dosing; participants received a high‐fat breakfast on days 1, 2, 3, 13, and 14 and a standard breakfast on other dosing days. In the b.i.d. dosing panels, the second dose of study drug was administered ~ 12 h after the morning dose.
Dosing in MAD panels occurred after the safety and tolerability data from at least the first three SAD panels were assessed and deemed safe. Additionally, MAD panels did not start until after safety data from SAD panels were available for a dose at least three‐fold greater than the starting dose in the MAD panels and until PK data were available from at least the first two SAD panels to model exposures in MAD panels. Dose escalation in MAD panels occurred only after safety and tolerability data through day 17 (day 16 for panel 7) of the preceding dose panel were assessed for at least six participants and deemed safe by the study investigator and the sponsor’s medical monitor.
Participants
Eligible participants included men and women of nonchildbearing potential, 18 to 55 years old with a body mass index (BMI) of 18 to 30 kg/m2, who were healthy as determined by medical history, physical examination, electrocardiogram (ECG), and clinical laboratory evaluations. Participants were excluded if they had significant acute or chronic medical illness; evidence of coagulopathy, prolonged or unexplained clinically significant bleeding, or frequent unexplained bruising or thrombus formation; current or recent gastrointestinal disease that could increase the risk of gastrointestinal bleeding or interfere with absorption of study drug (e.g., peptic or gastric ulcer disease, severe gastritis, or history of gastrointestinal surgery); any major surgery within 12 weeks of study drug administration; history of significant head injury within the last 2 years; history of keloids or other excessive scar formation; donation of blood within 4 weeks (or 2 weeks for plasma only) of study drug administration; blood transfusion within 3 months of study drug administration; smoking more than 10 cigarettes per day; or recent (within 6 months) drug or alcohol abuse.
Participants were prohibited from taking nonsteroidal anti‐inflammatory drugs, aspirin or other antiplatelet agents, anticoagulants, fish oil capsules, and ginkgo within 2 weeks of study drug administration. Use of prescription or over‐the‐counter acid controllers was prohibited within 4 weeks of study drug administration. Use of all other drugs, including strong CYP3A4 inhibitors or activators, over‐the‐counter medications, vitamins, iron supplements, nutritional supplements, and herbal preparations was prohibited within 2 weeks of study drug administration. Use of oral, injectable, or implantable hormonal contraceptive agents was prohibited within 3 months of study drug administration.
Study assessments
Safety
Safety and tolerability were evaluated based on a medical review of adverse event (AE) reports and the results of clinical laboratory tests, vital sign measurements, ECGs, and physical examinations up to 48 h postdose. Events of special interest included clinically and nonclinically significant bleeding. AEs were coded according to the Medical Dictionary for Regulatory Activities (MedDRA), version 20.0.
Pharmacokinetic and pharmacodynamic assessments
Plasma samples collected in all panels were analyzed for milvexian using a validated liquid chromatography/mass spectrometry (LC/MS) assay at LabConnect (Johnson City, TN, USA). Urine samples were collected in SAD panels 4 to 6 and in MAD panels 9, 10, and 12 to measure renal excretion of milvexian and were analyzed using a qualified LC/MS assay at LabConnect. PD parameters aPTT, prothrombin time, and FXI clotting activity were measured with validated assays at LabCorp Colorado Coagulation (Englewood, CO, USA). Sample collection details are provided in Tables [Link], [Link], [Link].
The PK properties of milvexian were derived from plasma concentration versus time and urinary excretion data. Assessed parameters included maximum observed plasma concentration (Cmax), area under the plasma concentration‐time curve from time zero to the time of last quantifiable concentration (AUC[0‐T]; SAD only), AUC from time 0 extrapolated to infinity (AUC[INF]; SAD only), AUC in one dosing interval (AUC[TAU]; MAD only), terminal plasma half‐life (T1/2), time of maximum observed plasma concentration (Tmax), and amounts of milvexian excreted in urine. Milvexian accumulation indices (ratios of AUC[TAU] and Cmax at steady‐state to after the first dose, respectively) were measured in MAD panels.
Statistical analyses
Safety was assessed for all treated participants who received at least one dose of study drug. The PK population was defined as all participants who received milvexian and had any available concentration‐time data. The evaluable PK population was defined as all participants who had adequate PK profiles. The PD population was defined as all participants who received any study medication and had PD biomarker data available.
Study sample size determination was not based on statistical power considerations. However, administration of milvexian to six participants in each panel was expected to provide ~ 80% probability of observing at least one occurrence of any AE that would occur with ~ 24% incidence in the population from which the sample was drawn.
Safety outcomes, including AEs, serious AEs (SAEs), deaths, AEs leading to discontinuation of study therapy, and AEs of clinically significant bleeding were summarized descriptively for all treated participants. AEs starting within 48 h postdose were considered treatment‐emergent AEs (TEAEs). Values for ECG parameters, vital signs, and clinical laboratory tests that were outside prespecified criteria were listed and summarized.
All plasma and urine milvexian PK data were summarized by part, panel, and period (where applicable), and visit day/time using descriptive statistics. Dose proportionality of a single dose of milvexian was assessed by estimating the slope of the linear regression of Log(AUC[INF]), Log(AUC[0‐T]), and Log(Cmax) on Log(Dose) for data from SAD panels 1 to 6 under fasted conditions using a power model. 33 Similarly, dose proportionality of multiple doses of milvexian was assessed by estimating the slope of the linear regression of Log(AUC[TAU]) and Log(Cmax) on Log(Dose) using data from the last dose (day 14) from MAD panels.
To assess the effect of food on the single‐dose PK of milvexian in panels 4 and 6, linear mixed model analyses were performed on Log(Cmax), Log(AUC[0‐T]), and Log(AUC[INF]) of milvexian, with treatment (fed or fasted) as a fixed effect and measurements within subject as repeated measures.
All statistical calculations were performed by SGS LS using the SAS (SAS Institute Inc., Cary, NC, USA; version 9.2 or higher) and/or Phoenix WinNonlin (Pharsight Corporation, Palo Alto, CA, USA; version 6.2 or higher) software.
RESULTS
Participants
A total of 104 participants were randomized and treated. Forty‐eight of these participants were treated in SAD panels (8 participants in each of 6 panels), and 56 participants were treated in MAD panels (8 participants in each of 7 panels). All 48 participants completed the SAD panels, and 47 of 56 participants completed the MAD panels; one participant in panel 7 discontinued treatment because of an AE (macular rash) after receiving one dose of milvexian 5 mg and all eight participants in panel 12 (who were to receive milvexian 35 mg b.i.d. or matched placebo) discontinued the study because of certification issues at the site of study drug manufacture. This resulted in incomplete PD data for participants in panel 12. Therefore, PD data from panel 12 is not included in these figures. Baseline demographic characteristics of all randomized and treated participants are summarized in Table 1.
TABLE 1.
Baseline demographic characteristics
| SAD | Pooled placeboa fasted (n = 8) | Pooled placebob fasted/fed (n = 4) | Panel 1 milvexian 4 mg fasted (n = 6) | Panel 2 milvexian 20 mg fasted (n = 6) | Panel 3 milvexian 60 mg fasted (n = 6) | Panel 4 milvexian 200 mg fasted/fed (n = 6) | Panel 5 milvexian 300 mg fasted (n = 6) | Panel 6 milvexian 500 mg fasted/fed (n = 6) | All participants (N = 48) |
|---|---|---|---|---|---|---|---|---|---|
| Sex, n (%) | |||||||||
| Female | 2 (25.0) | 0 | 1 (16.7) | 1 (16.7) | 1 (16.7) | 0 | 1 (16.7) | 1 (16.7) | 7 (14.6) |
| Male | 6 (75.0) | 4 (100) | 5 (83.3) | 5 (83.3) | 5 (83.3) | 6 (100) | 5 (83.3) | 5 (83.3) | 41 (85.4) |
| Age, y | |||||||||
| Median (range) | 42.0 (33–54) | 33.5 (29–54) | 34.0 (22–50) | 41.5 (33–54) | 47.5 (32–53) | 53.0 (25–55) | 45.5 (35–52) | 44.5 (39–55) | 43.5 (22–55) |
| Race, n (%) | |||||||||
| White | 8 (100) | 3 (75.0) | 6 (100) | 6 (100) | 5 (83.3) | 6 (100) | 6 (100) | 6 (100) | 46 (95.8) |
| American Indian or Alaska Native | 0 | 1 (25.0) | 0 | 0 | 0 | 0 | 0 | 0 | 1 (2.1) |
| Asian | 0 | 0 | 0 | 0 | 1 (16.7) | 0 | 0 | 0 | 1 (2.1) |
| BMI, kg/m2 | |||||||||
| Median (range) | 24.5 (23.0–27.1) | 23.4 (21.6–28.1) | 24.7 (21.6–26.2) | 23.6 (21.9–27.4) | 26.9 (24.4–29.4) | 26.4 (22.6–29.0) | 25.1 (22.2–28.6) | 24.7 (24.0–26.5) | 24.6 (21.6–29.4) |
| MAD | Pooled placebo (n = 14) | Panel 7 milvexian 5 mg q.d. fasted (n = 6) | Panel 8 milvexian 20 mg q.d. fasted (n = 6) | Panel 9 milvexian 70 mg q.d. fasted (n = 6) | Panel 12 milvexian 35 mg b.i.d. fasted (n = 6) | Panel 10 milvexian 200 mg q.d. fasted (n = 6) | Panel 13 milvexian 200 mg b.i.d. fasted (n = 6) | Panel 11 milvexian 500 mg q.d. fed (n = 6) | All participants (N = 56) |
|---|---|---|---|---|---|---|---|---|---|
| Sex, n (%) | |||||||||
| Female | 1 (7.1) | 0 | 0 | 1 (16.7) | 0 | 2 (33.3) | 2 (33.3) | 0 | 6 (10.7) |
| Male | 13 (92.9) | 6 (100) | 6 (100) | 5 (83.3) | 6 (100) | 4 (66.7) | 4 (66.7) | 6 (100) | 50 (89.3) |
| Age, y | |||||||||
| Median (range) | 38.0 (21–54) | 44.0 (38–52) | 52.0 (35–53) | 32.0 (28–51) | 47.0 (28–53) | 45.0 (31–54) | 49.5 (32–55) | 41.5 (24–53) | 44.5 (21–55) |
| Race, n (%) | |||||||||
| White | 14 (100) | 5 (83.3) | 4 (66.7) | 6 (100) | 5 (83.3) | 6 (100) | 6 (100) | 6 (100) | 52 (92.9) |
| Black or African American | 0 | 1 (16.7) | 0 | 0 | 1 (16.7) | 0 | 0 | 0 | 2 (3.6) |
| Asian | 0 | 0 | 2 (33.3) | 0 | 0 | 0 | 0 | 0 | 2 (3.6) |
| BMI, kg/m2 | |||||||||
| Median (range) | 24.4 (22.2–28.7) | 23.5 (21.7–28.0) | 24.1 (19.8–27.5) | 23.2 (20.1–28.9) | 25.2 (21.0–29.0) | 24.5 (20.0–27.9) | 26.9 (23.7–29.3) | 24.6 (22.8–28.2) | 24.3 (19.8–29.3) |
Abbreviations: BMI, body mass index; MAD, multiple ascending dose; SAD, single ascending dose.
Panels 1, 2, 3, and 5.
Panels 4 and 6.
Safety and tolerability
Administration of SADs of milvexian up to 500 mg and of MADs of milvexian up to 200 mg b.i.d. or 500 mg q.d. were generally safe and well‐tolerated. No deaths or other SAEs occurred during the study. All TEAEs were mild in severity.
Among fasted participants in SAD panels, one participant (8.3%) who received placebo and three participants (8.3%) who received milvexian reported TEAEs. These TEAEs included back pain (n = 1, placebo), headache (n = 2, milvexian 300 mg), and epistaxis (n = 1, milvexian 60 mg). In SAD panels 4 and 6, comparing fed versus fasting conditions, one participant (16.7%) in the milvexian 500‐mg group reported a TEAE of nausea after a high‐fat breakfast, and one participant in the placebo group (25.0%) reported a TEAE of back pain after fasting for 10 h. In MAD panels, TEAEs were reported for eight participants (57.1%) who received placebo and 21 participants (50.0%) who received milvexian (Table 2). The most frequently reported TEAEs in the MAD panels were headache (placebo, 7.1% and milvexian, 14.3%) and diarrhea (placebo, 21.4% and milvexian, 11.9%).
TABLE 2.
TEAEs in MAD panels reported in greater than one participant treated with milvexian
| AE, n (%) | Pooled placebo (n = 14) | Panel 7 milvexian 5 mg q.d. fasted (n = 6) | Panel 8 milvexian 20 mg q.d. fasted (n = 6) | Panel 9 milvexian 70 mg q.d. fasted (n = 6) | Panel 12 milvexian 35 mg b.i.d. fasted (n = 6) | Panel 10 milvexian 200 mg q.d. fasted (n = 6) | Panel 13 milvexian 200 mg b.i.d. fasted (n = 6) | Panel 11 milvexian 500 mg q.d. fed (n = 6) | All participants treated with milvexian (N = 42) |
|---|---|---|---|---|---|---|---|---|---|
| Any TEAE | 8 (57.1) | 4 (66.7) | 1 (16.7) | 5 (83.3) | 2 (33.3) | 4 (66.7) | 2 (33.3) | 3 (50.0) | 21 (50.0) |
| Headache | 1 (7.1) | 2 (33.3) | 1 (16.7) | 2 (33.3) | 0 | 1 (16.7) | 0 | 0 | 6 (14.3) |
| Diarrhea | 3 (21.4) | 1 (16.7) | 0 | 3 (50.0) | 0 | 1 (16.7) | 0 | 0 | 5 (11.9) |
| Dizziness | 1 (7.1) | 0 | 0 | 1 (16.7) | 1 (16.7) | 0 | 1 (16.7) | 0 | 3 (7.1) |
| Nausea | 1 (7.1) | 0 | 0 | 1 (16.7) | 0 | 2 (33.3) | 0 | 0 | 3 (7.1) |
| Abdominal discomfort | 0 | 1 (16.7) | 0 | 1 (16.7) | 0 | 0 | 0 | 0 | 2 (4.8) |
| Abdominal pain | 1 (7.1) | 0 | 0 | 1 (16.7) | 0 | 1 (16.7) | 0 | 0 | 2 (4.8) |
| Oropharyngeal pain | 0 | 0 | 0 | 0 | 0 | 1 (16.7) | 1 (16.7) | 0 | 2 (4.8) |
| Pain in extremity | 0 | 0 | 0 | 1 (16.7) | 0 | 1 (16.7) | 0 | 0 | 2 (4.8) |
Abbreviations: AE, adverse event; MAD, multiple ascending dose; TEAE, treatment‐emergent adverse event.
Bleeding events reported in the study included epistaxis, gingival bleeding, and petechia. Epistaxis was reported in one participant in the milvexian 60‐mg SAD group and one participant in the milvexian 5‐mg q.d. MAD group. Gingival bleeding was reported in one participant in the milvexian 5‐mg q.d. MAD group. Petechia was reported in one participant in the milvexian 70‐mg q.d. MAD group. All bleeding events were mild in severity, not serious, and did not lead to discontinuation of treatment, and the gingival bleeding and petechial events were considered to be treatment‐related. A decrease in platelet count from 191 × 109/L at baseline (predose day 1) to 81 × 109/L predose on day 2 was reported in one participant in the milvexian 35‐mg b.i.d. group. This event was considered to be treatment‐related, mild in severity, not serious, and did not lead to discontinuation of treatment (at the next sampling time 2 days later, the participant’s platelet count was within normal limits at 184 × 109/L).
Regarding the incidence of laboratory and vital sign abnormalities, no relevant differences were observed between the placebo and milvexian groups, and no clear milvexian dose effect was observed. There were no clinically significant ECG abnormalities.
Pharmacokinetics
SAD
Mean milvexian plasma concentration‐time profiles in fasted and fasted/fed panels are shown in Figure 2a, and mean PK parameters are summarized in Table 3. After single oral doses of milvexian ranging from 4 to 500 mg in a fasted state, median Tmax occurred at 3 h postdose in all panels, indicating a similar rate of absorption. T1/2 ranged from 8.3 to 13.8 h across all SAD panels. Under fasted conditions, Cmax and exposure appeared generally dose‐proportional at exposures ranging from 20 to 200 mg, whereas exposures consistent with saturable absorption behavior were observed at the higher doses of 300 and 500 mg.
FIGURE 2.
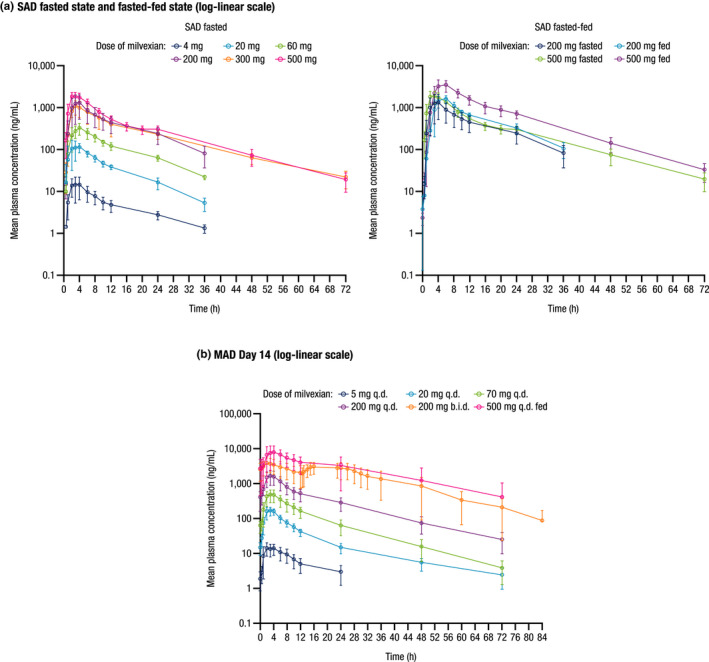
Mean (±SD) milvexian plasma concentration profiles over time in the (a) SAD and (b) MAD panels. SD, standard deviation; SAD, single ascending dose; MAD, multiple ascending dose. Mean profiles are presented on a log‐linear scale
TABLE 3.
SAD PK parameters a
| Panel 1 milvexian 4 mg fasted (n = 6) | Panel 2 milvexian 20 mg fasted (n = 6) | Panel 3 milvexian 60 mg fasted (n = 6) | Panel 4 milvexian 200 mg fasted (n = 6) | Panel 4 milvexian 200 mg fed (n = 6) | Panel 5 milvexian 300 mg fasted (n = 6) | Panel 6 milvexian 500 mg fasted (n = 6) | Panel 6 milvexian 500 mg fed (n = 6) | Dose proportionality, slope (90% CI)b | |
|---|---|---|---|---|---|---|---|---|---|
| Cmax, ng/ml | 13.3 (68.6) | 126 (36.7) | 337 (23.6) | 1068 (98.6) | 1639 (20.9) | 1017 (34.1) | 1853 (32.0) | 3359 (39.6) | 0.9886 (0.8990, 1.0783) |
| AUC(0‐T), ng·h/ml | 125 (67.7) | 1220 (20.3) | 3793 (13.7) | 12,471 (68.0) | 17,811 (19.4) | 14,588 (29.9) | 20,991 (26.8) | 44,330 (25.0) | 1.0536 (0.9777, 1.1295) |
| AUC(INF), ng·h/ml | 210 (22.7)c | 1284 (20.2) | 4104 (11.9) | 13,759 (61.8) | 19,106 (22.0) | 15,041 (29.1) | 21,326 (26.7) | 44,796 (25.3) | 0.9491 (0.8811, 1.0170) |
| T1/2, h | 10.3 (25.7) | 8.3 (17.0) | 9.9 (13.7) | 10.5 (28.8) | 9.0 (16.1) | 13.8 (21.0) | 12.2 (14.4) | 10.7 (12.1) | N/A |
| Tmax, h | 3.00 (2.00–4.00) | 3.00 (1.00–4.00) | 3.00 (2.00–4.00) | 3.00 (2.00–4.00) | 4.00 (3.00–6.00) | 3.00 (2.00–4.00) | 3.00 (2.00–4.00) | 4.00 (4.00–6.00) | N/A |
Abbreviations: AUC, area under the plasma concentration‐time curve; AUC(0‐T), area under the plasma concentration‐time curve from time zero to the time of last quantifiable concentration; AUC(INF), area under the plasma concentration‐time curve from time 0 extrapolated to infinity; CI, confidence interval; Cmax, maximum observed plasma concentration; CV%, coefficient of variation; max, maximum; min, minimum; N/A, not applicable; PK, pharmacokinetic; SAD, single ascending dose; T1/2, terminal phase half‐life; Tmax, time of maximum observed plasma concentration.
The table provides descriptive statistics of milvexian PK parameters. Values are geometric mean (geometric CV%) for Cmax and AUCs, arithmetic means (CV%) for T1/2, and median (min‐max) for Tmax.
Dose proportionality was assessed by estimating the slope of the linear regression of Log(Cmax), Log(AUC[0‐T]), and Log(AUC[INF]) on Log(dose) using the data from panels 1 to 6 under fasted conditions.
n = 3.
Under fed conditions, milvexian median Tmax was 4 h at doses of 200 and 500 mg (panels 4 and 6). Elimination rate was increased when milvexian was administered with food (mean T1/2 shortened by 1.5 h). Food intake increased the bioavailability of milvexian by ~ 1.4‐fold in the 200‐mg panel and by ~ 2‐fold in the 500‐mg panel.
Urinalysis results from samples collected in SAD panels 4 (milvexian 200 mg fasted/fed), 5 (milvexian 300 mg fasted), and 6 (milvexian 500 mg fasted/fed) showed that the mean total amount of milvexian excreted from 0 to 24 h postdose ranged from 6.9% to 17.8% of the administered dose. Urinary excretion was lower in the fasted state (6.9%–12.3% of the administered dose) than in the fed state (16.3%–17.8% of the administered dose), likely due to the higher observed bioavailability in the fed state.
MAD
Mean milvexian plasma concentration‐time profiles on day 14 of the MAD analysis are shown in Figure 2b, and mean PK parameters are summarized in Table 4. Based on visual inspection of trough plasma concentration profiles, a steady‐state concentration of milvexian was reached after 1 dosing day (i.e., on day 2) for the 5‐mg q.d. panel, after 3 dosing days (i.e., on day 4) in the 20‐, 70‐, 200‐, and 500‐mg q.d. dosing panels, and after 6 dosing days (i.e., on day 7) for the 200‐mg b.i.d. panel (data not shown). Mean T1/2 ranged from 11.4 to 18.1 h and was not correlated with dose level or dosing regimen. On day 14, increases in exposure were greater than dose‐proportional over the dose range studied (5–500 mg). Specifically, Cmax increased by 513‐fold, and AUC(TAU) increased 651‐fold as doses increased 100‐fold. As with the SAD panels, between doses of 20 and 200 mg in the fasted state, a generally dose‐proportional relationship was observed.
TABLE 4.
MAD PK Parameters a
| Panel 7 milvexian 5 mg q.d. fasted (n = 6) | Panel 8 milvexian 20 mg q.d. fasted (n = 6) | Panel 9 milvexian 70 mg q.d. fasted (n = 6) | Panel 10 milvexian 200 mg q.d. fasted (n = 6) | Panel 11 milvexian 500 mg q.d. fed (n = 6) | Panel 12 milvexian 35 mg b.i.d. fastedb (n = 6) | Panel 13 milvexian 200 mg b.i.d. fasted (n = 6) | Dose effect, PE (90% CI)c | |||
|---|---|---|---|---|---|---|---|---|---|---|
| AM | PM | AM | PM | |||||||
| Cmax, ng/ml | 14.8 (37.7)d | 171 (23.1) | 434 (57.2) d | 1512 (55.0) | 7595 (45.7) | 199 (29.1) | 159 (25.3) | 3579 (38.5) | 3001 (29.0) | 12,943 (1.2013, 1.3872) |
| AUC(0‐T), ng·h/ml | 146 (47.6)d | 1662 (22.3) | 5059 (48.5)c | 18,868 (51.0) | 142,452 (67.7) | 1362 (25.4) | 1354 (25.6) | 30,597 (41.2) | 69,778 (49.1) | N/A |
| AUC(TAU), ng·h/ml | 146 (47.6)d | 1373 (22.8) | 4224 (49.0)d | 14,634 (50.2) | 95,110 (51.9) | NE | NE | 30,734 (41.2) | 29,780 (35.4) | 13,475 (1.2555, 14,396) |
| T1/2, h | NE | 18.1 (61.7) | 11.4 (23.4)d | 13.2 (12.2) | 13.5 (30.9) | NE | NE | NE | 11.8 (11.8) | N/A |
| Tmax, h | 2.03 (1.00–4.00)d | 2.50 (2.00–4.00) | 3.00 (2.00–4.00)d | 3.50 (2.00–4.00) | 4.00 (2.00–6.00) | 3.00 (2.00–3.00) | 6.50 (3.00–10.00) | 2.00 (2.00–4.00) | 4.03 (3.00–12.00) | N/A |
| AI_AUC | 1.07 (12.1)d | 1.14 (18.8) | 1.06 (27.9)d | 1.50 (53.9) | 1.75 (28.8) | N/A | N/A | N/A | 3.99 (49.4) | N/A |
| AI_Cmax | 1.05 (15.3)d | 1.17 (18.3) | 0.93 (30.3)d | 1.27 (59.7) | 1.41 (16.1) | N/A | N/A | N/A | 2.68 (55.0) | N/A |
Abbreviations: AI, accumulation index; AI_AUC, area under the plasma concentration‐time curve accumulation index; AI_Cmax, area under the plasma concentration‐time curve Cmax accumulation index; AUC, area under the plasma concentration‐time curve; AUC(0‐T), area under the plasma concentration‐time curve from time zero to the time of last quantifiable concentration; AUC(TAU), area under the plasma concentration‐time curve in one dosing interval; Cmax, maximum observed plasma concentration; CV%, coefficient of variation; MAD, multiple ascending dose; max, maximum; min, minimum; N/A, not applicable; NE, not estimated; PE, point estimate; PK, pharmacokinetic; T1/2, terminal phase half‐life; Tmax, time of maximum observed plasma concentration.
The table provides descriptive statistics of milvexian PK parameters. Values are geometric mean (geometric CV%) for Cmax, AUCs, and AIs, arithmetic means (CV%) for T1/2, and median (min‐max) for Tmax.
Day 1.
Dose effect was assessed by estimating the slope of the linear regression of Log(Cmax) and Log(AUC[TAU]) on Log(dose) using the data after the last dose on day 14. All q.d. panels and the 200 mg b.i.d. panel were included.
n = 5.
No accumulation of milvexian was observed for the three lowest q.d. dosing panels (5, 20, and 70 mg; Cmax and AUC[TAU] accumulation ratios of 0.93–1.17). Accumulation ratios were higher at doses of 200 and 500 mg q.d. (1.27–1.75 for both Cmax and AUC[TAU]) and 200 mg b.i.d. (3.99 for AUC[TAU] and 2.68 for Cmax; Table 4).
Urinalysis results from samples collected in panel 9 (milvexian 70 mg q.d.) and panel 10 (milvexian 200 mg q.d.) showed that the mean total amounts of milvexian excreted from 0 to 24 h postdose appeared higher on day 14 than on day 1 (17.3% vs. 14.8% of the administered dose for the 70‐mg q.d. panel, and 14.1% vs. 10.4% of the administered dose for the 200‐mg q.d. panel, respectively).
Pharmacodynamics
Mean baseline aPTT values ranged from 31.8 to 36.3 s. After SADs and MADs of milvexian, there was a clear trend that aPTT was prolonged, and the magnitude of change was related to drug exposure (Figure S1a; see Figure 2 for drug exposure). Generally, timing for the maximum prolongation of aPTT for each participant coincided with Tmax, with 11.9%, 36.7%, 85.2%, 147.1%, 140.8%, and 188.3% maximal mean percent increase from baseline in aPTT for fasted SAD panels 4 mg, 20 mg, 60 mg, 200 mg, 300 mg, and 500 mg, respectively. The aPTT profiles over time during MAD panels are shown in Figure 3. Prothrombin time was not increased at any time (data not shown).
FIGURE 3.
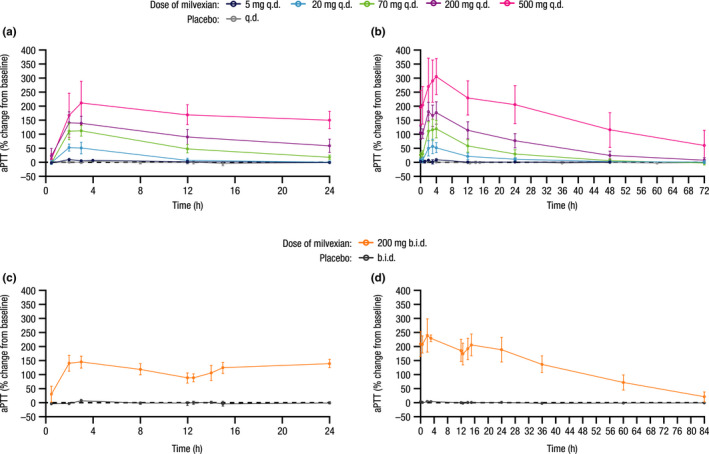
Mean (±SD) percent change from baseline in aPTT profiles over time for MAD panels at (a) day 1 q.d., (b) day 14 q.d., (c) day 1 b.i.d., and (d) day 14 b.i.d. SD, standard deviation; aPTT, activated partial thromboplastin time; MAD, multiple ascending dose. Data from participants dosed in the discontinued panel (panel 12, milvexian 35 mg b.i.d.) are not shown
Mean baseline FXI clotting activity ranged from 101.8% to 117.7%. Milvexian reduced FXI clotting activity, and the magnitude of the reduction was related to drug exposure (Figure S1b). The maximal observed effect occurred at 3 to 4 h postdose, which was generally consistent with Tmax, with –5.8%, –14.5%, –29.7%, –65.0%, –63.2%, and –77.4% maximal mean percent decrease from baseline in clotting activity for fasted SAD panels 4 mg, 20 mg, 60 mg, 200 mg, 300 mg, and 500 mg, respectively. FXI clotting activity profiles over time during MAD panels is shown in Figure S2.
DISCUSSION
The current study evaluated the safety, tolerability, PK, and PD of milvexian, one of the first oral FXIa inhibitors to be investigated in clinical trials. This novel small molecule was designed using innovative chemistry to achieve high affinity and selectivity for FXIa versus related proteases and to optimize its pharmaceutical properties. 28 The oral route of administration and once‐ or twice‐daily dosing may be preferable and more convenient for patients compared with injectable agents. The therapeutic potential of targeted FXIa inhibition to prevent arterial and venous thrombosis coupled with an anticipated improved bleeding risk profile compared with current standards of care support investigation of milvexian in several indications.
Results of this first‐in‐human study demonstrated that milvexian was safe and well‐tolerated in healthy volunteers following administration of SADs of 4 to 500 mg and MADs of up to 200 mg b.i.d. and 500 mg q.d. All observed TEAEs were mild in intensity, and there were no clinically significant bleeding events. There were no relevant differences between the placebo and milvexian groups in the overall incidence of TEAEs, and the milvexian safety profile was similar in fasted and fed participants.
The PK profile of milvexian showed rapid absorption (Tmax of 2–4 h), and an observed terminal elimination half‐life ranging from 8.3 to 13.8 h across SAD panels and from 11.4 to 18.1 h across MAD panels, supporting b.i.d. or q.d. dosing. Following administration of single oral doses of 4 to 500 mg, the rate of milvexian absorption was similar across all doses studied. Food intake modestly slowed the rate of absorption, increased the rate of elimination, and increased the bioavailability of milvexian. Based on preclinical studies, which have been conducted but are yet to be published, the primary excretion pathways are based on hepatic metabolism via cytochrome P450 enzymes. In general, exposure parameters demonstrated moderate to low variability.
In the MAD panels, increases in exposure (Cmax and AUC) were greater than dose proportional. This observation was likely due to a combination of formulation (i.e., spray dried dispersion) and food effects at doses greater than or equal to 200 mg. No accumulation of milvexian was observed with doses from 5 to 70 mg q.d., but accumulation was observed at higher doses (200 and 500 mg q.d. and 200 mg b.i.d.). Given the increasing bioavailability at doses of 200 and 500 mg, food would likely need to be administered to attain higher concentration targets in a predictable manner using a q.d. regimen. However, based on the observation that there was a greater than dose proportional increase at the 200 mg b.i.d. dose, this may represent a residual food effect due to enterohepatic recycling that consistently increases the bioavailability of the drug on multiple dosing. Therefore, in the case of b.i.d. dosing, co‐administration of milvexian with food may not further increase exposures at doses above 200 mg.
After SADs and MADs, there was a clear trend that aPTT was prolonged by milvexian administration. In MAD panels, the maximal PD effect was observed between 2 and 4 h postdose. Over time, the magnitude of aPTT prolongation decreased with decreasing milvexian plasma concentration. Consistent with the aPTT results, FXI clotting activity showed an exposure‐dependent decrease in activity, with the maximal effect observed between 3 and 4 h postdose. When the milvexian plasma concentration levels decreased, the magnitude of decrease in FXI clotting activity was also reduced. Based on internal preclinical models (unpublished), which derived target trough concentrations in addition to assessment of the aPTT and FXI clotting activity ratio, a minimum dose associated with milvexian activity could possibly be in the range of 10 mg and above. Overall, these observations are consistent with milvexian being a direct and reversible FXIa inhibitor.
The current study was limited by the small sample sizes in each panel. Furthermore, the PK parameters are primarily derived from male participants and thus may not fully describe variability resulting from sex differences. However, the two‐part sequential SAD and MAD design allowed for the stepwise evaluation of a broad range of milvexian doses. The safety, PK, and PD results of this study were used to inform dosing decisions for further phase I investigation in addition to subsequent dosing decisions in phase II clinical trials that are currently ongoing.
In conclusion, the results of this first‐in‐human study showed that milvexian was generally safe and well‐tolerated in healthy volunteers. The PK and PD profiles of milvexian, including dose‐proportional plasma concentration from 20 to 200 mg, a half‐life suitable for q.d. or b.i.d. dosing, low renal excretion, and low variability in PK and PD parameters, demonstrate that it is suitable for further clinical development.
CONFLICTS OF INTEREST
All authors were employees of Bristol Myers Squibb (BMS), Princeton, NJ, USA, at the time of the study. All authors are stockholders of BMS.
AUTHOR CONTRIBUTIONS
V.P., Z.W., J.L., D.L., M.D., M.C., and D.S. wrote the manuscript, designed the research, and analyzed the data. Research was performed by Robert Lins, MD, PhD, of SGS Life Sciences, a contract research organization.
Supporting information
Fig S1
Fig S2
Table S1
Table S2
Table S3
ACKNOWLEDGEMENTS
The authors thank Jian Wang for his contributions to the design of the study and interpretation of the results and Robert Lins, MD, PhD, for his contributions to performing the research. Medical writing support was provided by Cherie Koch, PhD, of Cello Health Communications/MedErgy and was funded by Bristol Myers Squibb and Janssen Global Services, LLC.
Perera V, Wang Z, Luettgen J, et al. First‐in‐human study of milvexian, an oral, direct, small molecule factor XIa inhibitor. Clin Transl Sci. 2022;15:330–342. 10.1111/cts.13148
Funding information
This study was sponsored by Bristol Myers Squibb. Medical writing support was provided by Cherie Koch, PhD, of Cello Health Communications/MedErgy, and was funded by Bristol Myers Squibb and Janssen Global Services, LLC.
REFERENCES
- 1. Cavallari I, Patti G. Efficacy and safety of oral anticoagulation in elderly patients with atrial fibrillation. Anatol J Cardiol. 2018;19:67‐71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kapil N, Datta YH, Alakbarova N, et al. Antiplatelet and anticoagulant therapies for prevention of ischemic stroke. Clin Appl Thromb Hemost. 2017;23:301‐318. [DOI] [PubMed] [Google Scholar]
- 3. Gurbel PA, Fox KAA, Tantry US, Ten Cate H, Weitz JI. Combination antiplatelet and oral anticoagulant therapy in patients with coronary and peripheral artery disease. Circulation. 2019;139:2170‐2185. [DOI] [PubMed] [Google Scholar]
- 4. Eisen A, Giugliano RP, Braunwald E. Updates on acute coronary syndrome: a review. JAMA Cardiol. 2016;1:718‐730. [DOI] [PubMed] [Google Scholar]
- 5. Bracey A, Shatila W, Wilson J. Bleeding in patients receiving non‐vitamin K oral anticoagulants: clinical trial evidence. Ther Adv Cardiovasc Dis. 2018;12:361‐380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ten Cate H, Hackeng TM, Garcia de Frutos P. Coagulation factor and protease pathways in thrombosis and cardiovascular disease. Thromb Haemost. 2017;117:1265‐1271. [DOI] [PubMed] [Google Scholar]
- 7. Lowenberg EC, Meijers JC, Monia BP, Levi M. Coagulation factor XI as a novel target for antithrombotic treatment. J Thromb Haemost. 2010;8:2349‐2357. [DOI] [PubMed] [Google Scholar]
- 8. Emsley J, McEwan PA, Gailani D. Structure and function of factor XI. Blood. 2010;115:2569‐2577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Puy C, Rigg RA, McCarty OJ. The hemostatic role of factor XI. Thromb Res. 2016;141(Suppl 2):S8‐S11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Seligsohn U. Factor XI deficiency in humans. J Thromb Haemost. 2009;7(Suppl 1):84‐87. [DOI] [PubMed] [Google Scholar]
- 11. Rosenthal RL, Dreskin OH, Rosenthal N. Plasma thromboplastin antecedent (PTA) deficiency; clinical, coagulation, therapeutic and hereditary aspects of a new hemophilia‐like disease. Blood. 1955;10:120‐131. [PubMed] [Google Scholar]
- 12. Franchini M, Veneri D, Lippi G. Inherited factor XI deficiency: a concise review. Hematology. 2006;11:307‐309. [DOI] [PubMed] [Google Scholar]
- 13. Lee SE, Choi YJ, Chi SI, Kim HJ, Seo KS. Factor XI deficiency and orthognathic surgery: a case report on anesthesia management. J Dent Anesth Pain Med. 2015;15:25‐29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Minnema MC, Friederich PW, Levi M, et al. Enhancement of rabbit jugular vein thrombolysis by neutralization of factor XI. In vivo evidence for a role of factor XI as an anti‐fibrinolytic factor. J Clin Invest. 1998;101:10‐14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wang X, Smith PL, Hsu M‐Y, et al. Effects of factor XI deficiency on ferric chloride‐induced vena cava thrombosis in mice. J Thromb Haemost. 2006;4:1982‐1988. [DOI] [PubMed] [Google Scholar]
- 16. Preis M, Hirsch J, Kotler A, et al. Factor XI deficiency is associated with lower risk for cardiovascular and venous thromboembolism events. Blood. 2017;129:1210‐1215. [DOI] [PubMed] [Google Scholar]
- 17. Salomon O, Steinberg DM, Koren‐Morag N, Tanne D, Seligsohn U. Reduced incidence of ischemic stroke in patients with severe factor XI deficiency. Blood. 2008;111:4113‐4117. [DOI] [PubMed] [Google Scholar]
- 18. Salomon O, Steinberg DM, Zucker M, Varon D, Zivelin A, Seligsohn U. Patients with severe factor XI deficiency have a reduced incidence of deep‐vein thrombosis. Thromb Haemost. 2011;105:269‐273. [DOI] [PubMed] [Google Scholar]
- 19. Meijers JC, Tekelenburg WL, Bouma BN, Bertina RM, Rosendaal FR. High levels of coagulation factor XI as a risk factor for venous thrombosis. N Engl J Med. 2000;342:696‐701. [DOI] [PubMed] [Google Scholar]
- 20. Yang DT, Flanders MM, Kim H, Rodgers GM. Elevated factor XI activity levels are associated with an increased odds ratio for cerebrovascular events. Am J Clin Pathol. 2006;126:411‐415. [DOI] [PubMed] [Google Scholar]
- 21. Merlo C, Wuillemin WA, Redondo M, et al. Elevated levels of plasma prekallikrein, high molecular weight kininogen and factor XI in coronary heart disease. Atherosclerosis. 2002;161:261‐267. [DOI] [PubMed] [Google Scholar]
- 22. Berliner JI, Rybicki AC, Kaplan RC, Monrad ES, Freeman R, Billett HH. Elevated levels of Factor XI are associated with cardiovascular disease in women. Thromb Res. 2002;107:55‐66. [DOI] [PubMed] [Google Scholar]
- 23. Pollack CV Jr, Kurz MA, Hayward NJ. EP‐7041, a Factor XIa inhibitor as a potential antithrombotic strategy in extracorporeal membrane oxygenation: a brief report. Crit Care Explor. 2020;2:e0196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sakimoto S, Hagio T, Yonetomi Y, et al. ONO‐8610539, an injectable small‐molecule inhibitor of blood coagulation Factor XIa, improves cerebral ischemic injuries associated with photothrombotic occlusion of rabbit middle cerebral artery. Stroke. 2017;48:Abstract WP286. [Google Scholar]
- 25. Thomas D, Kanefendt FS, Schwers S, Unger S, Yassen A, Boxnick S. First evaluation of the safety, pharmacokinetics and pharmacodynamics of BAY 2433334 a small molecule targeting coagulation Factor XIa in healthy young male participants. Res Pract Thromb Haemost. 2020;4:PB0243. [Google Scholar]
- 26. Weitz JI, Bauersachs R, Becker B, et al. Effect of osocimab in preventing venous thromboembolism among patients undergoing knee arthroplasty: the FOXTROT randomized clinical trial. JAMA. 2020;323:130‐139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gailani D, Gruber A. Factor XI as a therapeutic target. Arterioscler Thromb Vasc Biol. 2016;36:1316‐1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ewing WR. Discovery of BMS‐986177, an inhibitor of FXIa in phase 2 studies for antithrombotic therapy. Presented at: American Chemical Society (ACS) Fall 2020 Virtual Meeting & Expo; August 17‐20, 2020.
- 29. Wang X, Qiu L, Du F, Shukla N, Nawrocki A, Chintala M. Antithrombotic effects of a novel small molecule FXIa inhibitor BMS‐986177/JNJ‐70033093 in a rabbit AV‐shunt model of thrombosis. Presented at: International Society on Thrombosis and Haemostasis (ISTH) 2020 Virtual Congress; July 12–14, 2020. Poster PB0179.
- 30. Wong PC, Crain E, Dilger A, et al. Small‐molecule Factor XIa inhibitor, BMS‐986177/JNJ‐70033093, prevents and treats arterial thrombosis in rabbits at doses that preserve hemostasis. Presented at: International Society on Thrombosis and Haemostasis (ISTH) 2020 Virtual Congress; July 12–14, 2020. Poster PB0121.
- 31. ClinicalTrials.gov . A study on BMS‐986177 for the prevention of stroke in patients receiving aspirin and clopidogrel (AXIOMATIC‐SSP) <https://clinicaltrials.gov/ct2/show/NCT03766581>. Accessed July 15, 2020.
- 32. ClinicalTrials.gov. NCT03891524 . A study of JNJ‐70033093 (BMS‐986177) versus subcutaneous enoxaparin in participants undergoing elective total knee replacement surgery (AXIOMATIC‐TKR) <https://clinicaltrials.gov/ct2/show/NCT03891524>. Accessed July 15, 2020.
- 33. Gough K, Hutchison M, Keene O, et al. Assessment of dose proportionality: report from the Statisticians in the Pharmaceutical Industry/Pharmacokinetics UK Joint Working Party. Drug Information J. 1995;29:1039‐1048. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1
Fig S2
Table S1
Table S2
Table S3