

Abstract
Filgotinib, a preferential Janus Kinase‐1 inhibitor, is approved in Europe and Japan for treatment of rheumatoid arthritis and is being developed for treatment of other chronic inflammatory diseases. Three drug‐drug interactions studies were conducted in healthy subjects to evaluate the effect of P‐glycoprotein (P‐gp) modulation (study 1: P‐gp inhibition by itraconazole and study 2: P‐gp induction by rifampin) on filgotinib pharmacokinetics and the potential of filgotinib to impact exposure of metformin, an organic cation transporter (OCT) 2 and multidrug and toxin extrusion (MATE) 1/2K substrate (study 3). Co‐administration of filgotinib with itraconazole increased filgotinib exposure (maximum concentration [C max] by 64% and area under the curve to infinity [AUCinf] by 45%) but had no effect on the exposure of GS‐829845, filgotinib’s primary metabolite. Rifampin moderately reduced exposures of filgotinib and GS‐829845 (C max by 26% and AUCinf by 27% for filgotinib; C max by 19% and AUCinf by 38% for GS‐829845). The data confirmed that filgotinib is a P‐gp substrate. However, the magnitude of change in filgotinib/GS‐829845 exposure by P‐gp modulators is not deemed to be clinically relevant based on filgotinib exposure‐response analyses in subjects with rheumatoid arthritis. Filgotinib did not alter metformin exposures, indicating that filgotinib and GS‐829845 do not inhibit OCT2 and MATE1/2K at the clinical doses. Filgotinib was generally well‐tolerated when administered alone or with the co‐administered drugs in the studies. Results from these studies were the basis to enable the use of P‐gp modulators and substrates of OCT2, MATE1, and MATE2K with filgotinib without the need for dose modifications in the current approved rheumatoid arthritis population.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
Filgotinib is approved for treatment of patients with rheumatoid arthritis in Europe and Japan. Polypharmacy is common in patients with inflammatory diseases, such as rheumatoid arthritis.
WHAT QUESTION DID THIS STUDY ADDRESS?
The study evaluated the drug‐drug interaction potential for filgotinib with P‐gp modulators or substrates of OCT2, MATE1, and MATE2K to inform on the dosing recommendations.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
Filgotinib and GS‐829845 exposures are not impacted by P‐gp inhibitors or inducers to any clinically relevant extent. Additionally, filgotinib and GS‐829845 are not inhibitors of OCT2, MATE1, and MATE2K at the clinically relevant exposures.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
Concomitant use of P‐gp modulators or substrates of OCT2, MATE1, and MATE2K with filgotinib does not require dose modifications.
INTRODUCTION
Filgotinib (brand name: Jyseleca®) is an oral small molecule inhibitor of Janus Kinase (JAK)‐1. Filgotinib is approved in Europe for treatment of moderate‐to‐severe active rheumatoid arthritis in adult patients who have responded inadequately to, or who are intolerant to, one or more disease‐modifying anti‐rheumatic drugs (DMARDs) and in Japan for treatment of rheumatoid arthritis in patients who had an inadequate response to conventional therapies (including prevention of structural damage to joints). 1 In the rheumatoid arthritis phase III studies, both filgotinib 200 mg and 100 mg once‐daily doses significantly improved signs and symptoms and physical function in patients with active rheumatoid arthritis who had an inadequate response or intolerance to one or more biological DMARDs or patients with active rheumatoid arthritis and limited or no prior methotrexate exposure. 2 , 3 Filgotinib 200 mg is the therapeutic dose approved in Europe for rheumatoid arthritis, as it provides a more favorable benefit/risk profile over the 100 mg dose in this patient population. In the ulcerative colitis phase IIb/III studies, filgotinib 200 mg dose met all the primary end points in the study, inducing clinical remission at week 10 and maintaining clinical remission at week 58. Filgotinib 100 mg dose did not meet the primary end point in the induction studies. 4 Filgotinib is currently under regulatory review by the European Medicines Agency for treatment of ulcerative colitis. Additionally, filgotinib is currently under evaluation for treatment of Crohn’s disease. 5
Filgotinib inhibits JAK1 with greater than five‐fold higher potency over JAK2, JAK3, tyrosine kinase 2, and other kinases as measured in whole blood and cellular assays. 6 Filgotinib’s major metabolite, GS‐829845, which circulates in plasma at ~ 16‐ to 21‐fold the parent exposure (adjusted for differences in molecular weight), 7 is also a preferential inhibitor of JAK1 with ~ 1/10 the potency of filgotinib and it has been shown to contribute to overall pharmacodynamic effects of JAK1 inhibition following filgotinib dosing. 8
Filgotinib is extensively metabolized with ~ 9% and 5% of the orally administered dose recovered as unchanged filgotinib in urine and feces, respectively. Filgotinib is primarily metabolized by carboxylesterase 2 (CES2), and to a lesser extent by CES1, to form GS‐829845. Approximately 87% of the administered dose was eliminated in the urine as filgotinib and its metabolites, whereas about 15% of the dose was eliminated in the feces. GS‐829845 accounted for ~ 54% and 9% of dose recovered in urine and feces, respectively. 6 In vitro and in vivo data indicate that the drug‐drug interactions (DDIs) liability of filgotinib is relatively low with cytochrome P450 enzymes. 9 In vitro data suggests that both filgotinib and GS‐829845 are substrates of P‐glycoprotein. 6 Furthermore, in vitro studies indicate that filgotinib and GS‐829845 are not inhibitors of P‐gp, breast cancer resistance protein, organic cation transporter 1, bile salt export pump, or organic anion transporter 1, 3, and 4 at clinically relevant concentrations. 6 , 9 In vitro data also suggested that filgotinib inhibited organic cation transporter 2 (OCT2) and multidrug and toxin extrusion 1 and 2K (MATE1 and MATE2K), and organic anion transporting polypeptide 1B1 and 1B3 (OATP1B1/3). 6
Therefore, clinical evaluations were designed to assess whether filgotinib and GS‐829845 could be victims of P‐gp mediated drug interactions and to evaluate any clinically relevant interactions between filgotinib and drugs substrates of OCT2, MATE1, and MATE2K. Clinical evaluation of DDIs between filgotinib and substrates of OATP1B1/1B3 is ongoing.
This report summarizes the results from three phase I DDI studies. Study 1 assessed the effect of P‐gp inhibition on filgotinib pharmacokinetics using a P‐gp inhibitor, itraconazole. Study 2 evaluated the effect of P‐gp induction on filgotinib pharmacokinetics using a P‐gp inducer, rifampin. Study 3 evaluated the effect of filgotinib on metformin exposure, a probe substrate for OCT2, MATE1, and MATE2K.
Data from these drug interaction studies were used to inform the dosing recommendations of filgotinib when co‐administered with P‐gp modulators or with OCT2, MATE1, and MATE2K substrates.
METHODS
Study population
For all studies, eligible subjects included males (surgically sterile; studies 2 and 3 only) and non‐pregnant, non‐lactating females of 18–45 years of age with a body mass index between 19 and 30 kg/m2. Major inclusion criteria included healthy subjects based on medical history/physical examinations/laboratory evaluations, 12‐lead electrocardiogram without clinically significant abnormalities, creatinine clearance greater than 90 ml/min, no evidence of human immunodeficiency virus, hepatitis B virus or hepatitis C virus infection, and use of at least two forms of contraception, including an effective barrier method. Exclusion criteria included plasma and blood donation within 7 and 56 days of study entry, respectively, active medical illness, use of prescription drugs within 28 days of study drug dosing (except vitamins, acetaminophen, ibuprofen, and/or hormonal contraceptives).
Study design
Study design schemas are presented in Figure S1. For each study, the study protocol and informed consent were approved by the study center’s Institutional Review Board (Schulman Associates IRB, Cincinnati, OH for Study 1, and Advarra Institutional Review Board, Columbia, MD, for Studies 2 and 3), and subjects provided written consent before study participation.
Study 1 was an open‐label, fixed sequence, single‐center evaluation in healthy subjects at SeaView Jacksonville LLC (Jacksonville, FL). A single 100 mg dose of filgotinib was administered in the morning on day 1. This was followed by a washout period from days 2–9. On day 10, subjects received a single dose of itraconazole 200 mg, followed by a single 100 mg dose of filgotinib 1 h after itraconazole administration in the morning.
Study 2 was an open‐label, fixed sequence, single‐center study in healthy subjects at Quotient Sciences (Miami, FL). Eligible subjects received a single dose of 200 mg of filgotinib on day 1 in the morning, followed by a washout period from days 2–5. Then subjects received rifampin 600 mg once daily in the evening from days 6–17. On day 13, a single dose of 200 mg filgotinib was administered in the morning.
Study 3 was an open‐label, fixed sequence, single‐center study in healthy subjects at Quotient Sciences. Eligible subjects received a single dose of 850 mg of metformin in the morning on day 1. This was followed by a washout period from days 2–3. Afterward, subjects received 200 mg filgotinib once daily in the morning from days 4–13. Metformin 850 mg was administered in the morning on day 11.
All study drugs were administered under fasting conditions.
Pharmacokinetic sampling
Serial pharmacokinetic samples were collected at the following time points.
Study 1
On days 1 and 10 predose (≤5 min before dose), and at 0.5, 1, 2, 3, 4, 6, 8, 12, 18, 24, 36, 48, 72, 96, and 120 h postdose.
Study 2
On days 1 and 13 predose (≤5 min before dose), and at 0.5, 1, 2, 3, 4, 6, 8, 12, 18, 24, 36, 48, 72, 96, and 120 h postdose.
Study 3
On days 1 and 11 predose (≤5 min before dose), and at 0.5, 1, 2, 2.5, 3, 4, 6, 8, 12, 18, 24, 36, 48, and 72 h postdose.
Bioanalytical procedures
Plasma concentrations of filgotinib, GS‐829845, and metformin were determined using a validated high‐performance liquid chromatography‐tandem mass spectrometry (LC‐MS/MS). All plasma samples were analyzed within the timeframe supported by frozen sample stability storage data. Sample analyses for plasma concentrations of filgotinib and GS‐829845 were performed at QPS, LLC, as previously described. 10
Analysis of metformin concentrations in human plasma was performed and validated using an LC‐MS/MS method that is commercially available from QPS. The plasma samples were prepared for analysis by placing a 25‐µl aliquot into a 96‐well plate followed by the additional 50‐µl of metformin‐d6 (Cayman Chemicals) internal standard. The samples were extracted using 2% ammonium hydroxide in water using an Oasis WCX 96‐well solid‐phase extraction plate 30‐µm (Waters). The eluent in the plate was evaporated to dryness under nitrogen steam at 50°C and then reconstituted in water. After vortex‐mixing and centrifugation, 10‐µl of the supernatant was injected onto an Acquity UPLC HSS PFP (2.1 × 50 mm, 1.8 µm) analytical column (Waters). An SIL‐30ACMP autosampler (Shimadzu Scientific Instruments) was linked to a LC‐30AD pump (Shimadzu), coupled with an API 4000 mass spectrometer (Sciex) for sample analysis. The mobile phases were composted of acetonitrile: 10 mM ammonium acetate, 70:30 (v:v). The method used an isocratic elution with a flow rate of 0.6 ml/min and a total run time of 1.5 min. Data were acquired using multiple reaction monitoring in positive ion electrospray mode, with an operating source temperature of 600°C. The transitions used for metformin and its internal standard were m/z 130.0 to greater than 59.9 and m/z 136.0 to greater than 59.9, respectively. The lower and upper limits of quantitation for the assay of metformin were 2 and 2000 ng/ml.
Pharmacokinetic analyses
Pharmacokinetic parameters were estimated by noncompartmental methods using WinNonlin software (version 6.3; Pharsight). Pharmacokinetic parameters included area under the concentration versus time curve (AUC) from time zero to the last quantifiable concentration (AUClast), AUC extrapolated to infinity (AUCinf), maximum observed plasma concentration (C max), time to reach maximum concentration (T max), terminal elimination half‐life (t1/2), and apparent oral clearance (CL/F). Samples that were below the lower limit of quantitation after T max were treated as missing data in the noncompartmental analyses. As both filgotinib and GS‐829845 contribute to efficacy, effective AUC (AUCeff) was derived by accounting for the potency and the molecular weight difference between filgotinib and GS‐829845 using the following equation: AUCeff = filgotinib AUCinf + GS‐829845 AUCinf *1/10 * (425.51/357.43), where 425.51 and 357.43 are the molecular weights of filgotinib and GS‐829845, respectively.
Statistical analyses
The pharmacokinetic parameters were summarized statistically from the test and reference treatments. Furthermore, an analysis of variance using a mixed‐effects model with treatment as a fixed effect and subject as a random effect was fitted to the natural logarithmic transformation of AUC and C max of each analyte. Two‐sided 90% confidence intervals were calculated for the ratio of geometric least‐squares means of primary pharmacokinetic parameters (AUClast, AUCinf, and C max) of the test versus reference treatments. A reference treatment represents the victim drug alone. A test treatment represents the victim in the presence of the perpetrator.
Safety assessments
Safety assessments included clinical laboratory tests (hematology profile, chemistry profile, and urinalysis), physical examinations, vital signs, serum pregnancy tests (female subjects), and review of concomitant medications performed at screening, at baseline (day before first study dose), on the days before the pharmacokinetic blood sampling, and at various times during the study. Subjects were monitored for adverse events (AEs) throughout the study and follow‐up.
RESULTS
Subject demographics
Thirteen subjects enrolled in study 1 and 12 subjects completed the study. One subject discontinued from study 1 due to withdrawal of consent. All subjects that enrolled in studies 2 (14 subjects) and 3 (12 subjects) completed the studies. Demographics and baseline characteristics by study are summarized in Table 1.
TABLE 1.
Demographics and baseline characteristics of study participants
|
Study 1 N = 13 |
Study 2 N = 14 |
Study 3 N = 12 |
|
|---|---|---|---|
| Age (years) | 36 (25, 45) | 38 (22, 46) | 31 (20, 42) |
| Sex (M/F) | 9/4 | 0/14 | 0/12 |
| Race |
Black: 6 White: 7 |
Black: 2 White: 12 |
Black: 5 White: 7 |
| Weight (kg) | 78.9 (62.0, 96.3) | 68.5 (55.4, 85.4) | 65.3 (52.1, 88.1) |
| Height (cm) | 170 (154, 181) | 160 (149, 169) | 163 (152, 179) |
| Body mass index (kg/m2) | 27.4 (21.8, 29.9) | 26.7 (21.9, 30.1) | 24.5 (19.9, 30.0) |
| CLcr (ml/min) | 119 (93.2, 148) | 120 (90.8, 160) | 117 (84.9, 169) |
Data are presented as mean (minimum, maximum).
Abbreviation: CLcr, creatinine clearance calculated by the Cockcroft‐Gault equation.
Pharmacokinetics
Effect of P‐gp inhibition on filgotinib pharmacokinetics
Figure 1 shows plasma concentrations versus time profiles of filgotinib and GS‐829845 after administration of a single 100‐mg dose of filgotinib alone or 1 h after a single dose of itraconazole 200 mg, under fasting conditions. A summary of filgotinib and GS‐829845 pharmacokinetic parameters with and without itraconazole is presented in Table 2. Itraconazole increased filgotinib C max by 64% and AUCinf by 45% but did not affect filgotinib Tmax or t1/2. GS‐829845 C max, AUCinf, T max, and t 1/2 were similar with and without itraconazole. The combined exposure (AUCeff) of filgotinib and GS‐829845 was 21% higher following administration of filgotinib with itraconazole relative to administration of filgotinib alone.
FIGURE 1.
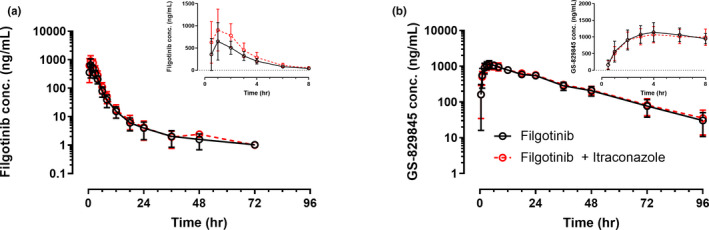
Mean (SD) plasma concentrations versus time profiles of filgotinib (a) and GS‐829845 (b) after a single dose of filgotinib administered alone or 1 h after itraconazole . Insets are mean (SD) plasma concentrations versus time profiles in a linear scale for the first 8 h
TABLE 2.
Filgotinib and GS‐829845 plasma pharmacokinetic parameters and statistical comparisons following a single dose of filgotinib 100 mg with or without itraconazole
| Pharmacokinetic parameters |
Filgotinib N = 13 |
Filgotinib + itraconazole N = 12 |
%GLSM ratio (90% CI) |
|---|---|---|---|
| Filgotinib | |||
| AUCinf (ng*h/ml) | 2190 (27.6) | 3130 (21.1) | 145 (133, 157) |
| AUClast (ng*h/ml) | 2170 (27.8) | 3110 (21.2) | 145 (133, 158) |
| C max (ng/ml) | 756 (44.7) | 1170 (24.7) | 164 (129, 208) |
| T max (h) | 1.00 (1.00, 2.00) | 1.0 (0.80,1.50) | |
| t 1/2 (h) | 7.4 (4.70, 8.30) | 6.26 (5.20, 8.10) | |
| CL/F (L/h) | 48.6 (25.0) | 33.3 (21.8) | |
| GS‐829845 | |||
| AUCinf (ng*h/ml) | 31,200 (18.4) | 32,400 (16.5) | 107 (104,110) |
| AUClast (ng*h/ml) | 30,800 (18.0) | 31,800 (15.8) | 106 (103,109) |
| C max (ng/ml) | 1160 (24.8) | 1090 (22.8) | 94.4 (88.5,101) |
| T max (h) | 4.00 (4.00, 4.00) | 4.00 (3.50, 5.00) | |
| t 1/2 (h) | 17.0 (14.0,19.5) | 17.0 (14.4, 22.5) | |
| Combined exposure | |||
| AUCeff (ng*h/ml) | 121 (108, 136) | ||
Data are presented as mean (percentage of coefficient of variation) except for t 1/2 and T max which are presented as median (first quartile and third quartile).
Abbreviations: AUCeff, effective area under the concentration versus time curve; AUCinf, area under the curve to infinity; AUClast, area under the concentration versus time curve from time zero to the last quantifiable concentration; CI, confidence interval; CL/F, total apparent clearance; Cmax, maximum concentration; GLSM ratio, geometric least square mean ratio; t1/2, terminal half‐life; Tmax, time to maximum concentration.
Effect of P‐gp induction on filgotinib pharmacokinetics
Figure 2 shows the plasma concentrations versus time profiles of filgotinib and GS‐829845 after administration of filgotinib alone or following multiple doses of rifampin 600 mg once daily. A summary of filgotinib and GS‐829845 pharmacokinetic parameters with and without rifampin is presented in Table 3. Filgotinib C max and AUCinf were reduced by 26% and 27%, respectively, following administration of filgotinib on day 8 of a 12‐day once‐daily regimen of rifampin, compared with administration of filgotinib alone. Filgotinib T max and t 1/2 values were similar following both treatments. GS‐829845 AUCinf and C max were lower by 38% and 19%, respectively, following administration of filgotinib after multiple doses of rifampin, compared with administration of filgotinib alone. GS‐829845 Tmax was similar following both treatments. The combined exposure (AUCeff) of filgotinib and GS‐829845 was reduced by 33% following administration of filgotinib after multiple doses of rifampin relative to administration of filgotinib alone.
FIGURE 2.
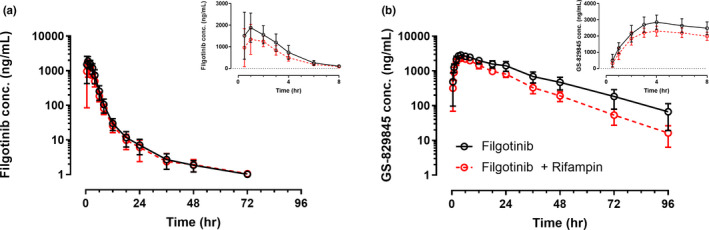
Mean (SD) filgotinib and GS‐829845 plasma concentrations versus time profiles following administration of filgotinib alone or after multiple doses of rifampin. Insets are mean (SD) plasma concentrations versus time profiles in a linear scale for the first 8 h
TABLE 3.
Filgotinib and GS‐829845 plasma pharmacokinetic parameters and statistical comparisons following administration of filgotinib alone or after multiple doses of rifampin
| Pharmacokinetic parameters |
Filgotinib N = 14 |
Filgotinib + rifampin N = 14 |
%GLSM ratio (90% CI) |
|---|---|---|---|
| Filgotinib | |||
| AUCinf (ng*h/ml) | 6960 (24.7) | 5070 (22.8) | 72.7 (69.1, 76.5) |
| AUClast (ng*h/ml) | 6980 (24.6) | 5050 (22.7) | 72.7 (69.1, 76.5) |
| C max (ng/ml) | 2190 (31.7) | 1640 (34.3) | 74.3 (83.4, 86.4) |
| Tmax (h) | 1.00 (1.00, 2.00) | 1.50 (1.00, 2.00) | — |
| t1/2 (h) | 8.40 (6.70, 10.0) | 7.70 (5.90, 8.60) | — |
| CL/F (L/h) | 30.1 (21.0) | 41.3 (21.2) | — |
| GS−829845 | |||
| AUCinf (ng*h/ml) | 77,400 (25.3) | 47,200 (17.1) | 61.9 (57.9, 66.1) |
| AUClast (ng*h/ml) | 76,700 (24.8) | 47,100 (17.0) | 62.3 (58.4, 66.4) |
| C max (ng/ml) | 2900 (15.1) | 2340 (12.8) | 81.0 (76.8, 85.4) |
| Tmax (h) | 4.00 (3.00, 4.00) | 4.00 (3.00, 4.00) | — |
| t1/2 (h) | 15.9 (14.8, 18.9) | 12.4 (11.7, 13.9) | — |
| Combined exposure | |||
| AUCeff (ng*h/ml) | 66.6 (58.6, 75.7) | ||
Data are presented as mean (percentage coefficient of variation) except for t 1/2 and T max which are presented as median (first quartile and third quartile).
Abbreviations: AUCeff, effective area under the concentration versus time curve; AUCinf, area under the concentration versus time curve to infinity; AUClast, area under the concentration versus time curve from time zero to the last quantifiable concentration; CI, confidence interval; CL/F, total apparent clearance; Cmax, maximum concentration; GLSM ratio, geometric least square mean ratio; t1/2, terminal half‐life; Tmax, time to maximum concentration.
Effect of filgotinib on exposure of metformin, an OCT2 and MATE1/2K substrate
Figure 3 shows the plasma concentrations versus time profiles of metformin after administration alone or on day 8 of an 11‐day regimen of filgotinib 200 mg once daily. Metformin plasma pharmacokinetic parameters following administration of metformin alone or with filgotinib are presented in Table 4. Filgotinib had no effect on metformin AUCinf or C max.
FIGURE 3.
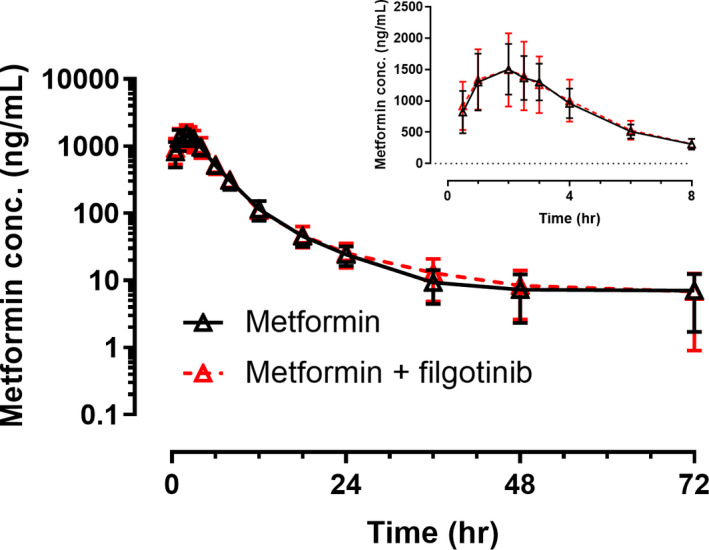
Mean (SD) metformin plasma concentrations versus time profiles following administration of metformin alone or with filgotinib. Insets are mean (SD) plasma concentrations versus time profiles in a linear scale for the first 8 h
TABLE 4.
Metformin plasma pharmacokinetic parameters and statistical comparisons following administration of metformin alone or with filgotinib
| Pharmacokinetic parameters |
Metformin N = 12 |
Metformin + filgotinib N = 12 |
%GLSM ratio (90% CI) |
|---|---|---|---|
| Metformin | |||
| AUCinf (ng*h/ml) | 8720 (22.9) | 9020 (27.1) | 102 (85.3, 122) |
| AUClast (ng*h/ml) | 8640 (22.8) | 8890 (27.3) | 102 (84.9, 122) |
| C max (ng/ml) | 1550 (22.6) | 1610 (31.0) | 102 (85.4, 121) |
| T max (h) | 2.00 (1.30, 2.00) | 2.00 (1.00, 2.00) | — |
| t 1/2 (h) | 7.90 (6.00, 15.10) | 9.70 (8.10, 17.5) | — |
| CL/F (L/h) | 103 (23.8) | 102 (30.5) | — |
Data are presented as mean (percentage coefficient of variation) except for t 1/2 and T max which are presented as median (first quartile and third quartile).
Abbreviations: AUCinf, area under the concentration versus time curve to infinity; AUClast, area under the concentration versus time curve from time zero to the last quantifiable concentration; CI, confidence interval; CL/F, total apparent clearance; Cmax, maximum concentration; GLSM ratio, geometric least square mean ratio; t1/2, terminal half‐life; Tmax, time to maximum concentration.
Safety and tolerability
During study 1, AEs were reported by two subjects (15%) (filgotinib: 1 subject [7.7%]; filgotinib + itraconazole: one subject [8.3%]). All of the AEs reported were grade 1 and were not related to study drug. A total of seven subjects (54%) (filgotinib: 2 subjects [15%]; filgotinib + itraconazole: seven subjects [58%]) had at least one graded laboratory abnormality, the majority of which were grade 1 or 2 in severity. Grade 3 hypertriglyceridemia was reported for one subject (8.3%) during the filgotinib + itraconazole treatment period. The subject did not experience any AE during the study. No grade 4 laboratory abnormalities were reported.
During study 2, AEs were reported for two subjects (14%) during the rifampin treatment period. All of the AEs reported were grade 1. Overall, AEs considered to be related to study drug by the investigator were reported for one subject (7.1%) who had nausea during the rifampin treatment period. A total of 13 subjects (93%) had at least one graded laboratory abnormality, the majority of which were grade 1 or 2 in severity (filgotinib: 2 subjects [14%]; rifampin: 13 subjects [93%]; filgotinib + rifampin: 11 subjects [79%]). A grade 3 laboratory abnormality was reported for two subjects (14%) during the rifampin treatment period which were occult blood in urine. No grade 4 laboratory abnormalities were reported.
During study 3, AEs were reported for three subjects (25%; filgotinib: 1 subject [8.3%]; metformin: 3 subjects [25%]; filgotinib + metformin: 1 subject [8.3%]). All of the AEs reported were grade 1. Overall, AEs considered to be related to study drug by the investigator were reported for one subject (8.3%). This subject had an AE considered related to study drug in each of the treatment phases (metformin: nausea + vomiting; filgotinib: headache; filgotinib + metformin: vomiting). A total of nine subjects (75%) during the filgotinib treatment period had at least one graded laboratory abnormality, the majority of which were grade 1 or 2 in severity. Grade 3 laboratory abnormalities were reported for three subjects (25%) during the filgotinib treatment period, which were as follows: occult blood in urine (2 subjects [17%]), and decreased lymphocytes and neutrophils (1 subject [8.3%]). No grade 4 laboratory abnormalities were reported.
For all three studies, study drug treatments were generally well‐tolerated. No deaths, serious AEs, or AEs leading to discontinuation of the study drug were reported. Additionally, there were no clinically relevant laboratory abnormalities, changes in vital signs, or electrocardiogram abnormalities.
DISCUSSION
Filgotinib is approved in Europe for treatment of patients with rheumatoid arthritis who have responded inadequately to, or who are intolerant to, one or more DMARDs and in Japan for treatment of rheumatoid arthritis in patients who had an inadequate response to conventional therapies. Additionally, filgotinib is under evaluation for treatment of other inflammatory diseases, including ulcerative colitis and Crohn’s disease. Polypharmacy is common in patients with inflammatory diseases. As such, it is important to understand any potential DDIs with filgotinib to provide adequate concomitant use recommendations.
Filgotinib is classified as a Biopharmaceutical Classification System (BCS) II compound as it has low solubility and high permeability. In human colon carcinoma‐derived (Caco‐2) cells, it showed active efflux. Since in vitro data suggested that filgotinib and GS‐829845 were substrates of P‐gp, we wanted to assess whether modulation of P‐gp activity by a P‐gp perpetrator may alter the pharmacokinetics of filgotinib and GS‐829845.
The 200 mg dose is the therapeutic dose approved for rheumatoid arthritis and is the proposed dose for treatment of ulcerative colitis. In study 1, filgotinib 100 mg was evaluated to account for any potential increase in filgotinib exposure by itraconazole. The pharmacokinetics of filgotinib are linear (both dose proportional and time‐independent) from 50 to 200 mg. 10 As such, a single dose of filgotinib was appropriate to evaluate the victim liabilities in studies 1 and 2. 11
Itraconazole is a strong CYP3A inhibitor and a clinical P‐gp inhibitor. A single dose of 200 mg itraconazole was administered in study 1; this dose was used in other drug interaction studies utilizing itraconazole as the P‐gp inhibitor in healthy subjects and has been shown to substantially increase the plasma exposures of probe P‐gp substrates. 12 , 13 Of note, the effect of itraconazole on the exposure of the probe P‐gp substrate, fexofenadine, was comparable among days 1, 3, and 6 of itraconazole dosing, 12 indicating that a single dose of itraconazole is adequate to maximize P‐gp inhibition. In vitro data indicate that filgotinib is primarily metabolized by CES2 and to a lesser degree by CES1 and filgotinib is not a substrate of the cytochrome P450 enzymes. As such, any interaction between filgotinib and itraconazole is expected to be primarily driven by P‐gp inhibition.
Itraconazole moderately increased the exposure of filgotinib by 45% without impacting the half‐life. This indicated that the modest effect is likely mediated through intestinal P‐gp inhibition. Furthermore, itraconazole did not affect the pharmacokinetics of GS‐829845. When the combined exposure and potency of filgotinib and GS‐829845 were taken into account, the increase in the effective exposure of filgotinib (AUCeff) when administered with itraconazole was small (~ 20%).
Rifampin 600 mg once daily was selected as the perpetrator to evaluate the impact of P‐gp induction in study 2. P‐gp induction by multiple 600 mg once‐daily doses of rifampin has been previously established. 14 In the current study, rifampin moderately reduced the AUC of filgotinib and GS‐829845 (by 27% and 38%, respectively). It is of note that rifampin causes moderate induction of both CES1 and CES2 in human hepatocytes. 15 Therefore, it is possible that the observed effect of rifampin in the current study is a result of the combined effect of CES and P‐gp induction. Overall, multiple doses of rifampin reduced the effective exposure of filgotinib (combined AUC of filgotinib and GS‐829845, adjusted by the molecular weight and potency ratio) by only 33%, compared with administration of filgotinib alone.
Exposure‐efficacy analyses in subjects with rheumatoid arthritis from the phase II and phase III clinical studies consistently revealed high response rates across the exposure range for filgotinib 200 mg and 100 mg doses. Additionally, no relationships were observed between filgotinib or GS‐829845 exposures and the most frequent treatment emergent AEs (TEAEs), grade 3/4 laboratory abnormalities, serious TEAEs, or serious infections up to week 52. 16 Overall, the effect of itraconazole or rifampin on filgotinib exposures was not deemed to be clinically meaningful based on the exposure‐response relationships observed in subjects with rheumatoid arthritis. The clinical relevance of P‐gp perpetrators in other inflammatory diseases will be re‐assessed based on the emerging data collected in the respective populations.
In vitro data suggested that filgotinib may inhibit OCT2, MATE1, and MATE2K, whereas GS‐829845 may inhibit OCT2 and MATE2K . The ratios of maximum unbound plasma concentration in patients with rheumatoid arthritis over the in vitro half‐maximal inhibitory concentration (IC50) against OCT2, MATE1, and MATE2K were 0.26, 0.26, and 0.44, respectively, for filgotinib and 0.5, less than 0.1, and 0.69, respectively, for GS‐829845 (data on file at Gilead Sciences, Inc.). These values were greater than the US Food and Drug Administration (FDA) recommended cutoff value of 0.1 that warrants clinical assessment.
In study 3, a single dose of metformin 850 mg was evaluated as a substrate for MATE1/2K and OCT2. Daily doses of up to 2550 mg metformin can be used clinically; however, due to gastrointestinal side effects, the typical daily dose of metformin does not exceed 2000 mg. 17 Metformin (single or multiple) doses up to 1000 mg are typically utilized in drug interactions studies. 18 Available data shows that the maximal increase of metformin exposure via OCT2, MATE1, and MATE2K inhibition is ~ 2.6‐fold. 11 , 17 Therefore, an 850 mg dose of metformin was utilized for evaluation in the present study.
Steady‐state concentrations of filgotinib and GS‐829845 are achieved in ~ 2 days and 4 days, respectively. Filgotinib demonstrates no accumulation following multiple doses, whereas GS‐829845 demonstrates approximately twofold accumulation. 8 Accordingly, filgotinib was administered as 200 mg once daily for 7 days prior to administration with metformin. This dosing regimen is representative of the highest anticipated clinical exposures of filgotinib and GS‐829845 and is in agreement with recommendations from regulatory agencies for assessing the maximum potential of a drug to be a perpetrator of DDIs. 11
Following administration of multiple doses of filgotinib, the exposure of metformin, a sensitive OCT2, MATE1, and MATE2K substrate, remained unchanged, compared with administration of metformin alone. These data confirm that filgotinib and GS‐829845 do not inhibit OCT2, MATE1, and MATE2K at the clinically relevant dose. Substrates of OCT2, MATE1, and MATE2K can be concomitantly administered with filgotinib without need for dose modifications.
Collectively, results from these studies indicate that filgotinib and GS‐829845 exposures are not impacted by P‐gp inhibitors or inducers to any clinically relevant extent, despite being in vitro P‐gp substrates. Both filgotinib and GS‐829845 are not inhibitors of OCT2, MATE1, and MATE2K at the clinically relevant exposures. Data from these studies were the basis to allow the concomitant use of P‐gp modulators or substrates of OCT2, MATE1, and MATE2K with filgotinib without the need for dose modifications in the current approved rheumatoid arthritis population. 6
CONFLICT OF INTEREST
Chia‐Hsiang Hsueh, Kacey Anderson, Gong Shen, Chohee Yun, Ann Qin, and Ahmed A. Othman are employees and may hold stocks of Gilead Sciences, Inc.
AUTHOR CONTRIBUTIONS
C.‐H.H., K.A., A.Q., and A.A.O. wrote manuscript. C.‐H.H., K.A., G.S., C.Y., and A.A.O. designed study and analyzed data. C.‐H.H., K.A., G.S., and C.Y. performed research. A.Q. contributed to new reagents/analytical tools.
Supporting information
Figure S1
ACKNOWLEDGEMENTS
The authors would like to thank Sunila Reddy, PharmD, for providing medical writing support for this manuscript.
Hsueh C‐H, Anderson K, Shen G, Yun C, Qin A, Othman AA. Evaluation of the potential drug interactions mediated through P‐gp, OCT2, and MATE1/2K with filgotinib in healthy subjects. Clin Transl Sci. 2022;15:361–370. 10.1111/cts.13152
Funding information
These studies were funded by Gilead Sciences, Inc.
REFERENCES
- 1. Dhillon S, Keam SJ. Filgotinib: first approval. Drugs. 2020;80:1987‐1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Genovese MC, Kalunian K, Gottenberg J‐E, et al. Effect of filgotinib vs placebo on clinical response in patients with moderate to severe rheumatoid arthritis refractory to disease‐modifying antirheumatic drug therapy: the FINCH 2 randomized clinical trial. JAMA. 2019;322:315‐325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Westhovens R, Rigby WF, van der Heijde D, et al. Filgotinib in combination with methotrexate or as monotherapy versus methotrexate monotherapy in patients with active rheumatoid arthritis and limited or no prior exposure to methotrexate: the phase 3, randomised controlled FINCH 3 trial. Ann Rheum Dis. 2021;80:727‐738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gilead Sciences, Inc Gilead and Galapagos announce positive topline results of phase 2b/3 trial of filgotinib in moderately to severely active ulcerative colitis [Press release]. 2020. Accessed July 20, 2021. https://www.gilead.com/news‐and‐press/press‐room/press‐releases/2020/5/gilead‐and‐galapagos‐announce‐positive‐topline‐results‐of‐phase‐2b3‐trial‐of‐filgotinib‐in‐moderately‐to‐severely‐active‐ulcerative‐colitis
- 5. National Library of Medicine (U.S.) O. Combined Phase 3, double‐blind, randomized, placebo‐controlled studies evaluating the efficacy and safety of filgotinib in the induction and maintenance of remission in subjects with moderately to severely active Crohn's disease identifier: NCT02914561. 2016. https://clinicaltrials.gov/ct2/show/NCT02914561
- 6. Jyseleca (Filgotinib) [package insert], County Cork, Ireland: Gilead Sciences Ireland UC. 2020. Accessed July 20, 2021. https://www.ema.europa.eu/documents/product‐information/jyseleca‐epar‐product‐information_en.pdf
- 7.European Medicines Agency: EMA/424374/2020 ‐ Jyseleca : EPAR ‐ Public assessment report 2020. Accessed July 20, 2021. https://www.ema.europa.eu/documents/assessment‐report/jyseleca‐epar‐public‐assessment‐report_en.pdf
- 8. Namour F, Diderichsen PM, Cox E, et al. Pharmacokinetics and pharmacokinetic/pharmacodynamic modeling of filgotinib (GLPG0634), a selective JAK1 inhibitor, in support of Phase IIB dose selection. Clin Pharmacokinet. 2015;54:859‐874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Namour F, Desrivot J, Van der Aa A, Harrison P, Tasset C, van't Klooster G. Clinical confirmation that the selective JAK1 inhibitor filgotinib (GLPG0634) has a low liability for drug‐drug interactions. Drug Metab Lett. 2016;10:38‐48. [DOI] [PubMed] [Google Scholar]
- 10. Anderson K, Zheng H, Kotecha M, et al. The relative bioavailability and effects of food and acid‐reducing agents on filgotinib tablets in healthy subjects. Clin Pharmacol Drug Dev. 2019;8:585‐594. [DOI] [PubMed] [Google Scholar]
- 11. Oh J, Chung H, Park S‐I, et al. Inhibition of the multidrug and toxin extrusion (MATE) transporter by pyrimethamine increases the plasma concentration of metformin but does not increase antihyperglycaemic activity in humans. Diabetes Obes Metab. 2016;18:104‐108. [DOI] [PubMed] [Google Scholar]
- 12. Shimizu M, Uno T, Sugawara K, Tateishi T. Effects of single and multiple doses of itraconazole on the pharmacokinetics of fexofenadine, a substrate of P‐glycoprotein. Br J Clin Pharmacol. 2006;62:372‐376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jalava KM, Partanen J, Neuvonen PJ. Itraconazole decreases renal clearance of digoxin. Ther Drug Monit. 1997;19:609‐613. [DOI] [PubMed] [Google Scholar]
- 14. Härtter S, Koenen‐Bergmann M, Sharma A, et al. Decrease in the oral bioavailability of dabigatran etexilate after co‐medication with rifampicin. Br J Clin Pharmacol. 2012;74:490‐500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wang D, Zou L, Jin Q, Hou J, Ge G, Yang L. Human carboxylesterases: a comprehensive review. Acta Pharm Sin B. 2018;8:699‐712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Meng A, Anderson K, Nelson C, et al. SAT0149 exposure‐response relationships for efficacy and safety of filgotinib and its metabolite gs‐829845 in subjects with rheumatoid arthritis based on phase 2 and phase 3 studies. Ann Rheum Dis. 2020;79:1013‐1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. GLUCOPHAGE® (metformin hydrochloride) [package insert]. New Brunswick, NJ: Bristol‐Myers Squibb; 2018. Accessed September 7, 2021. https://packageinserts.bms.com/pi/pi_glucophage.pdf
- 18. Stage TB, Brosen K, Christensen MM. A comprehensive review of drug‐drug interactions with metformin. Clin Pharmacokinet. 2015;54:811‐824. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1