

Abstract
Difelikefalin, a selective kappa‐opioid receptor agonist with limited central nervous system penetration, is being developed for the treatment of chronic pruritic conditions. This randomized, double‐blind, active‐ and placebo‐controlled, four‐way crossover study was designed to evaluate the abuse potential of difelikefalin in healthy recreational polydrug users. Using a 4 × 4 Williams design, nondependent adult users of opioids and hallucinogens (N = 44) were randomized to receive single intravenous (i.v.) injections of difelikefalin at supratherapeutic doses (5 and 15 mcg/kg); pentazocine (0.5 mg/kg), a schedule IV mu‐opioid partial agonist and kappa‐opioid receptor agonist; and placebo. The abuse potential of difelikefalin was compared with pentazocine and placebo using the maximal score (maximum effect [Emax]) of the Drug Liking visual analog scale (VAS; primary end point), along with multiple secondary end points of subject‐rated measures and pupillometry. Difelikefalin produced significantly lower Drug Liking VAS Emax, and lower peak positive, sedative, and perceptual effects compared with pentazocine. These effects of difelikefalin were small, brief, and not dose‐dependent, although marginally greater than those observed with placebo. Neither dose of difelikefalin elicited significant negative or hallucinogenic effects. On end‐of‐session measures of overall drug liking and willingness to take the drug again, difelikefalin did not differ from placebo, indicating subjects neither liked nor disliked the effects overall and did not feel motivated to take the drug again. Consistent with its lack of mu agonist activity, difelikefalin did not induce miosis compared with pentazocine. All treatments were generally well‐tolerated. This study indicates that difelikefalin presents a low potential for abuse.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
Difelikefalin is a selective kappa‐opioid receptor agonist with limited central nervous system penetration being developed for the treatment of chronic pruritus. Difelikefalin has no affinity for other opioid receptors and therefore is different from opioid analgesics that predominantly bind to mu opioid receptors.
WHAT QUESTION DID THIS STUDY ADDRESS?
This randomized, double‐blind, active‐ and placebo‐controlled, four‐way crossover study addressed whether difelikefalin has abuse potential in healthy recreational polydrug users.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
Intravenous difelikefalin, at supratherapeutic doses, had an abuse potential profile that was significantly lower than the schedule IV opioid pentazocine, and not meaningfully different from placebo. Difelikefalin did not elicit significant negative or hallucinogenic effects.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
These findings suggest that difelikefalin should not be a target for diversion for recreational use. This has important clinical implications for patients who may require this treatment, and it demonstrates that peripherally restricted kappa‐opioid receptor exhibit fewer adverse effects and toxicities related to abuse over typical opioids.
INTRODUCTION
Difelikefalin is a highly selective and full agonist at human kappa‐opioid receptors (KORs; Ki 0.32 nM) 1 and is being developed for the treatment of chronic pruritic conditions as an intravenous (i.v.) and oral formulation. 2 , 3
To avoid the characteristic psychotomimetic and dysphoric effects observed with some centrally acting KOR agonists, 4 , 5 , 6 the chemical structure of difelikefalin was selected based on preclinical studies demonstrating that tetrapeptides composed of non‐natural D‐amino acids can exhibit high potency and selectivity for KORs with essentially no detectable penetration into the central nervous system (CNS). 7 , 8 , 9 Difelikefalin does not undergo significant metabolism and is not a substrate for drug uptake transporters; its high hydrophilicity and polar surface area and charge at physiological pH result in negligible membrane permeability by passive diffusion. 1
Difelikefalin has no activity at receptors other than KORs, including mu‐opioid receptors (MORs), 1 which are associated with the rewarding, dependence‐producing, and, at high doses, potentially fatal respiratory depressant effects of commonly abused opioids. 10 , 11 , 12 , 13 , 14 Difelikefalin does not induce respiratory depression in healthy volunteers, 15 and dysphoria and abuse have not been reported in any clinical studies 2 , 3 thus far, which is consistent with its limited CNS penetration and lack of activity at MORs. 1
Whereas difelikefalin binds selectively to KORs, has limited CNS penetration, and has no active metabolites or chemical similarity to controlled substances (unpublished data), it is a new molecular entity belonging to the opioid pharmacological class. 1 Therefore, a human abuse potential study in recreational drug users with opioid and hallucinogen experience was conducted to compare difelikefalin to placebo and pentazocine, a centrally acting opioid analgesic that is controlled in schedule IV (C‐IV) of the Controlled Substances Act in the United States. 16 Pentazocine was selected as a positive control because it is a KOR agonist with only slightly lower affinity for human KOR than difelikefalin (Ki range: 2.2 to 16.8 nM), 17 , 18 with partial agonist properties at MORs 17 , 18 and low but meaningful abuse potential. 19 , 20 , 21
This is the first study in humans to examine the abuse potential of a selective KOR agonist with limited CNS penetration.
METHODS
Subjects
This study was conducted at PRA Health Sciences (Salt Lake City, UT, USA) in accordance with the US Food and Drug Administration (FDA) guidance on Assessment of Abuse Potential of Drugs. 22 No study procedures were initiated prior to written informed consent of all subjects. The protocol, study site, and informed consent form were approved by the New England Institutional Review Board (Newton, MA, USA). All subjects were compensated for their participation in the study according to local guidelines.
Subjects were healthy male and female nondependent, recreational (i.e., nontherapeutic) polydrug users, 18 to 55 years of age, with a history of opioid use (≥10 occasions within the previous year and ≥1 use within 12 weeks of the screening visit) and recent experience (within 60 days) with hallucinogenic substances (e.g., ketamine, phencyclidine, peyote, psilocybin, and Salvia). Subjects had a body mass index (BMI) within 19 to 33 kg/m2 and weight greater than 55 kg.
Subjects with positive alcohol breathalyzer or urine drug of abuse test results (except for tetrahydrocannabinol) were excluded, as well as subjects with a history or current diagnosis of drug or alcohol dependence (excluding nicotine and/or caffeine) based on the Diagnostic and Statistical Manual of Mental Disorders, 4th Edition. 23 Subjects were also excluded if they smoked more than 20 cigarettes, eight pipefuls, or eight cigars daily, and were unwilling to refrain from using nicotine products at specified times during the study. No concomitant medications were allowed, except for the treatment of adverse events (AEs) or the continuous use of hormonal contraceptives.
Study design and treatment
This was a single‐center, randomized, double‐blind, active‐ and placebo‐controlled, four‐way crossover design conducted between July and October 2014. The study included screening, qualification, treatment, and follow‐up phases. Within 21 days of the screening visit, eligible subjects were confined to the clinic from the day before the qualification phase until 24 h after final treatment, for a total of 12 days. A telephone follow‐up occurred 7 to 10 days postdischarge.
The qualification phase included a naloxone challenge and a drug discrimination test to confirm the absence of opioid physical dependence and the ability to tolerate and like the subjective effects of pentazocine, respectively. In the naloxone challenge, subjects received i.v. bolus naloxone HCl (Hospira, Lake Forest, IL, USA; 0.2 mg followed by 0.6 mg if no withdrawal signs were observed within 30 s), and were assessed for opiate withdrawal signs using the 11‐item Clinical Opiate Withdrawal Scale (COWS). 24 , 25 Subjects with no withdrawal symptoms (i.e., COWS score <5) proceeded to the drug discrimination test after a 12‐h washout and were administered pentazocine (0.5 mg/kg i.v.) or placebo on 2 successive days in a two‐way crossover, double‐blind, randomized design. Drug liking (“Do you like the drug effect you are feeling now?”) was assessed with a bipolar 100‐point visual analog scale (VAS) anchored with “strong disliking” (0), “neither like nor dislike” (50), and “strong liking” (100). The ability to discriminate between placebo and pentazocine was defined by a peak Drug Liking VAS score greater than or equal to 70 in response to pentazocine, a placebo response greater than or equal to 40 and less than or equal to 60, and a difference between pentazocine and placebo greater than or equal to 15 during the first 2 h following drug administration.
Qualifying subjects entered the treatment phase (after a 24‐h washout), which consisted of four treatment periods, with single i.v. bolus injections of difelikefalin (5 or 15 mcg/kg), pentazocine (0.5 mg/kg), or placebo. Subjects received each treatment separated by a 48‐h washout period according to a 4 × 4 Williams square randomization scheme. 26 The doses of difelikefalin were selected based on the anticipated therapeutic and supratherapeutic doses (3‐fold the therapeutic dose) for a previously explored indication of postoperative pain, 27 and represent doses 10 and 30 times higher than the intended therapeutic dose (0.5 mcg/kg) for the treatment of moderate‐to‐severe pruritus in patients undergoing hemodialysis. 2 , 3 The dose of pentazocine was based on a previous study. 21 Difelikefalin solution (10 mg/ml in 0.04 M isotonic acetate buffer, pH 4.5) was supplied by Cara Therapeutics, Inc., and diluted with an appropriate volume of sterile 0.9% sodium chloride. Pentazocine was supplied as Talwin Lactate Injection (30 mg/1 ml ampoule; Hospira). Placebo was the vehicle used for difelikefalin. The dosing volume of difelikefalin, pentazocine, or placebo was 3 ml in all treatment groups.
Pharmacodynamic assessments
Pharmacodynamic (PD) assessments to evaluate drug effects consisted of multiple questionnaires completed by each subject using electronic data capture tablets (Cambridge Cognition Ltd., Cambridge, UK; FDA 21 CFR Part 11 compliant), with the exception of one paper form, the Hallucinogen Rating Scale (HRS). Measures were taken prior to dosing (for measures not referring to the drug) and at various prespecified times throughout 8 h postdose.
Drug Liking VAS was the primary outcome measure. Secondary PD measures included a drug effect questionnaire (DEQ) assessing whether subjects felt any drug effects, good effects, bad effects, feeling high, feeling sick, nauseous, sleepy, dizzy, spaced‐out, floating, detached, or hallucinating at the time the questions were asked. Each item was assessed using a unipolar VAS anchored by “not at all” (0) and “extremely” (100). A bipolar VAS was used to assess alertness/drowsiness, with anchors of “very drowsy” (0), “normal” (50), and “very alert” (100).
Hallucinogenic/psychedelic effects were assessed over a 4‐h period (0 to 4 h postdose) using the 99‐item HRS (version 3.06), a validated self‐reported questionnaire sensitive to centrally active KOR agonists. 28 , 29 , 30 The HRS assesses six aspects of hallucinogenic drug experiences: intensity (strength of the overall experience), somaesthesia (interoceptive, visceral, and tactile effects), affect (emotional and affective responses), perception (visual, auditory, gustatory, and olfactory effects), cognition (alterations in thought processes or content), and volition (capacity to willfully control one’s body movements to interact with the environment). 31 Most items were rated using a five‐point scale (0 = not at all, 1 = slightly, 2 = moderately, 3 = very much, and 4 = extremely).
Global retrospective assessments of Overall Drug Liking, Take Drug Again, and Drug Similarity VAS and the Price Value Assessment Questionnaire (PVAQ) were assessed at 8 h postdose. The Overall Drug Liking VAS assesses the subject’s global drug liking by responding to “overall, my liking for this drug is” using a bipolar VAS anchored with “strong disliking” (0), “neither like nor dislike” (50), or “strong liking” (100). Subjects also responded to “Would you want to take the drug you just received again, if given the opportunity?” by marking a bipolar VAS anchored with “definitely would not” (0), “do not care” (50), or “definitely would” (100). Similarity of the study drugs to up to 11 drug classes was assessed using a unipolar Drug Similarity VAS anchored with “not at all similar” (0) to “very similar” (100). The PVAQ asked subjects to estimate the most they would pay for the same dose of the drug they received if illicitly available, with a $0–$100 scale divided into $5 increments.
Pupillometry
Pupillary constriction (miosis) was measured using a NeurOptic VIP 200 pupillometer in a dimly lit room (minimum 1 min dark adaptation).
Pharmacokinetic evaluation
Serial blood samples were collected prior to dosing and up to 8 h postdose (i.e., at 5 min and 0.25, 0.5, 0.75, 1, 1.5, 2, 3, 4, and 8 h postdose) to determine the plasma concentrations of difelikefalin or pentazocine. Samples were analyzed according to validated methods (AIT Biosciences, Indianapolis, IN, USA), using tandem mass spectrometric detection.
Safety evaluation
Safety assessments included recording of AEs, supine and standing vital signs, 12‐lead electrocardiogram, clinical laboratory tests (hematology, chemistry, and urinalysis), physical examinations, and continuous pulse oximetry monitoring (on dosing days from predose to 8 h postdose). Because of the aquaretic properties of difelikefalin, a class effect of KOR agonists, 32 additional measurements of serum sodium were made predose and at 12 and 24 h postdose, and fluid balance was monitored throughout the treatment period.
Statistical analysis
The analysis populations, hypothesis testing, and equivalence margins described below were based on revised FDA guidance 22 issued after the study was completed and thus PD end points were re‐analyzed post hoc accordingly. Results of original prespecified analyses are consistent with these post hoc results.
Drug Liking maximum effect (Emax; i.e., maximum VAS score or peak effect) was the primary end point. All other PD parameters were secondary. The primary analysis population (completer set) consisted of subjects who completed all treatment periods and had sufficient data for evaluation of Drug Liking Emax (i.e., at least 1 observation within 2 h of time to maximum observed plasma concentration [Tmax] for each treatment). The modified completer set consisted of all subjects in the completer set, excluding subjects with similar Drug Liking Emax scores (difference within 5 points) across all study treatments (including placebo), or subjects with a Drug Liking Emax for placebo greater than 60 and a difference between Drug Liking Emax for placebo and pentazocine less than or equal to 5. The primary PD parameter, Drug Liking Emax, was analyzed using the completer and modified completer sets, whereas the secondary PD parameters were analyzed using the completer set only.
Analysis of derived PD parameters was conducted with SAS version 9.4 using procedures appropriate for the particular analysis. Model and test selection (i.e., mixed effects model, paired t‐test, or Sign test) for each primary and secondary end point were determined through a stepwise procedure that included normality testing, homogeneity of variance testing, first‐order carryover effect evaluation, and assessment of skewness of the distribution of paired differences.
The analysis of the primary end point was conducted as one‐sided hypothesis tests at a significance level of α = 0.05, with predefined equivalence margins (δ; Table 1). To assess study validity, a margin of 15 was selected based on previous studies, 33 , 34 , 35 with a lower limit of the one‐sided 95% confidence interval (CI; equivalent to 2‐sided 90% CI) greater than 15 for the difference in Drug Liking Emax between pentazocine and placebo indicating that pentazocine had a clinically relevant higher Drug Liking VAS score than placebo. To identify absolute abuse potential of difelikefalin, a margin of 11 was selected based on Chen and Bonson (2013), 36 with the upper limit of the 95% CI (equivalent to 2‐sided 90% CI) greater than or equal to 11 for the difference in Drug Liking Emax between difelikefalin and placebo indicating that difelikefalin had a clinically relevant higher Drug Liking VAS score than placebo.
TABLE 1.
Drug Liking VAS Emax by treatment (top panel; primary end point) and treatment comparison (bottom panel)
| Drug liking VAS Emax | Placebo | Difelikefalin 5 mcg/kg | Difelikefalin 15 mcg/kg | Pentazocine 0.5 mg/kg |
|---|---|---|---|---|
| Mean (SD) | 52.6 (7.81) | 66.3 (13.34) | 67.3 (13.95) | 88.2 (12.55) |
| Median (min, max) | 50.0 (50, 85) | 64.0 (50, 99) | 69.0 (50, 100) | 91.0 (49, 100) |
| Inferential analysis results | |||
|---|---|---|---|
| Pairwise comparisons | LS mean difference | 90% CI | p value |
| Pentazocine 0.5 mg/kg vs. placebo | 35.50 | (31.41, 39.59) | <0.0001 a |
| Pentazocine 0.5 mg/kg vs. difelikefalin 5 mcg/kg | 21.95 | (17.85, 26.04) | <0.0001 b |
| Pentazocine 0.5 mg/kg vs. difelikefalin 15 mcg/kg | 20.92 | (16.83, 25.01) | <0.0001 b |
| Difelikefalin 5 mcg/kg vs. placebo | 13.55 | (9.46, 17.64) | 0.8485 c |
| Difelikefalin 15 mcg/kg vs. placebo | 14.58 | (10.49, 18.67) | 0.9251 c |
Least‐squares mean differences were estimated from a mixed‐effect model having treatment, period, sequence as fixed effects and subject nested within sequence as a random effect.
A 90% CI for a two‐sided test was derived for these comparisons, which provides the equivalent lower limit (or upper limit) to that of a 95% CI for a one‐sided test.
Abbreviations: CI, confidence interval; Emax, maximum effect; LS, least‐squares; SD, standard deviation; VAS, visual analog scale.
Completer set, N = 39.
One‐sided, α = 0.05; H0: µD−µC ≤ 15 vs. H1: µD−µC > 15.
One‐sided, α = 0.05; H0: µD−µB ≤ 0 vs. H1: µD−µB > 0, and H0: µD−µA ≤ 0 vs. H1: µD−µA > 0.
One‐sided, α = 0.05; H0: µB−µC ≥ 11 vs. H1: µB−µC < 11 and H0: µA−µC ≥ 11 vs. H1: µA−µC < 11.
Where A = difelikefalin 5 mcg/kg, B = difelikefalin 15 mcg/kg, C = placebo, D = pentazocine 0.5 mg/kg
Confidence limits of interest and statistically significant p values are bolded.
No specific margins were selected for secondary end points, as there is no literature to support selection of such margins. On these end points, for comparisons of pentazocine relative to placebo and each dose of difelikefalin, the null hypothesis was that the difference was less than or equal to 0 (1‐sided tests at α = 0.05). For comparisons between each dose of difelikefalin and placebo, the null hypothesis was that the difference was equal to zero (2‐sided tests at α = 0.10). Drug Similarity VAS scores were summarized descriptively only.
Demographics and safety results were summarized descriptively. Pharmacokinetic (PK) parameters were derived using standard linear, trapezoidal noncompartmental methods (Phoenix WinNonlin, version 6.3), and summarized descriptively.
RESULTS
Disposition and demographics
A total of 69 subjects out of 72 qualified after the naloxone challenge test. Of 69 subjects who entered the drug discrimination test, 44 qualified for the treatment phase, and 39 completed the study and were included in the completer set (Figure S1). Of the five subjects who did not complete the treatment phase, three discontinued due to AEs (see Safety Results), and two withdrew consent (Figure S1). Most subjects were men (79.5%), white (86.4%) and non‐Hispanic (88.6%), with a mean (standard deviation [SD]) age of 28.0 (7.72) years. All subjects had used opioids recreationally (with a minimum of 3 and 6 occasions for men and women, respectively, in the last 12 weeks) and hallucinogens (1 to 25 occasions within the past 60 days, with 59% of subjects having ≥10 times use in the last 12 months). Additional drug classes most frequently taken in the past 12 weeks were cannabinoids and stimulants.
Pharmacodynamics
Drug Liking
Mean Drug Liking VAS scores for pentazocine were in the “liking range” (i.e., >60 points) and higher than the other treatments over the first 3 h, whereas mean scores remained close to neutral (50 points) for placebo. Mean scores for difelikefalin 5 and 15 mcg/kg were notably lower than for pentazocine and within the acceptable placebo response range (≤60) with no differences between doses (Figure 1a).
FIGURE 1.
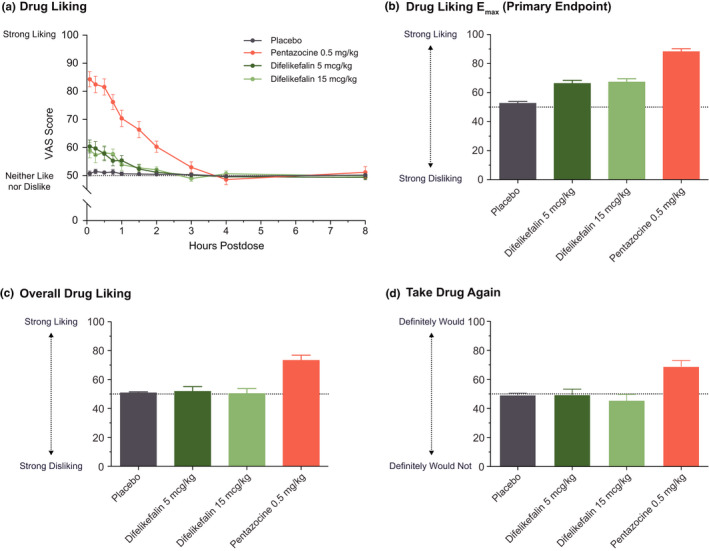
Mean (SE) drug liking VAS scores profile following administration of placebo, difelikefalin 5 and 15 mcg/kg, and pentazocine 0.5 mg/kg (a); Drug Liking VAS Emax (b); Overall Drug Liking VAS (c); and Take Drug Again VAS (d) at 8 h postdose. Completer set, N = 39. Each VAS was administered using a bipolar scale, where 50 was a neutral score. SE, standard error; VAS, Visual Analog Scale
Mean (SD) Drug Liking Emax (primary end point, completer set) was highest for pentazocine (88.2 [12.55]) and lowest for placebo (52.6 [7.81]), with mean scores for difelikefalin 5 mcg/kg (66.3 [13.34]) and 15 mcg/kg (67.3 [13.95]) showing no dose dependence (Figure 1b, Table 1). The least‐squares (LS) mean difference in Drug Liking Emax between pentazocine and placebo was 35.50 and the lower limit of the two‐sided 90% CI was 31.41 (i.e., greater than the margin of 15; p < 0.0001), thereby validating the study. The LS mean differences between pentazocine and difelikefalin 5 and 15 mcg/kg were 21.95 and 20.92, respectively, with lower limits of the 90% CI of 17.85 and 16.83, indicating that difelikefalin produced significantly less at‐the‐moment drug liking compared with pentazocine (p < 0.0001 for both comparisons). The LS mean differences between difelikefalin 5 and 15 mcg/kg and placebo were 13.55 and 14.58, respectively, with upper limits of the 90% CI of 17.64 and 18.67 (i.e., greater than the upper limit equivalence margin of 11, p > 0.8), indicating that responses to both doses of difelikefalin were greater than response to placebo (Table 1). Inferential analysis results of Drug Liking VAS Emax for the modified completer set (n = 37) were similar to those of the completer set (data not shown).
Results of the analyses of area under the effect curve from time zero to 4 or 8 h (AUE0–4; AUE0–8) for Drug Liking VAS followed the same pattern as that of Emax, with AUEs for difelikefalin significantly higher than placebo but with small mean differences (e.g., ranging from 5.19 to 8.13) compared with the mean difference between pentazocine and placebo (48.85; Table S1).
Global effects
Mean 8‐h scores for Overall Drug Liking and Take Drug Again VAS were greater than or equal to 69 points for pentazocine and significantly greater than placebo (p < 0.0001), whereas scores were at or near neutral for both doses of difelikefalin and not different from placebo (p > 0.47; Figure 1c,d, Table S1).
Mean values of PVAQ were lower for difelikefalin ($2.70 and $4.00 for 5 and 15 mcg/kg, respectively) and lowest for placebo ($1.20) compared with pentazocine ($9.60). However, median values for both difelikefalin doses and placebo were $0.00, whereas that of pentazocine was $10.00. Both doses of difelikefalin were valued significantly higher compared with placebo (p = 0.0632 and p < 0.0001), but significantly lower compared with pentazocine (p < 0.0001 for both doses; Table S1), indicating that subjects would be willing to pay more for pentazocine.
Positive, negative, and other effects
The Emax for the Good Effects, High, Any Effects, and Dizzy VAS exhibited a pattern similar to Drug Liking VAS Emax, with significantly higher Emax for pentazocine and both doses of difelikefalin relative to placebo, but Emax values for both doses of difelikefalin were significantly lower than for pentazocine (Figure 2a–d, Table S1). For the Bad Effects VAS, Emax values for pentazocine and both doses of difelikefalin were significantly higher than that of placebo, with no significant differences between pentazocine and difelikefalin and low scores overall (< 20 points), indicating relatively few negative effects (Figure 2e, Table S1). A similar pattern was observed for other negative effects measures (Feel Sick and Nausea VAS), with low scores reported overall (<15 points; Table S1). For the Alertness/Drowsiness VAS, Emax indicated minimal sedative effects (≤9 points; Figure 2f, Table S1); Emax was significantly higher for pentazocine compared with placebo and for pentazocine compared with difelikefalin 15 mcg/kg, but no significant differences were observed between pentazocine and difelikefalin 5 mcg/kg or between placebo and difelikefalin (Table S1). Pentazocine was associated with significantly higher Sleepy VAS Emax compared with placebo and both doses of difelikefalin, but only difelikefalin 15 mcg/kg had a statistically higher Emax compared with placebo (Table S1).
FIGURE 2.
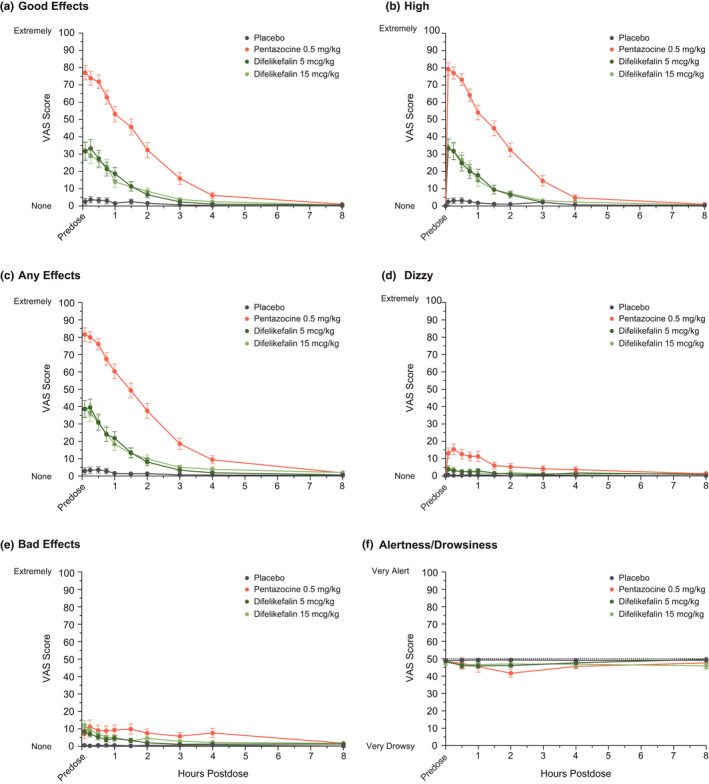
Time course of mean (SE) scores from selected unipolar VAS of the drug effects questionnaire (DEQ) (a–e) and Bipolar VAS for Alertness/Drowsiness (f). Completer Set, N = 39. SE, standard error; VAS, visual analog scale
Overall, the comparative profile of the DEQ indicates that both doses of difelikefalin have significantly fewer positive and other effects compared with pentazocine (Figure 3, Table S1) with low scores for negative effects.
FIGURE 3.
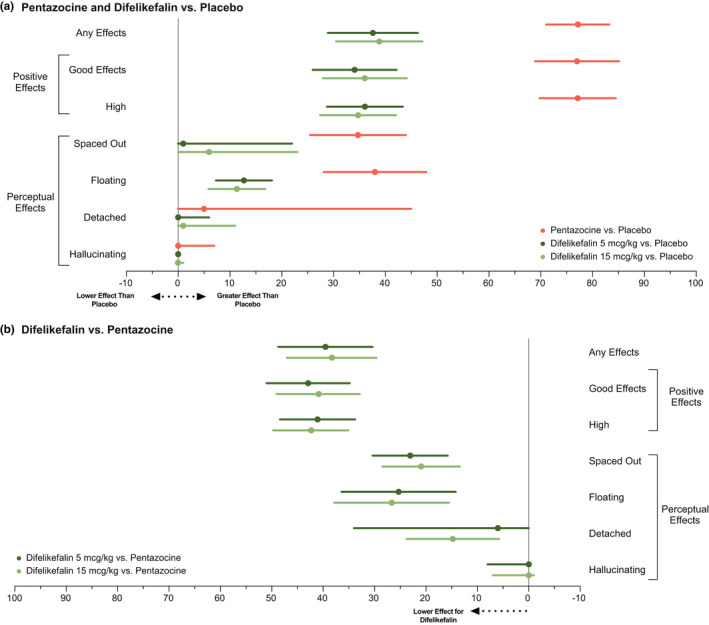
Differences in drug effects questionnaire (DEQ) VAS scores for pentazocine and difelikefalin treatment relative to placebo (a) and for pentazocine treatment relative to difelikefalin (b) for any effects, positive effects, and perceptual effects. Data represent the central value of treatment differences analysis in peak effect (Emax) with lower/upper limit for selected VAS item of the Drug Effects Questionnaire (completer set, N = 39). Depending on the test statistic used to compare treatment groups, the central value represented in the plots is either the mean or the median and the limits are either the upper/lower value of the 90% CI around the mean or the first and third quartile of the distribution. CI, confidence interval; VAS, visual analog scale
Perceptual effects
Emax values were significantly higher for pentazocine relative to placebo for the Spaced Out, Floating, and Detached VAS, whereas those for difelikefalin significantly separated from placebo but were intermediate and did not show dose dependence (Figure 3, Table S1). For the Hallucinations VAS, the Emax was low but significantly higher for pentazocine compared with placebo, with no other statistical differences on this endpoint (Figure 3, Table S1). For the 4‐h HRS subscales of Affect, Cognition, Intensity, Perception and Somaesthesia, scores were significantly higher for pentazocine and both doses of difelikefalin compared with placebo, but values for difelikefalin were significantly lower than those for pentazocine. For the HRS Volition subscale, scores were higher for pentazocine and both doses of difelikefalin compared with placebo, but no statistical difference was observed between pentazocine and either dose of difelikefalin (Figure S2, Table S1).
Drug similarity
Subjects rated pentazocine to be most similar to opioids (median: 65.0), followed by benzodiazepines (22.5), and not similar to cannabinoids, hallucinogens, or stimulants (≤2.0). Subjects’ ratings for difelikefalin were much lower compared with pentazocine for opioids (median: 14.5 and 10.5 for 5 and 15 mcg/kg, respectively) and benzodiazepines (8.0 and 1.5, respectively) and showed a decrease with increasing dose. Subjects did not rate difelikefalin as similar to cannabinoids, hallucinogens, or stimulants (median: 0.0). Subjects did not perceive placebo as similar to any drug class (Figure S3).
Pupillometry
Pupil diameter measured over the 8‐h postdose period indicated pentazocine induced miosis (Figure 4), with mean Emax reduction in pupil diameter of 2.5 mm from baseline (predose), whereas peak reductions for difelikefalin 5 mcg/kg (0.6 mm) and 15 mcg/kg (0.4 mm) were comparable to placebo (0.4 mm).
FIGURE 4.
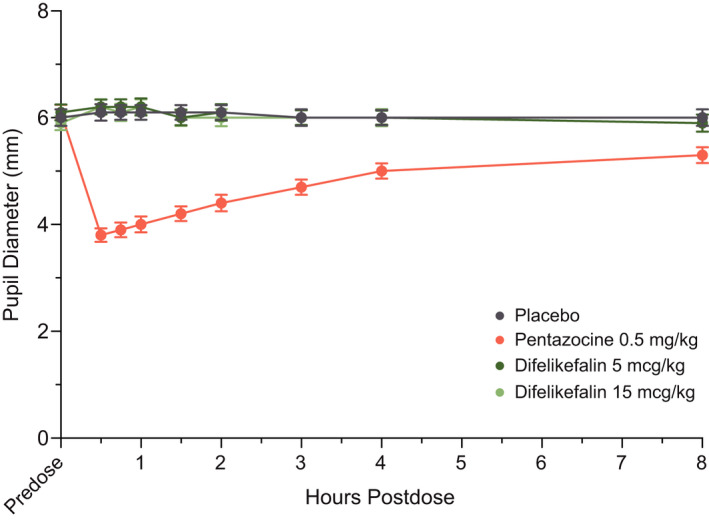
Mean (±SE) pupil diameter over time. Completer set, N = 39. SE, standard error
Pharmacokinetics
Difelikefalin plasma concentrations increased approximately dose proportionally, with a median Tmax of ~5 min and apparent plasma terminal elimination half‐life of 2 h (t1/2, Table 2). The PK parameters for pentazocine were generally consistent with previously reported data. 37 , 38 , 39 Of note, the apparent volume of distribution (Vd) for difelikefalin was substantially lower than that for pentazocine, consistent with its hydrophilic properties.
TABLE 2.
PK parameters following single intravenous dose of difelikefalin or pentazocine
| Parameter mean (SD) | Difelikefalin 5 mcg/kg (n = 41) | Difelikefalin 15 mcg/kg (n = 40) | Pentazocine 0.5 mg/kg (n = 40) |
|---|---|---|---|
| Cmax (ng/ml) | 61.9 (23.9) | 168.8 (52.6) | 296.0 (197.7) |
| Tmax (h) a | 0.1 [0.1, 0.3] | 0.1 [0.1, 0.3] | 0.1 [0.1, 0.8] |
| AUC0–t (h*ng/ml) | 94.0 (23.8) | 246.0 (40.1) | 487.4 (133.5) |
| t 1/2 (h) | 2.0 (0.2) | 2.0 (0.2) | 3.7 (1.2) |
| V d (ml/kg) | 155 (39.3) | 171 (28.0) | 4280 (1079.8) |
PK population = all randomized subjects who received ≥1 dose of study drug in the treatment phase and had adequate plasma concentration‐time data to allow for meaningful PK analysis.
Each treatment was separated by a 48‐hour washout period, which largely exceeded each treatment half‐life (t1/2) of 2 to 4 hours.
Abbreviations: AUC0–t, area under the plasma concentration‐time curve from time zero to the last quantifiable concentration; Cmax, maximum observed plasma concentration; PK, pharmacokinetic; SD, standard deviation; Tmax, time to maximum observed plasma concentration; t1/2, apparent plasma terminal elimination half‐life; Vd, apparent volume of distribution.
Median [range].
Safety results
All treatments were generally well‐tolerated, and all treatment‐emergent AEs (TEAEs) were mild in severity, with the exception of a moderate TEAE of anxiety in one subject that resulted in discontinuation. Three subjects discontinued from the study due to TEAEs following difelikefalin 5 mcg/kg (anxiety; injection‐site phlebitis) or pentazocine (muscle spasms). The most common TEAEs (>10%) were nausea and vomiting with pentazocine, and dizziness with difelikefalin 15 mcg/kg (Table 3).
TABLE 3.
Treatment‐emergent adverse events
| Subjects, n (%) | Placebo | Difelikefalin 5 mcg/kg | Difelikefalin 15 mcg/kg | Pentazocine 0.5 mg/kg |
|---|---|---|---|---|
| n = 41 | n = 41 | n = 40 | n = 40 | |
| Any TEAE | 6 (14.6) | 10 (24.4) | 16 (40.0) | 13 (32.5) |
| Any severe TEAE | 0 | 0 | 0 | 0 |
| Any serious TEAE | 0 | 0 | 0 | 0 |
| Discontinuation due to TEAE | 0 | 2 (4.9) | 0 | 1 (2.5) |
| TEAEs reported in ≥2 patients during any treatment | ||||
| Abdominal pain | 0 | 1 (2.4) | 2 (5.0) | 0 |
| Abdominal pain upper | 0 | 0 | 2 (5.0) | 0 |
| Constipation | 0 | 0 | 2 (5.0) | 1 (2.5) |
| Dyspepsia | 0 | 2 (4.9) | 1 (2.5) | 0 |
| Nausea | 0 | 1 (2.4) | 1 (2.5) | 5 (12.5) |
| Vomiting | 1 (2.4) | 0 | 2 (5.0) | 5 (12.5) |
| Chills | 0 | 1 (2.4) | 0 | 2 (5.0) |
| Groin pain | 0 | 2 (4.9) | 1 (2.5) | 0 |
| Dizziness | 0 | 1 (2.4) | 5 (12.5) | 2 (5.0) |
| Headache | 1 (2.4) | 3 (7.3) | 4 (10.0) | 4 (10.0) |
| Hypoesthesia | 0 | 2 (4.9) | 1 (2.5) | 1 (2.5) |
| Hot flush | 0 | 0 | 0 | 3 (7.5) |
Safety Population = all randomized subjects who received ≥1 dose of study medication in the treatment phase. Although 44 patients began the randomized treatment phase of the study, not all patients received all four treatments due to discontinuations.
Abbreviation: TEAE, treatment emergent adverse event.
No clinically significant changes from baseline in clinical laboratory or vital sign values were observed. Despite an increased volume of urine excretion following difelikefalin administration, proactive water consumption contributed to maintaining fluid balance, with overall fluid balance remaining close to neutral.
DISCUSSION
This study evaluated the human abuse potential of single i.v. doses of difelikefalin, a novel KOR agonist with limited CNS penetration, relative to pentazocine and placebo. The overall pattern of results suggests that difelikefalin has a significantly lower abuse potential than the C‐IV pentazocine in recreational polydrug users with opioid and hallucinogen experience. Study validity was confirmed by a significantly higher peak drug liking for pentazocine relative to placebo, and other measures.
In the primary analysis, doses of difelikefalin that are 10 and 30 times the planned therapeutic dose for pruritus in patients undergoing hemodialysis, 2 were associated with significantly lower peak Drug Liking VAS scores when compared with pentazocine but higher than placebo. However, difelikefalin did not differ from placebo on the measures of overall drug liking and willingness to take the drug again, suggesting limited reinforcing potential. These end‐of‐session measures have the advantage of assessing the overall drug experience and provide a valuable index of the likelihood of future abuse as they require the subjects to consider the entire drug experience and are less affected by potential acute drug effects at earlier assessments. 40 The transient elevations of at‐the‐moment drug liking with difelikefalin may be due to previous drug experience generating expectancy. Of note, an approximately three‐fold increase in difelikefalin exposure produced no further increase in drug liking, no effect on the duration of the response, and no effects on other measures, which is inconsistent with the profile of a drug with abuse potential. In addition, difelikefalin did not induce miosis, a physiological indicator of CNS MOR activity, 40 and was not rated as being similar to a typical opioid, results that are consistent with its lack of MOR agonist activity, in contrast to pentazocine.
Similarly, across other secondary end points, both pentazocine and difelikefalin have low negative effects overall but difelikefalin had significantly lower positive, sedative, and perceptual effects when compared with pentazocine, although marginally higher than those observed with placebo.
In contrast with the dose‐dependent hallucinogenic and dysphoric effects of centrally acting KOR agonists, 4 , 5 , 6 , 29 , 30 difelikefalin produced only marginal subjective effects (e.g., low HRS scores) despite its high potency at the KOR and the use of supratherapeutic doses. The lack of hallucinogenic effects, a basis for potential abuse of a KOR agonist, 29 , 30 is consistent with limited CNS penetration and low abuse potential.
Difelikefalin had an acceptable safety profile with TEAEs that were generally mild in severity. The overall incidence of TEAEs increased with increasing dose of difelikefalin. The incidence of AEs that are more typical of MOR agonists, such as nausea and vomiting, were lower for difelikefalin compared with pentazocine, consistent with its selectivity for KORs. Consistent with the low HRS scores, there were no TEAEs related to psychotomimesis. These findings are in line with the lack of reports of euphoric mood or dysphoria in controlled phase III clinical studies in patients with hemodialysis with pruritus. 2 Furthermore, no AEs suggesting misuse, abuse, diversion, or dependence were reported across more than 4000 subjects and healthy volunteers exposed to i.v. or oral difelikefalin thus far (data on file).
The FDA Guidance for abuse potential recommends evaluating the highest proposed therapeutic dose, along with doses at least two to three times greater. 22 In this study, difelikefalin doses were 10‐ and 30‐fold the proposed therapeutic dose. 2 Because difelikefalin did not produce meaningful negative subjective effects, with Emax less than 20 on unipolar 100‐point VAS, the selected doses are relevant to the abuse potential of difelikefalin and allowed assessment of potential rewarding effects of a drug with limited CNS penetration.
CONCLUSIONS
Considering the overall pattern of results across the measures evaluated in this study, it appears unlikely that difelikefalin will be regarded as an attractive drug of abuse for recreational drug users. Although it did differentiate marginally from placebo, difelikefalin did not present evidence of meaningful abuse potential with overall signals not only small but also limited in duration, and not dose dependent. The dose‐effect ceiling is in line with limited CNS penetration, and indicates there would be little incentive for drug abusers to increase the dose of difelikefalin to achieve a greater effect. The pharmacological profile of difelikefalin, including the findings of this study, suggest that it should not be a target for diversion for recreational use. Difelikefalin was approved by the FDA in August 2021 for the treatment of moderate‐to‐severe pruritus in adults undergoing hemodialysis and is not considered a controlled substance. 41
CONFLICTS OF INTEREST
F.M., R.H.S., J.Q., and C.L.M. are employees of Cara Therapeutics, Inc. F.M., R.H.S., J.Q., C.L.M., and M.E.L. are shareholders in Cara Therapeutics, Inc. M.J.S. and J.E.H. report consultant fees from Cara Therapeutics, Inc. L.W. was an employee and shareholder of PRA Health Sciences during the conduct of the study.
AUTHOR CONTRIBUTIONS
All authors wrote the manuscript. F.M., L.W., and R.H.S. designed the research. L.W. performed the research. M.J.S., J.Q., and C.M. analyzed the data.
DATA
Please contact Cara Therapeutics for data inquiries.
Supporting information
Fig S1
Fig S2
Fig S3
Table S1
ACKNOWLEDGEMENTS
The authors wish to give posthumous thanks to Reginald V. Fant, PhD, and Robert A. Medve, MD, for their invaluable contributions to the design and execution of this study. The authors also thank Rick Strassman, MD, for his advice and interpretation of the HRS results; Jessica Alvey (PRA HealthSciences) for her invaluable statistical guidance; and Amy Shaberman, PhD (Peloton Advantage, LLC, an OPEN Health company, Parsippany, NJ), who provided medical writing and editorial support, which was funded by Cara Therapeutics, Inc., under the direction of the authors. We thank the subjects who participated in this study.
Shram MJ, Spencer RH, Qian J, et al. Evaluation of the abuse potential of difelikefalin, a selective kappa‐opioid receptor agonist, in recreational polydrug users. Clin Transl Sci. 2022;15:535–547. 10.1111/cts.13173
Clinical trial number and registry URL: Not applicable (NA); not mandatory at the time of the study.
Funding information
This study was sponsored by Cara Therapeutics.
REFERENCES
- 1. Albert‐Vartanian A, Boyd MR, Hall AL, et al. Will peripherally restricted kappa‐opioid receptor agonists (pKORAs) relieve pain with less opioid adverse effects and abuse potential? J Clin Pharm Ther. 2016;41(4):371‐382. [DOI] [PubMed] [Google Scholar]
- 2. Fishbane S, Jamal A, Munera C, Wen W, Menzaghi F. A phase 3 trial of difelikefalin in hemodialysis patients with pruritus. N Engl J Med. 2020;382(3):222‐232. [DOI] [PubMed] [Google Scholar]
- 3. Fishbane S, Mathur V, Germain MJ, et al. Randomized controlled trial of difelikefalin for chronic pruritus in hemodialysis patients. Kidney Int Rep. 2020;5(5):600‐610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Pfeiffer A, Brantl V, Herz A, Emrich HM. Psychotomimesis mediated by kappa opiate receptors. Science. 1986;233(4765):774‐776. [DOI] [PubMed] [Google Scholar]
- 5. Pande AC, Pyke RE, Greiner M, Wideman GL, Benjamin R, Pierce MW. Analgesic efficacy of enadoline versus placebo or morphine in postsurgical pain. Clin Neuropharmacol. 1996;19(5):451‐456. [DOI] [PubMed] [Google Scholar]
- 6. Walsh SL, Strain EC, Abreu ME, Bigelow GE. Enadoline, a selective kappa opioid agonist: comparison with butorphanol and hydromorphone in humans. Psychopharmacology. 2001;157(2):151‐162. [DOI] [PubMed] [Google Scholar]
- 7. Vanderah TW, Schteingart CD, Trojnar J, Junien JL, Lai J, Riviere PJ. FE200041 (D‐Phe‐D‐Phe‐D‐Nle‐D‐Arg‐NH2): A peripheral efficacious kappa opioid agonist with unprecedented selectivity. J Pharmacol Exp Ther. 2004;310(1):326‐333. [DOI] [PubMed] [Google Scholar]
- 8. Vanderah TW, Largent‐Milnes T, Lai J, et al. Novel D‐amino acid tetrapeptides produce potent antinociception by selectively acting at peripheral kappa‐opioid receptors. Eur J Pharmacol. 2008;583(1):62‐72. [DOI] [PubMed] [Google Scholar]
- 9. Dooley CT, Ny P, Bidlack JM, Houghten RA. Selective ligands for the mu, delta, and kappa opioid receptors identified from a single mixture based tetrapeptide positional scanning combinatorial library. J Biol Chem. 1998;273(30):18848‐18856. [DOI] [PubMed] [Google Scholar]
- 10. Boom M, Niesters M, Sarton E, Aarts L, Smith TW, Dahan A. Non‐analgesic effects of opioids: opioid‐induced respiratory depression. Curr Pharm Des. 2012;18(37):5994‐6004. [DOI] [PubMed] [Google Scholar]
- 11. Valentino RJ, Volkow ND. Untangling the complexity of opioid receptor function. Neuropsychopharmacology. 2018;43(13):2514‐2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Darcq E, Kieffer BL. Opioid receptors: drivers to addiction? Nat Rev Neurosci. 2018;19(8):499‐514. [DOI] [PubMed] [Google Scholar]
- 13. Aldrich JV, McLaughlin JP. Peptide kappa opioid receptor ligands: potential for drug development. AAPS J. 2009;11(2):312‐322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dhaliwal A, Gupta M. Physiology, Opioid Receptor. StatPearls. StatPearls Publishing; 2021. [PubMed] [Google Scholar]
- 15. Viscusi ER, Torjman MC, Munera CL, Stauffer JW, Setnik BS, Bagal SN. Effect of difelikefalin, a selective kappa opioid receptor agonist, on respiratory depression: a randomized, double‐blind, placebo‐controlled trial. Clin Transl Sci. 2021;14:1886‐1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lists of: Scheduling Actions Controlled Substances Regulated Chemicals. 2020. https://www.deadiversion.usdoj.gov/schedules/orangebook/orangebook.pdf. Accessed September 11, 2020.
- 17. Zhu J, Luo LY, Li JG, Chen C, Liu‐Chen LY. Activation of the cloned human kappa opioid receptor by agonists enhances [35S]GTPgammaS binding to membranes: determination of potencies and efficacies of ligands. J Pharmacol Exp Ther. 1997;282(2):676‐684. [PubMed] [Google Scholar]
- 18. Toll L, Berzetei‐Gurske IP, Polgar WE, et al. Standard binding and functional assays related to medications development division testing for potential cocaine and opiate narcotic treatment medications. NIDA Res Monogr. 1998;178:440‐466. [PubMed] [Google Scholar]
- 19. Wood AJ, Moir DC, Campbell C, et al. Medicines evaluation and monitoring group: central nervous system effects of pentazocine. Br Med J. 1974;1(5903):305‐307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Preston KL, Bigelow GE, Liebson IA. Comparative evaluation of morphine, pentazocine and ciramadol in postaddicts. J Pharmacol Exp Ther. 1987;240(3):900‐910. [PubMed] [Google Scholar]
- 21. Zacny JP, Hill JL, Black ML, Sadeghi P. Comparing the subjective, psychomotor and physiological effects of intravenous pentazocine and morphine in normal volunteers. J Pharmacol Exp Ther. 1998;286(3):1197‐1207. [PubMed] [Google Scholar]
- 22. Assessment of abuse potential of drugs: Guidance for industry. 2017. https://www.fda.gov/regulatory‐information/search‐fda‐guidance‐documents/assessment‐abuse‐potential‐drugs. Accessed September 10, 2020.
- 23. American Psychiatric Association . Diagnostic and Statistical Manual of Mental Disorders: DSM‐IV‐TR. American Psychiatric Association; 2000. [Google Scholar]
- 24. Tompkins DA, Bigelow GE, Harrison JA, Johnson RE, Fudala PJ, Strain EC. Concurrent validation of the Clinical Opiate Withdrawal Scale (COWS) and single‐item indices against the Clinical Institute Narcotic Assessment (CINA) opioid withdrawal instrument. Drug Alcohol Depend. 2009;105(1–2):154‐159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wesson DR, Ling W. The Clinical Opiate Withdrawal Scale (COWS). J Psychoactive Drugs. 2003;35(2):253‐259. [DOI] [PubMed] [Google Scholar]
- 26. Williams EJ. Experimental designs balanced for the estimation of residual effects of treatments. Aust J Sci Res. 1949;2(2):149‐168. [Google Scholar]
- 27. Menzaghi F, Spencer R, Abrouk N, Lewis M, Chalmers D. CR845, a peripheral kappa opioid, provides better pain relief with less nausea and vomiting than placebo in patients after bunionectomy [abstract 422]. J Pain. 2015;16(4 suppl):S81. [Google Scholar]
- 28. Strassman RJ, Qualls CR, Uhlenhuth EH, Kellner R. Dose‐response study of N, N‐dimethyltryptamine in humans. II. Subjective effects and preliminary results of a new rating scale. Arch Gen Psychiatry. 1994;51(2):98‐108. [DOI] [PubMed] [Google Scholar]
- 29. González D, Riba J, Bouso JC, Gómez‐Jarabo G, Barbanoj MJ. Pattern of use and subjective effects of Salvia divinorum among recreational users. Drug Alcohol Depend. 2006;85(2):157‐162. [DOI] [PubMed] [Google Scholar]
- 30. Addy PH. Acute and post‐acute behavioral and psychological effects of salvinorin A in humans. Psychopharmacology. 2012;220(1):195‐204. [DOI] [PubMed] [Google Scholar]
- 31. Riba J, Rodríguez‐Fornells A, Strassman RJ, Barbanoj MJ. Psychometric assessment of the Hallucinogen Rating Scale. Drug Alcohol Depend. 2001;62(3):215‐223. [DOI] [PubMed] [Google Scholar]
- 32. Inan S. Kappa opioid agonist‐induced diuresis: characteristics, mechanisms, and beyond [published online ahead of print January 23, 2021]. Handb Exp Pharmacol. 1‐17. 10.1007/164_2020_399. [DOI] [PubMed] [Google Scholar]
- 33. Wilbraham D, Berg PH, Tsai M, et al. Abuse potential of lasmiditan: a phase 1 randomized, placebo‐ and alprazolam‐controlled crossover study. J Clin Pharmacol. 2020;60(4):495‐504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pathak S, Vince B, Kelsh D, et al. Abuse potential of samidorphan: a phase I, oxycodone‐, pentazocine‐, naltrexone‐, and placebo‐controlled study. J Clin Pharmacol. 2019;59(2):218‐228. [DOI] [PubMed] [Google Scholar]
- 35. Schoedel KA, Szeto I, Setnik B, et al. Abuse potential assessment of cannabidiol (CBD) in recreational polydrug users: A randomized, double‐blind, controlled trial. Epilepsy Behav. 2018;88:162‐171. [DOI] [PubMed] [Google Scholar]
- 36. Chen L, Bonson KR. An equivalence test for the comparison between a test drug and placebo in human abuse potential studies. J Biopharm Stat. 2013;23(2):294‐306. [DOI] [PubMed] [Google Scholar]
- 37. Beckett AH, Taylor JF, Kourounakis P. The absorption, distribution and excretion of pentazocine in man after oral and intravenous administration. J Pharm Pharmacol. 1970;22(2):123‐128. [DOI] [PubMed] [Google Scholar]
- 38. Berkowitz BA, Asling JH, Shnider SM, Way EL. Relationship of pentazocine plasma levels to pharmacological activity in man. Clin Pharmacol Ther. 1969;10(3):320‐328. [DOI] [PubMed] [Google Scholar]
- 39. Agurell S, Boréus LO, Gordon E, Lindgren JE, Ehrnebo M, Lönroth U. Plasma and cerebrospinal fluid concentrations of pentazocine in patients: assay by mass fragmentography. J Pharm Pharmacol. 1974;26(1):1‐8. [DOI] [PubMed] [Google Scholar]
- 40. Comer SD, Zacny JP, Dworkin RH, et al. Core outcome measures for opioid abuse liability laboratory assessment studies in humans: IMMPACT recommendations. Pain. 2012;153(12):2315‐2324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Cara Therapeutics, Inc . Korsuva (difelikefalin) [package insert]. Cara Therapeutics, Inc.; 2021. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1
Fig S2
Fig S3
Table S1