

Abstract
Pharmacokinetic drug interactions precipitated by botanical and other natural products (NPs) remain critically understudied. Investigating these complex interactions is fraught with difficulties due to the methodologic and technical challenges associated with the inherently complex chemistries and product variability of NPs. This knowledge gap is perpetuated by a continuing absence of a harmonized framework regarding the design of clinical pharmacokinetic studies of NPs and NP‐drug interactions. Accordingly, this Recommended Approach, the fourth in a series of Recommended Approaches released by the Center of Excellence for Natural Product Drug Interaction Research (NaPDI Center), provides recommendations for the design of clinical pharmacokinetic studies involving NPs. Building on prior Recommended Approaches and data generated from the NaPDI Center, such a framework is presented for the design of (1) phase 0 studies to assess the pharmacokinetics of an NP and (2) clinical pharmacokinetic NP‐drug interaction studies. Suggestions for NP sourcing, dosing, study design, participant selection, sampling periods, and data analysis are presented. With the intent to begin addressing the gap between regulatory agencies’ guidance documents about drug‐drug interactions and contemporary NPDI research, the objective of this Recommended Approach is to propose methods for the design of clinical pharmacokinetic studies of NPs and NP‐drug interactions.
INTRODUCTION
Pharmacokinetic interactions between pharmaceutical medications and botanical or other natural products (NPs) are uniquely difficult to predict and evaluate. However, the need for such assessments is compelling. The rationale is that the practice of self‐medicating with NPs is increasingly widespread and concentrated among individuals potentially at high risk for adverse NP‐drug interactions (NPDIs). A large survey of American adults (n = 26,157) reported that approximately one‐third used NPs. 1 NP usage was higher among individuals taking prescription medications and those afflicted with cardiovascular disease, diabetes, or cancer. 1 These data indicated that NP use is common in populations treated with drugs with narrow therapeutic indices; in fact, NP‐chemotherapeutic agent interactions have been a longstanding concern. 2 The collective data highlight that NP use is high among individuals with health risks, probably because of beliefs surrounding a benefit of “naturalness” and a desire to avoid toxicity from prescription medications. 3 , 4
Textbook examples of pharmacokinetic NPDIs include those involving the NP precipitants (aka perpetrators) St. John’s wort, a cytochrome P450 (CYP) 3A inducer, and grapefruit juice, a CYP3A inhibitor. Co‐consuming object (aka victim) drugs that are CYP3A substrates (e.g., cyclosporine, tacrolimus, hepatitis C protease inhibitors, HIV protease inhibitors, and several anti‐cancer agents) with St. John’s wort can lead to increased drug clearance and potentially reduced efficacy, whereas co‐consuming these drugs with grapefruit juice can lead to decreased drug clearance and potential toxicity. 5 , 6 Unlike NPs, the potential for concomitant medications to precipitate pharmacokinetic interactions with object drugs is well‐established, and multiple regulatory agencies have produced guidance documents about how to assess such drug‐drug interactions (DDIs). 7 , 8 , 9 , 10 , 11 Because DDI liability is well‐established for pharmaceutical drugs, DDIs can be averted by avoiding concomitant medications that are known inhibitors/inducers of relevant drug metabolizing enzymes and/or transporters or minimized via dosage adjustment of the precipitant and/or object drug. However, with few exceptions (e.g., St. John’s wort and grapefruit juice), whether a pharmacokinetic NPDI adversely impacts the outcomes (i.e., decreases effectiveness and/or increases toxicity) of an object drug typically is not known. Thus, perturbation of object drug pharmacokinetics, and in turn drug response, by an NP in the population at large and especially in vulnerable patient groups who report the highest use of NPs, constitutes an unmonitored and ongoing public health concern.
Recognizing the need to mitigate potential NPDI risk, the National Institutes of Health National Center for Complementary and Integrative Health established the Center of Excellence for Natural Product Drug Interaction Research (NaPDI Center), which was tasked to develop a set of Recommended Approaches to guide optimal NPDI research studies. 12 These Recommended Approaches provide frameworks for the (i) selection and prioritization of NPs as precipitants of potential pharmacokinetic NPDIs 13 ; (ii) sourcing and characterization of NPs for in vitro and in vivo pharmacokinetic studies 14 ; and (iii) evaluation of an NP as a precipitant of pharmacokinetic NPDIs using predictive static and/or physiologically‐based pharmacokinetic (PBPK) models 15 (Figure 1). These predictive models are based on in vitro studies assessing the potential of an NP to inhibit or induce drug metabolizing enzymes (e.g., CYPs) or transporters (e.g., organic anion transporting polypeptides) at pharmacologically relevant unbound plasma or intestinal concentrations of the NP constituent(s). Typically, these studies are conducted with individually expressed recombinant enzymes or transporter‐expressing cell lines/vesicles, followed with human liver microsomes/cytosol (for enzymes) or human hepatocytes (for enzymes and transporters). If the static and/or PBPK models predict a potential NPDI with the object drug, a clinical pharmacokinetic NPDI study will be triggered.
FIGURE 1.
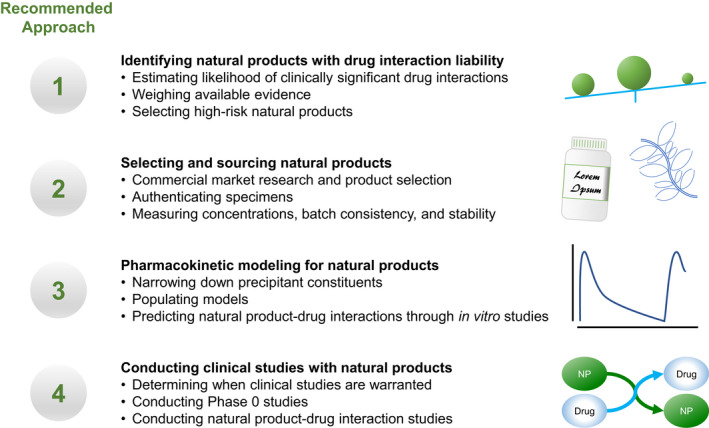
The National Center for Complementary and Integrative Health established the Center of Excellence for Natural Product Drug Interaction Research (NaPDI Center), whose primary goal is to develop guidelines for natural product‐drug interaction research studies. The current work is the fourth in a series of Recommended Approaches released by the NaPDI Center. These recommendations cover fundamental topics such as determining which natural products (NPs) or constituents are priorities for study, 13 sourcing NPs, 14 predicting pharmacokinetic NP‐drug interactions, 15 and conducting clinical pharmacokinetic studies when warranted (present work)
This Recommended Approach, the fourth in the series from the NaPDI Center, builds upon prior publications by presenting a strategy for conducting clinical pharmacokinetic studies involving NPs. Two types of studies are discussed: (i) phase 0 studies designed to determine the pharmacokinetics of a given NP for the first time, and (ii) clinical NPDI studies designed to determine the effects of an NP on the pharmacokinetics of (an) object drug(s).
WHEN ARE CLINICAL NATURAL PRODUCT‐DRUG INTERACTION STUDIES WARRANTED?
Due to the intensive resource and time requirements, conducting clinical studies for multiple NPs with unknown drug interaction liability is not feasible. Consequently, the NaPDI Center developed decision trees to guide the determination of whether clinical pharmacokinetic studies are warranted. 12 Briefly, if there are no in vitro data available but an NPDI is suspected, mechanistic in vitro studies involving established probe drugs and model systems should be conducted as described 13 , 15 (Figure 2). The advantage of these studies is that results can be extrapolated to other drugs metabolized or transported by the probed enzymes and transporters. For example, an in vitro‐in vivo extrapolation approach involving a mechanistic static model predicted 12 of 15 CYP3A‐mediated grapefruit juice‐drug interactions to within 25% of the observed change in object drug AUC. 16 More recently, mechanistic static models predicted that mitragynine, a major alkaloid in kratom (Mitragyna speciosa), would increase systemic exposure to midazolam and other CYP3A substrates by 1.55‐fold (zolpidem) to 14‐fold (buspirone) 17 and that the major cannabinoids in cannabis (Cannabis sativa), cannabidiol and (−)‐trans‐Δ9‐tetrahydrocannabinol, would increase systemic exposure to various CYP probe drugs by up to 24‐fold. 18 Regarding transporter‐mediated NPDIs, basic models were used to predict whether an interaction would occur between goldenseal and the transporter probe drugs furosemide, metformin, and rosuvastatin. 19 Additional examples are summarized in the NaPDI Center’s most recent Recommended Approach. 15
FIGURE 2.
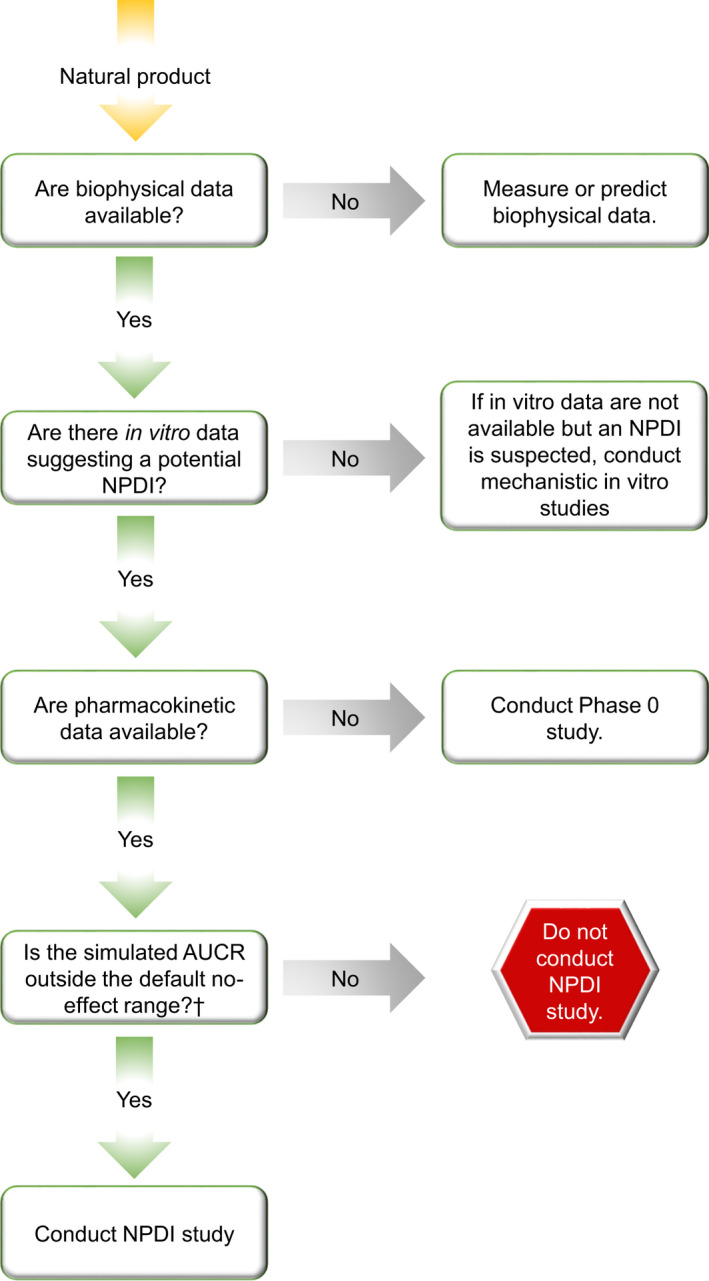
Decision tree for determining whether to conduct clinical pharmacokinetic phase 0 or natural product‐drug interaction (NPDI) studies. Phase 0 studies should be conducted when the pharmacokinetics of the phytoconstituents composing a natural product are unknown. These studies should yield fundamental metrics such as area under the blood or plasma concentration versus time curve (AUC), AUC ratio (AUCR), maximum concentration (Cmax), time to reach Cmax (tmax), and terminal half‐life (t1/2). Powered NPDI studies should be conducted if both of the following are true: (i) a phase 0 (or equivalent initial pharmacokinetic study) has been conducted and fundamental metrics are available, and (ii) an NPDI is predicted based on an AUCR laying outside the no‐effect range. †Default no‐effect range is 0.80–1.25 7
If biophysical and in vitro NPDI data are available and suggest an NPDI, a phase 0 study is merited to describe pharmacokinetic behavior of select phytoconstituents (targeted based on abundance and plausibility of biological effects) and recover fundamental metrics (e.g., area under the blood or plasma concentration versus time curve [AUC], maximum concentration [Cmax], time to reach Cmax [tmax], terminal half‐life ]t1/2], and renal clearance if urine is collected). Finally, if robust clinical pharmacokinetic information is available, in vitro data are available suggesting an NPDI, and static and/or PBPK models predict object drug AUC ratios (i.e., AUC of the object drug in the presence to absence of an NP) that lay outside the default no effect range of 0.80–1.25, 7 a powered clinical pharmacokinetic NPDI study in healthy volunteers may be warranted.
NATURAL PRODUCT SELECTION
To ensure the results of a clinical pharmacokinetic NPDI study will be generalizable, the NP to be studied should be a formulation that is commercially available and commonly used. The specific product should be selected and characterized in detail according to the NaPDI Center’s previous Recommended Approach. 14 Details that should be reported include genus and species, batch numbers, manufacturers, and both the labeled and measured concentrations of key phytoconstituents present in the NP. Additionally, for a clinical NPDI study, the product should be chosen to best reveal the interaction at the suspected site. For example, if the site of a suspected NPDI is the intestine, and the typical administration route for the commercially available formulation is oral, an ideal approach would be to study multiple oral formulations. This task can be daunting and may necessitate multiple studies with different products and probe drugs. For example, cannabis products can be administered orally, topically, or via inhalation and may precipitate NPDIs in the intestine, liver, and/or lung. 20 As aforementioned, cannabinoids have the potential to inhibit multiple CYPs, 18 , 20 but the site and magnitude of cannabinoid‐drug interactions likely will depend on the route of cannabis administration and associated peak tissue concentrations.
REDUCING PRE‐ANALYTICAL ERROR
If the stability of the NP phytoconstituent(s) in biofluids (e.g., plasma and urine) is unknown, a subset of initial pharmacokinetic samples should be processed in real time. Preferably, stability studies should precede the design of the pharmacokinetic study. Alternatively, a larger volume of blood/plasma can be collected per time point such that aliquots can be diverted to stability analysis.
ANTICIPATING UNEXPECTED RESULTS
As with any clinical pharmacokinetic study, an unexpected NPDI may stem from interindividual differences, including pharmacogenomics, race/ethnicity, sex, age, and dietary habits. An initial clinical NPDI study can be designed to anticipate the need for post hoc analyses of these effects. For example, participants may be presented the option to consent to collection of additional bio‐banking samples, which can be used for follow‐up metabolomics, metagenomics, or pharmacogenomics analyses if necessary.
PHASE 0 CLINICAL STUDY DESIGN
Study design and participants
The purpose of a phase 0 clinical study of an NP is to generate initial pharmacokinetic data to guide simulation of potential NPDIs. These studies should involve a typical consumer dose of the NP and a small number of participants, typically 5–10 healthy adult volunteers. Healthy volunteers are preferred to minimize potential adverse effects. Patients with any known health conditions are not preferred, as perturbations in drug pharmacokinetics may have adverse effects and alter the disposition of comedications. The healthy volunteers should be nonsmoking, should not be taking concomitant medications known to alter the activity of a drug metabolizing enzyme/transporter, and should not have known health problems. The cohort should be equally divided between men and nonpregnant, nonlactating women and should be ethnically and racially representative of the population. The protocol should include a predose period (e.g., 24 h) during which participants abstain from the following:
Caffeine due to the potential for acute interactions with CYP1A2 and having diuretic effects;
Alcohol;
Grapefruit juice and juices prepared from other related citrus fruits (e.g., pomelo) due to the potential for time‐dependent inhibition of intestinal CYP3A4;
Fruit juices (e.g., grapefruit, apple, and orange) and green tea due to the potential for acute interactions with intestinal uptake transporters, such as organic anion transporting peptides (OATPs);
Any other NPs, including dietary supplements;
Heavy exercise due to changes in xenobiotic clearance from acutely increased cardiac output and blood flow rate; and
Cannabis products (for at least 3 weeks prior to the study) due to the potential for interactions with various drug metabolizing enzymes and transporters.
Dosing and sample collection
Dosing regimens for a phase 0 study to determine NP phytoconstituent(s) pharmacokinetics differ from principles set forth by the US Food and Drug Administration (FDA) for exploratory investigational new drug studies. 21 Generally, sub‐pharmacological doses for NPs are nonexistent, and microdosing is not feasible. Rather, a fixed single‐dose regimen using a typical consumer dose of the NP is preferred to ensure that exposure is minimized and results are generalizable. The time of day the NP is administered should be defined based on common usage. The pharmacokinetics of an NP selected for a phase 0 study often are completely uncharacterized, but some pharmacokinetic end points and even concentration‐time profiles may be predicted from biophysical data using contemporary software, 15 , 22 allowing researchers to align sampling windows with the predicted maximum concentration of an uncharacterized phytoconstituent. This technique also can be used to derive estimates of the clearance of a given phytoconstituent to guide the duration of sample collection during the terminal elimination phase.
Clinical laboratory tests
Because the safety profiles of object drugs are known, select safety data can generally be collected for NPDI studies. 23 However, when predictive static or PBPK models or preclinical data indicate that the magnitude of an NPDI is likely to exceed the cutoff (i.e., producing an area under the curve ratio [AUCR] outside of the 0.80–1.25 range), 7 a more complete set of safety data may be warranted. For example, hematology and clinical chemistry should be included in the study schedule of events.
Data reporting
Study results should include the data elements presented in Table 1.
TABLE 1.
Data to report from clinical pharmacokinetic studies involving natural products
| Metric for specific natural product constituent(s) | Description |
|---|---|
| AUC | Geometric mean and 90% confidence interval |
| Cmax | Geometric mean and 90% confidence interval |
| tmax | Median and range |
| t1/2 | Geometric mean and 90% confidence interval |
| ClR (if urine and blood collected) | Geometric mean and 90% confidence interval |
| Ae (if urine collected) | Geometric mean and 90% confidence interval |
| Treatment compliance | Frequency tables of missed doses |
| Treatment duration | Average days of participation per subject, reported as mean and SD |
| Participant demographics | Age, sex, ethnicity/race, body mass index, vital signs |
| Bioanalytical variables | Lower limits of detection, percentage of samples outside limits of detection |
| Natural product data | Source (vendor information, lot and batch numbers), genus and species, formulation, preparation, measured and labeled concentration(s) of phytoconstituent(s), dose, administration route |
| Safety outcomes | Adverse events, serious adverse events, and corresponding rates; detection of any safety signals |
Abbreviations: Ae, amount excreted into the urine; AUC, area under the plasma or blood concentration versus time curve; ClR, renal clearance; Cmax, maximum concentration; t1/2, half‐life; tmax, time to reach Cmax.
CLINICAL PHARMACOKINETIC NATURAL PRODUCT‐DRUG INTERACTION STUDY DESIGN
A clinical pharmacokinetic NPDI study is warranted when static or PBPK models produce an estimated AUCR for the object drug that lay outside the standard no‐effect range of 0.80–1.25. 7 All of the same study design characteristics for phase 0 studies apply with the following exceptions.
Study design and participants
Whereas phase 0 studies are designed to initially characterize the pharmacokinetics of NP phytoconstituents, clinical NPDI studies are designed to evaluate the effects of NPs on the pharmacokinetics of one or more object drugs. Because object drug pharmacokinetics must be evaluated under two conditions (absence and presence of the NP), a crossover study design is recommended to maximize statistical power with a small number of participants. Arms of the crossover study should be separated by a washout period of at least five of the estimated half‐lives of either the NP precipitant phytoconstituent(s), if known, or the object drug, whichever is longer. If induction of a drug metabolizing enzyme or transporter is the suspected mechanism of the interaction, a fixed sequence study design is recommended.
Exclusion criteria should rule out drug regimens that may confound interpretation of the observed NPDI (e.g., current use of hormonal contraception, antibiotics, or strong inducers or inhibitors of the drug metabolizing enzyme[s] or transporter[s] believed to mediate the NPDI). A list of index inhibitors and inducers is curated by the FDA. 8
Dose of the precipitant natural product
NP dose selection for clinical studies evaluating NPDIs are akin to that for clinical DDI studies, except that the usual consumer dose of the NP should be used, rather than the maximum dose. 6 The following dosing regimens are suggested in order of preference.
As for phase 0 studies, fixed single‐dose regimens using a typical dose of the NP are recommended.
-
If one or both of the following conditions are met, chronic dosing may be considered:
Induction of a drug metabolizing enzyme/transporter is suspected, or
The precipitant NP constituent(s) are time‐dependent inhibitors, and the likelihood of toxicity is low.
Dose escalation may be considered if the likelihood of toxicity is low and a potential NPDI is suspected based on static or PBPK models but is not observed using a low initial typical consumer dose of the precipitant NP. The dose escalation schedule should remain within the typical consumer dose range of the NP.
Dose of the object drug
A small dose of the object drug may be considered to reduce the likelihood of toxicity if an NP is suspected to reduce clearance of the object drug. 24 Established probe drugs that can be used as object drugs are curated by the FDA 8 and should be dosed according to approved labeling. If the NP is believed to increase clearance of the object drug, careful consideration should be given to sampling intervals and to the lower limits of detection of the bioanalytical analysis method, which typically involves liquid chromatography tandem mass spectrometry.
Data reporting
Study results should include the data elements listed in Table 1 and Table 2. Pharmacodynamic indicators of a large pharmacokinetic perturbation (e.g., pupil diameter for opioid agonists) may be collected to correlate with changes in object drug exposure.
TABLE 2.
Additional data to report from clinical pharmacokinetic natural product‐drug interaction studies
| Metric | Description |
|---|---|
| AUC, Cmax, and t1/2 |
Natural product constituent(s): geometric mean and 90% confidence interval Object drug: geometric mean and 90% confidence interval in both the presence and absence of the natural product, as well as the treatment/baseline ratio |
| Renal clearance a |
Natural product constituent(s): geometric mean and 90% confidence interval Object drug: geometric mean and 90% confidence interval in both the presence and absence of the natural product, as well as the treatment/baseline ratio a |
| Renal excretion a |
Natural product constituent(s): geometric mean and 90% confidence interval Object drug: geometric mean and 90% confidence interval in both the presence and absence of the natural product, as well as the treatment/baseline ratio a |
| Clinical response (if available) | The clinical effect of the object drug (e.g., pupil diameter for centrally acting opioids) may be measured in the presence and absence of the natural product to supplement the observed change(s) in the pharmacokinetic data |
Abbreviations: AUC, area under the plasma or blood concentration versus time curve; Cmax, maximum concentration; t1/2, terminal half‐life.
Report if urine is collected.
FUTURE RESEARCH
The crux of the decision to conduct a clinical pharmacokinetic NPDI study is rooted in the results obtained from predictive static and/or PBPK models. 15 This decision point is considered crucial due to the substantive resources required to conduct clinical studies. Additional factors contribute to the uncertainty of these pharmacokinetic models. First, modeling NPDIs for which the site of interaction is the intestinal lumen or intestinal cells (enterocytes) is complicated by the presence of an unstirred water layer immediately adjacent to the mucosa. Second, pharmacokinetic models do not currently account for the increasingly apparent contribution of the microbiota to drug disposition. 25 , 26 The former may be explored with more sophisticated pharmacokinetic models. Regarding the latter, researchers should consider collecting fecal samples and conducting RNA sequencing studies to identify populations of microorganisms that may contribute to an NPDI.
SUMMARY
This Recommended Approach proposes that clinical pharmacokinetic studies involving NPs can be conducted similarly to those involving pharmaceutical drugs using available regulatory guidelines with appropriate modifications. Clinical studies with NPs can be conducted akin to (i) phase 0 studies if no pharmacokinetic data for the NP phytoconstituent(s) are available and/or (ii) pharmacokinetic NPDI studies when an NPDI is suspected (Figure 2). Because the guidelines for these types of clinical investigations do not account for several distinguishing characteristics of NPs, such as their complex chemical composition, inconsistent formulation, and unstandardized dosing, several modifications are proposed. First, the suspected site of the NPDI (e.g., gut, liver, and lungs) should inform the route of administration of the NP because some NPs may be administered by multiple routes (e.g., topically, orally, and inhaled). Second, a typical consumer dose of the NP should be used. Third, the NP should be sourced carefully in accordance with a previous Recommended Approach, 14 and the NP source, genus, species, formulation, route of administration, and measured and labeled concentration(s) of constituent(s) should be reported. Finally, all studies should have pre‐study washout periods during which participants abstain from other NPs, including fruit juices and cannabis products. These and other proposed modifications detailed in this Recommended Approach will enable NP researchers to determine the appropriate type of clinical investigation and follow the relevant established guidelines as closely as possible.
CONFLICT OF INTEREST
J.D.U. is a paid consultant for Esperion Therapeutics, Inc. and Schrödinger, Inc. and receives financial support from Takeda, Amgen, Janssen, and Gilead for his University of Washington Research Affiliate Program on Transporters (UWRAPT) research. E.J.C. is a paid medical writer for the Center of Excellence for Natural Product Drug Interaction Research. All other authors have no conflicts of interest to disclose.
Cox EJ, Rettie AE, Unadkat JD, Thummel KE, McCune JS, Paine MF. Adapting regulatory drug‐drug interaction guidance to design clinical pharmacokinetic natural product‐drug interaction studies: A NaPDI Center recommended approach. Clin Transl Sci. 2022;15:322–329. 10.1111/cts.13172
Funding information
This work was supported by the National Institutes of Health National Center for Complementary and Integrative Health and the Office of Dietary Supplements, specifically the Center of Excellence for Natural Product Drug Interaction Research (U54 AT008909).
REFERENCES
- 1. Rashrash M, Schommer JC, Brown LM. Prevalence and predictors of herbal medicine use among adults in the United States. J Patient Exp. 2017;4(3):108‐113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. McCune JS, Hatfield AJ, Blackburn AA, Leith PO, Livingston RB, Ellis GK. Potential of chemotherapy‐herb interactions in adult cancer patients. Support Care Cancer. 2004;12(6):454‐462. [DOI] [PubMed] [Google Scholar]
- 3. Boon H, Kachan N, Boecker A. Use of natural health products: how does being “natural” affect choice? Med Decis Making. 2013;33(2):282‐297. [DOI] [PubMed] [Google Scholar]
- 4. Tsui T, Boon H, Boecker A, Kachan N, Krahn M. Understanding the role of scientific evidence in consumer evaluation of natural health products for osteoarthritis an application of the means end chain approach. BMC Complement Altern Med. 2012;12:198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Nicolussi S, Drewe J, Butterweck V, Meyer zu Schwabedissen HE. Clinical relevance of St. John's wort drug interactions revisited. Br J Pharmacol. 2020;177(6):1212‐1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Koziolek M, Alcaro S, Augustijns P, et al. The mechanisms of pharmacokinetic food‐drug interactions ‐ A perspective from the UNGAP group. Eur J Pharm Sci. 2019;134:31‐59. [DOI] [PubMed] [Google Scholar]
- 7. FDA . Clinical Drug Interaction Studies ‐ Cytochrome P450 Enzyme‐ and Transporter‐Mediated Drug Interactions Guidance for Industry. U.S. Food and Drug Administration; 2020. https://www.fda.gov/media/134581/download. Accessed July 5, 2021. [Google Scholar]
- 8. FDA . Drug Development and Drug Interactions: Table of Substrates, Inhibitors and Inducers. U.S. Food and Drug Administration; 2020. https://www.fda.gov/drugs/drug‐interactions‐labeling/drug‐development‐and‐drug‐interactions‐table‐substrates‐inhibitors‐and‐inducers. Accessed July 5, 2021. [Google Scholar]
- 9. FDA . In Vitro Drug Interaction Studies — Cytochrome P450 Enzyme‐ and Transporter‐Mediated Drug Interactions: Guidance for Industry. U.S. Food and Drug Administration; 2020. https://www.fda.gov/media/134582/download. Accessed July 5, 2021. [Google Scholar]
- 10. EMA . Guideline on the Investigation of Drug Interactions (CPMP/EWP/560/95/Rev. 1 Corr. 2). Committee for Human Medicinal Products (CHMP); 2012. https://www.ema.europa.eu/en/documents/scientific‐guideline/guideline‐investigation‐drug‐interactions‐revision‐1_en.pdf. Accessed July 5, 2021. [Google Scholar]
- 11. Ministry of Labor and Welfare . Guideline on Drug Interaction for Drug Development and Appropriate Provision of Information, Notification No.0723‐4, Pharmaceutical Evaluation Division, Pharmaceuticals Safety and Environmental Health Bureau, Japan. Ministry of Labor and Welfare; 2018. https://www.pmda.go.jp/files/000228122.pdf. Accessed July 5, 2021. [Google Scholar]
- 12. Paine MF, Shen DD, McCune JS. Recommended approaches for pharmacokinetic natural product‐drug interaction research: a NaPDI center commentary. Drug Metab Dispos. 2018;46(7):1041‐1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Johnson EJ, González‐Peréz V, Tian DD, et al. Selection of priority natural products for evaluation as potential precipitants of natural product‐drug interactions: A NaPDI center recommended approach. Drug Metab Dispos. 2018;46(7):1046‐1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kellogg JJ, Paine MF, McCune JS, Oberlies NH, Cech NB. Selection and characterization of botanical natural products for research studies: a NaPDI center recommended approach. Nat Prod Rep. 2019;36(8):1196‐1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cox EJ, Tian DD, Clarke JD, et al. Modeling pharmacokinetic natural product‐drug interactions for decision‐making: a NaPDI center recommended approach. Pharmacol Rev. 2021;73(2):847‐859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ainslie GR, Wolf KK, Li Y, et al. Assessment of a candidate marker constituent predictive of a dietary substance‐drug interaction: case study with grapefruit juice and CYP3A4 drug substrates. J Pharmacol Exp Ther. 2014;351(3):576‐584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tanna RS, Tian DD, Cech NB, et al. Refined prediction of pharmacokinetic kratom‐drug interactions: time‐dependent inhibition considerations. J Pharmacol Exp Ther. 2021;376(1):64‐73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bansal S, Maharao N, Paine MF, Unadkat JD. Predicting the potential for cannabinoids to precipitate pharmacokinetic drug interactions via reversible inhibition or inactivation of major cytochromes P450. Drug Metab Dispos. 2020;48(10):1008‐1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Nguyen JT, Tian DD, Tanna RS, et al. Assessing transporter‐mediated natural product‐drug interactions via in vitro‐in vivo extrapolation: clinical evaluation with a probe cocktail. Clin Pharmacol Ther. 2021;109(5):1342‐1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cox EJ, Maharao N, Patilea‐Vrana G, et al. A marijuana‐drug interaction primer: Precipitants, pharmacology, and pharmacokinetics. Pharmacol Ther. 2019;201:25‐38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. FDA . Guidance for industry, investigators, and reviewers: Exploratory IND studies. 2006.
- 22. Johnson EJ, Won CS, Köck K, Paine MF. Prioritizing pharmacokinetic drug interaction precipitants in natural products: application to OATP inhibitors in grapefruit juice. Biopharm Drug Dispos. 2017;38(3):251‐259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. FDA . Determining the Extent of Safety Data Collection Needed in Late Stage Premarket and Postapproval Clinical Investigations. U.S. Food and Drug Administration; 2016. https://www.fda.gov/media/82664/download. Accessed July 5, 2021. [Google Scholar]
- 24. Park GJ, Bae SH, Park WS, et al. Drug‐drug interaction of microdose and regular‐dose omeprazole with a CYP2C19 inhibitor and inducer. Drug Des Devel Ther. 2017;11:1043‐1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wilson ID, Nicholson JK. Gut microbiome interactions with drug metabolism, efficacy, and toxicity. Transl Res. 2017;179:204‐222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nichols RG, Peters JM, Patterson AD. Interplay between the host, the human microbiome, and drug metabolism. Hum Genomics. 2019;13(1):27. [DOI] [PMC free article] [PubMed] [Google Scholar]