

Abstract
It is known that interleukin‐6 (IL‐6) can significantly modulate some key drug‐metabolizing enzymes, such as phase I cytochrome P450s (CYPs). In this study, a physiologically‐based pharmacokinetic (PBPK) model was developed to assess CYPs mediated therapeutic protein drug interactions (TP‐DIs) in patients with immune‐mediated inflammatory diseases (IMIDs) with elevated systemic IL‐6 levels when treated by anti‐IL‐6 therapies. Literature data of IL‐6 levels in various diseases were incorporated in SimCYP to construct respective virtual patient populations. The modulation effects of systemic IL‐6 level and local IL‐6 level in the gastrointestinal tract (GI) on CYPs activities were assessed. Upon blockade of the IL‐6 signaling pathway by an anti‐IL‐6 treatment, the area under plasma concentration versus time curves (AUCs) of S‐warfarin, omeprazole, and midazolam were predicted to decrease by up to 40%, 42%, and 46%, respectively. In patients with Crohn’s disease and ulcerative colitis treated with an anti‐IL‐6 therapy, the lowering of the elevated IL‐6 levels in the local GI tissue were predicted to result in further decreases in AUCs of those CYP substrates. The propensity of TP‐DIs under comorbidity conditions, such as in patients with cancer with IMID, were also explored. With further validation with relevant clinical data, this PBPK model may provide an in silico way to quantify the magnitude of potential TP‐DI in patients with elevated IL‐6 levels when an anti‐IL‐6 therapeutic is used with concomitant small‐molecule drugs. This model may be further adapted to evaluate the CYP modulation effect by other therapeutic modalities, which would significantly alter levels of proinflammatory cytokines during the treatment period.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
Interleukin‐6 (IL‐6) may significantly modulate some key drug‐metabolizing enzymes, including phase I cytochrome P450s (CYPs). A physiologically‐based pharmacokinetic (PBPK) model was developed to predict the impact of elevated IL‐6 level and anti‐IL‐6 mAb treatment on multiple CYP enzymes in patients with rheumatoid arthritis.
WHAT QUESTION DID THIS STUDY ADDRESS?
The aforementioned PBPK model was expanded to assess potential therapeutic protein drug interactions (TP‐DIs) between anti‐IL‐6 treatment and CYP substrate drugs in different immune‐mediated inflammatory disease (IMID) populations with elevated IL‐6 levels. For the inflammatory bowel disease (IBD) populations, modulation effects from elevated IL‐6 levels in the local gastrointestinal tract were taken into consideration. The potential additive modulation effect on CYPs from concomitant cancer‐IMID situation was also assessed. Furthermore, simulations at different hypothetical IL‐6 levels were performed to identify the IL‐6 levels, which would result in weak, moderate, and strong CYP modulation effects based on the definitions in the US Food and Drug Administration (FDA) drug‐drug interaction guidance.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
The PBPK platform model was expanded to assess the potential TP‐DIs during anti‐IL‐6 treatment in several IMIDs including systemic lupus erythematosus, ulcerative colitis, Crohn’s disease, type 1 diabetes, and cancer‐IMID comorbidity. The high local IL‐6 levels in patients with IBD were predicted to result in extra inhibition effect on the abundances of intestinal CYPs. Patients with cancer‐IMID manifested further decrease in systemic exposures of CYP substrate drugs compared with patients with IMID only. Cutoff values of IL‐6 level which would result in different levels of CYP modulation effect were identified.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
This PBPK model approach may serve as conceptual framework and workflow process to evaluate the modulation effect on CYPs in patients by therapeutic modalities which can significantly result in altered levels of proinflammatory cytokines during the treatment period.
INTRODUCTION
Drug‐drug interaction (DDI) characterization is an important element in optimizing the therapeutic benefits in polypharmacy conditions. Owing to divergent mechanisms of elimination, direct DDIs between therapeutic proteins (TPs) and small molecules are generally thought to be less likely. However, some disease conditions, such as inflammation, infections, and cancers, are known to modulate the expression of cytochrome P450 enzymes (CYPs) or some transporters 1 and then suppress/upregulate their enzymatic activity in the liver 2 and/or intestines. 3 The increased approvals and extensive use of TPs for treating such diseases has aroused unprecedented clinical needs and research interests in studying the CYP‐mediated TP‐drug interactions (TP‐DIs). In a recent review of approved TPs, 49 out of 150 TP labels contained pharmacokinetic (PK)‐related TP‐DI information from nonclinical and/or clinical DDI evaluations, more than half of the clinical PK TP‐DI evaluations showed no interactions, and no dose adjustment has been recommended for any of the remaining TPs. 4 Due to the innate deficiency of preclinical TP‐DI evaluation tools and high cost as well as long patient recruitment of clinical TP‐DI studies, a risk‐based strategy has been recommended to assess the TP‐DI propensity before a clinical TP‐DI study would be considered. 5 , 6 With that, a model‐informed approach would provide a useful tool to assess potential TP‐DIs, optimize clinical TP‐DI study design, or even avoid little value‐added clinical TP‐DI studies.
CYPs, a primary class of drug‐metabolizing enzymes distributed predominantly in the liver and intestines, have been reported to be regulated by certain proinflammatory cytokines released during inflammatory reactions. Cellular expressions of major CYPs, including CYP3A4/5, CYP2C9, CYP2C19, and CYP1A2, were downregulated upon exposure to high concentrations of IL‐6. 7 Exposure of the hepaRG hepatic cells to 10 ng/ml IL‐6 for 72 h decreased enzyme activity of CYP3A4 by greater than 80%, CYP1A2 by 60%, and CYP2B6 and 2C19 by 80%. 8 A 20% −70% downregulation in hepatic CYP3A4 enzyme activity modulated by several proinflammatory cytokines, such as IL‐6, interferon alpha (IFN‐α) and tumor necrosis factor alpha‐α (TNF‐α), was reported. 9 Dickmann et al. quantified the IL‐6 mediated regulation of multiple CYPs in hepatocytes, considering physiologically relevant levels, markers of acute phase response, and regulation of the IL‐6 receptor during in vitro treatment. 7
Patients with certain immune‐mediated inflammatory diseases (IMIDs), including Crohn’s disease (CD), ulcerative colitis (UC), rheumatoid arthritis (RA), systemic lupus erythematosus (SLE), and type 1 diabetes (T1D), have elevated serum levels of IL‐6. 10 , 11 , 12 , 13 , 14 The disease‐mediated IL‐6 increase interferes with the CYPs’ activities, consequently, may alter the PKs of certain victim drugs. Anti‐IL‐6/IL‐6R TPs used for inflammatory alleviation may restore drug metabolism by disrupting IL‐6’s modulation of CYPs. In addition, certain tumor‐associated inflammation was found to inhibit CYPs’ expression by 20–33% in liver and intestines in patients with cancer. 15 Of note, 10%–30% of patients with cancer also have comorbidity of IMIDs (i.e., cancer‐IMID). 16 Such comorbidities may further worsen CYP alternations.
Physiologically‐based pharmacokinetic (PBPK) model comprising compartments mimicking the anatomy and physiology of the human body has been a common tool to assess DDIs of small molecules. 17 , 18 , 19 Its utilization in supporting therapeutic protein drug development has been increasingly gaining more attention in recent years. In the recent draft guideline for TP‐DIs by the US Food and Drug Administration (FDA), sponsors are encouraged to discuss with the health authority on using the PBPK modeling approach to evaluate the DDI potential of a TP. 20 Machavaram et al. reported the first PBPK model to assess disease‐mediated TP‐DIs. 21 In their study, the developed PBPK model demonstrated the impact of IL‐6 and an anti‐IL‐6 receptor mAb, tocilizumab, on CYP3A4 activity measured by changes in systemic exposure of simvastatin in patients with RA. Jiang et al. developed a PBPK model to predict the impact of elevated IL‐6 level and an anti‐IL‐6 mAb, sirukumab, on multiple CYP enzymes in patients with RA. 22 In their study, the modulation effects of IL‐6 level on hepatic and intestinal CYPs’ activities were characterized using the data from in vitro studies and incorporated in a virtual RA patient population. The PBPK model was then applied to predict PKs of substrates of multiple CYPs, including CYP3A4, CYP2C9, CYP2C19, and CYP1A2, before and after sirukumab treatment in patients with RA. The model predictions were well aligned with the observed data from a clinical cocktails TP‐DI study. 10 The PBPK model’s predictive performance was further validated with the clinical data of tocilizumab on the PK profiles of simvastatin and omeprazole (an CYP2C19 substrate) in patients with RA.
In the current study, we expanded the aforementioned PBPK model developed by Jiang et al. 22 to assess potential TP‐DIs between anti‐IL‐6 treatment and CYP substrate drugs in different IMID populations with elevated IL‐6 levels, including SLE, UC, CD, and T1D. For CD and UC populations, additional modulation effects from elevated IL‐6 levels in the local gastrointestinal (GI) tract were taken into consideration. Similarly, we also examined the potential additive modulation effect on CYPs from concomitant cancer‐IMID situation. Furthermore, simulations at different hypothetical IL‐6 levels were performed to identify the IL‐6 levels which would result in weak, moderate, and strong CYP modulation effects based on the definitions in the FDA DDI guidance. 23
MATERIALS AND METHODS
Analysis of IL‐6 level in disease populations
Literature data of systemic baseline IL‐6 levels in healthy subjects 22 and in patients with UC, CD, SLE and T1D, 11 , 13 , 24 , 25 , 26 , 27 , 28 , 29 , 30 , 31 , 32 , 33 , 34 , 35 , 36 , 37 , 38 , 39 , 40 , 41 , 42 , 43 , 44 , 45 , 46 , 47 , 48 and baseline IL‐6 levels in the local GI tract of patients with UC and CD were collected and examined. 43 , 49 , 50 , 51 , 52 , 53 The systemic or local IL‐6 concentrations obtained from different studies were pooled and the average IL‐6 levels with associated variability was calculated with GraphPad (GraphPad Software, Inc.).
PBPK models
The SimCYP (version 17; SimCYP Limited, Sheffield, UK) population‐based absorption, distribution, metabolism, and excretion (ADME) simulator platform was used to develop the PBPK models for each individual victim drug (CYP index substrates: midazolam, omeprazole, S‐warfarin, and caffeine) in the virtual IMIDs’ patient, cancer‐IMIDs’ patient, and healthy populations. Virtual patient populations were characterized by incorporating the impact of different systemic IL‐6 levels on hepatic and intestinal expression of multiple CYPs in the healthy White population. CYP dynamics model incorporating mechanism‐based inhibition or induction was used to simulate the modulation effects of IL‐6 on the hepatic and intestinal metabolism of each victim drug. Enzyme regulation in a disease comorbidity situation was described in “Modeling of IL‐6 Profiles and CYP Enzyme Dynamics” section. Further details of the general aspects of the PBPK model properties, victim drug’s kinetics, and CYP dynamics in SimCYP simulator have been described previously. 54 In addition, the simulator built‐in library models of midazolam, omeprazole, S‐warfarin, and caffeine were used in the current PBPK model to predict concentration profiles of these CYPs’ substrates with modification through model optimization as described before. 22
Modeling of IL‐6 profiles and CYP enzyme dynamics
The systemic IL‐6 levels of SLE, UC, CD, and T1D used in the PBPK models were simulated with the same parameters as used in the previous PBPK model for the RA patient population with molecule weight (MW) = 21,000 g/mol, systemic clearance (CL i.v.) = 1.0 L/h, and volume of distribution at steady state (Vss) = 0.43 L/kg. 22 IL‐6 as driving force for the CYP regulation was introduced to the system via constant i.v. infusion at the rate of 0.012, 0.02, and 0.06 μg/h for 40 days, and the resultant steady‐state (SS) systemic IL‐6 levels were 12, 20, and 60 pg/ml, which represent the elevated IL‐6 levels in SLE, UC/CD, and T1D populations, respectively. The simulated IL‐6 concentrations were then linked to the modulation effects on the hepatic CYP enzyme abundances. The hepatic CYP activities were predicted to reach SS over the simulation period: 90% of the SS enzyme activities were predicted to achieve between 7 and 19 days after continuous exposure to elevated IL‐6 depending on the setting of turnover rate of each individual CYP isozyme within SimCYP. The CYP activities used in the current PBPK model were represented by CYP abundances in default SimCYP populations. A technical limitation of SimCYP in current PBPK model is that when i.v. infusion of IL‐6 was used to mimic the elevated IL‐6 level in a virtual disease population, intestines are not exposed to IL‐6 which prevents the use of the dynamic models for DDIs in intestines within SimCYP. To incorporate the effect on the CYPs in intestines, a pragmatic approach was used where the intestinal CYP abundance was manually calculated and adjusted in the population library to match the level of suppression of individual CYP simulated in the liver. For patients with UC and CD, the modulation effect of local IL‐6 levels on intestinal CYPs was also manually calculated and embedded with the modulation effect of systemic IL‐6 on hepatic CYPs for the simulation.
The modulation effects of IL‐6 on CYP2C9, CYP2C19, CYP3A4, CYP3A5, and CYP1A2 were modeled using Equation 1:
| (1) |
where, Enz,Liver/GI (t) is the level of the active hepatic or intestinal CYPs at any given time; Enz Liver/GI‐basal‐HV is the basal levels of hepatic or intestinal CYPs (EnzLiver/GI (t) = E 0 at t = 0) in healthy subjects; Emin/max is the minimum/maximal CYPs activity (i.e., maximum suppression/induction) expressed as a fraction of vehicle control; EC50 is the IL‐6 concentration that causes 50% of enzyme suppression/induction effect; and [IL‐6]t is the concentration of perpetrator IL‐6 at time t. The mean degradation rate constant (k deg, Liver/GI) of each CYP used for the simulations were the default values of SimCYP ADME simulator. 55
The values of Emin (CYP2C9: 0.053; CYP2C19: 0.214; and CYP3A5: 0.034) and EC50 (CYP2C9: 121.0 pg/ml; CYP2C19: 71.3 pg/ml; and CYP3A5: 51 pg/ml) were assigned based on in vitro study results. 7 The Emin for CYP3A4 (0.25), Emax for CYP1A2 (1.34), and EC50 values (CYP3A4: 75.2 pg/ml and CYP1A2: 8 pg/ml) were obtained by re‐analyzing the reported in vitro data as described previously. 22 It was reported that within the concentration range of 0–1000 pg/ml, concentration‐response profile of IL‐6 induction effect on CYP1A2 was bell‐shaped with peak effect reached at IL‐6 level of 100 pg/ml. Therefore, the data of the IL‐6 less than 100 pg/ml portion were used to calculate the regulation parameters. 22
In patients with cancer‐IMID, cancer induced CYPs reductions (20% for CYP1A2, 30% for CYP3A4, and 33% for CYP2C19 in the liver and 30% for CYP3A4 and 33% for CYP2C19 in the intestines) were incorporated as the baseline condition and additional modulation effect from IMIDs were assumed to assess the potential total TP‐DI in such comorbidity populations. 56 Hepatic (CYP1A2, 3A4, and 2C19) and intestinal (CYP3A4 and 2C19) basal CYP activities in patients with cancer only were calculated as below (Equation 2):
| (2) |
where, Enzliver/GI‐basal‐Cancer, X is the hepatic or intestinal basal activity in patients with cancer only for enzyme X (X = CYP1A2, CYP3A4, or CYP2C19); x% is the cancer‐induced reduction for enzyme X (20% for CYP1A2, 30% for CYP1A4, and 33% for CYP2C19). Finally, the modulation effects of further elevated IL‐6 resulted from IMIDs on hepatic and intestinal CYPs in patients with cancer‐IMID were modeled, as described in Equation 1 with the adjusted hepatic or intestinal basal CYP levels.
Of note, one assumption in this study is that in the absence of in vitro data describing the Emin and EC50 in enterocytes, it was assumed that the same modulation effects of systemic IL‐6 on the liver and intestines. 57 It was further assumed that systemic and local level of IL‐6 have a direct modulation effect on CYPs in the liver and intestines, as described in Equation 1. Moreover, the impact of IL‐6 on other enzymes or transporters was not considered in the current study.
Validation and application of PBPK model to assess TP‐DIs propensity between TPs and ‐CYP substrates
The previously developed PBPK model in patients with RA had been validated using the observed data from clinical TP‐DI studies, and the PBPK model predictions were aligned with the observed TP‐DI between tocilizumab and simvastatin, omeprazole, respectively. 6 , 58 Since that publication, additional clinical TP‐DI data related to IL‐6 modulation on CYPs in patients with RA have become available. Thus, we further validated the PBPK model by comparing the predicted changes of simvastatin exposure metrics after the treatment of an anti‐IL‐6R mAb sarilumab with the observed data from the clinical TP‐DI study in patients with RA. 59
To assess the IL‐6‐mediated TP‐DI effect in other IMIDs and patients with cancer‐IMIDs, PK profiles of CYP substrates before and after anti‐IL‐6 treatment in the developed virtual patient populations were simulated. Each scenario was simulated with 10 trials containing 10 subjects each with CYP substrates oral administration at the SS of regulatory effect of IL‐6 on the hepatic CYPs. The dose levels of CYP substrates used in the simulations were 0.03 mg/kg midazolam, 10 mg warfarin (plus 10 mg vitamin K), 20 mg omeprazole, and 100 mg caffeine. 10 Interindividual variabilities of model parameters were incorporated in this PBPK model using the SimCYP simulator default values. Cmax and AUC of simulated drugs’ PK profiles and 90% confidence intervals (CIs) of simulated values were calculated and summarized.
Classification of IL‐6 impact on TP‐DIs
According to the FDA DDI guidance, 23 a CYP modulator can be classified based on its magnitude of effect on an index CYP substrate: a weak, moderate, and strong inducer decreases the AUC of a CYP substrate by 20% to 50%, 50% to 80% and above 80%, respectively; whereas a weak, moderate, and strong inhibitor increases the AUC by 1.25‐ to 2‐, 2‐ to 5‐, and above fivefolds, respectively. Anti‐IL‐6 treatment alleviates the inhibitory effects of IL‐6 on CYP3A4, CYP2C9, and CYP2C19 so it acts as a “CYP inducer” in the virtual disease populations. IL‐6 has an inductive effect on CYP1A2 at concentration below 1000 pg/ml, thus anti‐IL‐6 acts as a CYP1A2 inhibitor in the virtual disease population. In order to have a more systemic assessment of modulation effect of IL‐6 level on CYPs activities and potential TP‐DIs by anti‐IL‐6 treatments, simulations at a wide range of baseline systemic IL‐6 concentration (from 10 to 1000 pg/ml) were performed and the IL‐6 level thresholds corresponding to weak, moderate, and strong impact were identified.
RESULTS
Additional validation of the PBPK model in RA
The previously developed PBPK model was able to adequately capture the impact of sirukumab treatment on systemic exposure of several CYP isozyme substrates in patients with RA and the model was successfully validated by comparing the model predictions to the reported PK data of simvastatin and omeprazole, before and after treatment of IL‐6R mAb tocilizumab in patients with RA. 22 In the current study, further model validation was conducted using data from a recent clinical study on the effect of sarilumab treatment on CYP substrates in patients with RA. 59 The observed AUC (mean ± SD) of simvastatin before sarilumab treatment was 82.7 ± 52.8 ng⋅h/ml and the value after sarilumab treatment was 47.9 ± 33.4 ng⋅h/ml, so the sarilumab treatment resulted in 42% decrease in simvastatin AUC (90% CI: 37%–53%). The model predicted simvastatin AUC was 95.7 ± 85.9 at IL‐6 level of 50 pg/ml (representing patients with RA before sarilumab treatment) and 48.9 ± 42.7 ng⋅h/ml at IL‐6 level of 0 pg/ml (representing patients with RA after sarilumab treatment). The predicted AUC of simvastatin was decreased by 49% (90% CI 42%–58%) after sarilumab treatment. Overall, the PBPK model predicted TP‐DI magnitude of effect following sarilumab treatment was consistent with the observed one. This additional validation, together with the previously reported model validation using the data from the sirukumab and tocilizumab studies, have demonstrated the utility of PBPK model in predicting disease‐mediated TP‐DIs in patients with IMID with elevated IL‐6 level.
Analysis of IL‐6 concentration in disease populations
The systemic IL‐6 levels collected from previous studies of various IMID populations were highly varied, as show in Figure 1, ranging from 3.6 to 30.2 pg/ml in SLE, 1.3 to 85 pg/ml in UC, 4.25 to 47 pg/ml in CD, and 2.1 to 187 pg/ml in T1D. 11 , 13 , 24 , 25 , 26 , 27 , 28 , 29 , 30 , 31 , 32 , 33 , 34 , 35 , 36 , 37 , 38 , 39 , 40 , 41 , 42 , 43 , 44 , 45 , 46 , 47 , 48 The calculated mean ± SD of systemic IL‐6 levels were 12.7 ± 8.5, 23.9 ± 27.6, 20.6 ± 19.8, 56.7 ± 75.6 pg/ml in SLE, UC, CD, and T1D patient populations, respectively. The reported local GI tissue IL‐6 concentration in patients with UC and CD were also highly variable: 917–5885 pg/ml for UC and 704–7649 pg/ml for CD based on patient‐derived cell culture. 43 , 49 , 50 , 51 , 52 , 53 The average local IL‐6 levels (3171 pg/ml for UC and 2674 pg/ml for CD) were used for assessing the additional regulation effect from high local GI IL‐6 level in the IBD patient populations.
FIGURE 1.
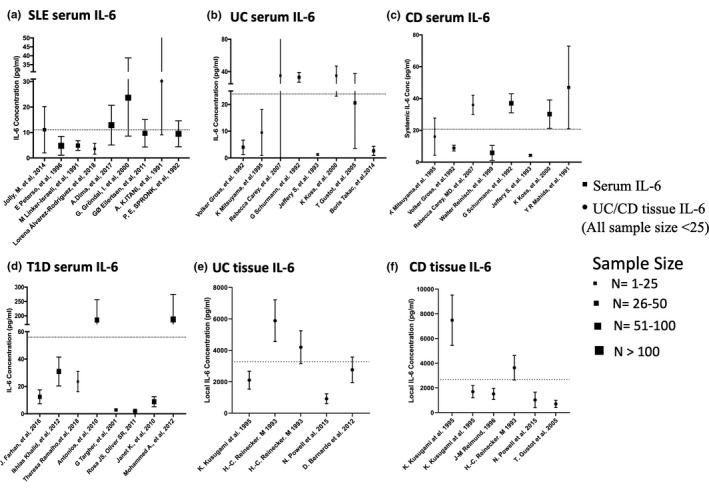
Pooled analysis of systemic interleukin (IL)‐6 levels. Systemic interleukin (IL)‐6 levels in patients with systemic lupus erythematosus (SLE) (a); patients with ulcerative colitis (UC) (b); patients with Crohn’s disease (CD) (c); patients with type 1 diabetes (T1D) (d); and also tissue IL‐6 levels in patients with UC (e) and patients with CD (f) based on published literature. Dotted lines represent the average systemic IL‐6 levels in SLE (12.7 ± 8.5 pg/ml), UC (23.9 ± 27.6 pg/ml), CD (20.6 ± 19.8 pg/ml), T1D (56.7 ± 75.6 pg/ml), and average tissue IL‐6 level in UC (3171 ± 1471 pg/ml), CD (2674 ± 1279 pg/ml) and the sample size of local IL‐6 studies were smaller than 25 patients. Symbols with SD bars represent observed population mean ± SD data from literatures
CYP enzymatic activity in patients with IMID, cancer/cancer‐IMID
The active hepatic CYP enzymatic activities in various IMIDs were simulated using SimCYP and were reflected by CYP abundances. The systemic (SLE and T1D) or local (UC and CD) IL‐6 levels were used to calculate the intestinal CYP abundances, which were then incorporated with specific systemic IL‐6 regulated hepatic CYP activity to assess the overall TP‐DIs in different IMID patient populations. The calculated intestinal CYP abundance in the healthy subject virtual population and in SLE, T1D, UC, and CD patient virtual populations are summarized in Table S1. As shown in Table S1, the high local IL‐6 levels in patients with UC and CD were predicted to result in extra inhibition effect on the abundances of CYP2C9, 2C19, and 3A4/5 in intestines in patients with UC and CD. The intestinal CYP3A4 abundance was predicted to decrease to 1.9 and 3.1 pmol/mg (pmols CYP isoform per mg of microsomal protein) in patients with UC and CD, respectively, compared to 66.2 pmol/mg in healthy subjects and 49.3 pmol/mg in patients with UC/CD when only inhibitory effect from systemic IL‐6 is considered.
In cancer‐IMID virtual patient populations, the adjusted basal hepatic and intestinal CYP abundances in the cancer‐only patient population and calculated intestinal CYP abundances in different cancer‐IMID patient populations are listed in Table S2. As shown in Table S2, the comorbidity may result in further inhibition in CYP3A4 and CYP2C19 abundances.
CYP substrates PK assessment in IMIDs and cancer‐IMIDs before versus after anti‐IL‐6 treatment
The PBPK model assessed the CYPs activities before‐ and after‐ anti‐IL‐6 treatment in the IMID patient populations by comparing the predicted PK profiles of CYP substrates in the presence of elevated systemic and/or local GI IL‐6 levels and in the absence of IL‐6 (represented by the profiles of healthy subjects). The model predicted plasma profiles for midazolam, omeprazole, S‐warfarin, and caffeine before and after anti‐IL‐6 treatment in SLE, UC/CD, and T1D patient populations are shown in Figure 2. The model predicted changes in drug exposures of the CYP substrates in different disease populations are listed in Table 1. As IL‐6 acts as an inhibitor for CYP3A4, CYP2C9, and CYP2C19 expression and activities, the simulation results suggested that the anti‐IL‐6 treatment may result in up to 28% and 46% decreases in midazolam Cmax and AUC, respectively. The model also predicted increases in S‐warfarin and omeprazole exposures, but to less extents, after anti‐IL‐6 treatment. For CYP1A2, IL‐6 acts as a mild inducer at IL‐6 level up to 1000 pg/ml, which is much higher than the systemic IL‐6 levels in these IMIDs. As expected, the model predicted up to 10% and 35% increases in caffeine Cmax and AUC, respectively, after anti‐IL‐6 treatment. The model predicted plasma profiles of the substrates in patients with CD with and without considering high local GI IL‐6 concentrations are displayed in Figure 3a and the corresponding exposure changes are listed in Table 2. The elevated IL‐6 levels at local GI tissues were predicted to contribute additional 11~16% decrease in AUC and Cmax of the CYP substrates compared to the situation where only systemic IL‐6 impact is considered.
FIGURE 2.
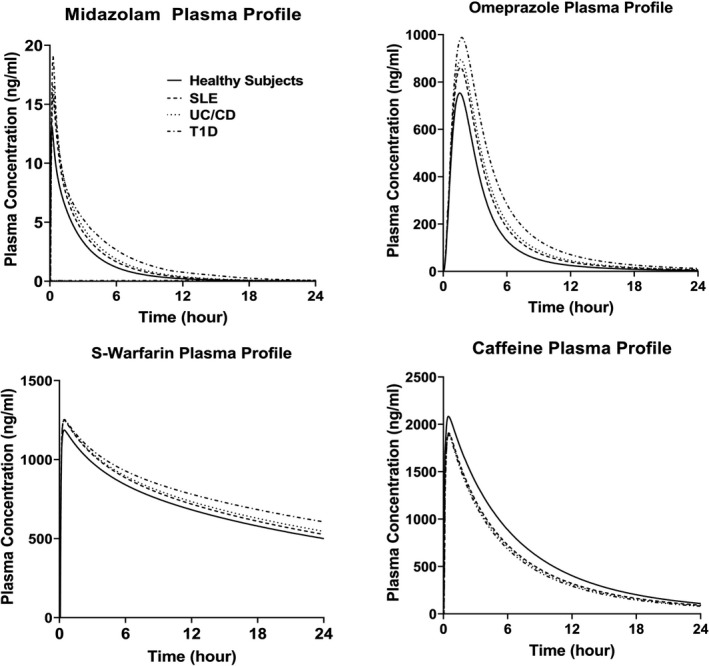
Predicted median plasma concentration profiles for CYP enzyme substrates in patients with IMID before and after anti‐IL‐6 treatment. Predicted median plasma concentration‐time profiles for midazolam, omeprazole, S‐warfarin, and caffeine in patients with SLE, UC/CD, and T1D before anti‐IL‐6 treatment, and in healthy subjects (after anti‐IL‐6 treatment condition). CD, Crohn’s disease; IMID, immune‐mediated inflammatory disease; SLE, systemic lupus erythematosus; T1D, type 1 diabetes; UC, ulcerative colitis
TABLE 1.
The Cmax and AUC changes of midazolam, omeprazole, S‐warfarin, and caffeine in patients with SLE, UC/CD, and T1D after anti‐IL‐6 treatment
| Substrates |
∆Cmax % 12 pg/ml SLE patients |
∆Cmax % 20 pg/ml UC/CD patients |
∆Cmax % 60 pg/ml T1D patients |
∆AUC % 12 pg/ml SLE patients |
∆AUC % 20 pg/ml UC/CD patients |
∆AUC % 60 pg/ml T1D patients |
|---|---|---|---|---|---|---|
| Caffeine (CYP1A2) | 8% ↑ | 9% ↑ | 10% ↑ | 27% ↑ | 31% ↑ | 35% ↑ |
| S‐Warfarin (CYP2C9) | 3% ↓ | 3% ↓ | 3% ↓ | 16% ↓ | 20% ↓ | 40% ↓ |
| Omeprazole (CYP2C19) | 13% ↓ | 16% ↓ | 24% ↓ | 18% ↓ | 22% ↓ | 42% ↓ |
| Midazolam (CYP3A) | 14% ↓ | 18% ↓ | 28% ↓ | 20% ↓ | 28% ↓ | 46% ↓ |
∆Cmax or ∆AUC = Cmax or AUC post‐anti‐IL‐6 treatment/Cmax or AUC pre‐anti‐IL‐6 treatment when Cmax or AUC increases after anti‐IL‐6 treatment.
∆Cmax or ∆AUC = 1 – (Cmax or AUC post‐anti‐IL‐6 treatment/Cmax or AUC pre‐anti‐IL‐6 treatment) when Cmax or AUC decreases after anti‐IL‐6 treatment.
Abbreviations: AUC, area under plasma concentration versus time curves; CD, Crohn’s disease; Cmax, maximum plasma concentration; SLE, systemic lupus erythematosus; T1D, type 1 diabetes (T1D); UC, ulcerative colitis.
↑: Increase in exposure metrics after anti‐IL‐6 treatment; ↓: Decrease in exposure metrics after anti‐IL‐6 treatment.
FIGURE 3.
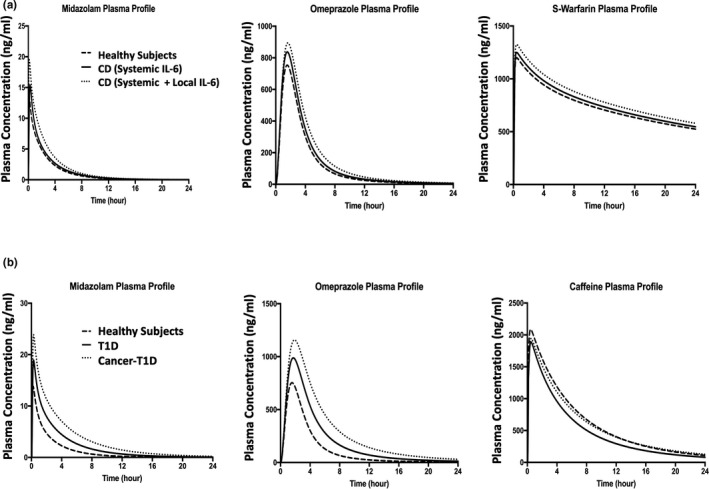
Predicted median plasma concentration profiles for CYP substrate compounds. Predicted median plasma concentration‐time profiles for midazolam, omeprazole, and S‐warfarin in patients with CD before versus after anti‐IL‐6 treatment (a). The solid lines represent patients with CD involving systemic IL‐6 only before anti‐IL‐6 treatment; the dotted lines represent patients with CD involving systemic and local IL‐6 before anti‐IL‐6 treatment; and the dashed lines represent healthy subjects (after anti‐IL‐6 treatment condition). Predicted median plasma concentration‐time profiles for midazolam, omeprazole, and caffeine in T1D/cancer‐T1D before versus after anti‐IL‐6 treatment (b). The solid lines represent T1D disease only before anti‐IL‐6 treatment; the dotted lines represent patients with cancer‐T1D before anti‐IL‐6 treatment; the dashed lines represent healthy subjects (after anti‐IL‐6 treatment condition). CD, Crohn’s disease; T1D, type 1 diabetes
TABLE 2.
The Cmax and AUC changes for midazolam, omeprazole, S‐warfarin, and caffeine in patients with UC and CD with or without local IL‐6 incorporation after anti‐IL‐6 treatment
| Substrates |
∆Cmax % UC/CD systemic IL‐6 |
∆Cmax % CD: systemic + local GI IL‐6 |
∆Cmax % UC: systemic + local GI IL‐6 |
∆AUC % UC/CD systemic IL‐6 |
∆AUC % CD: systemic + local GI IL‐6 |
∆AUC % UC: systemic + local GI IL‐6 |
|---|---|---|---|---|---|---|
| Caffeine (CYP1A2) | 9% ↑ | 9% ↑ | 9% ↑ | 31% ↑ | 31% ↑ | 31% ↑ |
| S‐Warfarin (CYP2C9) | 3% ↓ | 10% ↓ | 11% ↓ | 25% ↓ | 28% ↓ | 28% ↓ |
| Omeprazole (CYP2C19) | 16% ↓ | 23% ↓ | 23% ↓ | 28% ↓ | 32% ↓ | 33% ↓ |
| Midazolam (CYP3A) | 18% ↓ | 33% ↓ | 34% ↓ | 28% ↓ | 39% ↓ | 39% ↓ |
∆Cmax or ∆AUC = Cmax or AUC post‐anti‐IL‐6 treatment/Cmax or AUC pre‐anti‐IL‐6 treatment when Cmax or AUC increases after anti‐IL‐6 treatment.
∆Cmax or ∆AUC = 1 – (Cmax or AUC post‐anti‐IL‐6 treatment/Cmax or AUC pre‐anti‐IL‐6 treatment) when Cmax or AUC decreases after anti‐IL‐6 treatment.
Abbreviations: AUC, area under plasma concentration versus time curves; CD, Crohn’s disease; Cmax, peak plasma concentration; GI, gastrointestinal; UC, ulcerative colitis.
↑: Increase in exposure metrics after anti‐IL‐6 treatment; ↓: Decrease in exposure metrics after anti‐IL‐6 treatment.
The model predicted plasma profiles of the CYP substrates in healthy subjects, patients with T1D only, and patients with T1D‐cancer comorbidity before and after anti‐IL‐6 treatment are displayed in Figure 3b. The predicted exposure changes of the CYP substrates in different IMID‐cancer comorbidity patient populations are listed in Table 3. Compared with patients with IMIDs‐only, patients with cancer‐IMID manifested further decrease in systemic exposures of midazolam and omeprazole. For instance, anti‐IL‐6 treatment was predicted to result in 43% and 65% decreases in midazolam Cmax and AUC, respectively, in T1D‐cancer comorbidity population compared to 28% and 46% decreases in Cmax and AUC, respectively, in the T1D only patient population. Of note, in patients with cancer‐IMID, the inductive effect of elevated IL‐6 level on CYP1A2 was predicted to be offset by the cancer‐mediated downregulation effect on CYP1A2, as shown in Table 3.
TABLE 3.
The Cmax and AUC changes for midazolam, omeprazole, and caffeine in patients with cancer + SLE, cancer + UC/CD, and cancer + T1D patients after treatment
| Substrates |
∆Cmax % Cancer + SLE patients |
∆Cmax % Cancer + UC/CD patients |
∆Cmax % Cancer + T1D patients |
∆AUC % Cancer + SLE patients |
∆AUC % Cancer + UC/CD patients |
∆AUC % Cancer + T1D patients |
|---|---|---|---|---|---|---|
| Caffeine (CYP1A2) | 5% ↓ | 6% ↓ | 7% ↓ | 2% ↓ | 5% ↓ | 9% ↓ |
| Omeprazole (CYP2C19) | 27% ↓ | 29% ↓ | 35% ↓ | 47% ↓ | 51% ↓ | 60% ↓ |
| Midazolam (CYP3A) | 32% ↓ | 35% ↓ | 43% ↓ | 50% ↓ | 54% ↓ | 65% ↓ |
∆Cmax or ∆AUC = Cmax or AUC post‐anti‐IL‐6 treatment/Cmax or AUC pre‐anti‐IL‐6 treatment when Cmax or AUC increases after anti‐IL‐6 treatment.
∆Cmax or ∆AUC =1‐(Cmax or AUC post‐anti‐IL‐6 treatment/Cmax or AUC pre‐anti‐IL‐6 treatment) when Cmax or AUC decreases after anti‐IL‐6 treatment.
Abbreviations: AUC, area under plasma concentration versus time curves; CD, Crohn’s disease; Cmax, maximum plasma concentration; SLE, systemic lupus erythematosus; T1D, type 1 diabetes; UC, ulcerative colitis.
↑: Increase in exposure metrics after anti‐IL‐6 treatment; ↓: Decrease in exposure metrics after anti‐IL‐6 treatment.
Classification of IL‐6 impact on TP‐DIs
We have explored how disease‐elevated IL‐6 and anti‐IL‐6 TPs would modulate CYP activities in certain IMIDs and cancer‐IMIDs. To provide more systemic assessment of regulation effect of IL‐6 level on CYPs activities and potential TP‐DIs by anti‐IL‐6/6R treatments, simulations at a wide range of IL‐6 concentration were performed. The identified IL‐6 level cutoff values that were predicted to result in potential differential DDI effects during anti‐IL‐6/6R treatment are listed in Table 4. At baseline, IL‐6 level around 20 pg/ml or below, such as in patients with SLE, anti‐IL‐6/6R treatment was predicted to have minimum regulation effect on CYP3A4, CYP2C9, and CYP2C19. At higher baseline IL‐6 level but lower than 89, 81, or 69 pg/ml, anti‐IL‐6/6R treatment was predicted to act as a weak inducer for CYP3A4, CYP2C9, and CYP2C19, respectively. At IL‐6 level in the range of 89–279, 81–264, and 69–197 pg/ml, anti‐IL‐6/6R treatment was predicted to have moderate inductive effect on the three CYPs. At even higher IL‐6 levels, anti‐IL‐6 treatment may result in strong inductive effect on the CYPs. For CYP1A2, at IL‐6 levels below 1000 pg/ml, no or weak inhibitory effect was predicted during anti‐IL‐6 treatment. For this exercise, only systemic IL‐6 resulted CYP regulation effect was considered. Therefore, based on our current simulations, for certain IMIDs with much higher local IL‐6 level compared to systemic IL‐6 level, both systemic and local IL‐6 concentrations need to be taken into consideration for potential TP‐DIs.
TABLE 4.
IL‐6 threshold levels prediction for anti‐IL‐6 therapeutic proteins to cause significant CYP‐mediated TP‐DI based on FDA DDI classification
| Substrate |
IL‐6 level (pg/ml) Bioequivalence AUC ↓<20% |
IL‐6 level (pg/ml) Weak inducer 20%≤AUC ↓<50% |
IL‐6 level (pg/ml) Moderate inducer 50%≤UC ↓<80% |
IL‐6 level (pg/ml) Strong inducer AUC ↓≥80% |
|---|---|---|---|---|
| Omeprazole (CYP2C19) | <19 | 19–89 | 89–279 | >279 |
| S‐warfarin (CYP2C9) | <21 | 21–81 | 81–264 | >264 |
| Midazolam (CYP3A) | <12 | 12–69 | 69–197 | >197 |
| Substrate |
IL−6 level (pg/ml) Bioequivalence AUC↑<1.25‐fold |
IL−6 level (pg/ml) Weak inhibitor 1.25<AUC↑<2‐fold |
— | — |
| Caffeine (CYP1A2) | <10 | 10–1000 | — | — |
Abbreviations: AUC, area under plasma concentration versus time curves; DDI, drug‐drug interaction; FDA, US Food and Drug Administration; TP‐DI, therapeutic protein drug interaction; UC, ulcerative colitis.
↑: Increase in exposure metrics after anti‐IL‐6 treatment; ↓: Decrease in exposure metrics after anti‐IL‐6 treatment.
DISCUSSION
A previously developed PBPK platform for predicting TP‐DIs in patients with RA was further validated using additional clinical data and expanded to assess the potential TP‐DIs during anti‐IL‐6 treatment in several IMIDs, including SLE, UC, CD, T1D, and cancer‐IMID comorbidity.
As it has been discussed previously by Jiang et al., the accuracy of IL‐6 level in disease populations is critical for predicting the TP‐DIs using this PBPK modeling approach, because IL‐6 is assumed to be the driving force of multiple hepatic and intestinal CYPs modulations in vivo and it in turn influences the disease‐mediated TP‐DIs between the cytokine‐suppressing TPs and CYP substrates. One thing should be noted that IL‐6 binds to soluble and transmembrane IL‐6R and it is the IL‐6/IL‐6R complex that induces homodimerization of glycoprotein 130 (gp130), resulting in the activation of the signaling pathway which leads to certain CYP modulation. 60 Level of IL‐6 is a measurable marker indicating the activation of this pathway instead of the actual entity that regulates the CYPs by itself. Therefore, in this study, treatments neutralizing either IL‐6 or IL‐6R are assumed to have the same modulation effect on CYPs as both treatments block the IL‐6 signaling pathway (Figure S1). Indeed, the PBPK model originally developed using clinical TP‐DI data of sirukumab (against IL‐6) in patients with RA was able to predict the TP‐DI effects of tocilizumab and sarilumab (against IL‐6R), in the same disease population. 22 , 58 , 59
As shown in Figure 1, the reported systemic and local tissue IL‐6 levels are highly variable in a certain disease and more so across different IMIDs. Among SLE, UC, CD, and T1D, SLE has overall the lowest IL‐6 level with the averaged mean value of 12 pg/ml and the highest reported mean value less than 30 pg/ml. Based on the simulation results and identified IL‐6 cutoffs for different magnitude of potential TP‐DIs, anti‐IL‐6 treatment is unlikely to result in clinically meaningful TP‐DIs in patients with SLE. Reported IL‐6 level in T1D ranges from less than 10 pg/ml to around 200 pg/ml, results in the highest averaged mean value among all the IMIDs in this study. The PBPK model predicts more than 30% changes in AUCs of the substrates of all four CYPs including CYP3A4, CYP2C9, CYP2C19, and CYP1A2. For UC and CD, the averaged mean values of systemic IL‐6 levels are around 20 pg/ml and were predicted to have weak CYP regulation effect if not considering high IL‐6 levels in local tissue. The reported local GI tissue IL‐6 concentrations in patients with UC and CD are much higher than the systemic IL‐6 levels. Elevated IL‐6 mRNA in intestine mucosa and increased secretion of IL‐6 in colon specimens from patients with IBD patients were reported. 61 Mahavaram et al. predicted that the AUC of simvastatin decreased to a greater degree when suppression of CYP3A4 was considered both in the liver and intestines compared to the liver alone. 21 In their study, the simulation results suggest that to achieve the literature reported suppression magnitude of simvastatin AUC, IL‐6 level of 100 pg/ml in the liver and intestine model would be needed, whereas 1000 pg/ml IL‐6 would be needed in the liver only mode. Even at an IL‐6 level of 1000 pg/ml, the effect on maximum plasma concentration (Cmax) was still underestimated when suppression in the intestines was not taken into account. 21 In our study, modulation effect on both hepatic and intestinal CYPs were incorporated in the model: same inhibitory effect on hepatic and intestinal CYPs by systemic IL‐6 was assumed for SLE and T1D; and modulation effects on intestinal CYPs by local GI IL‐6 were incorporated for CD and UC. At the average local GI IL‐6 concentrations used for simulations in this study, the modulation effect on the intestinal CYPs were predicted to approach plateau. The high local GI IL‐6 levels were predicted to contribute additionally to the potential CYP modulation effect. The predicted drops in Cmax and AUCs of midazolam after anti‐IL‐6 treatment changed from 18% and 28% to 28% and 39%, respectively, after considering the additional induction effect from neutralizing IL‐6 in the GI tract. In patients with cancer‐IMID multimorbidity, the actual TP‐DIs from cancer and IL‐6 elevation might likely be much more complicated than the simple additive effect assumed in this study. The predictions in this study serve as a very initial attempt to assess potential effect from multimorbidity and more data and further exploration are needed to improve our understanding on TP‐DIs in these patients with cancer‐IMID.
In this study and the previously study by Jiang et al., complete blockade of the IL‐6 signaling pathway, including the part mediated by local GI IL‐6, were assumed after anti‐IL‐6/6R treatments (i.e., sirukumab, tocilizumab, and sarilumab). The reported plasma concentration of sirukumab on day 42 after a 300 mg subcutaneous dose is 27.3 nM. 10 Assuming GI tract biodistribution coefficient of 5–10%, 62 the tissue concentration of sirukumab would reach around 1365–2730 pM, which is still higher than the local IL‐6 concentration and would result in maximum regulation effect (local tissue IL‐6: 3000 pg/ml = 142 pM). Together with high binding affinity between sirukumab and IL‐6 (0.175 pM), a hypothesis of near‐complete neutralization of both systemic and local tissue IL‐6 until day 42 after sirukumab treatment is not unreasonable. There are several therapeutic mAbs targeting other inflammatory cytokines (e.g., IL‐17 and IL‐23) instead of directly targeting IL‐6 pathway have been approved or under development for treating various autoimmune inflammatory disorders. The regulations of these inflammatory cytokines and IL‐6 are related and their pathways interweave through the inflammation pathogenesis. 63 However, the IL‐6 neutralizing effects by these antibodies not directly targeting IL‐6 or IL‐6R are unlikely to be full blockade and consequently the less TP‐DIs would be expected. Data from two recent TP‐DI clinical trials showed that treatment of guselkumab (an anti‐IL‐23 mAb) or secukinumab (an anti‐IL‐17 mAb) did not influence the systemic exposure of the CYP substrates in patients with psoriasis. 64 , 65 The present PBPK model can be easily adapted to assess such TP‐DI potentials by incorporating the disease‐specific baseline IL‐6 level and assuming partial blockade of the IL‐6 signaling pathway.
This PBPK model approach may serve as conceptual framework and workflow process to evaluate the modulation effect on CYPs in patients by therapeutic modalities, which can significantly result in altered levels of proinflammatory cytokines during the treatment period. In this study, we have demonstrated the potential application of the PBPK modeling approach as an initial screening and assessment tool for TP‐DIs. Same as PBPK for small molecule, this approach is expected to help to design a proper TP‐DI study with the right dosing and sampling time to capture the maximum potential TP‐DI effects in the right patient populations. Further refinement and validation of the model may be possible once additional clinical TP‐DI data from IMID disease populations other than RA become available. Of note, we assessed the potential TP‐DIs in IMIDs which are chronic inflammatory diseases with elevated IL‐6 level at SS. For certain cancer indications, immunotherapies, such as T‐cell redirecting bispecific antibodies (bsAbs), may result in transient high level of IL‐6 along with the cytokine release syndrome (CRS) upon T cell’s activation, 66 and potentially lead to undesired TP‐DIs. Future work will include prospective evaluation of the CYP‐mediated TP‐DIs caused by such acute IL‐6 elevation.
CONFLICT OF INTEREST
Y.C., W.Z., X.M., and H.Z. are current employees and shareholders of Janssen. L.W. was an employee of Janssen at the time this work was done. All other authors declared no competing interests for this work.
AUTHOR CONTRIBUTIONS
L.W., Y.C., W.Z., X.M., and H.Z. wrote the manuscript. L.W., Y.C., and H.Z. designed the research. L.W., Y.C., W.Z., X.M., and H.Z. performed the research. L.W. and Y.C. analyzed the data. L.W., Y.C., and W.Z. contributed new reagents/analytical tools.
Supporting information
Figure S1
Table S1‐S2
ACKNOWLEDGEMENTS
The authors thank Dr. Xiling Jiang for her initial work on this PBPK model.
Wang L, Chen Y, Zhou W, Miao X, Zhou H. Utilization of physiologically‐based pharmacokinetic model to assess disease‐mediated therapeutic protein‐disease‐drug interaction in immune‐mediated inflammatory diseases. Clin Transl Sci. 2022;15:464–476. 10.1111/cts.13164
Lujing Wang and Yang Chen contributed equally to the manuscript.
REFERENCES
- 1. Bodeman CE, Dzierlenga AL, Tally CM, et al. Differential regulation of hepatic organic cation transporter 1, organic anion‐transporting polypeptide 1a4, bile‐salt export pump, and multidrug resistance‐associated protein 2 transporter expression in lymphocyte‐deficient mice associates with interleukin‐6 production. J Pharmacol Exp Ther. 2013;347:136‐144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Morgan ET. Impact of infectious and inflammatory disease on cytochrome P450‐mediated drug metabolism and pharmacokinetics. Clin Pharmacol Ther. 2009;85:434‐438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bertilsson PM, Olsson P, Magnusson KE. Cytokines influence mRNA expression of cytochrome P450 3A4 and MDRI in intestinal cells. J Pharm Sci. 2001;90:638‐646. [DOI] [PubMed] [Google Scholar]
- 4. Jing X, Ji P, Schrieber SJ, Fletcher EP, Sahajwalla C. Update on therapeutic protein‐drug interaction: information in labeling. Clin Pharmacokinet. 2020;59:25‐36. [DOI] [PubMed] [Google Scholar]
- 5. Zhou H, Davis HM. Risk‐based strategy for the assessment of pharmacokinetic drug‐drug interactions for therapeutic monoclonal antibodies. Drug Discov Today. 2009;14:891‐898. [DOI] [PubMed] [Google Scholar]
- 6. Evers R, Dallas S, Dickmann LJ, et al. Critical review of preclinical approaches to investigate cytochrome p450‐mediated therapeutic protein drug‐drug interactions and recommendations for best practices: a white paper. Drug Metab Dispos. 2013;41:1598‐1609. [DOI] [PubMed] [Google Scholar]
- 7. Dickmann LJ, Patel SK, Rock DA, Wienkers LC, Slatter JG. Effects of interleukin‐6 (IL‐6) and an anti‐IL‐6 monoclonal antibody on drug‐metabolizing enzymes in human hepatocyte culture. Drug Metab Dispos. 2011;39:1415‐1422. [DOI] [PubMed] [Google Scholar]
- 8. Rubin K, Janefeldt A, Andersson L, et al. HepaRG cells as human‐relevant in vitro model to study the effects of inflammatory stimuli on cytochrome P450 isoenzymes. Drug Metab Dispos. 2015;43:119‐125. [DOI] [PubMed] [Google Scholar]
- 9. Abdel‐Razzak Z, Loyer P, Fautrel A, et al. Cytokines down‐regulate expression of major cytochrome P‐450 enzymes in adult human hepatocytes in primary culture. Mol Pharmacol. 1993;44:707‐715. [PubMed] [Google Scholar]
- 10. Zhuang Y, de Vries DE, Xu Z, et al. Evaluation of disease‐mediated therapeutic protein‐drug interactions between an anti‐interleukin‐6 monoclonal antibody (sirukumab) and cytochrome P450 activities in a phase 1 study in patients with rheumatoid arthritis using a cocktail approach. J Clin Pharmacol. 2015;55:1386‐1394. [DOI] [PubMed] [Google Scholar]
- 11. Jolly M, Francis S, Aggarwal R, et al. Serum free light chains, interferon‐alpha, and interleukins in systemic lupus erythematosus. Lupus. 2014;23:881‐888. [DOI] [PubMed] [Google Scholar]
- 12. Gao SP, Mark KG, Leslie K, et al. Mutations in the EGFR kinase domain mediate STAT3 activation via IL‐6 production in human lung adenocarcinomas. J Clin Invest. 2007;117:3846‐3856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Snell‐Bergeon JK, West NA, Mayer‐Davis EJ, et al. Inflammatory markers are increased in youth with type 1 diabetes: the SEARCH Case‐Control study. J Clin Endocrinol Metab. 2010;95:2868‐2876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Atreya R, Neurath MF. Involvement of IL‐6 in the pathogenesis of inflammatory bowel disease and colon cancer. Clin Rev Allergy Immunol. 2005;28:187‐196. [DOI] [PubMed] [Google Scholar]
- 15. Coutant DE, Kulanthaivel P, Turner PK, et al. Understanding disease‐drug interactions in cancer patients: implications for dosing within the therapeutic window. Clin Pharmacol Ther. 2015;98:76‐86. [DOI] [PubMed] [Google Scholar]
- 16. Arenschield L. The trick of treating cancer in autoimmune diseases. Medscape Medical News. 2019. https://www.medscape.com/viewarticle/921742. [Google Scholar]
- 17. Bois FY. Physiologically based modelling and prediction of drug interactions. Basic Clin Pharmacol Toxicol. 2010;106:154‐161. [DOI] [PubMed] [Google Scholar]
- 18. Nestorov I. Whole‐body physiologically based pharmacokinetic models. Expert Opin Drug Metab Toxicol. 2007;3:235‐249. [DOI] [PubMed] [Google Scholar]
- 19. Gerlowski LE, Jain RK. Physiologically based pharmacokinetic modeling: principles and applications. J Pharm Sci. 1983;72:1103‐1127. [DOI] [PubMed] [Google Scholar]
- 20. U.S. Food & Drug Administration. Drug‐Drug Interaction Assessment for Therapeutic Proteins Guidance for Industry; Aug. 2020
- 21. Machavaram KK, Almond LM, Rostami‐Hodjegan A, et al. A physiologically based pharmacokinetic modeling approach to predict disease‐drug interactions: suppression of CYP3A by IL‐6. Clin Pharmacol Ther. 2013;94:260‐268. [DOI] [PubMed] [Google Scholar]
- 22. Jiang X, Zhuang Y, Xu Z, Wang W, Zhou H. Development of a physiologically based pharmacokinetic model to predict disease‐mediated therapeutic protein‐drug interactions: modulation of multiple cytochrome p450 enzymes by interleukin‐6. AAPS J. 2016;18:767‐776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. U.S. Food & Drug Administration. Clinical Drug Interaction Studies—Cytochrome P450 Enzyme‐ and Transporter‐Mediated Drug Interactions Guidance for Industry. Jan. 2020.
- 24. Carey R, Jurickova I, Ballard E, et al. Activation of an IL‐6:STAT3‐dependent transcriptome in pediatric‐onset inflammatory bowel disease. Inflamm Bowel Dis. 2008;14:446‐457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rosa JS, Oliver SR, Flores RL, et al. Altered inflammatory, oxidative, and metabolic responses to exercise in pediatric obesity and type 1 diabetes. Pediatr Diabetes. 2011;12:464‐472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kitani A, Hara M, Hirose T, et al. Autostimulatory effects of IL‐6 on excessive B cell differentiation in patients with systemic lupus erythematosus: analysis of IL‐6 production and IL‐6R expression. Clin Exp Immunol. 1992;88:75‐83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Reinisch W, Gasché C, Tillinger W, et al. Clinical relevance of serum interleukin‐6 in Crohn’s disease: single point measurements, therapy monitoring, and prediction of clinical relapse. Am J Gastroenterol. 1999;94:2156‐2164. [DOI] [PubMed] [Google Scholar]
- 28. Dima A, Jurcut C, Balanescu P, et al. Clinical significance of serum and urinary interleukin‐6 in systemic lupus erythematosus patients. Egypt Rheumatol. 2017;39:1‐6. [Google Scholar]
- 29. Grondal G, Gunnarsson I, Ronnelid J, et al. Cytokine production, serum levels and disease activity in systemic lupus erythematosus. Clin Exp Rheumatol. 2000;18:565‐570. [PubMed] [Google Scholar]
- 30. Linker‐Israeli M, Deans RJ, Wallace DJ, et al. Elevated levels of endogenous IL‐6 in systemic lupus erythematosus. A putative role in pathogenesis. J Immunol. 1991;147:117‐123. [PubMed] [Google Scholar]
- 31. Targher G, Zenari L, Bertolini L, Muggeo M, Zoppini G. Elevated levels of interleukin‐6 in young adults with type 1 diabetes without clinical evidence of microvascular and macrovascular complications. Diabetes Care. 2001;24:956‐957. [DOI] [PubMed] [Google Scholar]
- 32. Gross V, Andus T, Caesar I, Roth M, Scholmerich J. Evidence for continuous stimulation of interleukin‐6 production in Crohn’s disease. Gastroenterology. 1992;102:514‐519. [DOI] [PubMed] [Google Scholar]
- 33. Mahida YR, Kurlac L, Gallagher A, Hawkey CJ. High circulating concentrations of interleukin‐6 in active Crohn’s disease but not ulcerative colitis. Gut. 1991;32:1531‐1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ramalho T, Filgueiras L, Silva‐Jr IA, Pessoa AFM, Jancar S. Impaired wound healing in type 1 diabetes is dependent on 5‐lipoxygenase products. Sci Rep. 2018;8:14164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Takac B, Mihaljević S, Stefanić M, Glavas‐Obrovac L, Kibel A, Samardzija M. Importance of interleukin 6 in pathogenesis of inflammatory bowel disease. Coll Antropol. 2014;38(2):659–664. [PubMed] [Google Scholar]
- 36. AboElAsrar MA, Elbarbary NS, Elshennawy DE, Omar AM. Insulin‐like growth factor‐1 cytokines cross‐talk in type 1 diabetes mellitus: relationship to microvascular complications and bone mineral density. Cytokine. 2012;59:86‐93. [DOI] [PubMed] [Google Scholar]
- 37. Eilertsen GO, Nikolaisen C, Becker‐Merok A, Nossent JC. Interleukin‐6 promotes arthritis and joint deformation in patients with systemic lupus erythematosus. Lupus. 2011;20:607‐613. [DOI] [PubMed] [Google Scholar]
- 38. Farhan J, Al‐Shobaili HA, Zafar U, et al. Interleukin‐6: a possible inflammatory link between vitiligo and Type 1. Diabetes. 2014;71:151‐157. [DOI] [PubMed] [Google Scholar]
- 39. Koss K, Satsangi J, Welsh KI, Jewell DP. Is interleukin‐6 important in inflammatory bowel disease? Genes Immun. 2000;1:207‐212. [DOI] [PubMed] [Google Scholar]
- 40. Chatzigeorgiou A, Harokopos V, Mylona‐Karagianni C, et al. The pattern of inflammatory/anti‐inflammatory cytokines and chemokines in type 1 diabetic patients over time. Ann Med. 2010;42:426‐438. [DOI] [PubMed] [Google Scholar]
- 41. Álvarez‐Rodriguez L, Riancho‐Zarrabeitia L, Calvo‐Alen J, Lopez‐Hoyos M, Martinez‐Taboada V. Peripheral B‐cell subset distribution in primary antiphospholipid syndrome. Int J Mol Sci. 2018;19:589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Spronk PE, ter Borg EJ, Limburg PC, Kallenberg CG. Plasma concentration of IL‐6 in systemic lupus erythematosus; an indicator of disease activity? Clin Exp Immunol. 1992;90:106‐110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Gustot T, Lemmers A, Louis E, et al. Profile of soluble cytokine receptors in Crohn’s disease. Gut. 2005;54:488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hyams JS, Fitzgerald JE, Treem WR, Wyzga N, Kreutzer DL. Relationship of functional and antigenic interleukin 6 to disease activity in inflammatory bowel disease. Gastroenterology. 1993;104:1285‐1292. [DOI] [PubMed] [Google Scholar]
- 45. Peterson E, Robertson A, Emlen W. Serum and urinary interleukin‐6 in systemic lupus erythematosus. Lupus 1996;5:571‐575. [DOI] [PubMed] [Google Scholar]
- 46. Hammed IK, Rashid NF, Abed BA. Serum Interleukin‐6 level in children with type 1 diabetes mellitus. J Faculty Med Baghdad. 2012;54:228‐230. [Google Scholar]
- 47. Schurmann G, Betzler M, Post S, Herfarth C, Meuer S. Soluble interleukin‐2 receptor, interleukin‐6 and interleukin‐1 beta in patients with Crohn’s disease and ulcerative colitis: preoperative levels and postoperative changes of serum concentrations. Digestion. 1992;51:51‐59. [DOI] [PubMed] [Google Scholar]
- 48. Mitsuyama K, Toyonaga A, Sasaki E, et al. Soluble interleukin‐6 receptors in inflammatory bowel disease: relation to circulating interleukin‐6. Gut. 1995;36:45‐49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kusugami K, Fukatsu A, Tanimoto M, et al. Elevation of interleukin‐6 in inflammatory bowel disease is macrophage‐ and epithelial cell‐dependent. Dig Dis Sci. 1995;40:949‐959. [DOI] [PubMed] [Google Scholar]
- 50. Reinecker HC, Steffen M, Witthoeft T, et al. Enhanced secretion of tumour necrosis factor‐alpha, IL‐6, and IL‐1 beta by isolated lamina propria mononuclear cells from patients with ulcerative colitis and Crohn’s disease. Clin Exp Immunol. 1993;94:174‐181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Reimund J‐M, Wittersheim C, Dumont S, et al. Increased production of tumour necrosis factor‐alpha, interleukin‐1 beta, and interleukin‐6 by morphologically normal intestinal biopsies from patients with Crohn’s disease. Gut. 1996;39:684‐689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Powell N, Lo JW, Biancheri P, et al. Interleukin 6 increases production of cytokines by colonic innate lymphoid cells in mice and patients with chronic intestinal inflammation. Gastroenterology. 2015;149:456‐467.e415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Bernardo D, Vallejo‐Díez S, Mann ER, et al. IL‐6 promotes immune responses in human ulcerative colitis and induces a skin‐homing phenotype in the dendritic cells and Tcells they stimulate. Eur J Immunol. 2012;42:1337‐1353. [DOI] [PubMed] [Google Scholar]
- 54. Rowland Yeo K, Jamei M, Yang J, Tucker GT, Rostami‐Hodjegan A. Physiologically based mechanistic modelling to predict complex drug‐drug interactions involving simultaneous competitive and time‐dependent enzyme inhibition by parent compound and its metabolite in both liver and gut ‐ the effect of diltiazem on the time‐course of exposure to triazolam. Eur J Pharmaceut Sci. 2010;39:298‐309. [DOI] [PubMed] [Google Scholar]
- 55. Jamei M, Marciniak S, Feng K, et al. The Simcyp population‐based ADME simulator. Expert Opin Drug Metab Toxicol. 2009;5:211‐223. [DOI] [PubMed] [Google Scholar]
- 56. Schwenger E, Reddy VP, Moorthy G, et al. Harnessing meta‐analysis to refine an oncology patient population for physiology‐based pharmacokinetic modeling of drugs. Clin Pharmacol Ther. 2018;103:271‐280. [DOI] [PubMed] [Google Scholar]
- 57. Sanada H, Sekimoto M, Kamoshita A, Degawa M. Changes in expression of hepatic cytochrome P450 subfamily enzymes during development of adjuvant‐induced arthritis in rats. J Toxicol Sci. 2011;36:181‐190. [DOI] [PubMed] [Google Scholar]
- 58. Schmitt C, Kuhn B, Zhang X, Kivitz AJ, Grange S. Disease‐drug‐drug interaction involving tocilizumab and simvastatin in patients with rheumatoid arthritis. Clin Pharmacol Ther. 2011;89:735‐740. [DOI] [PubMed] [Google Scholar]
- 59. Lee EB, Daskalakis N, Xu C, et al. Disease‐drug interaction of sarilumab and simvastatin in patients with rheumatoid arthritis. Clin Pharmacokinet. 2017;56:607‐615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Tanaka T, Narazaki M, Kishimoto T. IL‐6 in inflammation, immunity, and disease. Cold Spring Harb Perspect Biol. 2014;6:a016295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Jones SC, Trejdosiewicz LK, Banks RE, et al. Expression of interleukin‐6 by intestinal enterocytes. J Clin Pathol. 1993;46:1097‐1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Shah DK, Betts AM. Antibody biodistribution coefficients: inferring tissue concentrations of monoclonal antibodies based on the plasma concentrations in several preclinical species and human. MAbs. 2013;5:297‐305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Tang C, Chen S, Qian H, Huang W. Interleukin‐23: as a drug target for autoimmune inflammatory diseases. Immunology. 2012;135:112‐124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Zhu Y, Xu Y, Zhuang Y, et al. Evaluating potential disease‐mediated protein‐drug interactions in patients with moderate‐to‐severe plaque psoriasis receiving subcutaneous guselkumab. Clin Transl Sci. 2020;13:1217‐1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Bruin G, Hasselberg A, Koroleva I, et al. Secukinumab treatment does not alter the pharmacokinetics of the cytochrome P450 3A4 substrate midazolam in patients with moderate to severe psoriasis. Clin Pharmacol Ther. 2019;106:1380‐1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Yuraszeck T, Kasichayanula S, Benjamin JE. Translation and clinical development of bispecific T‐cell engaging antibodies for cancer treatment. Clin Pharmacol Ther. 2017;101:634‐645. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1
Table S1‐S2