

Summary
Leishmaniasis is a neglected tropical disease endemic in over 90 countries. The disease has two main pathologies; cutaneous leishmaniasis (CL) that generally self-heals, and visceral leishmaniasis (VL) that is fatal if untreated. The majority of VL cases, concentrated on the Indian subcontinent (ISC) and East Africa, are caused by Leishmania donovani. However, recent foci of CL on the ISC have been attributed as an atypical phenotype of L. donovani including a recent outbreak in Himachal Pradesh, India. Whole genome sequencing and phylogenetic analysis was undertaken to investigate the origins and genetic factors leading to this pathology atypical of L. donovani. Here we demonstrate the isolate from Himachal Pradesh is derived from a genetic hybridization between two independent L. donovani parents from the ‘Yeti’ ISC1 divergent clade of parasites, identified in the Nepalese highlands. This reveals that intraspecies L. donovani hybrids can give rise to a novel strain associated with CL.
Subject areas: Biological sciences, Genetics, Genomics, Microbial genomics, Microbiology parasite
Graphical abstract
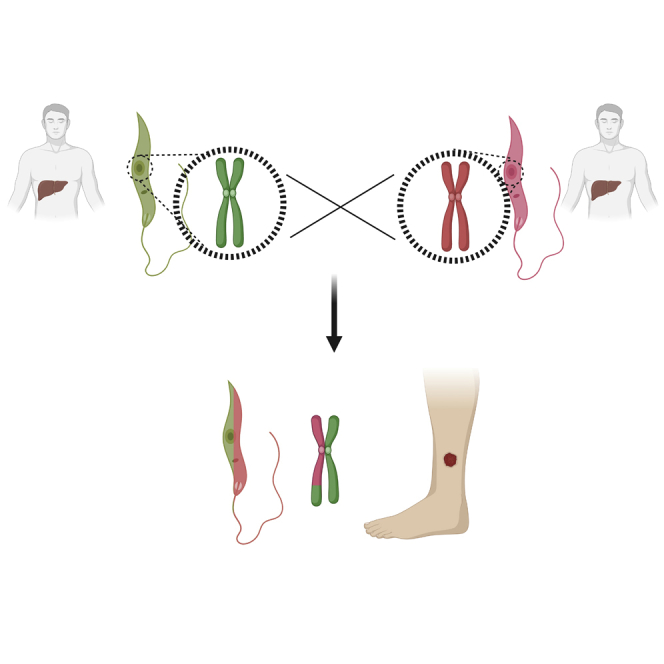
Highlights
-
•
A novel focus of cutaneous Leishmaniasis is emerging in Himachal Pradesh
-
•
The normally visceral Leishmania donovani parasite is responsible for the focus
-
•
The isolated parasite is an intraspecies genetic hybrid
-
•
Extensive genomic hybridization could explain this atypical phenotype
Biological sciences; Genetics; Genomics; Microbial genomics; Microbiology parasite
Introduction
Leishmaniasis is a disease complex with different parasite species associated with a gradation of clinical manifestations ranging from cutaneous leishmaniasis (CL) with skin lesions to mucocutaneous leishmaniasis (MCL) involving the respiratory mucous membrane to a lethal systemic visceral leishmaniasis (VL) (Burza et al., 2018). Among the 98 countries and territories endemic for leishmaniasis, 68 countries are endemic for both VL and CL, nine countries are endemic for VL alone, and 21 countries are endemic for CL alone (WHO, 2018a; 2018b). Although the Leishmania donovani complex is responsible for systemic VL, numerous Old World and New World Leishmania species cause CL including Leishmania tropica in the Indian subcontinent (ISC). The classical association between the infecting Leishmania species and their associated clinical outcomes is of interest to understand species-specific disease tropism, virulence, and pathogenesis.
During the last decade, this association has become blurred as parasite species known to cause VL are associated with CL and vice versa (Thakur et al., 2018). The atypical disease phenotype trend associated with novel Leishmania variants is reported in known and newer endemic regions of the world (Thakur et al., 2018, 2020; Siriwardana et al., 2019). In Sri Lanka, the atypical CL and the rare coexisting VL form, are caused by variants of L. donovani Mon-37 based on genetic characterization of L. donovani clinical isolates (Siriwardana et al., 2007; Karunaweera, 2009; Ranasinghe et al., 2012; Zhang et al., 2014; Kariyawasam et al., 2017; Lypaczewski et al., 2018; Samarasinghe et al., 2018). Whole genome based studies of the CL L. donovani isolates in comparison to VL isolates revealed discrete chromosomal and gene copy-number variations and single nucleotide polymorphisms (SNPs) as underlying genetic alterations giving rise to these atypical L. donovani variants (Zhang et al., 2014; Lypaczewski et al., 2018; Samarasinghe et al., 2018). More recently, genomic and phylogenic analysis of L. donovani clinical isolates from Sri Lanka identified interspecies genome hybridization and recombination between L. donovani and Leishmania major/L. tropica revealing multiple mechanisms contribute to the atypical CL phenotype of L. donovani (Lypaczewski and Matlashewski, 2021).
Concurrently, evidence that genetic hybridization between Leishmania species can generate exceptions to the classical species/disease associations is emerging (Ozbilgin et al., 2012; Iantorno et al., 2017). The occurrence of interspecies and intraspecies natural Leishmania hybrids has been recently demonstrated along with well documented experimentally derived hybrids exhibiting genome wide recombination events (Akopyants et al., 2009; Inbar et al., 2019; Cotton et al., 2020; Lypaczewski and Matlashewski, 2021). This phenomenon can lead to the emergence of novel Leishmania genotypes that may cause more than one clinical outcome, apart from contributing to the parasite genome diversity.
Recent developments in whole genome sequencing (WGS) with high resolution reference genomes have helped to unravel the genetic makeup of parasite species and strains providing insight into determinants driving differential disease phenotypes, atypical disease manifestations, drug resistance, and in defining the population structure circulating in the known and newer endemic regions (Imamura et al., 2016). A rigorous genome wide analysis of clinical isolates from the ISC has revealed a heterogeneous L. donovani population structure with discrete genetic lineages circulating in endemic lowlands of India, Nepal, and Bangladesh along with a newly emerging, genetically distinct ISC1 outlier subpopulation in the highlands of Nepal, now beginning to intrude into the low-lying regions (Cuypers et al., 2018; Seblova et al., 2019). The newer ISC1 L. donovani population harbors genetic variations in coding regions related to virulence and susceptibility to drug resistance (Cuypers et al., 2018; Seblova et al., 2019).
The L. donovani hybrid progenies in Sri Lanka appear to have originated from Ethiopia (Lypaczewski and Matlashewski, 2021), a known hot spot for intraspecific hybridization of L. donovani (Gelanew et al., 2010, 2014; Cotton et al., 2020) because of the coexistence of distinct clades of L. donovani. Similarly, there are at least 10 distinct groups of L. donovani circulating in the ISC (Imamura et al., 2016). It was therefore necessary to explore the possibility of genomic hybridization in the focus of CL cases caused by an atypical L. donovani in Himachal Pradesh, India (Thakur et al., 2020) through WGS of an L. donovani isolate derived from an indigenous CL patient. The results of this study provide a new perspective on the genetic basis for the dermotropic tropism and insight into the origin of this L. donovani variant (LdHP). The study will further contribute to the genetic surveillance of L. donovani populations indigenous to the ISC in support of the ongoing disease elimination program (Rijal et al., 2019).
Results
The Himachal Pradesh derived L. donovani isolate (LdHP) was retrieved from an indigenous CL patient, with a single erythematous and papular lesion above the upper lip diagnosed at 3 months after the initial appearance and there was no evidence of visceral disease (Figure S1A). The isolate was initially identified as an L. donovani variant based on species-specific sequence analysis of ITS1 and 6PGDH amplification products (Figure S1B).
Phylogeny of the atypical isolate
As previous analysis of samples originating from the Himachal Pradesh CL focus were reported to be distinct from the core ISC L. donovani strains (Thakur et al., 2020), we compared the whole genome sequence of the LdHP isolate to investigate its origins. As shown in Figure 1A, a phylogenetic analysis compared to the worldwide L. donovani isolates places LdHP in a distinct, yet related cluster in the ISC. Indeed, the LdHP isolate appears in the ISC1 ‘Yeti’ subgroup. As shown in Figure 1B, however, the ISC1 Yeti cluster consists of closely geographically related samples originating from the Nepalese highlands (red), whereas the LdHP sample was isolated from the distant Himachal Pradesh CL focus in India (Thakur et al., 2020) (green).
Figure 1.

Phylogeny of the atypical isolate
(A) Phylogeny of all L. donovani isolates showing the origin of the cutaneous Himachal isolate (LdHP, red) to be within the divergent ISC1 cluster of Indian L. donovani parasites. Genetic distance scale shown in the upper left corner.
(B) Geographical distribution of ISC1 isolates. The locations of sampling for all isolates grouped in ISC1 are shown in red markers and are clustered to a small region of the Nepalese highlands. The sampling location for the cutaneous Himachal isolate is shown in green. The Himachal isolate is genetically related but geographically distant from the ISC1 cluster. Scale bar representing 200 miles shown in the lower right corner.
Hybrid origins of the cutaneous Himachal isolate
All ISC1 isolates previously sequenced originated from VL patients, whereas LdHP was associated with a CL patient. We therefore performed a SNP analysis using the CL reference genome from Sri Lanka to examine potential genetic changes that could have contributed to the atypical phenotype of the LdHP strain. As shown in Figure 2A, WGS reveals multiple polymorphisms that were mostly heterozygous with frequencies around 50% as shown in this representative 10 kB section of chromosome 3. Consistent with previous observations, widespread heterozygosity could indicate a genetic hybridization between different genomes (Rogers et al., 2014; Lypaczewski and Matlashewski, 2021). We therefore compared the entire genome of LdHP, to the global population of L. donovani. A group of intraspecies hybrids between two distinct clades of L. donovani from Ethiopia (Cotton et al., 2020; Lypaczewski and Matlashewski, 2021) serves as a baseline for intraspecies hybridization and interspecies hybrids associated with CL in Sri Lanka as a baseline for hybridization across species (Lypaczewski and Matlashewski, 2021). As shown in Figure 2B, the heterozygosity ratio of LdHP was similar to that of intraspecific L. donovani hybrids suggesting that LdHP is an intraspecies hybrid parasite.
Figure 2.

Hybrid origins of the cutaneous Himachal isolate
(A) Representative alignment of a 10 kb window on chromosome 3 (x axis) for the cutaneous isolate (LdHP) showing the density of heterozygous polymorphisms. The individual reads (y axis) are aligned in the bottom panel. Perfect base matches are shown in gray, mismatches are colored according to the corresponding polymorphic base (ATCG). The upper panel shows the resulting coverage from all alignments on this region of the genome with the same color coding.
(B) The heterozygous polymorphism ratio of the Himachal cutaneous isolate is consistent with an intraspecies hybrid genotype. The frequency of heterozygous mutations in the cutaneous isolate was compared to those found in the worldwide population of L. donovani, and two baseline hybrid population frequencies: an intraspecies hybrid focus from distinct Ethiopian clades and an interspecies hybrid parasite focus in Sri Lanka previously characterized.
(C) Whole genome sequencing coverage depth to determine chromosomal copy number. Mean coverage of each gene is plotted along the chromosomes and colored according to median copy number (gray), decrease (red) or an increase in copy number (blues). The average of all chromosomes serves as the baseline for diploid levels, normally tetraploid chromosome 31 serves as the baseline for tetraploidy. Chromosomes one and six show increased copy number of all genes along their length consistent with a chromosomal amplification resulting in tetraploidy. Chromosomes 8,9,13–16,21,23,26 & 33 show an intermediate increase in copy numbers across the entire chromosomes consistent with an amplification resulting in triploidy (inner track). The location of all polymorphisms identified compared to the reference LdCL genome are shown in the middle track. Homozygous polymorphisms are colored in gray, heterozygous (likely hybrid) polymorphisms are colored in red. Heterozygous polymorphisms are spread evenly across the entire genome with the exception of chromosome 22 which appears mostly homozygous. The frequency of heterozygous SNP is plotted at every heterozygous loci (outer track) and shows polyploid chromosomes moving from an even ∼50/50 allele distribution to a bimodal (~33/66 or ∼25/75) distribution along the chromosomes with increased ploidy.
We investigated chromosomal dosage of this genome, as hybridization has been previously shown to be accompanied by extensive aneuploidy (Iantorno et al., 2017; Inbar et al., 2019). As shown for chromosomes 1 to 36 in Figure 2C (inner rings), the LdHP isolate has several triploid or tetraploid chromosomes as shown by the consistent increase in gene copy numbers across entire chromosomes. In addition, as seen in the outer ring, the overlaid frequencies of polymorphisms show a shift from unimodal distribution centered around the 50% as is expected in the case of a heterozygous diploid genome (for example, chromosomes 2–5) to bimodal distribution frequencies with peaks at 33/66% or 25/75% (example chromosomes one and 6), consistent with unequal heterozygous triploid and tetraploid chromosome numbers, respectively. Taken together, the relatively high level of chromosome aneuploidy is consistent with a hybrid strain.
Analysis of parental haplotypes
We previously demonstrated that polymorphisms present in hybrid isolates can be used to extrapolate the genetic makeup of the parental parasites (Lypaczewski and Matlashewski, 2021). As LdHP appeared to be consistent with an intraspecific hybridization event, we isolated the closest related genomes according to our phylogenetic analysis to filter heterozygous polymorphisms present in the putative hybrid to reconstruct the most likely genomes of the parental parasites. As shown in Figure 3A, the LdHP isolate (red) appear to be the progeny of an outcross event between an L. donovani parasite belonging to the core of the ISC1, and a parasite closely related to a unique LdBPK512 (Imamura et al., 2016; Seblova et al., 2019) isolate, itself an outlier in the ISC1 group, highlighting the novel and unique genetic makeup of the LdHP isolate.
Figure 3.

Analysis of parental haplotypes
(A) The likely parents of the hybrid strain both belong to the ISC1 L. donovani Indian subcontinent parasite group. The phylogeny analysis places the hybrid isolate (red) next to its closest parent proxy (LdBPK512). The reconstructed genotype for the second parent (blue) also falls with the ISC1 group. Genetic distance scale shown in the upper left corner.
(B) Species source and selection process used to collect and filter high quality whole genome sequencing data read sets from genomic origins expanded to Old World species of Leishmania. See also Figure S2.
As the exact parents of this hybrid progeny could not be identified, we further expanded our phylogeny analysis beyond the 684 isolates used prior (Lypaczewski and Matlashewski, 2021) to determine if a more precise parental strain could be identified. Further, as our previous analysis revealed the existence of mislabeled read sets in the NCBI sequencing read archive (SRA), we expanded our search to include whole genome sequencing experiments data from Old World species (L. donovani, L. infantum, L. tropica, and L. major) using similar criteria for high quality genome sequencing data as previously described (Lypaczewski and Matlashewski, 2021) (Figure 3B) resulting in 1908 genomes.
As the two parental strains leading to an intraspecies hybrid are more related, we were unable to perform read-based haplotype phasing as the distance between two consecutive polymorphisms exceeded the sequencing read-length in contrast to our previous observations (Lypaczewski and Matlashewski, 2021). However, because of the presence of extensive chromosomal aneuploidy, the unequal minor allele frequencies could be used to segregate and assign polymorphisms to a specific parent (Figure 2C). As shown in Figure S2A, we performed a phylogenetic analysis after assigning frequency-phased polymorphisms on chromosome 1 using 1265 isolates with at least 10X coverage on 90% of the chromosome 1. As shown, although the results of this haplotype phasing confirmed the whole genome phylogenetic analysis, an exact parental match remained elusive. In a further attempt to identify a closer parental strain, we examined the kDNA maxi circle sequences as they are more conserved and unilaterally inherited from a parent. 1503 isolates for which coverage over 10X could be obtained in the conserved region of the maxi circle were examined. As shown in Figure S2B, analysis of the core maxicircle region places the LdHP strain, and consequently one of its parents, within the ISC1 group but does not match any single parent.
Analysis of LdHP unique genetic features
As no exact parental match could be found, we examined the polymorphisms present in this hybrid progeny more closely to identify novel alleles. As all previous ISC1 isolates were derived from visceral infections, any polymorphism seen in the LdHP isolate at either heterozygous or homozygous frequencies were discarded if seen in any of the ISC1 sequencing sets. This analysis resulted in the identification of 1704 LdHP unique polymorphisms with coding change effects across 1299 genes as summarized in Figure 4A and Table S1.
Figure 4.

Analysis of LdHP unique genetic features
(A) Summary of LdHP unique genetic changes. Genes with LdHP unique polymorphisms are highlighted across the 36 chromosomes in the genome. Genes in the forward direction are labeled in blue, genes in the reverse direction are labeled in red.
(B) Annotation enrichment analysis of genes with unique polymorphisms in the cutaneous Himachal isolate along the chromosomes with increased ploidy and unequal parental contributions.
(C) Copy number analysis of the A2 gene family. Compared to both the baseline CL-SL and the increased SL-VL strain, the LdHP isolate shows a decrease in A2 gene copy numbers. Raw sequencing depth ratios are shown in the left column and copy numbers extrapolated using four A2 genes in the LdCL reference are shown in the middle column and represented visually in the right side. See also Table S1.
As chromosomes with bimodal distributions appear to favor contributions from one parent over the other, and previous observations revealed that Leishmania can benefit from aneuploidy modulation and unequal crossing over to drive its evolutionary program (Sterkers et al., 2014; Barja et al., 2017), we filtered polymorphisms in genes present on unequal chromosomes as potential sources of the atypical CL phenotype of LdHP. Briefly, we selected polymorphisms on aneuploid chromosomes with an increased contribution to the genome as reflected by a shift in the hybrid allele frequencies from ∼50% to ∼66%, 75%. As shown in Figure 4B, enrichment analysis performed both by pathway and ontology group highlights selection for carbohydrate pathway components.
Our previous observations identified a decreased copy number of the A2 virulence gene family in a cutaneous L. donovani isolate from Sri Lanka compared to a closely matched visceral isolate (Zhang et al., 2014). Therefore, we investigated the copy numbers of the A2 gene family in the LdHP isolate. As shown in Figure 4C, when the reference isolate is aligned to the reference sequence containing four A2 copies along with the related VL strain from Sri Lanka, the VL strain has a higher ratio of coverage in the A2 genes to the coverage of other genes on chromosome 22 as it contains more than the four copies placed in this reference genome. The closest available parental proxies, LdBPK512 and LdBPK026, have coverage ratios consistent with approximately four and three copies of A2, whereas the LdHP isolate is most consistent with three copies.
Discussion
This study characterizes the causative agent of CL, derived from a focus of CL in the Himachal Pradesh region of India as an atypical L. donovani strain. The major discovery within is that L. donovani intraspecies genomic hybridization is associated with a phenotypic shift from a visceral to a cutaneous parasite as the isolate analyzed was derived from a patient with typical papular skin lesion and no evidence of visceral disease (Figure S1). This is in contrast to a previous observation revealing interspecies L. tropica/L. donovani and L. major/L. donovani genomic hybridization associated with widespread transmission of CL in Sri Lanka (Lypaczewski and Matlashewski, 2021). This is the first example to our knowledge of an L. donovani intraspecies hybrid with an atypical phenotype associated with CL. However, in this case, it remains uncertain if this is a direct consequence of hybridization or the dominant phenotype of one of the parental strains. Moreover, in contrast to the previously described L. donovani hybrid parasites originating from the African continent and associated with CL in the ISC (Lypaczewski and Matlashewski, 2021), the LdHP isolate appears to have originated within the ISC and not imported from a hybrid ‘hotspot’ outside this region.
Although highly unlikely, a polyclonal infection remains a minor possibility. One clone always has a growth advantage and overtakes the culture rapidly even when both parasites are closely related (Domagalska et al., 2019). Therefore, it is extremely unlikely that two clonal lines have coexisted with an identical growth rate leading to a 50/50 mixture of alleles. Further, it is not possible to conclude whether the results reflect a hybrid genome adaptation to a wider population or whether this is a unique event. Analysis of additional patients from this region of Himachal will be required to address this question.
As shown in Figure 2C, the high density of segregating alleles is present on almost all the chromosomes, indicating a most likely equal and complete initial contribution of each genome from the parental parasites. Further, this isolate appears to be in the process of selecting parental genes as unequal genetic contributions can be seen in several aneuploid chromosomes, likely having selected genes allowing for a survival advantage in the skin. However, as shown in the frequency distributions on aneuploid chromosomes, alternative alleles (presumably linked to visceral organ survival) remain present in low amounts. As Leishmania has been shown to readily modulate its chromosome copy numbers through mosaic aneuploidy (Dumetz et al., 2017), the low copy genes which may convey a visceral organ survival advantage could reemerge. This is particularly worrying for the VL elimination program which largely targets symptomatic visceral disease cases (Rijal et al., 2019). These hybrid parasites causing CL could evade detection and become a hidden reservoir for VL cases if not considered in the ISC elimination program.
It is interesting that hybridization is used as a means for adaptation to the host cutaneous environment after years of pressure against visceral disease through the VL elimination program as parasites causing cutaneous disease may now have a survival advantage. This is similar to isolates from Sri Lanka where hybrids are also associated with the VL to CL shift. The advantage associated with this change has not been identified, but a strong pressure exists as multiple independent genetic conversions occurred in Sri Lankan CL isolates (Lypaczewski and Matlashewski, 2021) and it is possible that a similar environmental pressure may have recently emerged in Himachal Pradesh. It would be interesting to investigate the vector and its feeding habits in this environment as it may reveal a new animal reservoir driving this evolution.
Although future studies are required to confirm and focus on the mutated or amplified genes and pathways leading to the phenotypic conversion of this isolate, the carbohydrate metabolism pathways of L. donovani were enriched in chromosomes displaying abnormal ploidy. As modulation of glucose metabolism has been shown to contribute to resistance to oxidative stress (Ghosh et al., 2015), these changes could be related to the phenotypic changes displayed by this isolate. In addition, this selection would reflect an adaptation for the phagosomal environment of dermis macrophage compared to macrophages from the visceral organs.
Although the putative parental strains appear to belong to the ISC1 cluster (Figure 3A), their exact identity remains unknown. As the ISC1 cluster is sparsely populated, no exact genomic match could be found. Further, haplotype separation was inferred rather than determined through read-based phasing because of the low density of polymorphisms. As this hybrid is an intraspecies progeny of related parasites, the distance between two consecutive polymorphisms can be several multiples of the short Illumina read length and as such few polymorphisms can accurately be assigned to one parent. Although the extensive aneuploidy allows determination using haplotype frequency analysis on some chromosomes, accurate determination of the parental genotype would require a more extensive survey of ISC1 circulating strains.
Interestingly, although several polymorphisms could be assigned to a specific parent, over 15% of the genome contained entirely LdHP unique mutations, highlighting the genetic diversity of the missing ISC1 cluster isolates. Further investigation of these polymorphisms and perhaps a reduction in potential targets from a more extensive characterization of circulating ISC1 strains will be required to narrow down contributors to the atypical phenotype of the LdHP strain. In addition, although the polymorphism previously identified in a cutaneous isolate from Sri Lanka (Zhang et al., 2014; Lypaczewski et al., 2018) were not shared by the LdHP isolate, 18 of the 83 previously identified potential virulence factors in Sri Lanka also contained novel polymorphisms. Although 18 genes may not appear significant because of the large number of novel polymorphisms, these should nonetheless be prioritized in virulence studies. Similarly to the reduced A2 gene copy numbers in the cutaneous Sri Lankan isolates (Zhang et al., 2014; Lypaczewski et al., 2018), the LdHP isolate also has a low copy number of A2 genes although a direct visceral comparison is not possible and one of the isolates closely related to a parental strain (LdBPK026) also appears to have low copy numbers of A2 but is still associated with a visceral phenotype. However, it is possible that the combination of low A2 levels and certain polymorphisms acquired through hybridization was involved in the phenotype change of LdHP.
Limitations of the study
Although this report implicates genetic hybrids as a possible contributor to the CL foci, we report on a single isolate; therefore, it is unclear how prevalent these parasites are, because intraspecies L. donovani hybrids are not always easily detected by classical fingerprinting approaches and will require WGS of multiple isolates per CL focus to answer these and other outstanding questions.
Accession numbers
The sequencing data for this project has been deposited under the NCBI BioProject:PRJNA701770.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, peptides, and recombinant proteins | ||
| RPMI-1640 | Gibco | N/A |
| M199 | Gibco | N/A |
| Novy-MacNeal-Nicolle (NNN) rabbit blood agar (BA) | Himedia | N/A |
| Deposited data | ||
| L. donovani HP CL66 | This paper | BioProject:PRJNA701770 |
| Software and algorithms | ||
| samtools | Li et al., 2009 | N/A |
| VarScan2 | Koboldt et al., 2012 | N/A |
| TASSEL | Bradbury et al., 2007 | N/A |
| Bcftools | Li et al., 2009 | N/A |
| iToL | Letunic and Bork, 2019 | N/A |
| SRA-Toolkit | NCBI | N/A |
| Burrows Wheeler Aligner | Li, 2013 | N/A |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Greg Matlashewski (greg.matlashewski@mcgill.ca).
Materials availability
The LdHPCL66 isolate raw sequencing data have been deposited at NCBI SRA and are publicly available as of the date of publication.
Experimental model and subject details
The protocol was reviewed and approved by the Institutional Ethics Committee, Indira Gandhi Medical College (IGMC), Shimla, Himachal Pradesh Approval no. HFW (MS) G-5 (Ethics)/2014-10886 and Central University of Punjab, Approval no. CUPB/IEC/2016/034. Lesional biopsy sample was collected from the CL patient reporting at Department of Dermatology, Indira Gandhi Medical College, Shimla. Written consent was obtained before the sample collection, with record of baseline characteristics of the patient. Lesional 4 mm punch biopsy was taken from the indigenous CL patient, a 12-year-old girl. The patient was a resident from Rampur, a region with upcoming cases of CL along the river Sutluj in Himachal Pradesh, with no travel history outside the state.
Method details
Culture of the clinical isolate for WGS
Lesional tissue biopsy was minced into a fine homogenate and inoculated in the biphasic media containing M199 media (Gibco) in liquid phase and Novy-MacNeal-Nicolle (NNN) rabbit blood agar (BA) medium in solid phase in a sterile 48-well plate (Himedia) and in RPMI-1640 and M199 liquid media with 20% FBS, Penicillin (100 units/ml) and Streptomycin (100ug/ml). The inoculated plate was incubated at 25 ± 1°C in a shaker incubator and screened on alternate days. Once the promastigote form of the parasite was clearly visible with good growth, the culture was transferred to T-25 tissue culture flask. The promastigote culture was harvested at second passage and genomic DNA (gDNA) isolation was performed using the standard phenol-chloroform extraction method.
Genomic analysis of LdHP
Analysis of LdHP was performed as previously described (Lypaczewski and Matlashewski, 2021). Briefly, raw Illumina paired end sequencing reads were aligned to the reference LdCL genome (ASM371957v1) (Lypaczewski et al., 2018) using the Burrows Wheeler Aligner (Li, 2013). The alignments were tabulated using samtools mpileup (Li et al., 2009) and variant loci were identified using VarScan2 software (Koboldt et al., 2012). Genomic locations of SNPs and enrichment analyses were performed using TriTrypDB (Amos et al., 2022).
L. donovani global strains phylogeny
The phylogeny of L. donovani previously generated (Lypaczewski and Matlashewski, 2021) was merged to the SNV list generated for LdHP with BCFtools (Li et al., 2009) using the command ‘bcftools merge –missing-to-ref ’. The alignment was then transferred to TASSEL version 5.0 (Bradbury et al., 2007) to compute the distance matrix for a phylogenetic tree using the Neighbor-Joining algorithm. The phylogenetic tree was then saved in Newick format to be visualized using Interactive Tree of Life (IToL) (Letunic and Bork, 2019).
Heterozygosity of L. donovani isolates worldwide
As previously described (Lypaczewski and Matlashewski, 2021), variant loci from the VCF files were assigned a HET or HOM value based on the variant allele read frequency. The frequency of HET and HOM variant annotations across the entire genome was calculated on a per sample and plotted as the relative fraction (Het/Het + Hom). The baseline values for intraspecies and interspecies hybrids were derived from 10 previously characterized intraspecies hybrids and previously characterized Sri Lankan hybrids respectively. The remaining samples were used to calculate the normal L. donovani heterozygous polymorphism frequencies.
Reconstruction of hybrid parental genomes
The closest related L. donovani isolate based on phylogeny was used as potential Parent A (LdBPK512). Polymorphisms at homozygous levels in the Himachal hybrid isolate were considered to have been contributed by both parents. Loci with heterozygous polymorphisms in LdHP were segregated based on their presence in LdBPK512. The reconstructed genotype was then analyzed as described above. Approximate phasing (Legrand et al., 2008) of heterozygous SNPs on aneuploid chromosome one was performed by filtering minor allele frequencies (MAF) corresponding to three copies compared to one copy: MAF below 30% were assigned to one parent and MAF above 70% were assigned to a different parent. kDNA phylogeny was performed as above, using the Pasteur Strain L. donovani kDNA maxicircle sequence as a reference (CP022652.1) limited to the conserved positions 4,000-20,000. The Sankey diagram was designed with SankeyMATIC.
Coverage and ploidy
Mean coverage values were computed using the alignments generated described above. The depth of coverage was calculated for each gene using BEDtools with the command ‘bedtools coverage-mean-a [LdCLgenes.gff]-b [BWA alignment file]’ and plotted using Circos (Krzywinski et al., 2009) with a color-coded coverage depth with gray being average, red being below and blue being above. Additionally, the raw minor allele frequencies (MAF) values were obtained from the VarScan2 annotated VCF file for all heterozygous loci and plotted as an outer track.
A2 copy number determination
A2 copy numbers were estimated by sequencing depth coverage of A2 loci compared to the average sequencing depth on Chromosome 22 similarly to previous estimations (Zhang et al., 2014). The coverage of A2 loci was used with the known 4 copies in the LdCL reference genome to extrapolate an estimated gene copy number (Lypaczewski et al., 2018).
Acknowledgments
This research was supported by a grant from the Canadian Institutes of Health Research (CIHR) to G.M., a doctoral training award from the Fond de Recherche du Quebec en Santé (FRQS) to P.L., and a Collaborative Research Project Grant from ICGEB, Trieste (Project CRP/IND19-01) to M.J. The funding agencies had no role in the design, collection, analysis, or decision to publish this study. This research was enabled in part by support provided by Calcul Quebec and Compute Canada. The graphical abstract was created with BioRender.com.
Author contributions
Conceptualization, P.L.; Formal Analysis, P.L.; Methodology, P.L. and K.P.; Writing, P.L., G.M., and M.J.; Funding Acquisition, A.K., G.M., and M.J.; Resources, S.K. and L.T.; Project Administration, G.M. and M.J.
Declaration of interests
The authors declare no competing interests.
Published: February 18, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2022.103802.
Contributor Information
Greg Matlashewski, Email: greg.matlashewski@mcgill.ca.
Manju Jain, Email: manjujainmda@gmail.com.
Supplemental information
Data and code availability
-
•
LdHPCL66 sequencing data have been deposited at NCBI SRA and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Akopyants N.S., Kimblin N., Secundino N., Patrick R., Peters N., Lawyer P., Dobson D.E., Beverley S.M., Sacks D.L. Demonstration of genetic exchange during cyclical development of Leishmania in the sand fly vector. Science. 2009;324:265–268. doi: 10.1126/science.1169464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amos B., Aurrecoechea C., Barba M., Barreto A., Basenko E.Y., Bażant W., Belnap R., Blevins A.S., Böhme U., Brestelli J., et al. VEuPathDB: the eukaryotic pathogen, vector and host bioinformatics resource center. Nucleic Acids Res. 2022 doi: 10.1093/nar/gkab929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barja P.P., Pescher P., Bussotti G., Dumetz F., Imamura H., Kedra D., Domagalska M., Chaumeau V., Himmelbauer H., Pages M., et al. Haplotype selection as an adaptive mechanism in the protozoan pathogen Leishmania donovani. Nat. Ecol. Evol. 2017;1:1961–1969. doi: 10.1038/s41559-017-0361-x. [DOI] [PubMed] [Google Scholar]
- Bradbury P.J., Zhang Z., Kroon D.E., Casstevens T.M., Romdoss Y., Buckler E.S. TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics. 2007;23:2633–2635. doi: 10.1093/bioinformatics/btm308. [DOI] [PubMed] [Google Scholar]
- Burza S., Croft S.L., Boelaert M. Leishmaniasis. The Lancet. 2018;392:951–970. doi: 10.1016/S0140-6736(18)31204-2. [DOI] [PubMed] [Google Scholar]
- Cotton J.A., Durrant C., Franssen S.U., Gelanew T., Hailu A., Mateus D., Sanders M.J., Berriman M., Volf P., Miles M.A., et al. Genomic analysis of natural intra-specific hybrids among ethiopian isolates of leishmania donovani. PLoS Negl. Trop. Dis. 2020;14:1–26. doi: 10.1371/journal.pntd.0007143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuypers B., Berg M., Imamura H., Dumetz F., De Muylder G., Domagalska M.A., Rijal S., Bhattarai N.R., Maes I., Sanders M., et al. Integrated genomic and metabolomic profiling of ISC1, an emerging Leishmania donovani population in the Indian subcontinent. Infect. Genet. Evol. 2018;62:170–178. doi: 10.1016/j.meegid.2018.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domagalska M.A., Imamura H., Sanders M., Van den Broeck F., Bhattarai N.R., Vanaerschot M., Maes I., D’haenens E., Rai K., Rijal S., et al. Genomes of Leishmania parasites directly sequenced from patients with visceral leishmaniasis in the Indian subcontinent. PLoS Negl. Trop. Dis. 2019 doi: 10.1371/JOURNAL.PNTD.0007900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelanew T., Kuhls K., Hurissa Z., Weldegebreal T., Hailu W., Kassahun A., Abebe T., Hailu A., Schönian G. Inference of population structure of leishmania donovani strains isolated from different ethiopian visceral leishmaniasis endemic areas. PLoS Negl. Trop. Dis. 2010;4 doi: 10.1371/journal.pntd.0000889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumetz F., Imamura H., Sanders M., Seblova V., Myskova J., Pescher P., Vanaerschot M., Meehan C.J., Cuypers B., De Muylder G., et al. Modulation of aneuploidy in leishmania donovani during adaptation to different in vitro and in vivo environments and its impact on gene expression. mBio. 2017;8 doi: 10.1128/mBio.00599-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelanew T., Hailu A., Schőnian G., Lewis M.D., Miles M.A., Yeo M. Multilocus sequence and microsatellite identification of intra-specific hybrids and ancestor-like donors among natural ethiopian isolates of leishmania donovani. Int. J. Parasitol. 2014;44:751–757. doi: 10.1016/j.ijpara.2014.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh A.K., Sardar A.H., Mandal A., Saini S., Abhishek K., Kumar A., Purkait B., Singh R., Das S., Mukhopadhyay R., et al. Metabolic reconfiguration of the central glucose metabolism: a crucial strategy of Leishmania donovani for its survival during oxidative stress. FASEB J. 2015;29:2081–2098. doi: 10.1096/fj.14-258624. [DOI] [PubMed] [Google Scholar]
- Iantorno S.A., Durrant C., Khan A., Sanders M.J., Beverley S.M., Warren W.C., Berriman M., Sacks D.L., Cotton J.A., Grigg M.E. Gene expression in Leishmania is regulated predominantly by gene dosage. mBio. 2017;8 doi: 10.1128/mBio.01393-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imamura H., Downing T., van den Broeck F., Sanders M.J., Rijal S., Sundar S., Mannaert A., Vanaerschot M., Berg M., de Muylder G., et al. Evolutionary genomics of epidemic visceral leishmaniasis in the Indian subcontinent. eLife. 2016:5. doi: 10.7554/eLife.12613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inbar E., Shaik J., Iantorno S.A., Romano A., Nzelu C.O., Owens K., Sanders M.J., Dobson D., Cotton J.A., Grigg M.E., et al. Whole genome sequencing of experimental hybrids supports meiosis-like sexual recombination in leishmania. PLoS Genet. 2019;15 doi: 10.1371/journal.pgen.1008042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kariyawasam U.L., Selvapandiyan A., Rai K., Wani T.H., Ahuja K., Beg M.A., Premathilake H.U., Bhattarai N.R., Siriwardena Y.D., Zhong D., et al. Genetic diversity of Leishmania donovani that causes cutaneous leishmaniasis in Sri Lanka: a cross sectional study with regional comparisons. BMC Infect. Dis. 2017;17 doi: 10.1186/s12879-017-2883-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karunaweera N.D. Leishmania donovani causing cutaneous leishmaniasis in Sri Lanka: a wolf in sheep’s clothing? Trends Parasitol. 2009;25:458–463. doi: 10.1016/j.pt.2009.07.002. [DOI] [PubMed] [Google Scholar]
- Koboldt D.C., Zhang Q., Larson D.E., Shen D., McLellan M.D., Lin L., Miller C.A., Mardis E.R., Ding L., Wilson R.K. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012;22:568–576. doi: 10.1101/gr.129684.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krzywinski M., Schein J., Birol I., Connors J., Gascoyne R., Horsman D., Jones S.J., Marra M.A. Circos: an information aesthetic for comparative genomics. Genome Res. 2009;19:1639–1645. doi: 10.1101/gr.092759.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legrand M., Forche A., Selmecki A., Chan C., Kirkpatrick D.T., Berman J. Haplotype mapping of a diploid non-meiotic organism using existing and induced aneuploidies. PLoS Genet. 2008 doi: 10.1371/journal.pgen.0040001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letunic I., Bork P. Interactive Tree of Life (iTOL) v4: recent updates and new developments. Nucleic Acids Res. 2019;47 doi: 10.1093/nar/gkz239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv. 2013 http://arxiv.org/abs/1303.3997 1303. [Google Scholar]
- Lypaczewski P., Hoshizaki J., Zhang W.-W., McCall L.I., Torcivia-Rodriguez J., Simonyan V., Kaur A., Dewar K., Matlashewski G. A complete Leishmania donovani reference genome identifies novel genetic variations associated with virulence. Scientific Rep. 2018;8 doi: 10.1038/s41598-018-34812-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lypaczewski P., Matlashewski G. Leishmania donovani hybridisation and introgression in nature: a comparative genomic investigation. The Lancet Microbe. 2021;2:e250–e258. doi: 10.1016/S2666-5247(21)00028-8. [DOI] [PubMed] [Google Scholar]
- Ozbilgin A., Culha G., Zeyrek F.Y., Töz S., Gündüz C., Kurt Ö., Pratlong F., Ozbel Y., et al. Cutaneous and visceral tropisms of Leishmania tropica/Leishmania infantum hybrids in a murine model: first report of hybrid Leishmania strains isolated in Turkey. Int. J. Infect. Dis. 2012;16:e165. doi: 10.1016/j.ijid.2012.05.702. [DOI] [Google Scholar]
- Ranasinghe S., Zhang W.-W., Wickremasinghe R., Abeygunasekera P., Chandrasekharan V., Athauda S., Mendis S., Hulangamuwa S., Matlashewski G., Pratlong F. Leishmania donovani zymodeme Mon-37 isolated from an autochthonous visceral leishmaniasis patient in Sri Lanka. Pathog. Glob. Health. 2012;106:421–424. doi: 10.1179/2047773212Y.0000000054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rijal S., Sundar S., Mondal D., Das P., Alvar J., Boelaert M. Eliminating visceral leishmaniasis in South Asia: the road ahead. BMJ (Online) 2019 doi: 10.1136/bmj.k5224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers M.B., Downing T., Smith B.A., Imamura H., Sanders M., Svobodova M., Volf P., Berriman M., Cotton J.A., Smith D.F. Genomic confirmation of hybridisation and recent inbreeding in a vector-isolated leishmania population. PLoS Genet. 2014;10 doi: 10.1371/journal.pgen.1004092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samarasinghe S.R., Samaranayake N., Kariyawasam U.L., Siriwardana Y.D., Imamura H., Karunaweera N.D. Genomic insights into virulence mechanisms of Leishmania donovani: evidence from an atypical strain. BMC Genomics. 2018;19 doi: 10.1186/s12864-018-5271-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seblova V., Dujardin J.-C., Rijal S., Domagalska M.A., Volf P. ISC1, a new Leishmania donovani population emerging in the Indian sub-continent: vector competence of Phlebotomus argentipes. Infect. Genet. Evol. 2019;76 doi: 10.1016/j.meegid.2019.104073. [DOI] [PubMed] [Google Scholar]
- Siriwardana H.V.Y.D., Noyes H.A., Beeching N.J., Chance M.L., Karunaweera N.D., Bates P.A. Leishmania donovani and cutaneous leishmaniasis, Sri Lanka. Emerging Infect. Dis. 2007;13:476–478. doi: 10.3201/eid1303.060242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siriwardana Y., Zhou G., Deepachandi B., Akarawita J., Wickremarathne C., Warnasuriya W., Udagedara C., Ranawaka R.R., Kahawita I., Ariyawansa D., et al. Trends in recently emerged leishmania donovani induced cutaneous leishmaniasis, Sri Lanka, for the first 13 years. Biomed. Res. Int. 2019;2019 doi: 10.1155/2019/4093603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sterkers Y., Crobu L., Lachaud L., Pagès M., Bastien P. Parasexuality and mosaic aneuploidy in Leishmania: alternative genetics. Trends Parasitol. 2014:429–435. doi: 10.1016/j.pt.2014.07.002. [DOI] [PubMed] [Google Scholar]
- Thakur L., Singh K.K., Shanker V., Negi A., Jain A., Matlashewski G., Jain M. Atypical leishmaniasis: a global perspective with emphasis on the Indian subcontinent. PLoS Negl. Trop. Dis. 2018 doi: 10.1371/journal.pntd.0006659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thakur L., Singh K.K., Kushwaha H.R., Sharma S.K., Shankar V., Negi A., Verma G., Kumari S., Jain A., Jain M. Leishmania donovani infection with atypical cutaneous manifestations, Himachal Pradesh, India, 2014–2018. Emerging Infect. Dis. 2020;26:1864–1869. doi: 10.3201/eid2608.191761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO Status of endemicity of cutaneous leishmaniasis, worldwide, 2018. Geneva. 2018. https://www.who.int/leishmaniasis/burden/GHO_CL_2018.pdf?ua=1
- WHO Status of endemicity of visceral leishmaniasis, worldwide, 2018. Geneva. 2018. https://www.who.int/leishmaniasis/burden/GHO_VL_2018.pdf?ua=1
- Zhang W.W., Ramasamy G., McCall L.I., Haydock A., Ranasinghe S., Abeygunasekara P., Sirimanna G., Wickremasinghe R., Myler P., Matlashewski G. Genetic analysis of leishmania donovani tropism using a naturally attenuated cutaneous strain. PLoS Pathog. 2014;10:e1004244. doi: 10.1371/journal.ppat.1004244. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
LdHPCL66 sequencing data have been deposited at NCBI SRA and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.