

Abstract
Background:
Established prognostic indicators in rhabdomyosarcoma (RMS), the most common childhood soft tissue sarcoma, include several clinicopathologic features. Among pathologic features, anaplasia has been suggested as a potential prognostic indicator, but the clinical significance of anaplasia remains unclear.
Methods:
Patients enrolled on one of five recent Children’s Oncology Group clinical trials for RMS (D9602, n=357; D9802, n=80; D9803, n=462; ARST0331, n=335; and ARST0531, n = 414) with prospective central pathology review were included in this study. Clinicopathologic variables including demographic information, risk group, histologic subtype, and anaplasia were recorded along with overall survival (OS) and failure-free survival (FFS) with failure defined by recurrence, progression or death. The log-rank test was used to compare OS and FFS.
Results:
Anaplasia was more common in embryonal RMS (27% of all embryonal RMS) than other subtypes of RMS (11% for alveolar RMS, 7% for botryoid RMS, 11% for spindle cell RMS). On multivariate analyses, anaplasia was not an independent prognostic factor in RMS (OS: Hazard ratio (HR)=1.12, p=0.43; FFS: HR=1.07, p=0.56) across all subtypes or within embryonal RMS only (OS: HR=1.41, p=0.078; FFS: HR=1.25, p=0.16). Among tumors with TP53 mutations, 69% had anaplasia, while only 24% of tumors with anaplasia had a tumoral TP53 mutation.
Conclusions:
Anaplasia is not an independent indicator of adverse outcomes in RMS. Emerging information on the prognostic significance of TP53 mutations raises the possibility that anaplasia may be a surrogate marker of TP53 mutations in some cases. Tumoral TP53 mutation status may be investigated as a prognostic indicator in future studies.
Keywords: Rhabdomyosarcoma, anaplasia, TP53 Genes
Introduction:
Rhabdomyosarcoma (RMS) is the most common soft tissue sarcoma in childhood1. In addition to clinical features, including clinical group, stage, and age, certain histologic features have been important prognostic markers for RMS over the last decades2,3. It is now recognized, however, that histologic features may act as a surrogate for underlying biologic features. For instance, alveolar histology represents an approximate surrogate marker of FOXO1 fusion positivity, the latter of which is one of the most important negative prognostic factors in RMS, but roughly 20% of alveolar RMS are fusion-negative and have a better outcome4,5. Although histologic subtype is no longer part of risk stratification for patients with RMS, the significance of anaplastic histology has remained unclear.
Palmer et al first noted the presence of “Wilms tumor-like” anaplasia (characterized by pleomorphic cells and large atypical mitotic figures) in RMS, and in his series anaplasia was associated with a worse prognosis6,7. Subsequent small studies demonstrated in univariate analysis that anaplasia was associated with a worse prognosis in embryonal RMS (ERMS)8. The first large retrospective review of anaplasia9 demonstrated histologic anaplasia (with or without atypical mitotic figures) in 3% of RMS, but noted that anaplasia was more commonly seen in ERMS. This report distinguished focal (rare scattered cells) and diffuse anaplasia (cohesive clusters), with diffuse anaplasia being associated with worse outcome compared to no anaplasia. Qualman et al applied the same definition of anaplasia to a prospective series of 655 patients enrolled on International Rhabdomyosarcoma Study Group (IRSG) therapeutic trials between 1995 and 1998. This analysis demonstrated a higher rate of anaplasia (13%; 7% focal, 6% diffuse) with inferior outcome seen on univariate analysis for intermediate risk patients with ERMS, but not when controlled for other prognostic factors by multivariate analysis10.
We present a large analysis of 1648 patients with RMS enrolled on one of five Children’s Oncology Group (COG) RMS clinical trials studies between 1997 and 2013, and for whom central pathology assessment of anaplasia was performed prospectively, to determine whether anaplasia is an independent prognostic factor in RMS.
METHODS:
Clinicopathologic variables:
Patients diagnosed with RMS between 1997–2013 and enrolled on one of five COG clinical RMS studies (D9602, n=35711; D9802, n=8012; D9803, n=46213; ARST0331, n=33514; or ARST0531, n = 41415) with central pathology review were included in this analysis. Central pathology review was performed prospectively within 21 days of trial enrollment. One hundred sixty-seven (167) cases were excluded due to lack of central review data. Anaplasia was defined as the presence of large hyperchromatic nuclei (3x size of other tumor nuclei) and/or the presence of atypical mitoses16. Focal anaplasia was defined as scattered anaplastic cells and diffuse anaplasia was defined by the presence of “foci or large sheets” of anaplastic cells9. Clinicopathologic variables including age, race, sex, primary site, tumor invasiveness, regional lymph node involvement, tumor size IRSG stage, clinical group, and risk stratification as defined in the prior analysis by Qualman et al and used in COG trials as of 2013 (Supplemental Table 1)17 were analyzed with clinical outcome data and presence or absence of anaplasia18,19. For a subset of patients including in this analysis, data regarding tumor TP53 mutational status (based upon targeted panel sequencing) was available for correlation with the presence of anaplasia20.
Statistical methods:
Failure free survival (FFS) was defined as time from study entry to disease recurrence, progression, or death (from any cause) as a first event. Overall survival (OS) was defined as time from study entry to death from any cause or censored at the time of the last follow-up. Follow-up is current as of December 31, 2018. The log-rank test and Cox proportional hazards model were used to compare survival data. The chi-square test was performed to assess association between clinicopathologic variables. Software programs SAS and R were used for the analysis.
RESULTS:
A total of 1648 patients diagnosed with RMS were included in the study (Table 1). Seven hundred ninety-two patients had typical ERMS, with an additional 195 patients with botryoid RMS and 168 with spindle cell RMS. Four hundred thirty-three patients (26%) had alveolar RMS (ARMS). A FOXO1 fusion was present in 75% of ARMS (258 of 346 patients) for whom FOXO1 fusion status was available) (Table 2).
Table 1:
Clinicopathologic characteristics of patients with Rhabdomyoscarcoma with and without anaplasia (Children’s Oncology Group studies, 1997–2013)
| Characteristic | Anaplasia | p-value1 | ||
|---|---|---|---|---|
| None (n=1339) | Focal (n=133) | Diffuse (n=176) | ||
| Age, years | 0.084 | |||
| <1 | 67 (86%) | 4 (5%) | 7 (9%) | |
| 1–9 | 821 (80%) | 92 (9%) | 118 (11%) | |
| ≥10 | 451 (84%) | 37 (7%) | 51 (9%) | |
| Race | 0.15 | |||
| White | 979 (81%) | 103 (8%) | 130 (11%) | |
| Non-white | 235 (85%) | 18 (6%) | 25 (9%) | |
| Unknown | 125 (79%) | 2 (8%) | 21 (13%) | |
| Sex | 0.097 | |||
| Male | 807 (80%) | 86 (9%) | 116 (11%) | |
| Female | 532 (83%) | 47 (7%) | 60 (9%) | |
| Clinical Group | <0.0001 | |||
| I | 210 (71%) | 38 (13%) | 46 (16%) | |
| II | 229 (75%) | 22 (7%) | 56 (18%) | |
| III | 801 (86%) | 63 (7%) | 63 (7%) | |
| IV | 98 (84%) | 10 (8%) | 9 (8%) | |
| Unknown | 1 | - | - | |
| Stage | <0.0001 | |||
| 1 | 550 (76%) | 63 (9%) | 108 (15%) | |
| 2 | 247 (86%) | 17 (6%) | 22 (8%) | |
| 3 | 443 (85%) | 43 (8%) | 37 (7%) | |
| 4 | 98 (84%) | 10 (8%) | 9 (8%) | |
| Unknown | 1 | - | - | |
| Primary Site | <0.00012 | |||
| All favorable sites | 550 (76%) | 65 (9%) | 109 (15%) | |
| Orbit | 169 (82%) | 12 (6%) | 25 (12%) | |
| Head and neck/non-PM | 127 (84%) | 8 (5%) | 16 (11%) | |
| GU, nonbladder/prostate | 254 (69%) | 45 (12%) | 68 (19%) | |
| All unfavorable sites | 789 (86%) | 68 (7%) | 67 (7%) | |
| Parameningeal | 332 (89%) | 21 (6%) | 22 (6%) | |
| Bladder/prostate | 126 (88%) | 10 (7%) | 8 (6%) | |
| Extremity | 128 (81%) | 15 (9%) | 16 (10%) | |
| Other | 203 (83%) | 22 (9%) | 21 (8%) | |
| Tumor invasiveness | 0.0002 | |||
| T1 | 790 (78%) | 87 (9%) | 131 (13%) | |
| T2 | 546 (86%) | 46 (7%) | 45 (7%) | |
| Unknown | 3 | - | - | |
| Lymph node involvement | 0.059 | |||
| N0 | 1091 (80%) | 119 (9%) | 147 (11%) | |
| N1 | 237 (85%) | 14 (5%) | 27 (10%) | |
| Unknown | 11 | - | 2 | |
| Tumor size, cm | ||||
| ≤5 | 763 (82%) | 68 (7%) | 104 (11%) | 0.64 |
| >5 | 547 (81%) | 61 (9%) | 70 (10%) | |
| Unknown | 29 | 4 | 2 | |
| Risk Group | ||||
| High Risk | 98 (83%) | 10 (9%) | 9 (8%) | <0.0001 |
| Intermediate Risk | 573 (87%) | 50 (7%) | 37 (6%) | |
| Low Risk | 667 (77%) | 73 (8%) | 130 (15%) | |
| Unknown | 1 | - | ||
| Study | ||||
| D9602 | 278 (78%) | 41 (11%) | 38 (11%) | <0.0001 |
| D9802 | 71 (89%) | 5 (6%) | 4 (5%) | |
| D9803 | 366 (79%) | 52 (11%) | 44 (10%) | |
| ARST0331 | 252 (75%) | 18 (5%) | 65 (19%) | |
| ARST0531 | 372 (90%) | 17 (4%) | 25 (6%) | |
Chi-square test assessing association with anaplasia status (present or absent), after excluding “Unknown” if applicable.
Chi-square test assessing favorable vs unfavorable site with anaplasia status
Table 2:
Prevalence of Anaplasia by Histology1
| Anaplasia | p-value2 | |||
|---|---|---|---|---|
| None (n=1339) | Focal (n=133) | Diffuse (n=176) | ||
| Histology | <0.0001 | |||
| Embryonal | 581 (73%) | 88 (11%) | 123 (16%) | 0.00033 |
| Botryoid | 181 (93%) | 5 (2%) | 9 (5%) | |
| Spindle cell | 133 (79%) | 20 (12%) | 15 (9%) | |
| Alveolar | 386 (89%) | 19 (4%) | 28 (7%) | |
| FOXO1 + | 247 (96%) | 6 (2%) | 5 (2%) | |
| FOXO1 − | 73 (83%) | 6 (7%) | 9 (10%) | |
| FOXO1 unknown | 66 (76%) | 7 (8%) | 14 (16%) | |
| NOS4 | 58 (96%) | 1 (2%) | 1 (2%) | |
Determined by central histologic review
Chi-square test assessing association with anaplasia status (present or absent)
Chi-square test assessing association with anaplasia status (present or absent) with FOXO1 status in ARMS
NOS (Not otherwise specified) = insufficient sample to determine status
Anaplasia was seen across all age groups with no statistically significant difference between groups (Table 1). Anaplasia was more common in clinical groups I and II, favorable primary sites, Stage 1 and T1 (Table 1). Overall, there was no significant difference in FFS (5-year EFS of 68%, 69% and 76% for no, focal and diffuse anaplasia, respectively; p=0.22) or OS (5-year OS of 79%, 80% and 85% for no, focal and diffuse anaplasia, respectively; p=0.15) between non anaplastic RMS and anaplastic RMS (Figure 1 and 2). In multivariate analysis controlling for risk group and age, anaplastic RMS was not a significant indicator of clinical outcome (Supplemental Table 2). The median follow-up duration for surviving patients was 7 years (Range: 1 day – 14.8 years)
Figure 1:
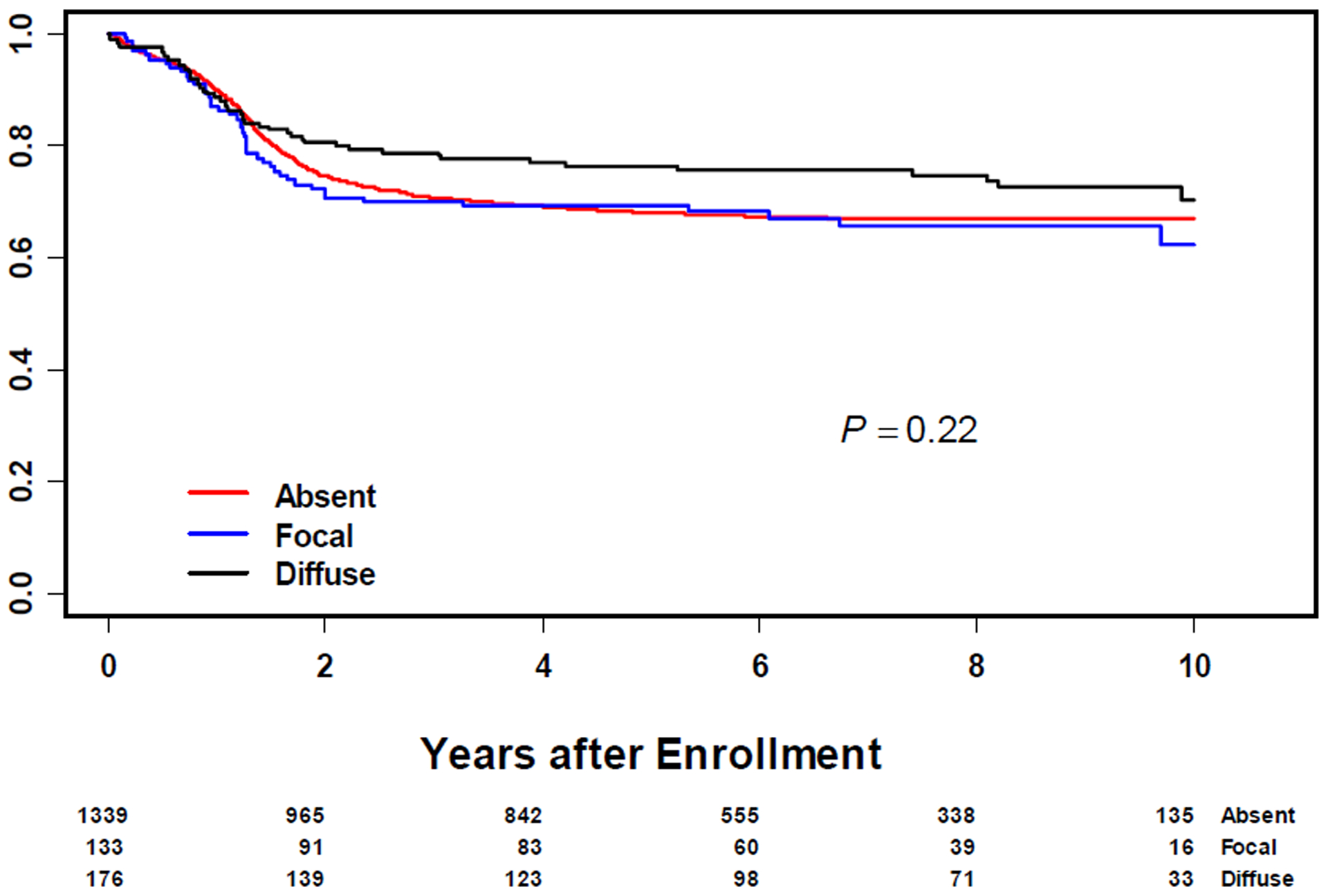
Failure Free Survival in All Patients with Rhabdomyosarcoma by Anaplasia Status
Figure 2:
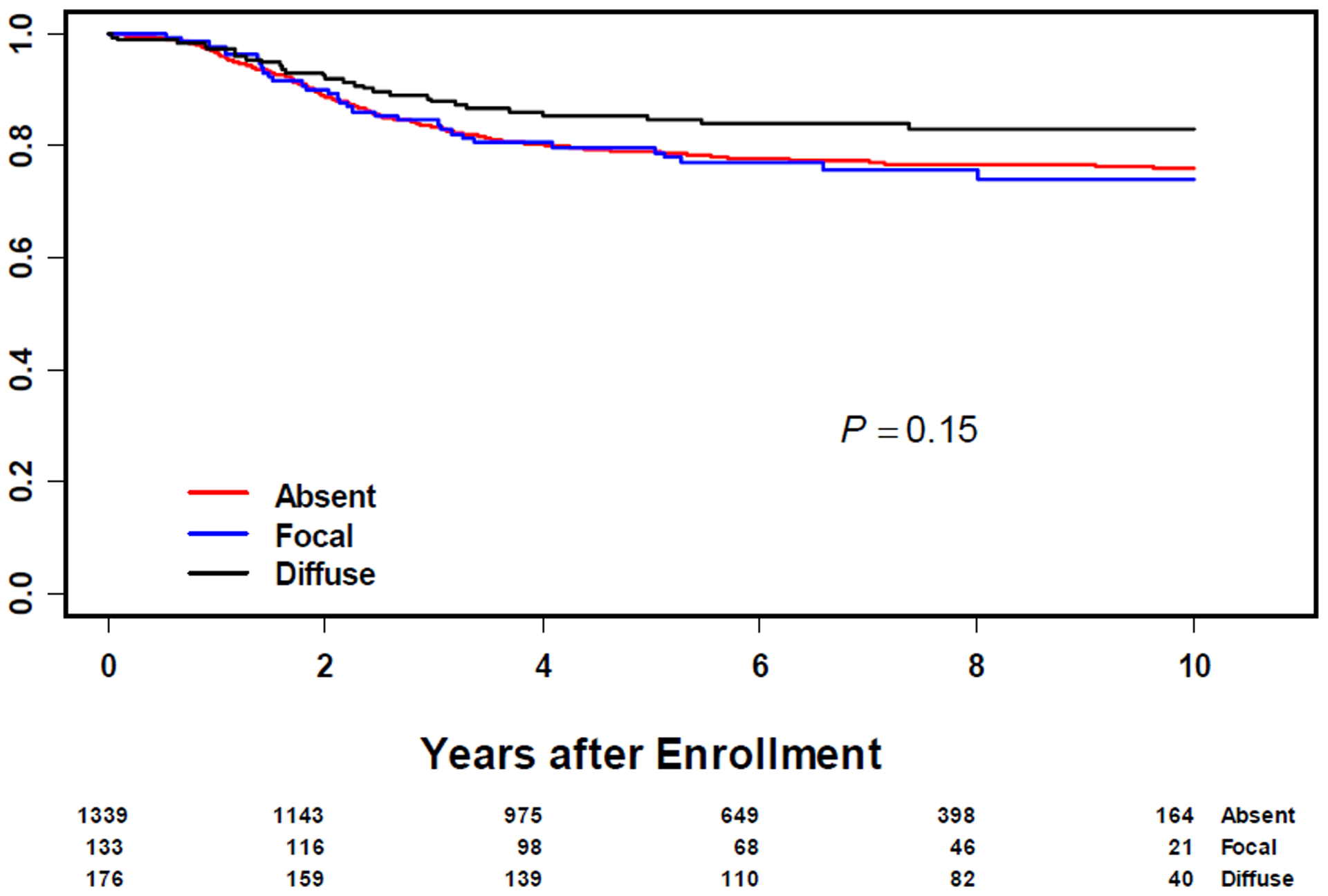
Overall Survival in All Patients with Rhabdomyosarcoma by Anaplasia Status
Alveolar RMS:
Anaplasia was less commonly documented in ARMS in comparison to ERMS (Table 2). Forty-seven patients with ARMS (of 433; 11%) had anaplasia, more frequently documented in FOXO1 fusion negative ARMS (p = 0.0003). On univariate analysis, OS and FFS was not significantly different between ARMS with or without anaplasia (Supplemental table 3).
Botryoid and Spindle cell RMS:
A majority of Botryoid RMS (93%) and Spindle cell RMS (79%) were not anaplastic. The presence of anaplasia did not significantly alter the OS and EFS for either histologic subtype.
Embryonal RMS:
The majority of RMS with histologic anaplasia were ERMS (n = 211 of 309 cases; 68%). Both by univariate and multivariate analysis, anaplastic morphology showed no statistically significant association with OS (hazard ratio (HR)=1.26, p=0.23 and HR=1.41, p=0.078) or FFS (HR=1.16, p = 0.34 and HR=1.25, p=0.16) (Table 3). In a sub-group analysis of the intermediate risk ERMS, anaplasia showed no significant association with OS (HR=1.58, p = 0.08) or EFS (HR=1.13, p=0.61) (Supplemental table 4).
Table 3:
Univariate and Multivariate Analysis of Prognostic Factors in all Patients with Embryonal Rhabdomyosarcoma
| Univariate Analysis | Multivariate Analysis | |||
|---|---|---|---|---|
| HR1 (95% CI) | P value | HR (95% CI) | p-value | |
| Failure Free Survival | ||||
| Intermediate risk | 1.72 (1.30–3.27) | 0.0001 | 2.37 (1.74–3.24) | <0.0001 |
| High risk | 3.55 (2.38–5.30) | <0.0001 | 5.35 (3.45–8.29) | <0.0001 |
| Age ≥ 10 years | 1.07 (0.79–1.46) | 0.66 | 1.30 (0.95–1.78) | 0.11 |
| Anaplastic Morphology | 1.16 (0.86–1.58) | 0.34 | 1.25 (0.92–1.70) | 0.16 |
| Overall Survival | ||||
| Intermediate risk | 2.28 (1.60–3.26) | <0.0001 | 4.33 (2.81–6.67) | <0.0001 |
| High risk | 5.57 (3.58–8.68) | <0.0001 | 12.05 (7.07–20.54) | <0.0001 |
| Age ≥ 10 years | 1.21 (0.82–1.76) | 0.34 | 1.64 (1.11–2.42) | 0.013 |
| Anaplastic Morphology | 1.26 (0.86–1.83) | 0.23 | 1.41 (0.96–2.05) | 0.078 |
Hazard ratio
TP53 mutation:
Tumor TP53 mutation analysis was available for 146 patients, of which thirteen (9%) had a TP53 pathogenic mutation (Table 4). Thirty-eight of tumors with known TP53 status demonstrated anaplasia. Nine of thirty-eight (24%) of the tumors with anaplasia harbored a TP53 mutation. In contrast, a vast majority of tumors with TP53 mutations demonstrated histologic anaplasia (9/13; 69%). In tumors with no anaplasia, TP53 mutations were rarely observed (4/108, 3.7%).
Table 4:
Subset of Rhabdomyosarcoma Patients with known TP53 Mutation Status
| TP53 mutation | No anaplasia (n=108) | Focal anaplasia (n=16) | Diffuse anaplasia (n=22) |
|---|---|---|---|
| Absent in tumor | 104 | 11 | 18 |
| Present in tumor | 4 | 5 | 4 |
DISCUSSION:
This study represents the largest prospective analysis of anaplasia in pediatric RMS. Focal or diffuse anaplasia was present in 19% of patients with RMS, with no statistically significant differences across age groups (Table 1). Anaplasia was observed more commonly in RMS occurring within favorable sites and tumors that are completely resected at diagnosis (Group I/II). In contrast to prior reports, anaplasia was not an independent prognostic indicator in RMS in either univariate or multivariate analyses.
Anaplasia was more common in ERMS, similar to what has been demonstrated in previous studies8–10. Although several previous studies have suggested differences in clinical outcome for patients with anaplastic ERMS on univariate analysis, no association with outcome was confirmed on multivariate analysis. Although we showed no significant difference in outcome by univariate or multivariate analyses, our results (Hazard ratio 1.25 for FFS and 1.41 for OS) are similar to those published by Qualman et al. (Hazard ratio 1.6 for FFS and 1.7 for OS).
Anaplasia was more frequently seen in IRS Clinical Group 1, Stage 1, favorable site, small tumors and overall low-risk tumors. It is possible that a subset of tumors in our study population (IRS Group 1) were completely resected at diagnosis. We acknowledge that a larger sample thus available for evaluation may represent a potential bias affecting our observations.
There are a few differences between our study population and those described in prior reports. Qualman et al, had a higher percentage of ARMS (30%) than in our study; however, the diagnosis of ARMS in the Qualman et al study was made based on International Classification of Rhabdomyosarcoma (ICR) criteria using histology alone, and the percentage of ARMS diagnoses was likely overestimated10. Our study integrated translocation information for ARMS and used re-review histologic diagnoses which likely explains the increased numbers of ERMS21.
There was an increased prevalence of anaplasia (19%; Focal- 8% and Diffuse – 11%) in our study, in contrast to the observations of Qualman et al (13%; Focal- 6% and Diffuse – 7%) or Kodet (~3%, 110/approximately 3000 cases, 58 focal and 52 diffuse). The reason for this is unclear. The definition of anaplasia did subtly shift between 1983 and 1993 from requiring the presence of atypical mitoses to allowing for nuclear enlargement with or without atypical mitotic figures although the definition has been standard since 19937,9. This is unlike the definition of anaplasia in Wilms tumor where presence of atypical mitoses is a requirement. Also, unlike anaplasia in Wilms tumor (assessed on resection specimen), anaplasia in RMS was mostly assessed on pre-treatment biopsies and assigned into focal and diffuse groups as defined above, upon central review.
There is limited literature on cytogenetic/molecular aberrations in anaplastic RMS. Earlier studies investigated chromosomal abnormalities in RMS, demonstrating genomic imbalance including chromosomal gains and losses across the different subtypes of RMS22,23. Subsequently, it has been shown that genomic amplification is more frequent in anaplastic ERMS and fusion positive ARMS23. Recently, in a small series of 87 cases of RMS, Casey et al reported that a subset of RMS harbors a high tumor mutation burden (TMB) and such tumors correlate with poor overall survival; they proposed that high TMB is an independent risk factor in the prognostication of RMS24. This observation has not yet been confirmed in a larger series of RMS.
Studies have also suggested that anaplasia is related to TP53 mutational status. Hettmer et al observed a high rate of germline TP53 pathogenic variants (73%; 11/15 patients) in anaplastic RMS25 and suggested anaplasia may be more common in patients with Li Fraumeni syndrome. In a series of 631 RMS with somatic mutational analysis, Shern et al demonstrated tumoral TP53 mutations in 12% of RMS, which was also associated with inferior outcome20. We were able to combine this tumor mutational data and central pathology review data for 146 patients in this study. In this subset of patients, tumor TP53 mutations were present in 9% (13/146). Tumor TP53 mutations were identified in only 24% (9/38) of all anaplastic RMS in our study; however, most tumors with TP53 mutations had anaplastic morphology (9/13; 69%). Casey et al identified a TP53 mutation in a subset of their patients with high TMB, and showed a significant correlation with poor OS24. Further analysis of TMB and TP53 mutational status may be useful in future prospective studies of RMS.
Conclusions:
We demonstrate that anaplasia is not an independent adverse prognostic factor in RMS but that the prevalence of the diagnosis has climbed in recent years, suggesting a shift in criteria. If on future investigation, tumor TP53 mutation is confirmed as an independent adverse prognostic factor, anaplasia could be used as to identify tumors with a higher probability of harboring a TP53 mutation. Future studies including both germline and somatic sequencing are needed to confirm the role of TP53 mutations in the risk stratification of RMS.
Supplementary Material
Highlights:
Anaplasia is commonly seen in embryonal rhabdomyosarcoma compared to other subtypes
Anaplasia is not an independent indicator of adverse prognosis in rhabdomyosarcoma
TP53 mutation status & association with adverse prognosis needs more investigation
Role of funding source:
Children’s Oncology Group Grants U10CA180886, U10CA180899, U10CA098543, U10CA098413 supported COG Operations and Statistics and Data Centers in the design, conduct, and analysis of the clinical trials used in this manuscript. St. Baldrick’s Foundation grant supported COG institutions who enrolled patients in the clinical trials used in this manuscript. Seattle Children’s Foundation grant from Kat’s Crew Guild through the Sarcoma Research Fund supported statisticians who prepared this manuscript.
Footnotes
Declaration of Interest statement:
Douglas S Hawkins has the following disclosures: Loxo Oncology, Bayer, Bristol Myers Squibb, Lilly: Clinical trial fees paid to Seattle Children’s to offset costs of study conduct; reimbursed for or provided travel, housing, and food to attend medial advisory board meetings; Celgene: Reimbursed for or provided travel, housing, and food to attend medial advisory board meetings; Eisai, Glaxo Smith Kline, Sanofi, Novartis, Amgen, Seattle Genetics, Jazz Pharmaceuticals, Incyte: Clinical trial fees paid to Seattle Children’s to offset costs of study conduct.
All other authors have no disclosures.
References:
- 1.Skapek SX, Ferrari A, Gupta AA, et al. Rhabdomyosarcoma. Nat Rev Dis Prim. 2019;5(1). doi: 10.1038/s41572-018-0051-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dantonello TM, Int-Veen C, Winkler P, et al. Initial patient characteristics can predict pattern and risk of relapse in localized rhabdomyosarcoma. J Clin Oncol. 2008;26(3):406–413. doi: 10.1200/JCO.2007.12.2382 [DOI] [PubMed] [Google Scholar]
- 3.Carli M, Colombatti R, Oberlin O, et al. European intergroup studies (MMT4–89 and MMT4–91) on childhood metastatic rhabdomyosarcoma: Final results and analysis of prognostic factors. J Clin Oncol. 2004;22(23):4735–4742. doi: 10.1200/JCO.2004.04.083 [DOI] [PubMed] [Google Scholar]
- 4.Skapek SX, Anderson J, Barr FG, et al. PAX-FOXO1 fusion status drives unfavorable outcome for children with rhabdomyosarcoma: A children’s oncology group report. Pediatr Blood Cancer. 2013;60(9):1411–1417. doi: 10.1002/pbc.24532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hibbitts E, Chi Y, Hawkins DS, et al. Refinement of risk stratification for childhood rhabdomyosarcoma using FOXO1 fusion status in addition to established clinical outcome predictors: A report from the Children’s Oncology Group. Cancer Med. 2019;8(14):6437–6448. doi: 10.1002/cam4.2504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Palmer NSN FM. Histology and prognosis in rhabdomyosarcoma (IRS-I) ; In: Proc Amer Soc Clin Oncol. ; 1982:1:170. [Google Scholar]
- 7.Palmer N Histopathology and prognosis in the second Intergroup Rhabdomyosarcoma Study (IRS II). In: Proc Amer Soc Clin Oncol. ; 1983:229. [Google Scholar]
- 8.Hawkins HK, Camacho-Velasquez JV. Rhabdomyosarcoma in children: Correlation of form and prognosis in one institution’s experience. Am J Surg Pathol. 1987;11(7):531–542. doi: 10.1097/00000478-198707000-00005 [DOI] [PubMed] [Google Scholar]
- 9.Kodet R, Newton WA, Hamoudi AB, Asmar L, Jacobs DL, Maurer HM. Childhood rhabdomyosarcoma with anaplastic (pleomorphic) features. A report of the Intergroup Rhabdomyosarcoma Study. Am J Surg Pathol. 1993;17(5):443–453. doi: 10.1097/00000478-199305000-00002 [DOI] [PubMed] [Google Scholar]
- 10.Qualman S, Lynch J, Bridge J, et al. Prevalence and Clinical Impact of Anaplasia in Childhood Rhabdomyosarcoma: A Report from the Soft Tissue Sarcoma Committee for the Children’s Oncology Group. Cancer. 2008;113(11):3242–3247. doi: 10.1002/cncr.23929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Beverly Raney R, Walterhouse DO, Meza JL, et al. Results of the Intergroup Rhabdomyosarcoma Study Group D9602 Protocol, Using Vincristine and Dactinomycin With or Without Cyclophosphamide and Radiation Therapy, for Newly Diagnosed Patients With Low-Risk Embryonal Rhabdomyosarcoma: A Report From the Soft. J Clin Oncol. 2011;29(10):1312–1318. doi: 10.1200/JCO.2010.30.4469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pappo AS, Lyden E, Breitfeld P, et al. Two Consecutive Phase II Window Trials of Irinotecan Alone or in Combination With Vincristine for the Treatment of Metastatic Rhabdomyosarcoma: The Children’s Oncology Group. J Clin Oncol. 2007;25(4):362–369. doi: 10.1200/JCO.2006.07.1720 [DOI] [PubMed] [Google Scholar]
- 13.Arndt CAS, Stoner JA, Hawkins DS, et al. Vincristine, actinomycin, and cyclophosphamide compared with vincristine, actinomycin, and cyclophosphamide alternating with vincristine, topotecan, and cyclophosphamide for intermediate-risk rhabdomyosarcoma: Children’s Oncology Group Study D9803. J Clin Oncol. Published online 2009. doi: 10.1200/JCO.2009.22.3768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Walterhouse DO, Pappo AS, Meza JL, et al. Shorter-duration therapy using vincristine, dactinomycin, and lower-dose cyclophosphamide with or without radiotherapy for patients with newly diagnosed low-risk rhabdomyosarcoma: A report from the soft tissue sarcoma committee of the Children’s Oncology. J Clin Oncol. Published online 2014. doi: 10.1200/JCO.2014.55.6787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hawkins DS, Chi YY, Anderson JR, et al. Addition of vincristine and irinotecan to vincristine, dactinomycin, and cyclophosphamide does not improve outcome for intermediate-risk rhabdomyosarcoma: A report from the children’s oncology group. In: Journal of Clinical Oncology. ; 2018. doi: 10.1200/JCO.2018.77.9694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Faria P, Beckwith JB, Mishra K, et al. Focal Versus Diffuse Anaplasia in Wilms Tumor—New Definitions with Prognostic Significance. Am J Surg Pathol. 1996;20(8):909–920. doi: 10.1097/00000478-199608000-00001 [DOI] [PubMed] [Google Scholar]
- 17.Hawkins DS, Spunt SL, Skapek SX. Children’s Oncology Group’s 2013 blueprint for research: Soft tissue sarcomas. Pediatr Blood Cancer. 2013;60(6):1001–1008. doi: 10.1002/pbc.24435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Meza JL, Anderson J, Pappo AS, Meyer WH, Children’s Oncology Group. Analysis of prognostic factors in patients with nonmetastatic rhabdomyosarcoma treated on intergroup rhabdomyosarcoma studies III and IV: the Children’s Oncology Group. J Clin Oncol. 2006;24(24):3844–3851. doi: 10.1200/JCO.2005.05.3801 [DOI] [PubMed] [Google Scholar]
- 19.Lawrence W, Anderson JR, Gehan EA, Maurer H. Pretreatment TNM staging of childhood rhabdomyosarcoma: a report of the Intergroup Rhabdomyosarcoma Study Group. Children’s Cancer Study Group. Pediatric Oncology Group. Cancer. 1997;80(6):1165–1170. doi: [DOI] [PubMed] [Google Scholar]
- 20.Shern JF, Patidar R, Song Y, et al. Targeted resequencing of pediatric rhabdomyosarcoma: report from the Children’s Oncology Group, the Children’s Cancer and Leukaemia Group, The Institute of Cancer Research UK, and the National Cancer Institute. J Clin Oncol. 2018;36(15_suppl):10515. doi: 10.1200/JCO.2018.36.15_suppl.10515 [DOI] [Google Scholar]
- 21.Rudzinski ER, Teot LA, Anderson JR, et al. Dense pattern of embryonal rhabdomyosarcoma, a lesion easily confused with alveolar rhabdomyosarcoma: A report from the soft tissue sarcoma committee of the children’s oncology group. Am J Clin Pathol. Published online 2013. doi: 10.1309/AJCPA1WN7ARPCMKQ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bridge JA, Liu J, Weibolt V, et al. Novel genomic imbalances in embryonal rhabdomyosarcoma revealed by comparative genomic hybridization and fluorescence in situ hybridization: an intergroup rhabdomyosarcoma study. Genes Chromosomes Cancer. 2000;27(4):337–344. doi: [DOI] [PubMed] [Google Scholar]
- 23.Bridge JA, Liu J, Qualman SJ, et al. Genomic gains and losses are similar in genetic and histologic subsets of rhabdomyosarcoma, whereas amplification predominates in embryonal with anaplasia and alveolar subtypes. Genes Chromosom Cancer. 2002;33(3):310–321. doi: 10.1002/gcc.10026 [DOI] [PubMed] [Google Scholar]
- 24.Casey DL, Wexler LH, Pitter KL, Samstein RM, Slotkin EK, Wolden SL. Genomic Determinants of Clinical Outcomes in Rhabdomyosarcoma. Clin Cancer Res. Published online November 7, 2019:clincanres.2631.2019. doi: 10.1158/1078-0432.CCR-19-2631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hettmer S, Archer NM, Somers GR, et al. Anaplastic rhabdomyosarcoma in TP53 germline mutation carriers. Cancer. 2014;120(7):1068–1075. doi: 10.1002/cncr.28507 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.