

Abstract
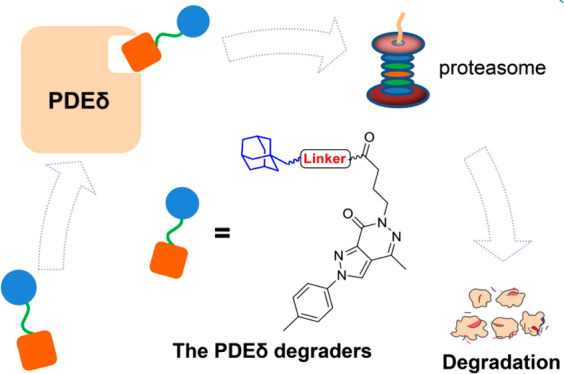
The development of KRAS–PDEδ protein–protein interaction (PPI) inhibitors is generally hampered by limited antitumor activity. Herein, the first hydrophobic tagging (HyT)-based PDEδ degraders were designed. Compound 17c efficiently bound to PDEδ and induced degradation of PDEδ in SW480 colon cancer cells. As compared with PDEδ inhibitor deltazinone, HyT-based degrader 17c exhibited improved antitumor activity toward KRAS mutant cancer cells. This study highlighted the potential of HyT as a valuable chemical tool for tumorigenic PDEδ knockdown, which could be developed into a promising strategy for antitumor drug discovery.
Keywords: KRAS−PDEδ PPI, HyT, PDEδ degrader, antitumor
Kirsten rat sarcoma 2 viral oncogene homologue (KRAS) protein is closely related to the occurrence and development of tumors.1−3 Notably, almost 86% of pancreatic cancers and 41% of colon cancers are associated with KRAS mutations. After translation, KRAS protein is modified in the C-terminal hypervariable region and then transported to the cell membrane, executing the function that binds it with the related effector proteins to activate the downstream signaling pathway.4,5 In cancer cells, the conformation of KRAS is changed and the KRAS mutants continue to bind with GTP to produce abnormal proliferation signals, resulting in the continuous proliferation of tumor cells. Therefore, KRAS and related signaling pathways were regarded as important targets for the development of antitumor therapeutics.6−9
KRAS–PDEδ protein–protein interaction (PPI) plays an important role in the functional regulation of KRAS.10 PDEδ regulates the localization of KRAS on the cell membrane.11 The hydrophobic cavity of PDEδ binds to the farnesylated KRAS, which enhances the dissolution and diffusion of KRAS in the cells and promotes the correct localization and enrichment of KRAS in the cytoplasm. The inhibition of PDEδ leads to abnormal transport and distribution of KRAS, thus interfering with KRAS signal transduction pathway and inhibiting proliferation of tumor cells.11−13
In recent years, a number of small molecule KRAS–PDEδ inhibitors (Figure 1), such as deltazinone (1),14−16 deltarasin (2),9,17 and deltasonamide (3),18 have been reported. We also discovered several classes of small molecule PDEδ inhibitors (4–6) through structure-based drug design.19,20 These inhibitors generally exhibited potent PDEδ binding activity and effectively blocked the KRAS–PDEδ PPI. However, most of them are limited by poor antitumor activity, because endogenous Arl2 could induce the fast release of inhibitors from PDEδ.18 Thus, novel modulation strategies are highly desirable to block the KRAS–PDEδ PPI.
Figure 1.

Representative KRAS–PDEδ PPI inhibitors and degraders
Hydrophobic tagging (HyT) has emerged as a promising strategy for targeted protein degradation through ubiquitination-proteasome system.21,22 The HyT approach induces a hydrophobic and bulky group (e.g., amantadine) to a small-molecule ligand of the target protein. The hydrophobic group of HyT is located on the surface of the target protein, which is regarded as a misfolded part (a partially unfolded protein) by the protein repair mechanism. The “unfolded” protein would then be folded by the chaperone protein. However, after failing to repeat folding, the target protein will be recognized and degraded by the ubiquitin-proteasome system to maintain the homeostasis of cell function (Figure 2A). As an alternative strategy of PROTACs23 (proteolysis targeting chimeras), the HyT technology has been successfully used to degrade various drug targets, such as HaloTag fusion proteins,24 erythroblastosis oncogene B3 (ERBB3),25 and androgen receptor (AR).26 However, as compared with PROTACs, the successful examples of HyT-based degraders are rather limited.
Figure 2.

HyT-mediated degraders of PDEδ. (A) Schematic diagram of the HyT technology. (B) Rational design of HyT-based PDEδ degraders by the prediction of binding model of compound 1 with PDEδ (PDB code 5E80). (C) Structure of target compounds 15a–17f.
Previously, we confirmed that targeted protein degradation was an effective strategy to interfere with the function of KRAS-PDEδ interaction and designed the first PDEδ degrader (7) using the PROTAC approach.27 Herein we designed the first HyT-mediated PDEδ degraders, which showed potent PDEδ degrading potency and antitumor activity.
Deltazinone (compound 1) is a highly potent small molecule KRAS–PDEδ inhibitor, which has been widely used as a lead compound or tool molecule in the studies of KRAS–PDEδ PPI.14 Starting from compound 1, we have successfully designed potent inhibitors and PROTAC-based degraders of PDEδ. Inspired by these results, herein, compound 1 was selected as a template molecule for the design of HyT-based degraders. The predicted binding model of compound 1 with PDEδ indicates that the terminal phenylamine is exposed to the protein surface (Figure 2B), offering a favorable site for inducing HyT groups. Moreover, structural modification of terminal phenylamine had little effect on the binding affinity with PDEδ. For example, replacement of the phenylamine group of compound 1 with amphetamine was tolerated (compound 8).14 Therefore, the terminal phenyl group was replaced by a linker and a HyT group (amantadine), resulting in a series of new PDEδ degraders (Figure 2C, compounds 15–17). Theoretically, the amantadine HyT could simulate the misfolding of the PDEδ protein and the unfolded protein is recognized and degraded by ubiquitin-proteasome system, thus blocking the KRAS-PDEδ PPI.
The procedures for the synthesis of target compounds were outlined in Schemes S1–S4. Commercially available 4-methylaniline (9) was transformed to compound 10 through diazotization and a coupling reaction. Following the cyclization reaction of compound 10, intermediate 11 was obtained, which was further cyclized to afford intermediate 12 under the conditions of hydrazine hydrate in EtOH. Compound 12 was reacted with methyl 4-bromobutanoate via a nucleophilic substitution reaction to give intermediate 13, which was subsequently subjected to ester hydrolysis reaction to afford the corresponding carboxylic acid 14. Target compounds 15a and 15b were prepared by condensing key intermediate 14 with amantadine and 1-adamantanemethylamine, respectively (Scheme S1). An amidation reaction between commercially available starting material 18 and different Boc-N-carboxylic acids obtained compounds 19a and 19b. The Boc group of intermediates 19a and 19b were deprotected to give compounds 16a and 16b, which were further condensed with intermediate 14 to yield compounds 16a and 16b. Compound 18 and various dicarboxylic monomethyl esters were reacted to afford compounds 21a–21c, which were further hydrolyzed and condensed with tert-butyl 4-aminophenethylcarbamate to afford intermediates 23a–23c. After deprotection, compounds 24a–24c were condensed with intermediate 14 in the presence of HATU and DIPEA to give target compounds 16c–16e (Scheme S2). Commercially available reagents 25a–25e were condensed with compound 14 to yield corresponding intermediates, which were subsequently deprotected to obtain compounds 26a–26e. Target compounds 17a–17e were afforded by reacting compounds 26a–26e with 1-adamantaneacetic acid under the condition of HBTU and DIPEA (Scheme S3). Similar to the protocols for the synthesis of intermediates 26a–26e, compound 29 was obtained via the condensation and hydrolysis reactions. Intermediate 29 was reacted with tert-butyl 4-aminophenethylcarbamate to afford compound 30, which was deprotected to afford compound 31. Finally, condensation between compound 31 and intermediate 14 yielded target compound 17f (Scheme S4).
Initially, a fluorescence polarization (FP) binding assay was performed to investigate the binding affinity of all target compounds with PDEδ using compound 1 as the positive control. As shown in Table 1, most of the target compounds exhibited good binding activity. Compound 15a with a short linker showed excellent PDEδ binding affinity (Ki = 13.6 nM). Compound 15b with the linker extension of one carbon atom exhibited slightly enhanced PDEδ binding activity (Ki = 9.3 nM) to improve the flexibility of the terminal amantadine hydrophobic group. Subsequently, compounds containing various linkers were further synthesized and evaluated. However, with an increase in the alkyl linker length, the binding activity of compounds 16a and 16b were reduced to 41 and 44.1 nM, respectively. Compounds 16c–16e were designed by introduction of the phenyl group to enhance the rigidity of the linker. However, their binding affinities were significantly decreased. These results demonstrated that the long alkyl linker might be unfavorable for PDEδ binding. Finally, the terminal amide group was reversed to obtain compounds 17a–17f. Among them, two compounds, 17c and 17d, exhibited excellent binding activity, with Ki values of 26 and 29 nM, respectively.
Table 1. PDEδ Binding Affinity and Antiproliferative Activities of Target Compoundsa.
| IC50 (μM) |
||||
|---|---|---|---|---|
| compds | linker | Ki (nM) | SW480 | HCT116 |
| 15a | n = 0 | 13.6 ± 9.4 | >100 | >100 |
| 15b | CH2 | 9.3 ± 1.7 | >100 | >100 |
| 16a | (CH2)3 | 41.0 ± 18.0 | 8.0 ± 1.2 | 11.4 ± 1.6 |
| 16b | (CH2)5 | 44.4 ± 6.8 | >100 | 32.5 ± 7.5 |
| 16c | (CH2)3 | 8.6 ± 1.3 | 19.8 ± 12.7 | 39.0 ± 1.5 |
| 16d | (CH2)5 | 45 ± 10.2 | >100 | >100 |
| 16e | (CH2)9 | >1000 | 14.0 ± 9.2 | >100 |
| 17a | (CH2)2 | 38.8 ± 1.5 | 10.0 ± 5.5 | 20.4 ± 8.7 |
| 17b | (CH2)4 | 49.3 ± 13.0 | 7.0 ± 1.3 | 50.7 ± 0.1 |
| 17c | (CH2)6 | 26.0 ± 2.6 | 4.8 ± 0.2 | 7.7 ± 2.0 |
| 17d | (CH2)8 | 29.0 ± 0.7 | 5.6 ± 2.9 | 17.7 ± 3.2 |
| 17e | CH2(CH2OCH2)2CH2 | 132.0 ± 5.1 | 5.4 ± 0.9 | 18.5 ± 5.8 |
| 17f | (CH2)6 | 146.5 ± 16.1 | >100 | >100 |
| deltazinone (1) | 8.0 ± 0.5 | 18.3 ± 8.3 | 82.4 ± 6.4 | |
Values represent a mean ± SD of at least three independent experiments. The values of Ki were obtained according to eqs S1–S6 in the Supporting Information.
In consideration of the binding activities, all the target compounds were further evaluated for the effects on PDEδ degradation in the SW480 cell line (KRAS mutant human colon cancer cells) by the Western blot analysis. As shown in Figure 3A, compounds 15a, 16a, and 17c–17e significantly induced PDEδ degradation in SW480 cells at a concentration of 10 μM. Particularly, compound 17c exhibited the best degrading activity (about an 86% degradation rate at 10 μM). However, no significant degradation was observed when the SW480 cells were treated with the other compounds. When the concentration was reduced to 1 μM (Figure 3B), compounds 15a, 16a, and 17c-d retained the degradation activity. Among them, compound 17c still achieved the best degradation activity (about 79% degradation rate at 1 μM). Furthermore, the dose-dependent effect of compound 17c was explored (Figure 3C). The results indicated that compound 17c exhibited significant PDEδ degradation in a concentration-dependent manner and achieved a DC50 (concentration causing 50% protein degradation) value of 11.4 μM in SW480 cells for 24 h. Next, the downstream proteins regulated by PDEδ were investigated (Figure 4A, B). Consistent with the positive drug, compound 17c could down-regulate p-Akt and p-Erk in a concentration-dependent manner, whereas it had little effect on the expression of t-Akt and t-Erk. These results suggested that compound 17c effectively degraded PDEδ in SW480 cancer cells and then caused the changes in the downstream pathways.
Figure 3.

Effects of target compounds on the degradation of PDEδ. (A, B) Target compounds induced the degradation of PDEδ at 10 μM and 1 μM. (C) Compound 17c dose-dependently degraded PDEδ. The protein expression contents of PDEδ were quantified by ImageJ. The data were confirmed at least three times (**P < 0.05 and ***P < 0.005 relative to the control group at indicated times).
Figure 4.

Antitumor mechanism of compound 17c. (A, B) Change in PDEδ downstream proteins in SW480 cancer cells induced by compound 1 and compound 17c for 24 h. The Western blot analysis was repeated at least three times. (C, D) Compound 17c induced apoptosis of SW480 cancer cells after 24 h of treatment. All bar graphs present the mean ± SD.
Given the potent PDEδ binding activity and degradation efficiency, the antiproliferative activities of the target compounds were investigated against two different KRAS mutant human colon cancer cells (SW480 and HCT116 cell lines) by a cell counting kit-8 (CCK8) assay (Table 1). The results revealed that most of the target compounds showed better antiproliferative activity toward SW480 cells than that of HCT116 cells. As compared with the PDEδ inhibitor deltazinone, improved antitumor activities were observed for several HyT-based degraders (e.g., compounds 16a, 16e, and 17a–17e). Particularly, compound 17c (SW480 IC50 = 4.8 μM; HCT116 IC50 = 7.7 μM) exhibited the best antiproliferative activity. Importantly, the in vitro antitumor efficacies of compounds 17c were generally consistent with the degradation efficiency and the binding activity of PDEδ. Moreover, its antiproliferative activity was further evaluated against MIAPACA-2 pancreatic cancer cells (Figure S1). The results indicated that the IC50 value of compound 17c was 17.5 μM, which was also significantly better than that of deltazinone (IC50 = 70.6 μM). Furthermore, the assay of flow cytometry with Annexin V/PI staining was performed to elucidate the antitumor mechanism of action of compound 17c. As shown in panels C and D in Figure 4, compound 17c significantly induced apoptosis of SW480 cells after 24 h of treatment. At a concentration of 40 μM, the apoptosis rate of compound 17c was 83.67%, which was better than that of deltazinone (5.4% apoptosis rate at 50 μM). However, the definite correlation between degradation activity and apoptosis rate could not be detected at the experimental concentrations. Taken together, compound 17c, the most potent PDEδ degrader, was proven to possess good antitumor activity.
In summary, the HyT technology was identified as a feasible strategy for targeted degradation of PDEδ. HyT-based degrader 17c exhibited a potent PDEδ binding affinity, PDEδ protein degradation activity, and antiproliferation activity, which represents a promising tool molecule or lead compound for further structural optimization and biological studies. This study also expands the application scope of the HyT approach and highlights the effectiveness of the HyT-based PDEδ degradation strategy in antitumor drug discovery.
Glossary
Abbreviations
- PPI
protein–protein interaction
- HyT
hydrophobic tagging
- KRAS
Kirsten rat sarcoma 2 viral oncogene homologue
- PROTACs
proteolysis targeting chimeras
- ERBB3
erythroblastosis oncogene B3
- AR
androgen receptor
- FP
fluorescence polarization
- DC50
concentration causing 50% protein degradation
- CCK8
cell counting kit-8
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.1c00670.
Additional information about the synthetic procedure, compound characterization and biological assays (PDF)
Author Contributions
† M.G. and S.H. contributed equally to this work. G.D. and C.S. designed the experiments and revised the manuscript. M.G., S.H., and J.C. synthesized evodiamine borate derivatives and carried out the biological experiments. M.G., S.H., and Y.L. assisted with the biological experiments and in the preparation of the manuscript. All authors discussed the results and analyzed the data. All authors read and approved the final manuscript.
This work was supported by the National Key Research and Development Program of China (grant 2020YFA0509100 to C.S.), the National Natural Science Foundation of China (grants 21738002 and 82030105 to C.S., and 82003567 to S.H.), and Shanghai Rising-Star Program (20QA1411700 to G.D.)
The authors declare no competing financial interest.
Supplementary Material
References
- Muzny D. M.; Bainbridge M. N.; Chang K.; Dinh H. H.; Drummond J. A.; Fowler G.; Kovar C. L.; Lewis L. R.; Morgan M. B.; Newsham I. F.; et al. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akbani R.; Akdemir K. C.; Aksoy B. A.; Albert M.; Ally A.; Amin S. B.; Arachchi H.; Arora A.; Auman J. T.; Ayala B.; et al. Genomic classification of cutaneous melanoma. Cell 2015, 161, 1681–1696. 10.1016/j.cell.2015.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguirre A. J.; Hruban R. H.; Raphael B. J.; Canc Genome Atlas Res N. Integrated genomic characterization of pancreatic ductal adenocarcinoma. Cancer Cell 2017, 32, 185–203. 10.1016/j.ccell.2017.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malumbres M.; Barbacid M. Timeline - ras oncogenes: The first 30 years. Nat. Rev. Cancer 2003, 3, 459–465. 10.1038/nrc1097. [DOI] [PubMed] [Google Scholar]
- Pylayeva-Gupta Y.; Grabocka E.; Bar-Sagi D. Ras oncogenes: Weaving a tumorigenic web. Nat. Rev. Cancer 2011, 11, 761–774. 10.1038/nrc3106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waters A. M.; Der C. J. Kras: The critical driver and therapeutic target for pancreatic cancer. Cold Spring Harbor Perspect. Med. 2018, 8, a031435 10.1101/cshperspect.a031435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox A. D.; Fesik S. W.; Kimmelman A. C.; Luo J.; Der C. J. Drugging the undruggable ras: Mission possible?. Nat. Rev. Drug Discovery 2014, 13, 828–851. 10.1038/nrd4389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessler D.; Gmachl M.; Mantoulidis A.; Martin L. J.; Zoephel A.; Mayer M.; Gollner A.; Covini D.; Fischer S.; Gerstberger T.; et al. Drugging an undruggable pocket on kras. Proc. Nat. Acad. Sci. U. S. A. 2019, 116, 15823–15829. 10.1073/pnas.1904529116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann G.; Papke B.; Ismail S.; Vartak N.; Chandra A.; Hoffmann M.; Hahn S. A.; Triola G.; Wittinghofer A.; Bastiaens P. I. H.; Waldmann H. Small molecule inhibition of the kras-pde delta interaction impairs oncogenic kras signalling. Nature 2013, 497, 638–642. 10.1038/nature12205. [DOI] [PubMed] [Google Scholar]
- Spiegel J.; Cromm P. M.; Zimmermann G.; Grossmann T. N.; Waldmann H. Small-molecule modulation of ras signaling. Nat. Chem. Bio. 2014, 10, 613–622. 10.1038/nchembio.1560. [DOI] [PubMed] [Google Scholar]
- Chen F.; Alphonse M. P.; Liu Y.; Liu Q. Targeting mutant kras for anticancer therapy. Curr. Top. Med. Chem. 2019, 19, 2098–2113. 10.2174/1568026619666190902151307. [DOI] [PubMed] [Google Scholar]
- Liu P.; Wang Y.; Li X. Targeting the untargetable kras in cancer therapy. Acta Pharm. Sin. B 2019, 9, 871–879. 10.1016/j.apsb.2019.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein C. H.; Truxius D. C.; Vogel H. A.; Harizanova J.; Murarka S.; Martin-Gago P.; Bastiaens P. I. H. Pde delta inhibition impedes the proliferation and survival of human colorectal cancer cell lines harboring oncogenic kras. Int. J. Cancer 2019, 144, 767–776. 10.1002/ijc.31859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Gago P.; Fansa E. K.; Wittinghofer A.; Waldmann H. Structure-based development of pde delta inhibitors. Bio. Chem. 2017, 398, 535–545. 10.1515/hsz-2016-0272. [DOI] [PubMed] [Google Scholar]
- Papke B.; Murarka S.; Vogel H. A.; Martin-Gago P.; Kovacevic M.; Truxius D. C.; Fansa E. K.; Ismail S.; Zimmermann G.; Heinelt K.; et al. Identification of pyrazolopyridazinones as pded inhibitors. Nat. Commun. 2016, 7, 11360. 10.1038/ncomms11360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murarka S.; Martin-Gago P.; Schultz-Fademrecht C.; Al Saabi A.; Baumann M.; Fansa E. K.; Ismail S.; Nussbaumer P.; Wittinghofer A.; Waldmann H. Development of pyridazinone chemotypes targeting the pde prenyl binding site. Chem.—Eur. J. 2017, 23, 6083–6093. 10.1002/chem.201603222. [DOI] [PubMed] [Google Scholar]
- Zimmermann G.; Schultz-Fademrecht C.; Kuechler P.; Murarka S.; Ismail S.; Triola G.; Nussbaumer P.; Wittinghofer A.; Waldmann H. Structure guided design and kinetic analysis of highly potent benzimidazole inhibitors targeting the pde delta prenyl binding site. J. Med. Chem. 2014, 57, 5435–5448. 10.1021/jm500632s. [DOI] [PubMed] [Google Scholar]
- Martin-Gago P.; Fansa E. K.; Klein C. H.; Murarka S.; Janning P.; Schuermann M.; Metz M.; Ismail S.; Schultz-Fademrecht C.; Baumann M.; Bastiaens P. I. H.; Wittinghofer A.; Waldmann H. A pde6 delta-kras inhibitor chemotype with up to seven h-bonds and picomolar affinity that prevents efficient inhibitor release by arl2. Angew. Chem., Int. Ed. 2017, 56, 2423–2428. 10.1002/anie.201610957. [DOI] [PubMed] [Google Scholar]
- Chen L.; Zhuang C.; Lu J.; Jiang Y.; Sheng C. Discovery of novel kras-pde delta inhibitors by fragment-based drug design. J. Med. Chem. 2018, 61, 2604–2610. 10.1021/acs.jmedchem.8b00057. [DOI] [PubMed] [Google Scholar]
- Jiang Y.; Zhuang C.; Chen L.; Lu J.; Dong G.; Miao Z.; Zhang W.; Li J.; Sheng C. Structural biology-inspired discovery of novel kras-pde delta inhibitors. J. Med. Chem. 2017, 60, 9400–9406. 10.1021/acs.jmedchem.7b01243. [DOI] [PubMed] [Google Scholar]
- Neklesa T. K.; Crews C. M. Chemical biology greasy tags for protein removal. Nature 2012, 487, 308–309. 10.1038/487308a. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Jiang X.; Feng F.; Liu W.; Sun H. Degradation of proteins by protacs and other strategies. Acta Pharm. Sin. B 2020, 10, 207–238. 10.1016/j.apsb.2019.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai A. C.; Crews C. M. Induced protein degradation: An emerging drug discovery paradigm. Nat. Rev. Drug Discovery 2017, 16, 101–114. 10.1038/nrd.2016.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neklesa T. K.; Tae H. S.; Schneekloth A. R.; Stulberg M. J.; Corson T. W.; Sundberg T. B.; Raina K.; Holley S. A.; Crews C. M. Small-molecule hydrophobic tagging-induced degradation of halotag fusion proteins. Nat. Chem. Bio. 2011, 7, 538–543. 10.1038/nchembio.597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie T.; Lim S. M.; Westover K. D.; Dodge M. E.; Ercan D.; Ficarro S. B.; Udayakumar D.; Gurbani D.; Tae H. S.; Riddle S. M.; Sim T.; Marto J. A.; Jaenne P. A.; Crews C. M.; Gray N. S. Pharmacological targeting of the pseudokinase her3. Nat. Chem. Bio. 2014, 10, 1006–1012. 10.1038/nchembio.1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustafson J. L.; Neklesa T. K.; Cox C. S.; Roth A. G.; Buckley D. L.; Tae H. S.; Sundberg T. B.; Stagg D. B.; Hines J.; McDonnell D. P.; Norris J. D.; Crews C. M. Small-molecule-mediated degradation of the androgen receptor through hydrophobic tagging. Angew. Chem., Int. Ed. 2015, 54, 9659–9662. 10.1002/anie.201503720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng J.; Li Y.; Wang X.; Dong G.; Sheng C. Discovery of novel pdeδ degraders for the treatment of kras mutant colorectal cancer. J. Med. Chem. 2020, 63, 7892–7905. 10.1021/acs.jmedchem.0c00929. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.