

Abstract
Introduction:
Sleep disturbances are a common symptom in patients with Huntington’s disease (HD). However, it is unclear when in the disease course of HD sleep disturbances become more frequent compared to the general population. This study investigated the frequency and odds of developing sleep disturbances between adults with HD or at-risk for HD and non-HD controls.
Methods:
Participants from the Enroll-HD study were split by both disease type and disease severity using CAG length, diagnostic confidence level, and total functional capacity score. Multivariate logistic regression was used to calculate odds ratios adjusted for age, sex, tobacco and alcohol use, depression and psychosis scores, and cognition to compare HD groups to non-HD controls. Cox proportional hazards models and Kaplan Meier curves were used to determine differences in probabilities of developing sleep disturbances and how sleep disturbances are related to age at motor onset.
Results:
The odds of sleep disturbance was greater in HD participants than non-HD controls (OR: 1.93, p < 0.001). Additionally, those with juvenile HD and late-stage disease had the greatest odds of sleep disturbance development. The development of a sleep disorder in manifest HD participants was observed to be around the time of disease onset.
Conclusions:
Sleep disturbances are more frequent in HD patients than those without HD. The frequency is also greater in those with juvenile HD and increases as the disease progresses. This is supplemented by the finding that the onset of sleep disturbances occurs near the time of motor onset of HD.
MeSH Key Words: Huntington Disease, Sleep, Sleep Wake Disorders, Disease Progression
INTRODUCTION
Huntington’s disease (HD) is a single-gene, autosomal dominant disorder in which a CAG repeat sequence is expanded beyond the length seen in the normal population [1]. The length of the CAG repeat sequence is inversely related to age at onset, which is generally between the ages of 30 and 50 years [2]. HD is characterized by motor symptoms, cognitive decline, and behavioral changes [3]. However, there are a variety of additional symptoms that can occur in HD that can contribute to the burden of this disease. Specifically, sleep disturbances are a common finding in HD and have been shown to impact up to 90% of patients [4, 5]. Sleep disturbances can be further classified as insomnia, earlier sleep onset, and delayed rapid eye movement sleep, among others [6–9].
Sleep disturbances have been reported in patients with the genetic mutation that causes HD prior to the onset of symptoms, as well as in patients with motor-manifest HD [6, 10–12]. Therefore, it is not clear when in the disease course of HD sleep disturbances become more frequent relative to the general population. Understanding the time course of sleep disruptions in HD is critically important because changes in sleep patterns have been associated with negative impacts on quality of life and functioning [8, 13]. Additionally, the treatment of sleep disturbances can be quite challenging for practitioners. Therefore, further our understanding of this significant HD comorbidity has the potential to significantly impact patient care. The primary aim of this study was to determine the frequency of sleep disturbances in patients with HD compared to non-HD controls by disease type and disease stage. The start of sleep disturbance symptoms relative to disease onset was also investigated.
MATERIALS AND METHODS
Description of Data
The fifth version of the Enroll-HD platform was used for these analyses (https://enroll-hd.org/). All Enroll-HD study sites are required to obtain and maintain local ethics committee approvals. Signed informed consent forms for each participant must also be obtained for their data to be included in the datasets. This platform contains information for 21,116 participants, including those with the genetic mutation who have the manifest form of disease (motor-manifest), those with the genetic mutation who have not yet received a motor diagnosis of HD (preHD), and those who do not have the genetic mutation for the disease (non-HD controls) [14]. Of those who do not have the genetic mutation, some are relatives of study participants with the genetic mutation (first or second degree blood relative) and others are family controls (not related by blood). The primary objective of this study was to determine whether there is a difference in frequency and odds of developing sleep disturbances between those with the genetic mutation for HD and non-HD controls. Three analyses were performed to address the primary objective: 1) a comparison of all HD participants and non-HD controls, 2) a comparison of HD participants by type of disease (incomplete penetrance [CAG of 36–39], adult onset [CAG of 40–59], and juvenile onset [CAG of 60 or above]) and non-HD controls [CAG < 36], and 3) a comparison of HD participants by disease stage (preHD or manifest HD) and non-HD controls. The secondary objective of this study was to describe at what point during disease course sleep disturbances are most commonly observed amongst participants with HD.
The appearance of a sleep disturbance was determined using comorbidity and medication data. Comorbidities and medication use are collected prospectively throughout the study and self-reported by participants at each Enroll-HD visit. At the initial Enroll-HD visit, data are also collected on past comorbidities or medication use. Comorbidities included as a sleep disturbance and indications for medications used to treat sleep disturbances can be found in Supplemental Table 1. The most common comorbidities included insomnia and sleep apnea. The most commonly reported indications for initiating a medication for a sleep disturbance were “insomnia” and “sleep disorder.” The first instance of a comorbidity or medication indicating a sleep disturbance with a listed start date was used for each participant. The age at which a sleep disturbance began was calculated by adding the difference in years between a participant’s age at their baseline visit and sleep disturbance start date to their age at baseline. Participants whose calculated sleep disturbance onset year began prior to birth were also excluded. Ages in the Enroll-HD database are reported after being rounded to the nearest whole number. Therefore, if a participant is 21.4 years old at day 0 (baseline visit), they will be reported as being 21 years old. If this participant reported that they have had sleep changes since birth, the start date of their sleep disturbance would be reported as −7811 (21.4*365), as this calculation is made using the participant’s actual birthdate prior to rounding the age to ensure participant anonymity. Using the reported age of 21, we would assume that the earliest day that the participant was alive would be −7665 (21*365), but the reported onset of sleep disturbances would be listed as −7811. This was a rare occurrence and only one participant was excluded for this reason.
Primary Analysis
The primary analysis of this study was a cross-sectional design using a participant’s baseline Enroll-HD visit (visit day 0). The genetic mutation status was determined using a participant’s CAG repeat length. Those with a repeat length of 36 or greater were considered to have the genetic mutation. Those with a repeat length less than 36 did not have the genetic mutation and were considered a non-HD control. A participant was excluded if they were missing age at their baseline visit. Any alcohol and tobacco use were determined using the current use indicator and the history of previous use indicator. History of previous use was collected at the baseline Enroll-HD visit whereas current use was updated at each visit. If a participant had a history of previous use or current use at their baseline, they were considered to have prior alcohol or tobacco use. Alcohol use and tobacco use have also been shown to affect sleep quality in healthy individuals [15, 16]. Participants were also excluded if they were missing scores on the depression and psychosis subscores for the Problem Behavior Assessment – Short Form or the Symbol Digit Modality Test at their baseline visit. Psychiatric disorders have also been associated with impaired sleep in both patients with HD and healthy patients [4, 17]. Participants with the mutation for HD were also excluded if they had a missing diagnostic confidence level (DCL) or total functional capacity (TFC) score from the Unified Huntington’s Disease Rating Scale [18]. For the analysis with HD participants stratified by type of disease, CAG repeat length was used to determine the groups. Those with CAG repeat lengths between 36 and 39 were considered to have incomplete penetrance HD, those with CAG repeat lengths between 40 and 59 were considered to have adult-onset HD, and those with CAG repeat lengths greater than 59 were considered to have juvenile onset HD. For the analysis with HD participants stratified by disease stage, the DCL and TFC score were used to determine groups. Participants with HD and a DCL less than four were considered to have premanifest HD, those with a DCL of four and a TFC score of seven or greater (Stages I and II) were considered to be early-stage, those with a DCL of four and a TFC score between three and six (Stage III) were considered to be mid-stage, and those with a DCL of four and a TFC score less than three were considered to be late-stage (Stage IV and V). The disease stages have been condensed from five commonly used stages [19] determined by TFC score to three stages for ease in comparison. A sleep disturbance had occurred for a participant if the age at which the disturbance started was less than the age of the participant at their baseline visit, given the cross-sectional nature of the primary analysis. Participants with no start day indicated for any sleep disturbance were also excluded as it could not be determined if the sleep disturbance was present prior to the baseline visit.
For the primary analysis, we first calculated the frequency for all participants with HD and non-HD controls and then calculated frequency by disease type and stage. Multivariable logistic regression using presence of a sleep disturbance as the dependent variable was then used to calculate odds ratios (ORs) for the three comparisons: HD vs Non-HD, HD Stage vs Non-HD, and HD Type vs Non-HD. The models were conducted using a binomial distribution and logit link function. Adjusted models included age, sex, history of alcohol use, history of tobacco use, depression subscore, psychosis subscore, and number correct on the symbol digit modality test as covariates in the model.
Secondary Analyses
For the secondary analyses, Kaplan Meier curves using age as the time scale were first plotted to compare all participants with HD to non-HD controls. All visits for each participant were used to identify a censor day of either the age of the last visit date for each participant if a sleep disturbance had not occurred or the age at sleep disturbance occurrence. In addition, participants had to be free of sleep disturbances at their baseline visit. Participants without a recorded start day for a sleep disturbance were excluded from these analyses. A log-rank test was used to determine if there was a difference in time to sleep disturbance occurrence between the groups and a left-truncated Cox proportional hazards model was used to obtain hazard ratios and confidence intervals. Cox models were adjusted for sex, SDMT score, psychosis subscore, and tobacco use and stratified on depression subscore and alcohol use to satisfy the proportional hazards assumption. Participants with HD were then stratified by disease type and the Kaplan Meier curves were recreated and new hazard ratios calculated using the previously described left-truncated Cox model. Next, we assessed the relationship between age at onset of motor symptoms of HD and the presence of sleep disturbances. This analysis could only be performed using participants with motor-manifest HD who had a recorded age at clinical onset. The age at which a sleep disturbance began was converted to time in years relative to age at clinical onset for participants with the genetic mutation. Those with the genetic mutation who did not have a listed age at clinical onset were excluded from this analysis. A Kaplan Meier curve was created without stratification to identify patterns of sleep disturbance initiation relative to disease onset. A second set of Kaplan Meier curves stratified by disease type were then created. Kaplan Meier curves stratified by disease stage were not created as disease stage is heavily dependent on the years relative to onset. The log-rank test was used to determine significant differences between the curves and an adjusted left-truncated Cox proportional hazards model as previously described was used to calculate hazard ratios and confidence intervals, though no stratification on depression subscore or alcohol use was necessary to meet the hazards assumption. A Cox proportional hazards model was also used to assess the association between CAG repeat length as a continuous value and development of a sleep disturbance. Results were considered significant at a p-value of less than 0.05. All analyses were performed using R software, version 3.6.3.
RESULTS
Primary Analysis
For the primary analysis there were 4,916 non-HD controls and 14,791 participants with HD (Figure 1). Of those participants with HD, 833 had incomplete penetrance HD, 13,862 had adult-onset HD, and 96 had juvenile-onset HD (Table 1). There were significant differences in age, sex, tobacco use, alcohol use, depression subscores, psychosis subscores, and SDMT score between the groups. The overall frequency of sleep disturbances in all participants was 13.1 percent (n = 2589). Frequency of sleep disturbances was lowest in the non-HD control group and increased with CAG length (Table 1). Without controlling for any potential confounding variables, the odds of having a sleep disturbance was 3.10 times higher in those with the genetic mutation for HD compared to those without (95% CI 2.73 – 3.54, p < 0.001; Supplemental Table 2). When controlling for potential confounding variables, the odds of having a sleep disturbance was greater in those with the genetic mutation for HD than in non-HD controls (OR 1.93, 95% CI 1.68 – 2.23, p < 0.001). When stratifying by disease type and controlling for potential confounding variables, all three types of HD had significantly higher odds than the non-HD control group (Supplemental Table 2). The odds were similar between the incomplete penetrance and adult onset groups (Incomplete Penetrance: OR 1.95, 95% CI 1.54 – 2.47, p < 0.001; Adult Onset: OR 1.94, 95% CI 1.68 – 2.24, p < 0.001) and higher in those with a CAG length of 60 or greater when compared to non-HD controls (OR 5.24, 95% CI 3.16 – 8.49, p < 0.001).
Figure 1: Participant Inclusion Flow Chart.
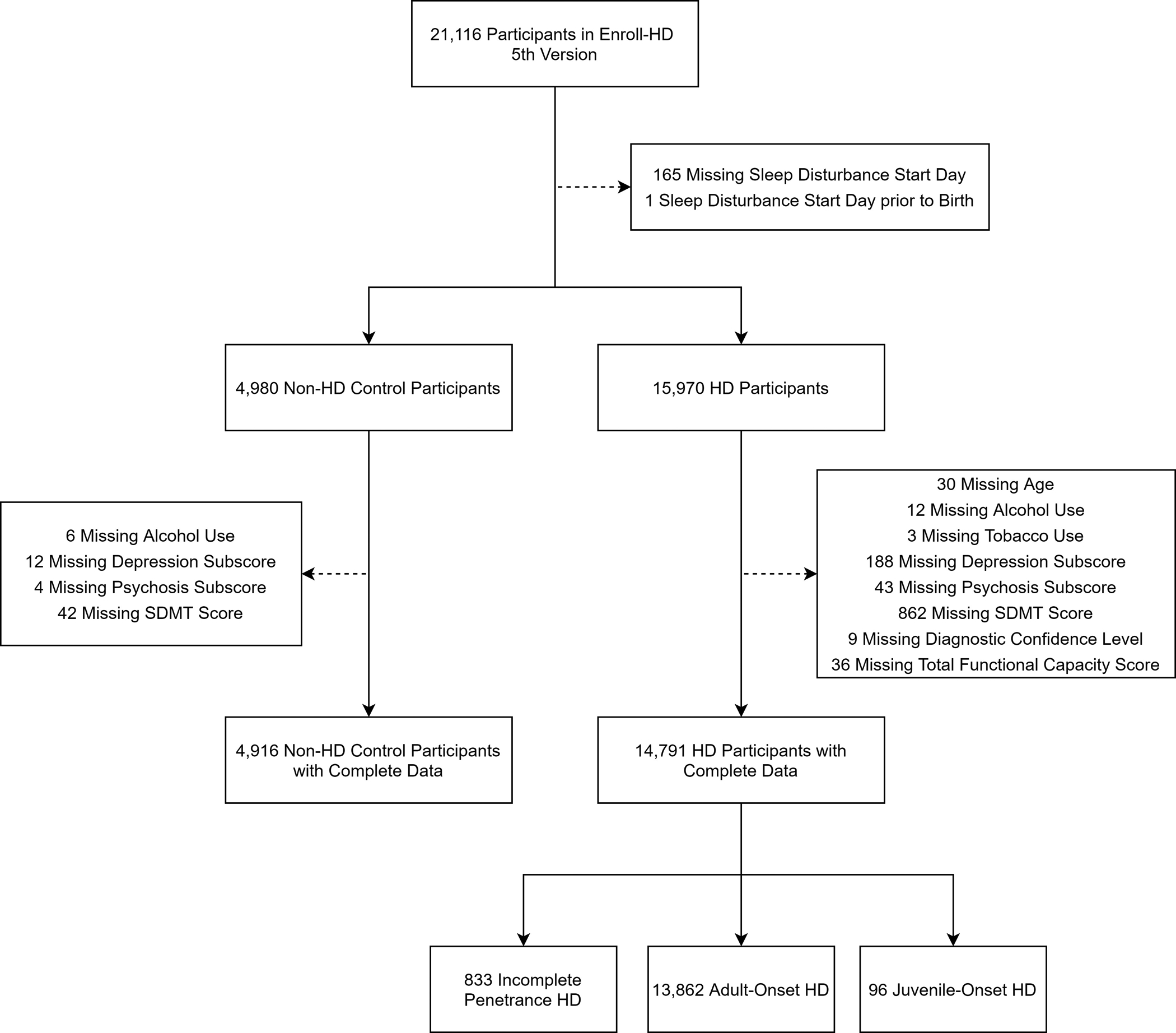
Figure 1 displays the implementation of exclusion and inclusion criteria for participants in this study. Participants are first placed into groups based on genetic mutation status (all participants with HD and non-HD controls). Participants with HD are then further divided into groups based on disease type.
Abbreviations:
HD: Huntington’s disease
Table 1.
Participant Characteristics by Disease Type and Stage
| HD Disease Type | HD Disease Stage | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Non-HD Controls (N = 4,958) |
All HD (N = 14,791) |
CAG 36–39 (N = 833) |
CAG 40–59 (N = 13,862) |
CAG ≥ 60 (N = 96) |
Premanifest (N = 5,612) |
Early Stage (N = 6,696) |
Mid Stage (N = 1,911) |
Late Stage (N = 572) |
|
| Sleep Disturbance Prevalence | 277 (5.6) | 2312 (15.6) | 119 (14.3) | 2166 (15.6) | 27 (28.1) | 472 (8.4) | 1124 (16.8) | 516 (27.0) | 200 (35.0) |
| Age (y) | 46.6 (14.8) | 48.2 (13.9) | 55.1 (15.3) | 47.9 (13.6) | 24.6 (4.2) | 40.5 (12.4) | 51.9 (12.3) | 54.8 (13.0) | 56.6 (13.4) |
| Sex (F) | 3006 (61.1) | 7994 (54.0) | 468 (56.2) | 7476 (53.9) | 50 (52.1) | 3318 (59.1) | 3310 (49.4) | 1034 (54.1) | 332 (58.0) |
| History of Alcohol Use (%) | 2761 (56.2) | 7696 (52.0) | 512 (61.5) | 7163 (51.7) | 21 (21.9) | 3524 (62.8) | 3348 (50.0) | 706 (36.9) | 118 (20.6) |
| History of Tobacco Use (%) | 2126 (43.2) | 7090 (47.9) | 379 (45.5) | 6672 (48.1) | 39 (40.6) | 2534 (45.2) | 3344 (49.9) | 936 (49.0) | 276 (48.3) |
| Depression Subscore | 3.4 (5.1) | 5.0 (6.3) | 3.9 (5.6) | 5.0 (6.3) | 3.9 (5.2) | 4.6 (6.1) | 5.2 (6.2) | 5.4 (6.8) | 5.4 (6.3) |
| Psychosis Subscore | 0.1 (0.7) | 0.3 (1.5) | 0.1 (0.5) | 0.3 (1.6) | 0.4 (1.6) | 0.1 (1.1) | 0.2 (1.4) | 0.5 (2.2) | 0.9 (3.0) |
| SDMT Score | 50.0 (12.1) | 32.2 (17.7) | 41.3 (17.3) | 31.7 (17.6) | 19.5 (11.4) | 48.0 (12.9) | 26.3 (11.2) | 14.3 (8.9) | 5.0 (6.5) |
Data are presented as mean (SD) for continuous variables and N (%) for categorical variables. The statistical difference in in sleep disturbance prevalence, sex, history of alcohol use, and history of tobacco use were determined using the chi-squared test while the one-way ANOVA test was used for all other parameters. In each comparison to the Non-HD Controls, all groups were statistically significantly different with p value < 0.001. SDMT – Symbol Digit Modality Test
When stratifying participants with HD by disease stage, there were 5,612 participants with premanifest HD, 6,696 participants with early-stage HD, 1,911 participants with mid-stage HD, and 572 participants with late-stage HD (Table 1). These groups significantly differed on age, sex, history of tobacco use, history of alcohol use, depression subscore, psychosis subscore, and SDMT score. The frequency of sleep disturbances increased as the disease worsened (Table 1). After controlling for potential confounding variables, the odds increased as the disease progressed with the premanifest group having an odds ratio of 1.56 (95% CI 1.34 – 1.83, p < 0.001), the early-stage group having an odds ratio of 2.60 (95% CI 2.21 – 3.07, p < 0.001), the mid-stage group having an odds ratio of 4.26 (95% CI 3.47 – 5.24, p < 0.001), and the late-stage group having an odds ratio of 5.81 (95% CI 4.44 – 7.62, p < 0.001) compared with controls (Supplemental Table 2).
Secondary Analyses
The Kaplan Meier curves for participants with HD and non-HD controls presented a better understanding at what point in time the two groups begin to differ (Figure 2A). Included in this analysis were 3,687 control participants and 9,937 participants with HD. The curves between the two groups were significantly different (p-value < 0.001) using the Log-Rank test. Visual inspection of Figure 2A shows that the curve of the HD group seems to deviate from the non-HD controls at approximately the age of 40, which would be an approximate age of onset of many participants. In addition, the adjusted hazard ratio for HD participants compared to non-HD controls using the left-truncated Cox model was 4.09 (95% CI 3.23 – 5.18, p < 0.001; Supplemental Table 3). Similarly, when plotting curves stratified by disease type, the deviation is earlier with larger CAG lengths, which would indicate that sleep disturbances increase in frequency with increasing disease burden (Figure 2B). This was also reflected in the hazard ratios calculated using the adjusted Cox model which increased with CAG length (Incomplete Penetrance: HR 2.63, 95% CI 1.72 – 4.01, p < 0.001; Adult Onset: HR 4.12, 95% CI 3.26 – 5.22, p < 0.001, Juvenile Onset: HR 20.65, 95% CI 8.82 – 48.32, p < 0.001). Therefore, we created a survival curve of only participants with HD centered around age at onset and stratified by their disease type (Figure 3). There were 7,058 participants from the previous analysis with a listed age at motor onset. After splitting the participants into groups based on their CAG length, there were no differences between the groups (p-value = 0.5). This curve presented a similar heightened risk starting around the age at onset for all groups. A Cox proportional hazards model also showed no significant difference between the hazard ratio comparing adult-onset HD to incomplete penetrance, though there was a significant difference between juvenile-onset HD and the incomplete penetrance group (Adult Onset: HR 1.17, 95% CI 0.76 – 1.79, p = 0.339; Juvenile Onset: HR 2.15, 95% CI 1.01 – 4.57, p = 0.046). In addition when using CAG length as a continuous predictor in the Cox proportional hazards model, the variable was significant with a hazard ratio of 1.036 (95% CI 1.02 – 1.05, p < 0.001).
Figure 2: Relationship between Presence of HD Mutation and Starting Age of a Sleep Disturbance.
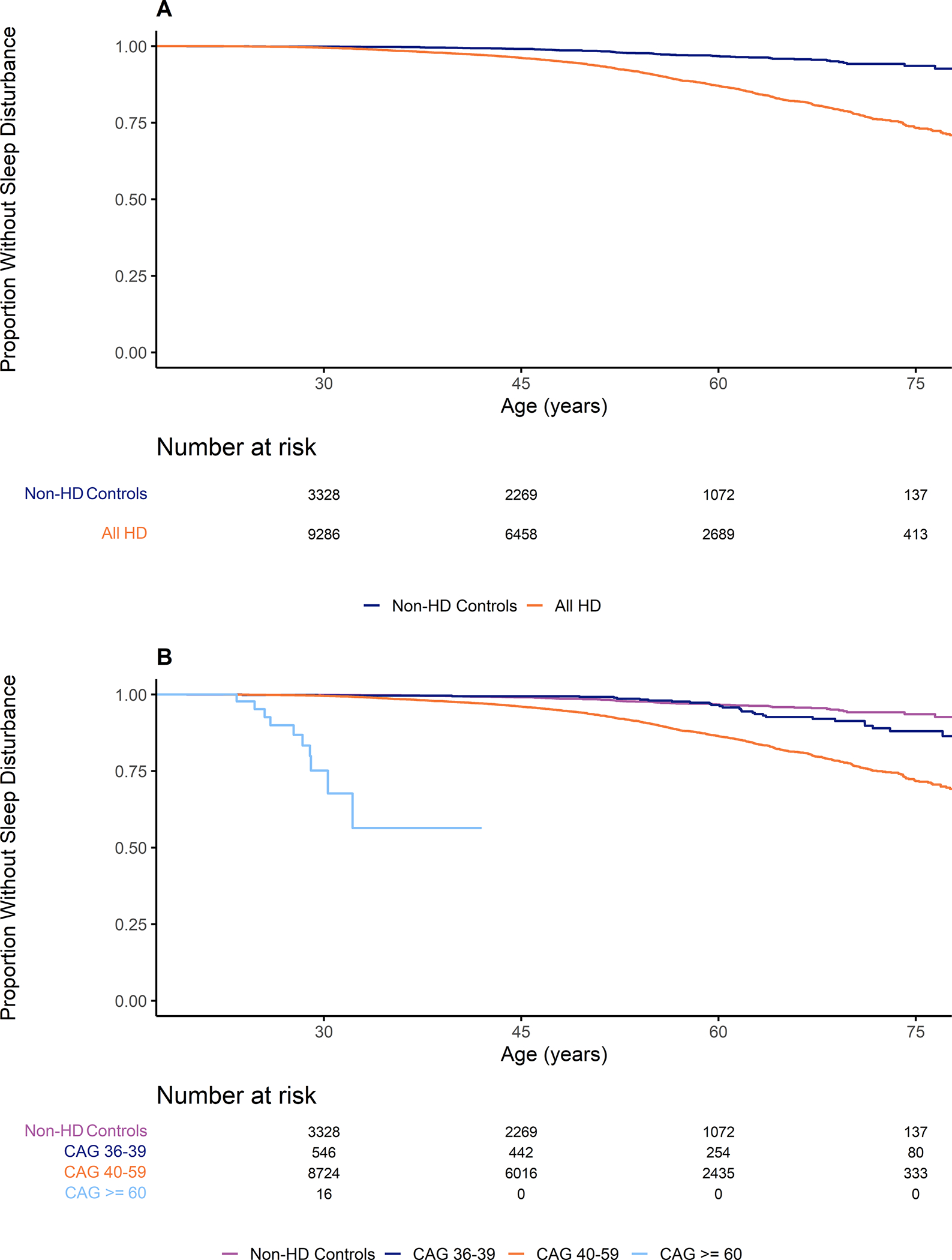
Figure 2A presents the relationship between presence of the mutation for HD and the age at which a sleep disturbance begins. These results include all participants with the mutation for HD regardless of disease state. Figure 2B presents the relationship between disease type and the age at which a sleep disturbance begins.
Abbreviations:
HD: Huntington’s disease
Figure 3: Relationship between CAG Length and Starting Age of Sleep Disturbance relative to Age at Onset.
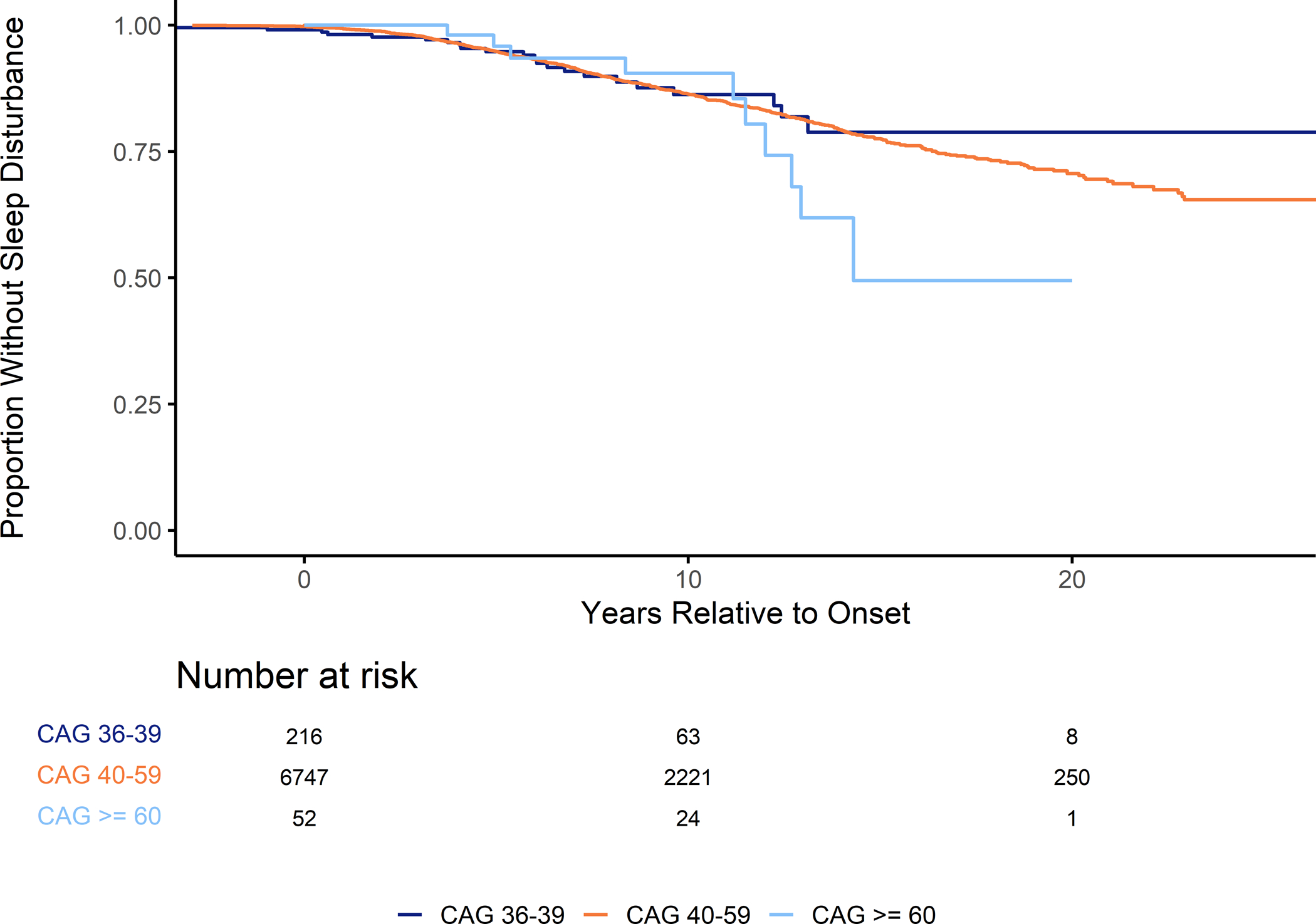
Figure 3 presents the relationship between CAG repeat length and age at the start of a sleep disturbance relative to age at motor-onset. These results only include participants with manifest HD who had a recorded age at clinical onset.
Abbreviations:
CAG: Cytosine-Adenine-Guanine
DISCUSSION
This study provides evidence that patients with HD have a significantly higher odds of having a reported sleep disturbance compared to controls and that the frequency of sleep disturbances increases with juvenile HD and with worsening disease stage. Individuals with the preHD had a significantly greater odds of having a sleep disturbance compared to non-HD controls which supports results from the largest study conducted in preHD patients that found abnormalities in sleep continuity using actigraphy, polysomnography and sleep latency tests [11]. The actual odds of having abnormalities in the sleep cycle may be larger as the abnormalities detected in the previous study may not have been clinically noticeable and warrant treatment or diagnosis. Our present analysis only included sleep disturbances that were documented through medication use or reported by a participant; therefore, this analysis is far less sensitive to subtle changes that may have been found in the previous study [11]. It is also possible that the actual odds of having a disturbance may be larger than the results seen here due to the controls used in this study. These controls consist largely of family member controls and one study showed that increased nighttime to daytime activity was mirrored in profiles of caregivers of patients with HD [20].
Our secondary analyses provide additional clarity to our primary analysis. Specifically, we have demonstrated that the emergence of sleep disturbances in HD seems to occur at approximately the time of motor onset and the likelihood of developing a disrupted sleep comorbidity increases with increasing disease burden. This could suggest that subclinical abnormalities occur prior to motor onset of disease and reach a threshold that warrants patient reporting near the time of motor diagnosis. These results help to explain why there was an increasing trend in frequency that continued from premanifest disease through late stage. Lastly, our results of CAG length being related to sleep disturbance development in relation to age at onset adds to the current literature, though conflicts with previous results [6].
There are many theories to describe the increase in sleep disturbances in manifest HD patients. One suggestion is that the REM sleep executive systems, located in the brainstem, are damaged which could occur prior to motor onset [6]. Mouse models have led to a potential explanation of abnormalities in the suprachiasmatic nucleus of the hypothalamus, which controls melatonin synthesis [21, 22]. Circadian melatonin secretion abnormalities have also been described in some HD sleep studies [23, 24]. One study also identified thalamic atrophy in individuals with manifest HD who had sleep problems, which is consistent with previous literature describing the role of the thalamus in nonrapid eye movement sleep [25]. The hypothalamus, which also plays an important role in the regulation of sleep and wakefulness, has also been identified as an area where there is reduced grey matter volume in preHD patients compared to healthy controls, however no differences in circadian markers were observed in that study [26]. Other hypothesized causes of sleep abnormalities in HD include involuntary movements [27] or the presence of psychiatric conditions, such as depression and anxiety [28]. Greater severity of depressive symptoms have been shown to be associated with sleep disturbances in both preHD patients and those with motor symptoms [29]; Medication used to treat common early symptoms of HD also have the potential to alter sleep structure which could lead to sleep abnormalities.
While this study provides evidence of the point in time during which sleep disturbances are most common in HD patients, there are limitations. First, all information regarding age at onset, type of sleep disturbance, start of sleep disturbance, and medication used for sleep disturbances are self-reported. Sleep disturbances were only captured if there was a medication or comorbidity associated with sleep, not using sleep questionnaires or evaluations. Many sleep disturbances may not require physician care and could be underreported. The increased number of healthcare visits of or medication use for psychiatric symptoms for the manifest HD patients could account for a greater capture of sleep symptoms in that group of patients. Another limitation of this study is the control group was comprised largely of family and spousal controls. Sleep patterns and disturbances may be more similar to HD patients and may not reflect differences between HD patients and the general population. Finally, medications may have been used that were not indicated for sleep disturbances, but still may have had an impact on the development of sleep disturbances. However, it is reasonable to believe that the effect of not including these medications would be equal across all participant groups and would cause the frequency across all groups to increase.
Sleep disturbances can be extremely troubling for patients with HD and their caregivers and may have a negative impact on overall quality of life. Disrupted sleep can also represent a clinical conundrum for practitioners as sleep disturbances can be resistant to treatment in HD. Additionally, many medications may be contraindicated in older adults or be associated with drug interactions with other medications commonly used in HD, such as tetrabenazine or antipsychotics. Therefore, gaining a better understanding of the time course of the development of sleep disturbances is crucial in improving the overall care that patients with HD receive. Here, we have shown that the onset of sleep disturbances may be closely related to the timing of motor onset. Furthermore, there may be an important pathologic or psychologic event around the time of onset that leads to deteriorating sleep in HD patients. Further research is needed to assess the potential pathologic pathways that lead to sleep abnormalities and when during disease progression disturbances can be noticed through diagnostic testing.
Supplementary Material
ACKNOWLEDGEMENTS
Enroll-HD is a clinical research platform and longitudinal observational study for Huntington’s disease families intended to accelerate progress towards therapeutics; it is sponsored by CHDI Foundation, a nonprofit biomedical research organization exclusively dedicated to collaboratively developing therapeutics for HD. Enroll-HD would not be possible without the vital contribution of the research participants and their families.
Funding:
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Footnotes
Declarations of interest: none
REFERENCES
- [1].A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. The Huntington’s Disease Collaborative Research Group, Cell 72(6) (1993) 971–83. [DOI] [PubMed] [Google Scholar]
- [2].CAG Repeat Not Polyglutamine Length Determines Timing of Huntington’s Disease Onset, Cell 178(4) (2019) 887–900. e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Walker FO, Huntington’s disease, Lancet (London, England) 369(9557) (2007) 218–28. [DOI] [PubMed] [Google Scholar]
- [4].Herzog-Krzywoszanska R, Krzywoszanski L, Sleep Disorders in Huntington’s Disease, Frontiers in psychiatry 10 (2019) 221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Taylor N, Bramble D, Sleep disturbance and Huntingdon’s disease, The British journal of psychiatry : the journal of mental science 171 (1997) 393. [DOI] [PubMed] [Google Scholar]
- [6].Arnulf I, Nielsen J, Lohmann E, Schiefer J, Wild E, Jennum P, Konofal E, Walker M, Oudiette D, Tabrizi S, Durr A, Rapid eye movement sleep disturbances in Huntington disease, Archives of neurology 65(4) (2008) 482–8. [DOI] [PubMed] [Google Scholar]
- [7].Hansotia P, Wall R, Berendes J, Sleep disturbances and severity of Huntington’s disease, Neurology 35(11) (1985) 1672–4. [DOI] [PubMed] [Google Scholar]
- [8].Aziz NA, Anguelova GV, Marinus J, Lammers GJ, Roos RA, Sleep and circadian rhythm alterations correlate with depression and cognitive impairment in Huntington’s disease, Parkinsonism & related disorders 16(5) (2010) 345–50. [DOI] [PubMed] [Google Scholar]
- [9].Wiegand M, Moller AA, Lauer CJ, Stolz S, Schreiber W, Dose M, Krieg JC, Nocturnal sleep in Huntington’s disease, Journal of neurology 238(4) (1991) 203–8. [DOI] [PubMed] [Google Scholar]
- [10].Goodman AO, Rogers L, Pilsworth S, McAllister CJ, Shneerson JM, Morton AJ, Barker RA, Asymptomatic sleep abnormalities are a common early feature in patients with Huntington’s disease, Current neurology and neuroscience reports 11(2) (2011) 211–7. [DOI] [PubMed] [Google Scholar]
- [11].Lazar AS, Panin F, Goodman AO, Lazic SE, Lazar ZI, Mason SL, Rogers L, Murgatroyd PR, Watson LP, Singh P, Borowsky B, Shneerson JM, Barker RA, Sleep deficits but no metabolic deficits in premanifest Huntington’s disease, Annals of neurology 78(4) (2015) 630–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Bellosta Diago E, Perez Perez J, Santos Lasaosa S, Viloria Alebesque A, Martinez Horta S, Kulisevsky J, Lopez Del Val J, Circadian rhythm and autonomic dysfunction in presymptomatic and early Huntington’s disease, Parkinsonism & related disorders 44 (2017) 95–100. [DOI] [PubMed] [Google Scholar]
- [13].Diago EB, Martinez-Horta S, Lasaosa SS, Alebesque AV, Perez-Perez J, Kulisevsky J, Del Val JL, Circadian Rhythm, Cognition, and Mood Disorders in Huntington’s Disease, Journal of Huntington’s disease 7(2) (2018) 193–198. [DOI] [PubMed] [Google Scholar]
- [14].Landwehrmeyer GB, Fitzer-Attas CJ, Giuliano JD, Gonçalves N, Anderson KE, Cardoso F, Ferreira JJ, Mestre TA, Stout JC, Sampaio C, Data Analytics from Enroll-HD, a Global Clinical Research Platform for Huntington’s Disease, Movement disorders clinical practice 4(2) (2017) 212–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Stein MD, Friedmann PD, Disturbed sleep and its relationship to alcohol use, Substance abuse 26(1) (2005) 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Amiri S, Behnezhad S, Smoking and risk of sleep-related issues: a systematic review and meta-analysis of prospective studies, Canadian journal of public health = Revue canadienne de sante publique 111(5) (2020) 775–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Freeman D, Sheaves B, Waite F, Harvey AG, Harrison PJ, Sleep disturbance and psychiatric disorders, The lancet. Psychiatry 7(7) (2020) 628–637. [DOI] [PubMed] [Google Scholar]
- [18].Unified Huntington’s Disease Rating Scale: reliability and consistency. Huntington Study Group, Movement disorders : official journal of the Movement Disorder Society 11(2) (1996) 136–42. [DOI] [PubMed] [Google Scholar]
- [19].Paulsen JS, Wang C, Duff K, Barker R, Nance M, Beglinger L, Moser D, Williams JK, Simpson S, Langbehn D, van Kammen DP, Challenges assessing clinical endpoints in early Huntington disease, Movement disorders : official journal of the Movement Disorder Society 25(15) (2010) 2595–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Morton AJ, Wood NI, Hastings MH, Hurelbrink C, Barker RA, Maywood ES, Disintegration of the sleep-wake cycle and circadian timing in Huntington’s disease, The Journal of neuroscience : the official journal of the Society for Neuroscience 25(1) (2005) 157–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Kudo T, Schroeder A, Loh DH, Kuljis D, Jordan MC, Roos KP, Colwell CS, Dysfunctions in circadian behavior and physiology in mouse models of Huntington’s disease, Experimental neurology 228(1) (2011) 80–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Rudenko O, Tkach V, Berezin V, Bock E, Detection of early behavioral markers of Huntington’s disease in R6/2 mice employing an automated social home cage, Behavioural brain research 203(2) (2009) 188–99. [DOI] [PubMed] [Google Scholar]
- [23].Aziz NA, Pijl H, Frölich M, Schröder-van der Elst JP, van der Bent C, Roelfsema F, Roos RA, Delayed onset of the diurnal melatonin rise in patients with Huntington’s disease, Journal of neurology 256(12) (2009) 1961–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Alders J, Smits M, Kremer B, Naarding P, The role of melatonin in sleep disturbances in end-stage Huntington’s disease, The Journal of neuropsychiatry and clinical neurosciences 21(2) (2009) 226–7. [DOI] [PubMed] [Google Scholar]
- [25].Baker CR, Domínguez D JF, Stout JC, Gabery S, Churchyard A, Chua P, Egan GF, Petersén Å, Georgiou-Karistianis N, Poudel GR, Subjective sleep problems in Huntington’s disease: A pilot investigation of the relationship to brain structure, neurocognitive, and neuropsychiatric function, Journal of the Neurological Sciences 364 (2016) 148–153. [DOI] [PubMed] [Google Scholar]
- [26].Bartlett DM, Domínguez DJ, Reyes A, Zaenker P, Feindel KW, Newton RU, Hannan AJ, Slater JA, Eastwood PR, Lazar AS, Ziman M, Cruickshank T, Investigating the relationships between hypothalamic volume and measures of circadian rhythm and habitual sleep in premanifest Huntington’s disease, Neurobiol Sleep Circadian Rhythms 6 (2019) 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Neutel D, Tchikviladze M, Charles P, Leu-Semenescu S, Roze E, Durr A, Arnulf I, Nocturnal agitation in Huntington disease is caused by arousal-related abnormal movements rather than by rapid eye movement sleep behavior disorder, Sleep medicine 16(6) (2015) 754–9. [DOI] [PubMed] [Google Scholar]
- [28].Videnovic A, Leurgans S, Fan W, Jaglin J, Shannon KM, Daytime somnolence and nocturnal sleep disturbances in Huntington disease, Parkinsonism & related disorders 15(6) (2009) 471–4. [DOI] [PubMed] [Google Scholar]
- [29].Baker CR, Dominguez JFD, Stout JC, Gabery S, Churchyard A, Chua P, Egan GF, Petersen A, Georgiou-Karistianis N, Poudel GR, Subjective sleep problems in Huntington’s disease: A pilot investigation of the relationship to brain structure, neurocognitive, and neuropsychiatric function, Journal of the Neurological Sciences 364 (2016) 148–153. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.