

Over the last two decades, advances in the field of epilepsy genetics have led to hope for precision treatments,[1] and certain genetic diagnoses can direct treatment strategies.[2,3] One of the most common genetic epilepsies is Dravet syndrome (DS), associated with the gene SCN1A, which encodes the sodium voltage-gated channel alpha subunit one.[4] Here we describe a boy with DS presenting as recurrent status epilepticus (SE). Early identification of a pathogenic SCN1A variant led to expedited implementation of fenfluramine via the Investigational New Drug (IND) mechanism prior to federal approval for use in DS.[5,6]
The patient is a five-year-old boy who presented at seven months with convulsive SE, starting with an unprovoked hemiclonic seizure and progressing to bilateral tonic-clonic activity for approximately 10 minutes, requiring rescue medication. EEG and MRI were normal. Given the length of the seizure and young age, levetiracetam was initiated. One month later, he was hospitalized for a second episode of convulsive SE lasting 30 minutes. An epilepsy gene panel identified a de novo heterozygous likely pathogenic variant in SCN1A (c.4162G>T, p.Glu1388Ter). The clinical picture, including subsequent development of myoclonic seizures, was consistent with DS.
Despite treatment with topiramate, clobazam, valproic acid, and the ketogenic diet, the patient continued with approximately monthly episodes of generalized convulsive SE. Most occurred with febrile illness and some required intubation and intensive care unit (ICU) admission. His convulsive seizure frequency did not meet criteria for the randomized clinical trial for fenfluramine; therefore, through the IND mechanism of the Food and Drug Administration (FDA), we initiated fenfluramine at age 28 months with the goal of reducing SE frequency and severity. The starting dose was 0.2 mg/kg/d, which was increased to 0.5 mg/kg/d over the course of two months; this was subsequently increased to a maximum dose of 0.7 mg/kg/d 14 months after initiation to optimize therapy for myoclonic seizures.
Prior to starting fenfluramine, the patient had 14 hospital admissions, including five to the ICU, over 20 months. Since initiation, the patient has had no admissions for SE in over two years despite several febrile illnesses (figure 1). He continues with daily clusters of myoclonic and atonic seizures. Stiripentol was trialed after initiation of fenfluramine for treatment of myoclonic seizures, but this was weaned after one month due to concerns about increasing myoclonic seizure frequency. Due to a sesame allergy, Epidiolex was not an option as it is delivered in sesame oil. Development was normal until the first episode of SE (seven months), with subsequent plateau and global delay. After initiation of fenfluramine and cessation of SE, he has made developmental progress, including walking by two years of age and development of verbal language with phrases. Parent-reported quality of life has improved. Side effects from fenfluramine included transient fatigue and loose stools, which resolved with probiotics. Monitoring with annual echocardiograms has been normal.
Figure 1.
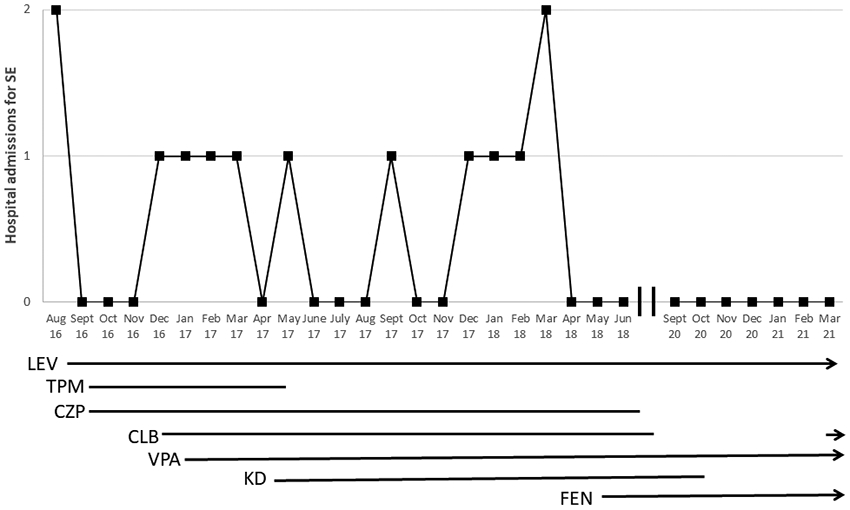
Per month hospital admissions for episodes of status epilepticus. As of February 2021, patient remained on LEV, VPA, FEN, and CLB. SE = status epilepticus; LEV = levetiracetam; TPM = topiramate; CZP = clonazepam; CLB = clobazam; VPA = valproic acid; KD = ketogenic diet; FEN = fenfluramine
We present a patient with DS whose initial convulsive seizures all resulted in SE, and whose diagnosis was corroborated by identification of a SCN1A variant within two months of presentation. He had a dramatic response to fenfluramine, with complete resolution of convulsive SE for over two years of follow-up. Early syndrome identification and genetic diagnosis allowed for rapid implementation of therapies that can be effective in DS [4], including fenfluramine under an individual IND initiated by our epilepsy genetics research team, through which a trial of fenfluramine was formally enrolling. Fenfluramine is thought to act primarily via the serotonergic system but also impacts excitatory/inhibitory balance via other pathways. Although the exact mechanism of anti-seizure activity is not fully understood, preclinical and clinical studies demonstrate efficacy in DS.[7]
We illustrate the potential for treatment decisions based on a genetic diagnosis to result in dramatically improved patient outcome. Even when patients do not meet criteria for enrollment in a clinical trial, close collaboration between clinical and research teams can lead to alternative opportunities, including use of the IND mechanism for access to experimental therapies. As genetically based epilepsy treatments emerge in the coming years, swift syndrome identification and genetic testing will yield precise diagnoses, guide treatment decisions, and improve patient outcomes.
Supplementary Material
Test Yourself.
-
What are some of the advantages of early genetic diagnosis in epilepsy?
Early diagnosis can help point towards gene-specific treatment approaches and help connect a patient with on-going disease-specific research trials.
-
If a patient does not qualify for a clinical trial, are there any alternative routes?
Yes; if a patient does not qualify for a clinical trial, access to an experimental drug may be obtained through the IND mechanism of the FDA in some cases.
-
What is a new therapy for DS that reduces episodes of SE?
Fenfluramine
Acknowledgement:
We thank Zogenix for their support in treating the described patient by IND and for review of this report.
References:
- 1.El Achkar CM, Olson HE, Poduri A, Pearl PL. The Genetics of the Epilepsies. Curr Neurol Neurosci Rep 2015;15:39. [DOI] [PubMed] [Google Scholar]
- 2.Perucca P, Perucca E. Identifying mutations in epilepsy genes: Impact on treatment selection. Epilepsy Res 2019;152:18–30. [DOI] [PubMed] [Google Scholar]
- 3.Striano P, Minassian BA. From Genetic Testing to Precision Medicine in Epilepsy. Neurotherapeutics 2020;17:609–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wirrell EC, Laux L, Donner E, Jette N, Knupp K, Meskis MA, et al. Optimizing the Diagnosis and Management of Dravet Syndrome: Recommendations From a North American Consensus Panel. Pediatr Neurol 2017;17:180–7. [DOI] [PubMed] [Google Scholar]
- 5.Lagae L, Sullivan J, Knupp K, Laux L, Polster T, Nikanorova M, et al. Fenfluramine hydrochloride for the treatment of seizures in Dravet syndrome: a randomised, double-blind, placebo-controlled trial. Lancet 2019;394:2243–54. [DOI] [PubMed] [Google Scholar]
- 6.Nabbout R, Mistry A, Zuberi S, Villeneuve N, Gil-Nagel A, Sanchez-Carpintero R, et al. Fenfluramine for Treatment-Resistant Seizures in Patients With Dravet Syndrome Receiving Stiripentol-Inclusive Regimens. JAMA Neurol 2020;77:300–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Balagura G, Cacciatore M, Grasso EA, Striano P, Verrotti A. Fenfluramine for the Treatment of Dravet Syndrome and Lennox–Gastaut Syndrome. CNS Drugs 2020;34:1001–7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.