

Abstract
Over the past decade, pathogenic variants in all members of the ASXL family of genes, ASXL1, ASXL2, and ASXL3, have been found to lead to clinically distinct but overlapping syndromes. Bohring-Opitz Syndrome (BOPS) was first described as a clinical syndrome and later found to be associated with pathogenic variants in ASXL1. This syndrome is characterized by developmental delay, microcephaly, characteristic facies, hypotonia, and feeding difficulties. Subsequently, pathogenic variants in ASXL2 were found to lead to Shashi-Pena Syndrome (SHAPNS) and in ASXL3 to lead to Bainbridge-Ropers Syndrome (BRPS). While SHAPNS and BRPS share many core features with BOPS, there also seem to be emerging clear differences. Here, we present 5 cases of BOPS, 1 case of SHAPNS, and 4 cases of BRPS. By adding our cohort to the limited number of previously published patients, we review the overlapping features of ASXL-related diseases that bind them together, while focusing on the characteristics that make each neurodevelopmental syndrome unique. This will assist in diagnosis of these overlapping conditions and allow clinicians to more comprehensively counsel affected families.
Keywords: ASXL, ASXL1, ASXL2, ASXL3, Bohring-Opitz Syndrome, Shashi-Pena Syndrome, Bainbridge-Ropers Syndrome
1. Introduction
Over the past 10 years, pathogenic variants in the additional sex combs-like (ASXL) genes have been found to lead to distinct neurodevelopmental syndromes (Bainbridge, et al., 2013; Shashi, et al., 2017; Hoischen, et al., 2011). The ASXL family is comprised of three genes in humans: ASXL1, ASXL2, and ASXL3. While these genes seem to have similar functions, and pathogenic variants cause similar clinical features, distinct features of each syndrome are starting to emerge. Here, we present 10 cases and focus on unique characteristics of each condition.
The ASXL family of genes are all involved in epigenetic and transcriptional regulation (reviewed in (Katoh, 2013)). ASXL1 and ASXL2 can bind to polycomb repressive complex 2 to promote histone methylation (Lai and Wang, 2013; Abdel-Wahab, et al., 2012). ASXL1, ASXL2, and ASXL3 promote histone deubiquitination through interaction with BRCA-1-associated protein 1 (Sahtoe, et al., 2016; Daou, et al., 2015; Srivastava, A., et al., 2016). In addition, ASXL2 has been found to promote histone deacetylation (Li, et al., 2017). Somatic pathogenic variants in ASXL1 and ASXL2 can lead to hematological cancers (Wang, et al., 2014; Li, et al., 2017), but this is less common with ASXL3 variants (Oak and Ohgami, 2017; Duployez, et al., 2016). While there is significant interest in the oncological role of the ASXL genes, this work focuses on germ-line pathogenic variants that have been recently described to lead to distinct neurodevelopmental disorders.
Germ-line pathogenic variants in ASXL1 are associated with Bohring-Opitz Syndrome (BOPS). BOPS was defined in 1999 as a distinct clinical syndrome characterized by microcephaly, frontal bulging/trigonocephaly, glabellar nevus flammeus, upslanted palpebral fissures, prominent eyes (exophthalmos), cleft lip and palate, thick hair, feeding difficulties, and flexion deformities of the upper limbs (Bohring, et al., 1999). The flexion deformities lead to the “Bohring-Opitz Syndrome posture” that is clinically recognizable. Neurological manifestations include completely penetrant intellectual disability, as well as epilepsy, contractures, and bulbar dysfunction to varying degrees (Bohring, et al., 2006). Individuals with BOPS may also present with agenesis of the corpus callosum, Dandy-Walker malformation, and ventriculomegaly (Bohring, et al., 2006). In 2011, BOPS was found to be due to de novo pathogenic variants in ASXL1 in 7 of 13 tested individuals, suggesting that clinically-recognized BOPS is not a monogenic disease (Hoischen, et al., 2011), though other monogenic causes have yet to be identified.
The growing accessibility of whole-exome sequencing led to the discovery that pathogenic variants in ASXL2 lead to a syndrome that manifests as macrocephaly, prominent eyes, arched eyebrows, hypertelorism, glabellar nevus flammeus, neonatal feeding difficulties, hypotonia, variable intellectual disability, and possible hypoglycemia, now known as Shashi-Pena Syndrome (SHAPNS) (Shashi, et al., 2017). MR imaging of the brain demonstrates cerebral atrophy (Shashi, et al., 2017). While developmental delay was universal, the degree of intellectual disability later was variable, and growth parameters were not markedly affected (Shashi, et al., 2017).
Pathogenic variants in the final member of the ASXL family, ASXL3, lead to Bainbridge-Ropers Syndrome (BRPS). BRPS is characterized by severe feeding difficulties, severe developmental delay and intellectual disability, poor expressive speech development, microcephaly, arched eyebrows, prominent forehead, hypertelorism with downslanted palpebral fissures, and hypotonia (Bainbridge, et al., 2013; Balasubramanian, et al., 2017). These children commonly have autistic features with hand-flapping and rocking behaviors and can sometimes be aggressive (Balasubramanian, et al., 2017). Approximately one-third of these individuals also experience seizures (Myers, et al., 2018). In contrast to BOPS, both BRPS and SHAPNS were only recognized as clinical syndromes after identification of pathogenic variants.
Here, we present 10 individuals with pathogenic variants in the ASXL family of genes (9 of these individuals are previously unreported). The clinical phenotypes of these individuals are consistent with the previously described phenotypes typical of each syndrome. Finally, we delve into the phenotypic spectrum of ASXL mutations with a focus on unifying and distinguishing features.
2. Case Reports
This study involved a retrospective review of the electronic medical record for the 10 presented individuals (Table 1). The study was reviewed by the Institutional Review Board at the Children’s Hospital of Philadelphia and granted an exemption.
Table 1:
Clinical features of 5 individuals with ASXL1 pathogenic variants, 1 individual with ASXL2 pathogenic variant, and 4 individuals with ASXL3 pathogenic variants
| Individual/Gene | Individual 1 ASXL1 |
Individual 2 ASXL1 |
Individual 3 ASXL1 |
Individual 4 ASXL1 |
Individual 5 ASXL1 |
Individual 6 ASXL2 |
Individual 7 ASXL3 |
Individual 8 ASXL3 |
Individual 9 ASXL3 |
Individual 10 ASXL3 |
|---|---|---|---|---|---|---|---|---|---|---|
| Variant | c.1867C>T (p.Q623X) | c.1517_1518del GA | c.4060G>T (p.E1354X) | c.2893 C>T (p.R965X) | c.2332C>T (p.Q788X) | c.4228T>G (p.C1410G) | c.4322C>G (p.S1441X) | c.1895dupC (p.Q633Tfs*14) | c. 3349C>T (p.R1117X) | c. 1990C>T (p.Q664X) |
| Inheritance | de novo | de novo | de novo | Mother in mosaic state | unknown | de novo | de novo | de novo | de novo | de novo |
| Age / Sex | 5y / Male | 13y / Male | 12y / Female | 5y / Female | 5y / Female | 13y / Male | 12y / Female | 6y / Male | 13y / Female | 16y / Male |
| Age at Diagnosis | 19 months | 7 years | 6 years | 12 months | 3 months | 7 years | 8 years | 3 years | 9 years | 12 years |
| Growth | ||||||||||
| Microcephaly | − (66th %ile at 5 years) | + (<0.01 %ile at 12 years) | − (57th %ile at 6 years) | − (3.4 %ile at 26 months) | − (1.5 %ile at 3 years) | − | − (55th %ile at 32 months) | + (0.23 %ile at 24 months) | + (1.6 %ile at 25 months) | − (34th %ile at 12 years) |
| Macrocephaly | − | − | − | − | − | + (50th %ile at 2 months, 90th %ile at 10 years when height was at 25th %ile) | − | − | − | − |
| Failure-to-thrive | − (17th %ile at 5 years) | + (<0.01 %ile at 12 years) | + (0.14 %ile at 5 years) | + (0.53 %ile at 26 months) | + (<0.01 %ile at 3 years) | (90th %ile at 8 years to 7th %ile at 12 years. Improvement to 30th %ile at 13 years) | + (0.55 %ile at 31 months) | + (15th %ile at 24 months) | + (<0.01 %ile at 25 months) | + (<0.01 %ile at 12 years) |
| Facial Features | ||||||||||
| Frontal prominence | + | + | + | |||||||
| Nevus flammeus | − | + (glabella) | + (forehead) | + | ||||||
| Ptosis | − | + | + | |||||||
| Upslanted eyes | − | + | − | + | − | − | ||||
| Downslanted eyes | + | − | + | − | + | + | ||||
| Eyes, other | Epicanthal folds | − | − | Prominent, hypertelorism | − | Prominent, hypertelorism, fullness of upper eyelid, prominent eyelashes | ptosis | hypertelorism | − | Ptosis, strabismus |
| Ear malformation | low-set ears | low set, posteriorly rotated | Squared superior portion helix, large lower lobes | posteriorly rotated ears, anteverted nares | ||||||
| Nose malformation | up-turned nasal tip | depressed nasal bridge, upturned nasal tip | anteverted nares, depressed nasal bridge | Low columella | ||||||
| Other facial features | High-arched palate, widely spaced teeth | Trigonocephaly, bitemporal narrowing, synophrys, high-arched palate, micrognathia | − | high palate | High-arched palate | Scaphocephaly, multiple areas of frontal upsweep hair with two posterior hair whorls, arched eyebrows, high palate, gingival overgrowth | − | Prominent forehead | Synophrys, high palate | − |
| Constitutional Features | ||||||||||
| Thick hair | + | + | ||||||||
| “BOPS posture” | + | + | ||||||||
| Other constitutional features | single palmar crease, high-arched feet | Hirsutism, widely spaced nipples, 5th digit brachydactyly | Slender fingers | − | Hirsutism, single palmar crease, contracture of left 3rd proximal interphalangeal joint, and left foot metatarsus adductus | Bifid uvula, toe 2 overriding 3 bilaterally, laterally deviated distal toes | Inverted nipples | − | − | − |
| Muscle tone | ||||||||||
| Hypotonia | + (severe) | + (severe) | + (axial) | + (before hypoxic event) | + | + (axial) | + | + (late) | + | + |
| Hypertonia | − | − | + (appendicular) | + (after hypoxic event) | − | + (appendicular) | − | + (early) | − | − |
| Seizures | ||||||||||
| Epilepsy | − | + | + | + | + | + | + | − | − | − |
| Medications | − | Levetiracetam | valproic acid | oxcarbazepine | Levetiracetam, phenobarbital | Lacosamide, phenobarbital | levetiracetam | − | − | − |
| Neurological – Other History | ||||||||||
| MRI Imaging | Normal (1 year of age) | Agenesis of the corpus callosum, cerebellar vermis hypoplasia, delayed white matter development (18 months of age) | dysmorphic appearance of the craniovertebral junction (4 months of age) | Periventricular leukomalacia, paucity of white matter, findings consistent with hypoxic-ischemic injury | short corpus callosum and possible Blake’s pouch cyst (fetal) | Normal (5.5 years of age) | normal | Normal (6 months of age) | thin corpus callosum, absent rostrum, and small frontal lobes | normal |
| Neurological - Other | cortical visual impairment | Nonverbal | Nonverbal | Optic atrophy, Pigmentary retinal dystrophy, hearing deficiency, excessive sleepiness, autonomic dysfunction | Exotropia s/p surgical correction, able to communicate with assistive device | Speech regression | diffusely decreased strength, strabismus s/p surgical correction | |||
| Developmental Delay/Intellectual Disability | ||||||||||
| Motor delay | + | + | + | + | + | + | + | + | + | + |
| Speech delay | + | + | + | + | + | + | + | + | + | + |
| Intellectual disability | + (profound) | + | + | + (profound) | + | + | + | + | + | |
| Gastrointestinal | ||||||||||
| Feeding difficulties | + | + | + | + | + | + | + | + | + | + |
| GERD | + | + | + | + | + | + | + | + | ||
| Feeding tube | G-tube with Nissen fundoplication | G-tube with Nissen fundoplication | G-tube with Nissen fundoplication | G-tube | GJ tube-dependence and TPN dependence | G-tube | G-tube with Nissen fundoplication | G-tube | ||
| Other, Gastrointestinal | hiatal hernia | chronic pancreatitis, microvesicular steatohepatitis, GI dysmotility | Alpha-1-antitrypsin deficiency | |||||||
| Endocrinology | ||||||||||
| Precocious puberty | − | + | + | + | − | − | + | − | ||
| Glucose | − | − | − | − | − | Ketotic hypoglycemia | − | Ketotic hypoglycemia | Hypoglycemia | |
| Other, Endocrinology | − | ectopic pituitary | ||||||||
| Respiratory | ||||||||||
| Obstructive sleep apnea | + | + (BiPap) | + | + (BiPap) | + (BiPap) | − | + | − | ||
| Surgical interventions | adenoidectomy | tonsillectomy and adenoidectomy | Supraglottoplasty, tracheostomy with ventilator support | − | tonsillectomy and adenoidectomy | − | ||||
| Other, Respiratory | breath-holding spells | difficult airway | Subglottic stenosis, choanal atresia, laryngomalacia | − | Laryngotracheomalacia | − | ||||
| Cardiac | Stills murmur, thickened aortic root | Left superior vena cava, Left ventricular hypertrophy | Noncompaction cardiomyopathy (attributed to TTN mutation) | Atrial septal defect | Bradycardia | Bradycardia, ventricular ectopy | − | Murmur | − | − |
| Musculoskeletal | − | Scoliosis (> 45° curve), brachydactyly | Scoliosis, abnormality of the craniovertebral junction | − | contracture of left 3rd proximal interphalangeal joint, left foot metatarsus adductus | Rhabdomyolysis, hip fracture, foot fracture | − | − | − | − |
| Genitourinary | Penile adhesions | Hypospadias, undescended testicle | Incontinence | − | − | Neurogenic bladder requiring vesicostomy | − | − | − | Enuresis |
| Sleep and Behavior | ||||||||||
| Irregular sleep/wake cycle | + | + | + | + | ||||||
| Excessive sleepiness | + | + | ||||||||
| Autism | + | + | + | + | ||||||
| Aggression | (possible) | + | ||||||||
| Hand flapping | + | + | ||||||||
| Other, behavior | Behavior disturbance | ADHD, pervasive developmental delay | Obsessive compulsive disorder | |||||||
2.1. ASXL1
2.1.1. Individual 1
Individual 1 is a male born full-term via vaginal delivery after a high-risk pregnancy requiring bedrest for the first 4.5 months of the pregnancy due to an enlarged ovarian cyst. He weighed between the 25-50th percentile at birth. His development was remarkable for gross motor delay in the setting of severe hypotonia, and physical therapy was initiated within the first year of life. Chromosomal microarray and Prader Willi methylation testing were negative. At 19 months of age, whole exome sequencing (WES) revealed a novel, heterozygous, de novo pathogenic variant in ASXL1 (NM_015338.5:c.1867C>T; p.Q623X). By 3 years of age, he was gaining weight but being managed for sleep apnea, breath-holding spells, behavioral disturbances, and syncopal episodes of unknown etiology. EEG revealed no seizures, but bifrontal spikes were noted to be present. He was able to speak in full sentences, count to 10, and recognize colors and animals. He was receiving physical, occupational, and speech therapy.
2.1.2. Individual 2
Individual 2 is a male born at 39 weeks’ gestation, during which fetal ultrasound revealed agenesis of the corpus callosum. After birth, he had feeding difficulties and was unable to suck and swallow. A G-tube was placed at 3 months of age, and obstructive sleep apnea was identified, requiring continuous positive airway pressure during sleep. He had failure-to-thrive with weight, height, and head circumference all <5 percentile. EEG demonstrated episodes of generalized discharges and subclinical seizures, so he was started on levetiracetam. He had precocious puberty, and further clinical details related to this were not available. At 7 years of age, WES demonstrated a novel, de novo, pathogenic frameshift variant in ASXL1 (NM_015338.5:c.1517_1518delGA). By 10 years of age, his respiratory status was worsening leading to a nearly continuous BiPap requirement. At 12 years of age he was not able to speak any words but was able to make choices with an eye gaze system at school. He was not able to use his hands for pointing or gestures.
2.1.3. Individual 3
Individual 3 is a female born with failure-to-thrive and severe gastroesophageal reflux disease, requiring G-tube with fundoplication by 1 year of age. She had global developmental delay. She experienced her first lifetime seizure at 6 years of age and was started on levetiracetam. She also had sleep difficulties managed with melatonin. Karyotype and chromosomal microarray were normal. At 7 years of age she was noted to have thelarche, pubarche, and body odor and diagnosed with central precocious puberty. WES by 10 years of age revealed a novel, de novo pathogenic variant in ASXL1 (NM_015338.5:c.4060G>T;p.E1354X), as well as a variant of unclear significance in TTN associated with noncompaction type cardiomyopathy. The TTN variant (c.66716delT;p.L22239X) was maternally inherited, but whether the mother also has cardiac disease remains unclear. At last evaluation at 12 years of age, she continued to have breakthrough seizures requiring transition to valproic acid. She was nonverbal but able to indicate her wishes by shaking her head ‘yes’ and ‘no’. She was able to walk with leg braces. Regarding her fine motor skills, she was not able to use utensils but able to eat with her hands.
2.1.4. Individual 4
Individual 4 is a female born prematurely at 34 weeks’ gestation after a pregnancy complicated by intrauterine growth retardation (IUGR). After birth, her early course was remarkable for gross global developmental delay, respiratory insufficiency due to laryngomalacia, and frequent emesis leading to growth restriction and eventual G-tube placement with Nissen fundoplication. By 5 months of age supraglottoplasty was performed. This did not sufficiently ameliorate her respiratory insufficiency, so tracheostomy with continuous ventilatory support was necessary by 8 months of age. Her examination was remarkable for microcephaly, nevus flammeus over glabella, prominent and upslanted eyes, hypertelorism, anteverted nares, depressed nasal bridge, choanal atresia, high palate, and “BOPS posture” (Figure 1a–b). In addition, pubic hair was noted at birth, and thelarche had occurred prior to 12 months of age. At 12 months of age, she experienced febrile status epilepticus secondary to sepsis with resulting hypoxic-ischemic encephalopathy. Expedited whole exome sequencing at this time revealed a recuring (Magini, et al., 2012), pathogenic variant in ASXL1 (NM_015338.5:c.2893 C>T;p.R965X). This variant was also noted in the mother, though in a mosaic state in blood with no overt features of BOPS. For her epilepsy, she was initially maintained on levetiracetam, but then transitioned to oxcarbazepine monotherapy for lack of efficacy. By 5 years of age, she was non-verbal, non-mobile, and unable to use her hands. This patient was previously reported at 3 years of age as the first description of BOPS inherited from an unaffected, germline mosaic parent (Bedoukian, et al., 2018).
Figure 1.

Representative images of individuals with ASXL1, ASXL2, and ASXL3 pathogenic variants. (a-b) Individual 4 with pathogenic variant in ASXL1 demonstrating frontal prominence, glabellar nevus flammeus, upslanted eyes, hypertelorism, clenched hands with upper extremity hypertonia, and with tracheostomy. (c-e) Individual 6 with pathogenic variant in ASXL2 demonstrating long face with narrow biparietal diameter, fullness of the superior eyelids, upslanted palpebral fissures, hypertelorism, squared superior portion helix with large lower earlobes, and low columella at 3.5 years of age (c) and 6.5 years of age (d-e). (f-h) Individual 7 with pathogenic variant in ASXL3 demonstrating ptosis and downslanted palpebral fissures.
2.1.5. Individual 5
Individual 5 is a female born at 38 weeks’ gestation after a pregnancy complicated by IUGR. After birth she was transferred to the Intensive Care Unit for management of feeding and respiratory difficulties. She was discharged home by day-of-life 10. By 2 months of age she was having significant feeding difficulty requiring nasogastric tube placement, axial and appendicular hypotonia, and purposeless eye movements. By 3 months of age, this individual was found to have a recurring (Hoischen, et al., 2011), pathogenic variant in the ASXL1 gene (NM_015338.5:c.2332C>T; p.Gln788X) of unknown inheritance. She presented with epilepsy at 6 months of age and started on levetiracetam and phenobarbital. Seizures were subclinical and focal, originating from the right temporal lobe. She required frequent hospitalizations for recurrent emesis leading to dehydration before 18 months of age. She required close follow up with Gastroenterology due to an inability to tolerate continuous G-tube feeds. She was non-verbal with no purposeful movements. She developed severe obstructive sleep apnea requiring BiPap when sleeping.
2.2. ASXL2
2.2.1. Individual 6
Individual 6 is a male born at 32 weeks’ gestation following placental abruption after maternal fever at 30 weeks’ gestation. Antenatal ultrasounds were reportedly normal. He spent the first 5 weeks of life in the Intensive Care Unit for respiratory distress, apnea, and hyperbilirubinemia. By 2 months of age, he was noted to have clenched fists with cortical thumbing and episodes of leg shaking concerning for seizures, for which he was started on topiramate and subsequently changed to phenobarbital and lacosamide for treatment of partial epilepsy with secondary generalization. He had normal early growth (length and weight) with progressive fall in BMI. Dysmorphic examination at 10 years, 8 months old was remarkable for mild relative acquired macrocephaly, long face with narrow biparietal diameter, tight sublingual frenulum limiting full tongue extrusion, small vertical chin crease, as well as other details listed in Table 1 (Figure 1c–e). He had global developmental delay, and excessive sleepiness that was partially attributed to phenobarbital. He had truncal weakness and achieved independent walking by 22 months of age. He experienced repeated episodes of ketotic hypoglycemia (lowest glucose 49 mg/dL, confirmed by diagnostic fast with nadir glucose 45 mg/dL and beta-hydroxybutyrate 2.2 mM) leading to repeated hospitalizations. He eventually required gastro-jejunal feeding with total parental nutrition (TPN) supplementation for severe gastrointestinal dysmotility with chronic intestinal pseudo-obstruction. He had kyphosis, and was prescribed IV bisphosphonate therapy for multiple pathological fractures, including a hip fracture at 9 years old after which he could no longer walk (dual-energy x-ray absorptiometry scans did not disclose areal bone mineral density Z-scores <−2.0, but assessments were limited based on instrumentation) as well as a foot fracture. WES performed on a clinical diagnostic basis on his nuclear family in blood at 7 years of age demonstrated he had a de novo, novel, likely pathogenic variant in ASXL2 (NM_018263.4:c.4228T>G;p.C1410G). He was also found to have mild intellectual disability, neurodevelopmental regression with lost ability to read and write, anxiety disorder, attention deficit hyperactivity disorder, and pervasive developmental delay. Apart from these clinical problems attributable to ASXL2-related disease, he also exhibited symptoms of a familial progressive myopathy that also manifest in his mother and younger sister with rhabdomyolysis; progressive spasticity, clonus, progressive resting tremor, and ataxia; chronic respiratory failure (requiring nighttime BiPap); chronic pain; pigmentary retinopathy; mild left optic atrophy identified by optical coherence tomography; autonomic dysfunction characterized by tachycardia, sweating, flushing, and body temperature fluctuations; central adrenal insufficiency treated with glucocorticoid replacement; hepatic microvesicular steatosis; chronic pancreatitis; sleep disorder; immune dysfunction; sideroblastic anemia; bone marrow failure; iron deficiency anemia; and platelet dysfunction. These additional features are not typical of ASXL2-related disease and as many of them also occur with variable severity in his affected mother and sister who do not have the ASXL2 variant appear likely attributable to a second, yet-to be identified, maternally-inherited genetic etiology. Further clinical information is provided in Supplementary Material.
2.3. ASXL3
2.3.1. Individual 7
Individual 7 is a female born full-term after a pregnancy remarkable for oligohydramnios. By 6 months of age, she was noted to have strabismus, axial hypotonia, and failure-to-thrive. Exotropia was later surgically corrected at 1 year of age. Development was grossly delayed; she walked at 34 months of age and was non-verbal at 4 years of age. Physical examination was remarkable for ptosis, downslanted palpebral fissures, bilateral inverted nipples, axial hypotonia, and hand flapping stereotypies (Figure 1f–h). Precocious puberty was also noted, with pubarche occurring at age 8. Thelarche was found to be normal at age 9.5. WES was sent at 8 years of age, revealing a novel, de novo pathogenic variant in the ASXL3 gene (NM_030632.1:c.4322C>G;p.S1441X). At this time, patient was making slow developmental progress and able to communicate with computer software in simple sentences. Seizures began at age 11 characterized by generalized tonic-clonic movements. By this time, she knew all of her letters and numbers, was able to read at a first-grade level, but continued to be non-verbal. She had no developmental regression and was able to walk and run independently. She had ongoing physical, occupational, and speech therapy.
2.3.2. Individual 8
Individual 8 is a male born full-term after an uncomplicated pregnancy. After birth, he began to have difficulty feeding and developed failure-to-thrive, and by 4 months of age, caretakers felt that he was stiff. He then experienced global developmental delay; he started rolling at 10 months of age, and was able to sit by 16 months of age. He developed stereotypies of hand twirling in front of his face and was diagnosed with autism by 2 years of age. By 3 years of age patient was referred for WES revealing a novel, de novo pathogenic variant in the ASXL3 gene (NM_030632.1:c.1895dupC;p.Q633Tfs*14). By 6 years of age, he was non-verbal with limited communication skills. He was unable to feed himself and was placed in special education at school.
2.3.3. Individual 9
Individual 9 is a female born after 39 weeks’ gestation. On the first day of life, she had intermittent episodes of duskiness, found to be due to laryngotracheomalacia. By 1 year of life she was noted to have global developmental delay, microcephaly, and failure to thrive. She was able to roll, but not sit independently or vocalize. In the setting of presumed gastroenteritis at age 3, she was found to have a ketotic hypoglycemia to blood glucose of 42. Her blood glucoses were then routinely monitored without recurrent episodes. After fasting for 20 hours, she was also able to mount a normal counter-regulatory hormonal response. Patient was able to walk with the assistance of a walker and say 1 word by 4 years of age. By age 8 routine blood glucose monitoring was discontinued. By 9 years of age WES was performed revealing a recurring (Zhang, et al., 2018), de novo pathogenic variant in ASXL3 (NM_030632.1:c. 3349C>T;p.R1117X). At 10 years of age psychological testing at this time revealed motors skills at 28-month level, communication skills less than 14-month level, and language at 27-month level. By age 13, she had a single unprovoked seizure and was not placed on anti-seizure medications. She continued to be non-verbal but was able to use an electronic speech device. She was able to walk independently.
2.3.4. Individual 10
Individual 10 is a male born at full-term after an uncomplicated pregnancy and delivery. He was noted to have global developmental delay with failure to thrive. Between the age of 2 to 3, patient presented to the Emergency Room twice with altered mental status and was found to have blood glucoses in the 30s, once with no ketones in the urine and the other with small ketones (15 mg/dl) in the urine. His blood glucoses were then monitored at home without recurrent episodes. Patient began walking independently at 3 years of age. By 6 years of age, he knew about 50 words and was able to create two word phrases. WES was obtained by 12 years of age revealing a recurring (Srivastava, S., et al., 2014), de novo pathogenic variant in ASXL3 (NM_030632.1:c.1990C>T;p.Q664X). At age 12 he had moderate intellectual disability; he was able to speak in short phrases, but most speech was not intelligible outside of the immediate family. He was able to read 3-word sentences. His hypotonia improved over time.
3. Discussion
The patients presented here provide further confirmation of the previously described phenotypes attributed to pathogenic variants in ASXL1, ASXL2, and ASXL3, respectively. As the numbers of individuals identified with pathogenic variants within each of these genes grows, the degree of overlap of the clinical phenotypes has become clearer. As depicted in Figure 2, children with pathogenic variants in ASXL1, ASXL2, and ASXL3 share several features including characteristic facial features (arched eyebrows and hypertelorism), feeding difficulties most prominent in early life, hypotonia, and developmental delay. Intellectual disability and epilepsy are also quite common, to variable degrees.
Figure 2.
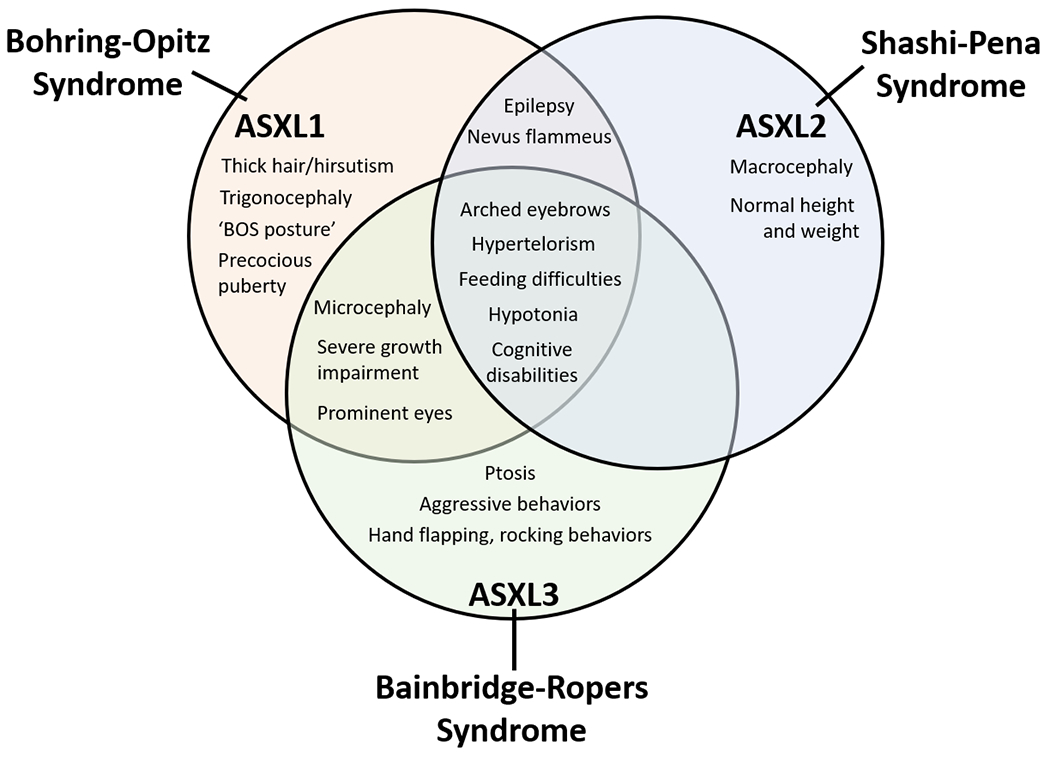
Bohring-Opitz Syndrome, Shashi-Pena Syndrome, and Bainbridge-Ropers Syndrome have an overlapping phenotype but can also be distinguished by unique features.
To better understand ASXL-related disease, we reviewed the history of these disorders and their reported clinical overlap. When ASXL3 pathogenic variants were first described to lead to Bainbridge-Ropers Syndrome (BRPS), overlap was noted with Bohring-Opitz Syndrome (BOPS) of non-specific features including developmental delay, feeding difficulties, and arched eyebrows, but without findings of trigonocephaly and prominent eyes (Bainbridge, et al., 2013). These similarities were attributed to overlapping regions of expression between ASXL1 and ASXL3 (Bainbridge, et al., 2013). Here, we find that in addition to these core features, patients with ASXL1 or ASXL3 pathogenic variants also are more likely to have microcephaly, severe growth impairment, and intellectual disability (Figure 2). Those with ASXL1 pathogenic variants are more likely to have the “BOPS posture” (2/5 in our cohort). Those with ASXL3 pathogenic variants tend to have a higher likelihood of having ptosis (2/4 in our cohort) and exhibiting hand flapping and aggressive behaviors (3/4 in out cohort). In our cohort, 4/5 patients with ASXL1 pathogenic variants (individuals 2-5) had a diagnosis of epilepsy and were on anti-seizure medications as compared to 1/4 of those with ASXL3 pathogenic variants (Individual 9). This is consistent with previous reports suggesting that epilepsy is not as common in individuals with ASXL3 pathogenic variants as compared to ASXL1 (Kuechler, et al., 2017; Srivastava, A., et al., 2016; Hori, et al., 2016; Myers, et al., 2018).
Children with ASXL2 pathogenic variants leading to Shashi-Pena Syndrome (SHAPNS) share multiple core features that characterize the ASXL-family of diseases including arched eyebrows, prominent eyes, hypertelorism, feeding difficulties, and developmental delay (Shashi, et al., 2017). However, these patients can be distinguished by macrocephaly, essentially normal height and weight parameters, and less severe cognitive impairment (Figure 2) as observed in our patient (individual 6). Like children with BOPS, there is an increased likelihood of having seizures (5/6 of previously published patients and 1/1 in our cohort). Our patient (Individual 6) with SHAPNS was also found to have multiple endocrine imbalances including ketotic hypoglycemia, pathological fractures, and adrenal insufficiency. Hypoglycemia has been previously described in 3/6 individuals with ASXL2 pathogenic variants, though it seemed to be persistent in only 2/6 individuals (Shashi, et al., 2017). In our patient, the etiology of the hypoglycemia was not clear, but given unexpectedly low extent of ketosis for the degree of hypoglycemia, non-specific impaired regulation of insulin signaling may contribute. His ketotic hypoglycemia responded to continuous GJ feeds that were ultimately supplemented then replaced with TPN. Additionally, 2/6 of previously reported individuals (Shashi, et al., 2017) with pathogenic variants in ASXL2 had bone disease manifest as decreased bone density and multiple fractures, similar to our patient. However, his bone health impairment was more severe, complicated by multiple fractures and requiring ongoing IV bisphosphonate therapy (Individual 6). Mouse models support a role for ASXL2 in bone metabolism, as mutation in Asxl2 leads to decreased osteoclast development and lower bone mineral density (Farber, et al., 2011; Izawa, et al., 2015). Despite the known association between pathogenic variants in ASXL2 and myeloid malignancies in mice and humans (Micol, et al., 2017; Li, et al., 2017), germ-line pathogenic variants in ASXL2 have not been found to date to lead to cancer in humans although it is possible this may change as the cohort of children with known ASXL2 disease grow older. Interestingly, our ASXL2 proband also demonstrated multiple clinical problems not reported in other published cases, including neurodevelopmental regression, myopathy, severe GI dysmotility with chronic pseudoobstruction, resting tremor, spasticity and clonus, contractures, progressive ataxia, chronic pain, adrenal insufficiency, pancreatic dysfunction, autonomic dysfunction, rhabdomyolysis, pigmentary retinopathy, optic atrophy, bone marrow and platelet dysfunction, iron deficiency anemia, immune dysfunction, and microvesicular steatohepatitis. Microvesicular steatohepatitis identified at 25 month of age was attributed to his TPN-dependence. However, a separate genetic cause that has not yet been identified is strongly suspected to underlie these more complex features, which have not been reported in other individuals with SHAPNS but are seen to variable degrees in his maternal family members. In contrast, his ASXL2 pathogenic variant is de novo, and explains his unique developmental disability, partial epilepsy, osteopenia, relative macrocephaly, and hypoglycemia that are not present in his family members.
While hypoglycemia does not seem to be a common feature shared by patients with BRPS (Bainbridge, et al., 2013; Kuechler, et al., 2017; Srivastava, A., et al., 2016; Hori, et al., 2016; Balasubramanian, et al., 2017), 2/4 individuals we present here (Individual 9 and 10) experienced hypoglycemia early in life. Individual 9 had a ketotic hypoglycemia in the setting of likely dehydration, which most likely was an appropriate physiological response. Individual 10 however had an inappropriate ketotic response to profound hypoglycemia early in life. The etiology of this was never confirmed and seems to have improved with age as the patient is now able to fast up to 14 hours overnight without hypoglycemia. Unlike Individual 6 with the pathogenic variant in ASXL2, the hypoglycemia in our two patients with pathogenic variants in ASXL3 improved with age. There is a report of a single child with BRPS with persistent hypoglycemia who presented on day-of-life 1 with hypoglycemia after being born to an insulin-dependent diabetic mother (Dinwiddie, et al., 2013). This child had 2 family members with hypoglycemia in childhood that evolved into insulin-dependent diabetes, while the patient herself had hypoglycemic seizures early in life (Dinwiddie, et al., 2013). This child was found to have a pathogenic variant in ABCC8 (ATP-Binding Cassette, Subfamily, Member 8) gene, thought to be consistent with a diagnosis of Familial Hyperinsulinemic Hypoglycemia Type 1 and separate from her pathogenic variant in ASXL3 (Dinwiddie, et al., 2013). While ASXL2 is thought to play a role in glucose regulation via insulin sensitivity (Izawa, et al., 2015), what role, if any, ASXL3 plays in glucose metabolism remains unknown.
Further dysfunction along the neuroendocrine axis occurs in individuals with BOPS and pathogenic variants in ASXL1. In our newly reported cohort, 3/5 children (Individuals 2, 3, and 4) with pathogenic variants in ASXL1 had premature onset of puberty. Individual 4 in particular was found to have pubic hair at birth and thelarche before 12 months of age. This is consistent with previous reports of 2 children with BOPS due to pathogenic variants in ASXL1, one of whom developed pubarche and thelarche by 10 years of age, and the other developing pubarche at seven months of age and thelarche at seven years of age (Russell, et al., 2015). In addition, 1/4 of our patients with pathogenic variants in ASXL3 (Individual 7) had premature pubarche. Precocious puberty was not initially described to be a cardinal feature of BOPS; however, since the identification that approximately 50% of those with BOPS have pathogenic variants in ASXL1 (Hoischen, et al., 2011), it remains possible that precocious puberty may be more likely in individuals with pathogenic variants in ASXL1 as opposed to those clinically diagnosed with BOPS. The mechanism of premature onset of puberty in this population is unclear, but given that ASXL1 is an important epigenetic and transcriptional regulator, mutation of this gene likely has pleotropic effects.
As summarized in Figure 2, pathogenic variants in the 3 members of the ASXL family of genes lead to distinct but overlapping phenotypes. While BOPS was initially described as a clinical syndrome with about 50% of cases due to a pathogenic variant in ASXL1, SHAPNS and BRPS are primarily defined by their underlying genetic etiologies. Over time, it is likely that further published cases of BOPS will be slanted towards those patients with known pathogenic variants in ASXL1. Comparing the patients with BOPS who do and do not have known pathogenic variants in ASXL1 could lead to the identification of subtle differences in phenotype. The BOPS cohort of patients who do not have pathogenic variants in ASXL1 may have another monogenic etiology yet to be discovered, or more simply, these patients may still have mutations in ASXL1 that are more difficult to identify, e.g. deep intronic mutations or mutations in regulatory elements. In this era of rapidly improving genetic technologies, the ASXL family of disorders continues highlight the distinctions between phenotype-driven and genotype-driven diagnoses.
Supplementary Material
Acknowledgements
This work was supported in part by the Research and Cure of Refractory Epilepsy fund, the Children’s Hospital of Philadelphia Mitochondrial Disease Frontier Program, and the National Institutes of Health, National Institutes of Neurological Disease and Stroke (R01NS082761 to EDM). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Conflict of Interest
The authors declare that they have no competing interests.
References
- Abdel-Wahab O, Adli M, LaFave LM, Gao J, Hricik T, Shih AH … Levine RL. (2012). ASXL1 mutations promote myeloid transformation through loss of PRC2-mediated gene repression. Cancer cell, 22, 180–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bainbridge MN, Hu H, Muzny DM, Musante L, Lupski JR, Graham BH … Ropers HH. (2013). De novo truncating mutations in ASXL3 are associated with a novel clinical phenotype with similarities to Bohring-Opitz syndrome. Genome medicine, 5, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balasubramanian M, Willoughby J, Fry AE, Weber A, Firth HV, Deshpande C … Tomkins S. (2017). Delineating the phenotypic spectrum of Bainbridge-Ropers syndrome: 12 new patients with de novo, heterozygous, loss-of-function mutations in ASXL3 and review of published literature. Journal of medical genetics, 54, 537–543. [DOI] [PubMed] [Google Scholar]
- Bedoukian E, Copenheaver D, Bale S, Deardorff M. (2018). Bohring-Opitz syndrome caused by an ASXL1 mutation inherited from a germline mosaic mother. American journal of medical genetics.Part A, 176, 1249–1252. [DOI] [PubMed] [Google Scholar]
- Bohring A, Oudesluijs GG, Grange DK, Zampino G, Thierry P. (2006). New cases of Bohring-Opitz syndrome, update, and critical review of the literature. American journal of medical genetics.Part A, 140, 1257–1263. [DOI] [PubMed] [Google Scholar]
- Bohring A, Silengo M, Lerone M, Superneau DW, Spaich C, Braddock SR … Opitz JM. (1999). Severe end of Opitz trigonocephaly (C) syndrome or new syndrome? American Journal of Medical Genetics, 85, 438–446. [DOI] [PubMed] [Google Scholar]
- Daou S, Hammond-Martel I, Mashtalir N, Barbour H, Gagnon J, Iannantuono NV … Affar el B. (2015). The BAP1/ASXL2 Histone H2A Deubiquitinase Complex Regulates Cell Proliferation and Is Disrupted in Cancer. The Journal of biological chemistry, 290, 28643–28663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinwiddie DL, Soden SE, Saunders CJ, Miller NA, Farrow EG, Smith LD … Kingsmore SF. (2013). De novo frameshift mutation in ASXL3 in a patient with global developmental delay, microcephaly, and craniofacial anomalies. BMC medical genomics, 6, 32–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duployez N, Micol JB, Boissel N, Petit A, Geffroy S, Bucci M … Preudhomme C. (2016). Unlike ASXL1 and ASXL2 mutations, ASXL3 mutations are rare events in acute myeloid leukemia with t(8;21). Leukemia & lymphoma, 57, 199–200. [DOI] [PubMed] [Google Scholar]
- Farber CR, Bennett BJ, Orozco L, Zou W, Lira A, Kostem E … Lusis AJ. (2011). Mouse genome-wide association and systems genetics identify Asxl2 as a regulator of bone mineral density and osteoclastogenesis. PLoS genetics, 7, e1002038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoischen A, van Bon BW, Rodriguez-Santiago B, Gilissen C, Vissers LE, de Vries P … de Vries BB. (2011). De novo nonsense mutations in ASXL1 cause Bohring-Opitz syndrome. Nature genetics, 43, 729–731. [DOI] [PubMed] [Google Scholar]
- Hori I, Miya F, Ohashi K, Negishi Y, Hattori A, Ando N … Saitoh S. (2016). Novel splicing mutation in the ASXL3 gene causing Bainbridge-Ropers syndrome. American journal of medical genetics.Part A, 170, 1863–1867. [DOI] [PubMed] [Google Scholar]
- Izawa T, Rohatgi N, Fukunaga T, Wang QT, Silva MJ, Gardner MJ … Zou W. (2015). ASXL2 Regulates Glucose, Lipid, and Skeletal Homeostasis. Cell reports, 11, 1625–1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh M (2013). Functional and cancer genomics of ASXL family members. British journal of cancer, 109, 299–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuechler A, Czeschik JC, Graf E, Grasshoff U, Huffmeier U, Busa T … Wieczorek D. (2017). Bainbridge-Ropers syndrome caused by loss-of-function variants in ASXL3: a recognizable condition. European journal of human genetics : EJHG, 25, 183–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai HL, Wang QT. (2013). Additional sex combs-like 2 is required for polycomb repressive complex 2 binding at select targets. PloS one, 8, e73983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, He F, Zhang P, Chen S, Shi H, Sun Y … Yang FC. (2017). Loss of Asxl2 leads to myeloid malignancies in mice. Nature communications, 8, 15456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magini P, Della Monica M, Uzielli ML, Mongelli P, Scarselli G, Gambineri E … Seri M. (2012). Two novel patients with Bohring-Opitz syndrome caused by de novo ASXL1 mutations. American journal of medical genetics.Part A, 158A, 917–921. [DOI] [PubMed] [Google Scholar]
- Micol JB, Pastore A, Inoue D, Duployez N, Kim E, Lee SC … Abdel-Wahab O. (2017). ASXL2 is essential for haematopoiesis and acts as a haploinsufficient tumour suppressor in leukemia. Nature communications, 8, 15429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers KA, White SM, Mohammed S, Metcalfe KA, Fry AE, Wraige E … Scheffer IE. (2018). Childhood-onset generalized epilepsy in Bainbridge-Ropers syndrome. Epilepsy research, 140, 166–170. [DOI] [PubMed] [Google Scholar]
- Oak JS, Ohgami RS. (2017). Focusing on frequent ASXL1 mutations in myeloid neoplasms, and considering rarer ASXL2 and ASXL3 mutations. Current medical research and opinion, 33, 781–782. [DOI] [PubMed] [Google Scholar]
- Russell B, Johnston JJ, Biesecker LG, Kramer N, Pickart A, Rhead W … Graham JM. (2015). Clinical management of patients with ASXL1 mutations and Bohring-Opitz syndrome, emphasizing the need for Wilms tumor surveillance. American journal of medical genetics.Part A, 167A, 2122–2131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahtoe DD, van Dijk WJ, Ekkebus R, Ovaa H, Sixma TK. (2016). BAP1/ASXL1 recruitment and activation for H2A deubiquitination. Nature communications, 7, 10292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shashi V, Pena LDM, Kim K, Burton B, Hempel M, Schoch K … Kortum F. (2017). De Novo Truncating Variants in ASXL2 Are Associated with a Unique and Recognizable Clinical Phenotype. American Journal of Human Genetics, 100, 179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava A, Ritesh KC, Tsan YC, Liao R, Su F, Cao X … Bielas SL. (2016). De novo dominant ASXL3 mutations alter H2A deubiquitination and transcription in Bainbridge-Ropers syndrome. Human molecular genetics, 25, 597–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava S, Cohen JS, Vernon H, Baranano K, McClellan R, Jamal L … Fatemi A. (2014). Clinical whole exome sequencing in child neurology practice. Annals of Neurology, 76, 473–483. [DOI] [PubMed] [Google Scholar]
- Wang J, Li Z, He Y, Pan F, Chen S, Rhodes S … Yang FC. (2014). Loss of Asxl1 leads to myelodysplastic syndrome-like disease in mice. Blood, 123, 541–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R, He XH, Lin HY, Yang XH. (2018). Bainbridge-Ropers syndrome with ASXL3 gene variation in a child and literature review. Zhonghua er ke za zhi = Chinese journal of pediatrics, 56, 138–141. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.