

Abstract
Aberrations in the Hedgehog (Hh) signaling pathway are responsible for a broad range of human cancers, yet only a subset rely on the activity of the clinical target, Smoothened (Smo). Emerging cases of cancers that are insensitive to Smo-targeting drugs demand new therapeutic targets and agents for inhibition. As such, we sought to pursue a recently discovered connection between the Hedgehog pathway transcription factors, the glioma-associated oncogene homologues (Glis), and protein kinase C (PKC) isozymes. Here, we report our assessment of a structurally diverse library of PKC effectors for their influence on Gli function. Using cell lines that employ distinct mechanisms of Gli activation up- and downstream of Smo, we identify a PKC effector that acts as a nanomolar Gli antagonist downstream of Smo through a mitogen-activated protein kinase kinase (MEK)-independent mechanism. This agent provides a unique tool to illuminate crosstalk between PKC isozymes and Hh signaling and new opportunities for therapeutic intervention in Hh pathway-dependent cancers.
Graphical Abstract
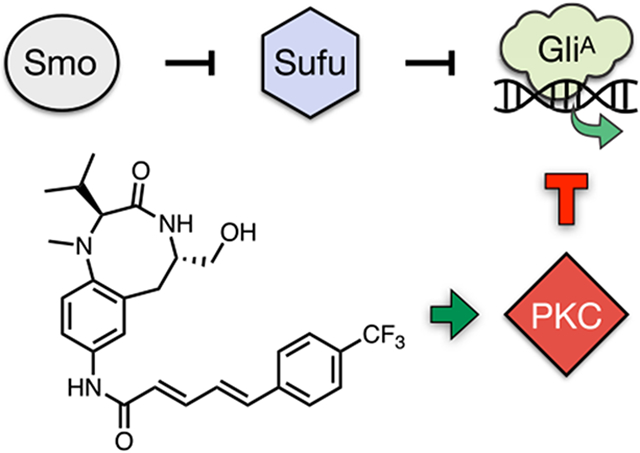
Aberrant activity of the glioma-associated (Gli) transcription factors (Glil, Gli2, and Gli3) within the Hedgehog (Hh) signaling pathway is a driving factor in several major human cancers.1-5 While the Hh pathway serves essential roles during embryogenesis,6 reactivation of Gli activity in adult tissue is oncogenic.7-9 In canonical Hh signaling, the pathway is stimulated by binding of the secreted Hh morphogen to its transmembrane receptor Patched 1 (Ptch1).10 This event releases the negative regulation of Ptch1 on the seven-transmembrane receptor Smoothened (Smo), transducing the signal across the membrane and triggering activation of the Gli transcription factors (Figure 1A). Alternatively, noncanonical Hh signaling, in which Gli transcription is activated independently of the Hh–Ptch1–Smo axis,11-13 can be initiated at multiple points upstream, downstream, or epistatic to Smo. Studies have traced transcriptional Gli amplification or post-translational Gli regulation to cross-talk with a number of signaling pathways, including MEK/ERK,14 PI3K/AKT,15 TGF-β,16 mTOR/S6K1,17 and others. Despite the diversity of Gli activation mechanisms, all FDA-approved drugs target the same site within Smo. Hence, no therapies are available for Gli tumorigenesis that is stimulated downstream of Smo or is resistant to these molecules. Chemical biology approaches to elucidate cellular mechanisms that govern Gli regulation are needed to identify next-generation drugs for Gli-driven cancers.18,19
Figure 1.

Hedgehog signaling pathway and PKC. (A) Mechanism of Hedgehog pathway activation. Binding of Hh to Ptch1 releases suppression of Smo, whereupon activated Smo promotes dissociation of Gli proteins from Sufu. Gli proteins are converted into their active forms (GliA), which translocate to the nucleus and induce Hh pathway target gene expression. Activation of MEK/ERK signaling is independently able to regulate the formation of GliA. PKCs are reported to act directly on both proteins in the Hh pathway and MAP kinases. (Hh, Hedgehog; Ptch1, Patched 1; Smo, Smoothened; Gli, glioma-associated oncogene homologue; Sufu, Suppressor of Fused; MAP, mitogen-activated protein kinase, MEK, mitogen-activated protein kinase kinase; PKC, protein kinase C). (B) Regulation of PKC isozymes. Comparison of the conventional, novel, and atypical classes of the PKC family showing the regulatory domain cofactors required for enzyme activation. (C) RNA-seq analysis of PKC isozymes in NIH-3T3 cells after 30 h treatment with SAG (200 nM) or control (DMSO). PKC isozymes from each class are expressed in NIH-3T3 cells at significant levels, which are unaffected by SAG treatment. Mean count and fold change for the Hh pathway target genes Gli1, Ptch1, Pgm5, and Angtpl4 are shown for comparison. Differential expression was analyzed using DESeq2 with false discovery rate (FDR)-corrected p-value <0.01. Biological triplicates were analyzed for each condition.
Several studies have identified events that intersect with Gli activity, including reports that show a regulatory role for protein kinase C (PKC) isozymes.20-27 Importantly, PKC isozymes are proposed to interact with Hh signaling at different points during pathway activation. For example, PKCι/λ directly phosphorylates Gli to upregulate Hh target genes,23 whereas PKCα has been shown to regulate Gli activity via the mitogen-activated protein kinase kinase (MEK/ERK) pathway.25,28 PKCδ possesses pro-apoptotic effects in numerous cancers;26 however, the effects of this isozyme on Gli are duration- and cell type-dependent.25,27 While crosstalk between the Hh pathway and PKC provides opportunities for small molecule intervention in both canonical and noncanonical Gli regulation, the activities of individual PKC effectors remain poorly understood, and only a limited set of PKC agonists have been employed in studies of Hh signaling.29-31 In this report, we identify a small molecule PKC effector that exhibits nanomolar inhibition of Gli activity downstream of Smo. These studies provide a valuable tool to investigate the PKC–Gli axis and new avenues for therapeutic development.
PKCs, like other families of kinases, contain a conserved catalytic domain responsible for phosphate transfer.32-34 In addition, specific classes of PKCs are susceptible to small molecule regulation through endogenous diacylglycerol (DAG) and/or calcium binding regulatory domains. Affinity for DAG and/or calcium can be further enhanced by phospholipid mediators such as phosphatidylserine. The ten human PKC isoforms are divided into three categories according to their sensitivity to DAG and interaction with calcium (Figure 1B). While conventional PKCs (α, βI, βII, γ) are sensitive to calcium and DAG, novel PKCs (δ, θ, η, ε) are sensitive to DAG but insensitive to calcium. By contrast, atypical PKCs (ζ, ι, λ) lack both calcium and C1 regulatory domains and are thus insensitive to both.
To identify a suitable cell-based model for evaluating PKC effectors, we first assessed the expression levels of various PKC isozymes in NIH-3T3 mouse embryonic fibroblasts (MEFs) using RNA-seq (Figure 1C). Five PKC isoforms were observed at significant levels: PKCα (conventional), PKCγ (conventional), PKCδ (novel), PKCε (novel), and PKCλ (atypical); therefore, PKCs from each class were represented. RNA-seq analysis in the presence of the Smo agonist SAG (Smoothened AGonist, 200 nM) demonstrated that expression of these isozymes did not change significantly upon Smo activation (Figure 1C).
To advance the potential of PKC effectors as Gli antagonists, we sought to define small molecule structures that could target specific aspects of Gli regulation within Hh-signaling cells.35 While the structural characteristics of each PKC class are well established, selective activation of PKC isozymes remains an unmet challenge.36 Hence, as opposed to classification based on PKC isozyme targets, PKC effectors are classified by the structural domains to which they bind.32 Regulatory domain modulators are a diverse collection of small molecules that influence PKC cellular localization, phosphorylation, and degradation37-40 These agents can modify the activity of conventional and novel PKC isozymes, which possess a DAG-binding site in the C1 regulatory domain. By contrast, kinase domain inhibitors, which have variable isozyme selectivity, are available for all three PKC classes.36 While each class of PKC effector has been studied in a multitude of biological contexts, only a handful of reports have studied PKC activity in Hh signaling.21-27 In addition, although numerous molecules including natural products, peptides, and synthetic agents can interact with PKCs, structural features within a given scaffold often lead to idiosyncratic isozyme preferences, potencies, and mechanisms of action.31,41 Because distinct PKC isozymes are emerging as critical regulators of Hh signaling, we sought to intercept Hh activity with specific PKC modulators. Given the untapped diversity of these agents, we established a compound library encompassing three types of PKC effectors: (1) DAG-based lipids, (2) regulatory domain activators of the C1-binding site, and (3) selective and unselective inhibitors of the catalytic domain (Figure S1).
We examined the effect of this structurally diverse library of PKC modulators on Gli activity in Shh-LIGHT2 cells, an NIH-3T3-derived cell line stably transfected with a Gli-dependent firefly luciferase reporter and a TK-driven Renilla luciferase control reporter for normalization.42 This cell line provides a well-established first assay to identify agents with nanomolar potency and significant inhibition of Gli-driven luciferase activity initiated at Smo (Figure 2). In each assay, SAG (200 nM) was coadministered to induce transcriptional Gli activity. After a 30 h exposure to SAG and each PKC effector, normalized Gli-driven luciferase activity was measured and compared to SAG alone as a control. Each compound was evaluated at a concentration of 1.5 μM to determine level of Gli-driven luciferase inhibition (y-axis, Figure 2A). The half-maximal inhibitory concentration (x-axis, Figure 2A) for each agent was evaluated in individual dose–response experiments. As a reference, the clinical Smo inhibitor vismodegib (gray circle, Figure 2A) was also evaluated.30,31,43,44
Figure 2.

Evaluation of PKC effectors in Shh-LIGHT2 cells treated with 200 nM SAG. (A) Comparison of compound potency versus magnitude of inhibition of Gli-driven luciferase: y-axis, relative Gli-driven luciferase activity at 1.5 μM dose of each compound; x-axis, half maximal inhibitory concentration (IC50) for each compound. Compounds: vismodegib (gray circle), regulatory domain C1-binding site activators (blue circles), DAG-based lipid activators (green circles), and catalytic domain inhibitors (red circles). (B) Structures of vismodegib and 1–5 with compound potency and Gli-driven luciferase reporter activity in the presence of 200 nM SAG and 1.5 μM of each compound. For panels A and B, Gli luciferase is calculated relative to Gli-driven luciferase activity induced by 200 nM SAG (100%). All values are the mean of n > 3 biological replicates ± SD.
PKC effectors from each domain-targeting group showed a range of inhibitory effects on Gli-driven luciferase activity (Figure S1, Table S1, and Figure S3). Kinase domain inhibitors selective for conventional PKCs (Gö-6976) or atypical PKCs (myristoylated pseudosubstrate inhibitor and PKC-9) displayed minimal differences versus control (red circles, Figure 2A). Likewise, DAG-based lipids caused mild inhibition of Gli-driven luciferase activity at micromolar to near-micromolar concentrations (green circles, Figure 2A).
Most significantly, the class of C1-binding site regulatory domain PKC activators consisted of compounds that strongly inhibited Gli-driven luciferase activity at nanomolar to near-nanomolar concentrations: TPPB (1), indolactam V (2), phorbol 12-myristate 13-acetate (PMA) (3), prostratin (4), and ceramide (5) (blue circles, Figure 2A). Dipeptide 145 was the most potent compound investigated, showing almost complete suppression of Gli-driven luciferase activity at double-digit nanomolar concentrations (IC50 = 20 ± 7 nM, Figure 2B). This effect was comparable to that of vismodegib, demonstrating that PKC effectors can act at therapeutically relevant concentrations. Significantly, 1 is unique among these compounds as a neuroprotective agent38,39,46,47 without significant tumorigenic activity,36 suggesting it as a lead for therapeutic development.
The regulatory domain activator and established tumor-promoter 348 also showed nanomolar inhibition of Gli-driven luciferase activity in this cell type. The structurally related but non-tumor-promoting 449 displayed a similar level of inhibition to 3 but reduced potency.37,50 While initial reports have suggested that 3 functions to positively regulate Gli,25 subsequent studies have identified an alternative mechanism of Gli antagonism via PKCδ.26 These conflicting observations may arise from differences in the time course of these studies, which can influence the dominant mechanism by which these agents act on PKC (Figure S2).
While clinical inhibitors have been successful at treating Gli-driven cancers initiated at or upstream of Smo, an increasing number of human cancers are recognized as originating from Smo-independent Gli activity.51,52 An elegant report by Toftgård and co-workers has demonstrated that the effector 3 can block Gli activity initiated downstream of Smo in MEFs.27 To examine the potential for PKC modulators to influence Smo-independent Gli-driven luciferase activity, we evaluated our five most potent inhibitors in Sufu-KO-LIGHT cells.53,54 Sufu-KO-LIGHT cells lack the Hh pathway component Sufu, a direct negative regulator of the Gli transcription factors, and thus exhibit constitutive Gli activity arising downstream of Smo. As in Shh-LIGHT2 cells, Sufu-KO-LIGHT cells express a stably integrated Gli-driven firefly luciferase reporter; in this assay, Gli-driven luciferase signal is normalized to cell viability.
Examination of 1–5 in Sufu-KO-LIGHT cells revealed that compounds 1, 2, 3, and 4 retained potent effects on Gli activity. As anticipated, the potency and maximum inhibition by the Smo inhibitor vismodegib decreased by more than 2 orders of magnitude in these cells as compared to Shh-LIGHT2 cells (22% inhibition at 1.5 μM, Figure 3A, Table S2, and Figure S3; IC50 > 5 μM, Figure 3C). By contrast, GANT-61, a downstream inhibitor of Gli,35 maintained a potency of IC50 = 2.2 ± 0.2 μM and the ability to significantly inhibit Gli activity (Figure 3B,C). Of the PKC effectors, 5 lost 3 orders of magnitude in potency, which might reflect a direct effect of this lipid on the function of Smo.
Figure 3.

Analysis of lead compounds in Sufu-KO-LIGHT cells. (A) Dose–response curves for vismodegib and 1 in Sufu-KO-LIGHT cells and Shh-LIGHT2 cells stimulated with 200 nM SAG. (B) Relative Gli-driven luciferase activity at 1.5 μM dose of vismodegib, GANT, or 1–5 in Sufu-KO-LIGHT cells. (C) Half maximal inhibitory concentration (IC50, nM) and percent Gli-driven luciferase activity relative to control at 1.5 μM dose of vismodegib, GANT, or 1–5 in Sufu-KO-LIGHT cells. For panels A and B, Gli luciferase is calculated relative to Gli-driven luciferase activity induced by 200 nM SAG in Shh-LIGHT2 cells (100%) or DMSO in Sufu-KO-LIGHT cells (100%). All values are the mean of n > 3 biological replicates ± SD.
The most potent inhibitor in Shh-LIGHT2 cells, 1, suppressed both hSHH-N and SAG activation (Figure S4) and was able to achieve near-complete inhibition of constitutive Gli-driven luciferase activity in Sufu-KO-LIGHT cells. Although 1 demonstrated significant inhibition of Gli-driven luciferase activity in Shh-LIGHT2 and Sufu-KO-LIGHT cells, control studies revealed that 1 caused an unexpected increase in CMV-driven firefly luciferase activity in NIH-3T3 cells at relevant concentrations (Figure S5). Because an increase in luciferase activity can be linked to ligand-based stabilization of the luciferase enzyme,55 we sought to validate the effect of 1 on Gli in orthogonal, non-luciferase-based assays. To directly measure the effect of 1 on Gli target gene expression, we assessed levels of Gli1 in both Shh-LIGHT2 and Sufu-KO-LIGHT cells treated with 1 after 30 h by qPCR. Co-incubation of varying concentrations of 1 with Shh-LIGHT2 cells stimulated with 200 nM SAG or Sufu-KO-LIGHT cells resulted in corresponding reduction of Gli1 mRNA in both cell types (Figure 4A,B). To examine activity in different cellular contexts, we measured the ability of 1 to inhibit Gli-dependent differentiation of C3H10T1/2 mesenchymal stem cells to alkaline phosphatase (ALP)-positive osteoblasts,56 which occurred at an IC50 of 45 ± 15 nM (versus 58 ± 32 nM for vismodegib). We also tested the activity of 1 against Gli-dependent proliferation of ASZ001 basal cell carcinoma cells and found that 1 inhibited mBCC proliferation with an IC50 of 0.16 ± 0.44 μM (versus 2.0 ±0.1 μM for vismodegib) (Figure 4C,D). Collectively, these observations place conformationally restricted dipeptide 1 in a select class of Smo-independent Gli inhibitors with nanomolar potency in diverse cell types.
Figure 4.

Characterization of Gli inhibition in diverse cell types and on Gli-driven luciferase activity in the presence of a MEK inhibitor. (A) Gli1 mRNA levels in Shh-LIGHT2 cells treated with 200 nM SAG and 1 at the concentrations indicated for 30 h. (B) Gli1 mRNA levels in Sufu-KO-LIGHT cells treated with 1 at the concentrations indicated for 30 h. In panels A and B, fold change is calculated relative to levels of B2M RNA using the ΔΔCt method. (C) Dose–response curves for 1 and vismodegib in C3H10T1/2 cells. Alkaline phosphatase activity (ALP) is calculated relative to Gli-driven ALP activity induced by 200 nM SAG (100%). (D) Dose–response curves for growth inhibition by 1 and vismodegib in ASZ001 mBCC cells treated at the concentrations indicated for 48 h, measured using the CellTiter assay. Viability is calculated relative to DMSO (100%). (E) Relative Gli-driven luciferase inhibition in Shh-LIGHT2 cells by 1 (1.5 μM) in the presence of SAG (200 nM) and MEK inhibitor CI-1040 (6, 100 nM). (F) Relative Gli-driven luciferase inhibition in Sufu-KO-LIGHT cells by 1 (1.5 μM) in the presence of MEK inhibitor 6 (100 nM). For panels E and F, Gli luciferase is calculated relative to Gli-driven luciferase activity induced by 200 nM SAG in Shh-LIGHT2 cells (100%) or DMSO in Sufu-KO-LIGHT cells (100%). All values are the mean of n > 3 biological replicates ± SD.
PKC effectors can influence diverse cellular processes that intersect with Gli both up- and downstream of Smo.24-27 Previous studies have suggested that the PKC activator 3 acts on Hh signaling downstream of Smo and by dual mechanisms dependent on and independent of MEK/ERK signaling.27 Given this connection, we sought to establish whether the MEK/ERK pathway contributed to the activity of compound 1. To ascertain whether the activity of 1 is dependent on MEK signaling, we examined the effect of 1 in Gli-driven luciferase activity in the presence of the selective MEK inhibitor CI-1040 (6, 100 nM).57 In Shh-LIGHT2 cells, 1 maintained an ability to inhibit Gli-driven luciferase activity with 200 nM SAG upon coapplication of 100 nM 6 (Figure 4F and Figure S6). Likewise, in Sufu-KO-LIGHT cells, 1 retained the ability to inhibit Gli in the presence of 100 nM 6 (Figure 4G and Figure S6). Taken together, these studies indicate that 1 can influence downstream Gli activity in a manner that does not require MEK signaling. While further studies are required to address potential PKC-independent effects of 1 at prolonged time periods, our findings provide further support for a PKC–Gli axis and illuminate significant new mechanisms to target oncogenic Gli activity.
Despite the clinical success of Smo-targeting drugs, a growing number of cancers are associated with Gli activity that is insensitive to Smo antagonism. In this study, we investigated a diverse library of PKC effectors for their ability to regulate endogenous Gli activity at multiple points within the Hh pathway. Our studies reveal structure-sensitive and highly potent effects of PKC agonists on Gli activity both up- and downstream of Smo. These new connections are inherently sensitive to small molecule intervention and can be exploited to address disease-specific dependencies. Importantly, our strategy established the PKC effector 1 as an antagonist of Smo-independent Gli activity with equal potency to clinical Smo-targeting drugs and a new agent to elucidate PKC–Gli crosstalk in Gli-driven cancers.
Supplementary Material
ACKNOWLEDGMENTS
We thank J. K. Chen (Stanford University) for Shh-LIGHT2, Sufu-KO-LIGHT, C3H10T1/2, and TM3-Gli-Luc cells and S. X. Atwood (UC Irvine) for ASZ001 mBCC cells. We thank N. V. Patel (CSU Fullerton) for use of a GloMax luminometer. G.C.Z. was supported through a training grant (NIH GM07616) provided by the National Institutes of Health. This research was supported by start up funds granted by the College of Natural Sciences and Mathematics at California State University, Fullerton (K.L.B.), and the Division of Chemistry and Chemical Engineering at the California Institute of Technology (A.E.O.).
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acschembio.0c00355.
Methods and additional tables and figures (PDF)
METHODS
Details of the experimental procedures are provided in the Supporting Information.
The authors declare no competing financial interest.
Contributor Information
UyenPhuong Tran, Department of Chemistry and Biochemistry, California State University Fullerton, Fullerton, California 92831, United States.
Grace C. Zhang, Division of Chemistry and Chemical Engineering California Institute of Technology, Pasadena, California 91125, United States.
Ryan Eom, Department of Chemistry and Chemical Biology, Cornell University, Ithaca, New York 14853, United States.
Kelvin L. Billingsle, Department of Chemistry and Biochemistry, California State University Fullerton, Fullerton, California 92831, United States.
Alison E. Ondrus, Division of Chemistry and Chemical Engineering, California Institute of Technology, Pasadena, California 91125, United States.
REFERENCES
- (1).Wu F, Zhang Y, Sun B, McMahon AP, and Wang Y (2017) Hedgehog signaling: From basic biology to cancer therapy. Cell Chem. Biol 24, 252–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Pak E, and Segal RA (2016) Hedgehog signal transduction: Key players, oncogenic drivers, and cancer therapy. Dev. Cell 38, 333–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Briscoe J, and Thérond PP (2013) The mechanisms of Hedgehog signalling and its roles in development and disease. Nat. Rev. Mol. Cell Biol 14, 416–429. [DOI] [PubMed] [Google Scholar]
- (4).Ruiz i Altaba A, Sánchez P, and Dahmane N (2002) Gli and hedgehog in cancer: tumours, embryos and stem cells. Nat. Rev. Cancer 2, 361–372. [DOI] [PubMed] [Google Scholar]
- (5).Matise MP, and Joyner AL (1999) Gli genes in development and cancer. Oncogene 18, 7852–7859. [DOI] [PubMed] [Google Scholar]
- (6).Varjosalo M, and Taipale J (2008) Hedgehog: Functions and mechanisms. Genes Dev. 22, 2454–2472. [DOI] [PubMed] [Google Scholar]
- (7).Rubin LL, and de Sauvage FJ (2006) Targeting the Hedgehog pathway in cancer. Nat. Rev. Drug Discovery 5, 1026–1033. [DOI] [PubMed] [Google Scholar]
- (8).Teglund S, and Toftgård R (2010) Hedgehog beyond medulloblastoma and basal cell carcinoma. Biochim. Biophys. Acta, Rev. Cancer 1805, 181–208. [DOI] [PubMed] [Google Scholar]
- (9).Galperin I, Dempwolff L, Diederich W, and Lauth M (2019) Inhibiting Hedgehog - An update on pharmacological compounds and targeting strategies. J. Med. Chem 62, 8392–8411. [DOI] [PubMed] [Google Scholar]
- (10).Kong JH, Siebold C, and Rohatgi R (2019) Biochemical mechanisms of vertebrate hedgehog signaling. Development 146, dev166892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Pietrobono S, Gagliardi S, and Stecca B (2019) Noncanonical Hedgehog signaling pathway in cancer: Activation of GLI transcription factors beyond Smoothened. Front. Genet 10, 556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Jenkins D (2009) Hedgehog signalling: Emerging evidence for non-canonical pathways. Cell. Signalling 21, 1023–1034. [DOI] [PubMed] [Google Scholar]
- (13).Lauth M, and Toftgård R (2007) Non-Canonical Activation of GLI Transcription Factors: Implications for Targeted Anti-Cancer Therapy. Cell Cycle 6, 2458–2463. [DOI] [PubMed] [Google Scholar]
- (14).Schnidar H, Eberl M, Klingler S, Mangelberger D, Kasper M, Hauser-Kronberger C, Regl G, Kroismayr R, Moriggl R, Sibilia M, and Aberger F (2009) Epidermal Growth Factor Receptor signaling synergizes with Hedgehog/GLI in oncogenic transformation via activation of the MEK/ERK/JUN pathway. Cancer Res. 69, 1284–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Riobo NA, Lu K, Ai X, Haines GM, and Emerson CP (2006) Phosphoinositide 3-kinase and Akt are essential for Sonic Hedgehog signaling. Proc. Natl. Acad. Sci. U. S. A 103, 4505–4510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Javelaud D, Alexaki VI, Dennler S, Mohammad KS, Guise TA, and Mauviel A (2011) TGFβ/SMAD/GLI2 signaling axis in cancer progression and metastasis. Cancer Res. 71, 5606–5610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Wang Y, Ding Q, Yen C-J, Xia W, Izzo JG, Lang J-Y, Li C-W, Hsu JL, Miller SA, Wang X, Lee D-F, Hsu J-M, Huo L, LaBaff AM, Liu D, Huang T-H, Lai C-C, Tsai F-J, Chang W-C, Chen C-H, Wu T-T, Buttar NS, Wang KK, Wu Y, Wang H, Ajani J, and Hung M-C (2012) The crosstalk of mTOR/S6K1 and Hedgehog pathways. Cancer Cell 21, 374–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Němec V, Hylsová M, Maier L, Flegel J, Sievers S, Ziegler S, Schröder M, Berger B-T, Chaikuad A, Valčíková B, Uldrijan S, Drápela S, Souček K, Waldmann H, Knapp S, and Paruch K (2019) Furo[3,2- b ]pyridine: A Privileged Scaffold for Highly Selective Kinase Inhibitors and Effective Modulators of the Hedgehog Pathway. Angew. Chem., Int. Ed 58, 1062–1066. [DOI] [PubMed] [Google Scholar]
- (19).Kremer L, Hennes E, Brause A, Ursu A, Robke L, Matsubayashi HT, Nihongaki Y, Flegel J, Mejdrová I, Eickhoff J, Baumann M, Nencka R, Janning P, Kordes S, Schöler HR, Sterneckert J, Inoue T, Ziegler S, and Waldmann H (2019) Discovery of the Hedgehog pathway inhibitor Pipinib that targets PI4KIIIß. Angew. Chem., Int. Ed 58, 16617–16628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Montagnani V, and Stecca B (2019) Role of protein kinases in Hedgehog pathway control and implications for cancer therapy. Cancers 11, 449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Li J, and Evers BM (2011) Hedgehog and Protein Kinase C Signaling, in Hedgehog signaling activation in human cancer and its clinical implications (Xie J, Ed.), Springer, New York, NY. [Google Scholar]
- (22).Brodie C, and Lomonaco SL (2010) Protein Kinase C in Cancer Signaling and Therapy, in PKCδ as a target for chemotherapeutic drugs (Kazanietz MG, Ed.), Springer, New York, NY. [Google Scholar]
- (23).Atwood SX, Li M, Lee A, Tang JY, and Oro AE (2013) GLI activation by atypical protein kinase C ι/λ regulates the growth of basal cell carcinomas. Nature 494, 484–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Neill GW, Ghali LR, Green JL, Ikram MS, Philpott MP, and Quinn AG (2003) Loss of protein kinase Cα expression may enhance the tumorigenic potential of Gli1 in basal cell carcinoma. Cancer Res. 63, 4692–4697. [PubMed] [Google Scholar]
- (25).Riobo NA, Haines GM, and Emerson CP (2006) Protein kinase C-delta and mitogen-activated protein/extracellular signal-regulated kinase-1 control GLI activation in hedgehog signaling. Cancer Res. 66, 839–845. [DOI] [PubMed] [Google Scholar]
- (26).Cai Q, Li J, Gao T, Xie J, and Evers BM (2009) Protein kinase C delta negatively regulates hedgehog signaling by inhibition of Gli1 activity. J. Biol. Chem 284, 2150–2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Lauth M, Bergström A, and Toftgård R (2007) Phorbol esters inhibit the Hedgehog signalling pathway downstream of Suppressor of Fused, but upstream of Gli. Oncogene 26, 5163–5168. [DOI] [PubMed] [Google Scholar]
- (28).Schnidar H, Eberl M, Klingler S, Mangelberger D, Kasper M, Hauser-Kronberger C, Regl G, Kroismayr R, Moriggl R, Sibilia M, and Aberger F (2009) Epidermal Growth Factor Receptor signaling synergizes with Hedgehog/GLI in oncogenic transformation via activation of the MEK/ERK/JUN pathway. Cancer Res. 69, 1284–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Newton AC, and Brognard J (2017) Reversing the paradigm: Protein kinase C as a tumor suppressor. Trends Pharmacol. Sci 38, 438–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Cooke M, Magimaidas A, Casado-Medrano V, and Kazanietz MG (2017) Protein Kinase C in cancer: The top five unanswered questions. Mol. Carcinog 56, 1531–1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Mochly-Rosen D, Das K, and Grimes KV (2012) Protein Kinase C, an elusive therapeutic target? Nat. Rev. Drug Discovery 11, 937–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Newton AC (1995) Protein Kinase C: Structure, function, and regulation. J. Biol. Chem 270, 28495–28498. [DOI] [PubMed] [Google Scholar]
- (33).Steinberg SF (2008) Structural basis of Protein Kinase C isoform function. Physiol. Rev 88, 1341–1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Lipp P, and Reither G (2011) Protein kinase C: The “masters” of calcium and lipid. Cold Spring Harbor Perspect. Biol 3, No. a004556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Lauth M, Bergström Å, Shimokawa T, and Toftgård R (2007) Inhibition of GLI-mediated transcription and tumor cell growth by small-molecule antagonists. Proc. Natl. Acad. Sci. U. S. A 104, 8455–8460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Wu-Zhang AX, and Newton AC (2013) Protein kinase C pharmacology: Refining the toolbox. Biochem. J 452, 195–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Kedei N, Lubart E, Lewin NE, Telek A, Lim L, Mannan P, Garfield SH, Kraft MB, Keck GE, Kolusheva S, Jelinek R, and Blumberg PM (2011) Some phorbol esters might partially resemble Bryostatin 1 in their actions on LNCaP prostate cancer cells and U937 leukemia cells. ChemBioChem 12, 1242–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Kelsey JS, Cataisson C, Chen J, Herrmann MA, Petersen ME, Baumann DO, McGowan KM, Yuspa SH, Keck GE, and Blumberg PM (2016) Biological activity of the bryostatin analog Merle 23 on mouse epidermal cells and mouse skin. Mol. Carcinog 55, 2183–2195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Ryckbosch SM, Wender PA, and Pande VS (2017) Molecular dynamics simulations reveal ligand-controlled positioning of a peripheral protein complex in membranes. Nat. Commun 8, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Szallasi Z, and Blumberg PM (1991) Prostratin, a nonpromoting phorbol ester, inhibits induction by phorbol 12-myristate 13-acetate of ornithine decarboxylase, edema, and hyperplasia in CD-1 mouse skin. Cancer Res. 51, 5355–5360. [PubMed] [Google Scholar]
- (41).Nakagawa Y (2012) Artificial analogs of naturally occurring tumor promoters as biochemical tools and therapeutic leads. Biosci., Biotechnol., Biochem 76, 1262–1274. [DOI] [PubMed] [Google Scholar]
- (42).Taipale J, Chen JK, Cooper MK, Wang B, Mann RK, Milenkovic L, Scott MP, and Beachy PA (2000) Effects of oncogenic mutations in Smoothened and Patched can be reversed by cyclopamine. Nature 406, 1005–1009. [DOI] [PubMed] [Google Scholar]
- (43).Von Hoff DD, LoRusso PM, Rudin CM, Reddy JC, Yauch RL, Tibes R, Weiss GJ, Borad MJ, Hann CL, Brahmer JR, Mackey HM, Lum BL, Darbonne WC, Marsters JC Jr., de Sauvage FJ, and Low JA (2009) Inhibition of the Hedgehog pathway in advanced basal-cell carcinoma. N. Engl. J. Med, 361, 1164–1172. [DOI] [PubMed] [Google Scholar]
- (44).Rudin CM, Hann CL, Laterra J, Yauch RL, Callahan CA, Fu L, Holcomb T, Stinson J, Gould SE, Coleman B, LoRusso PM, Von Hoff DD, de Sauvage FJ, and Low JA (2009) Treatment of medulloblastoma with hedgehog pathway inhibitor GDC-0449. N. Engl J. Med, 361, 1173–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Kozikowski AP, Nowak I, Petukhov PA, Etcheberrigaray R, Mohamed A, Tan M, Lewin N, Hennings H, Pearce LL, and Blumberg PM (2003) New amide-bearing benzolactam-based Protein Kinase C modulators induce enhanced secretion of the amyloid precursor protein metabolite sAPPalpha. J. Med. Chem 46, 364–373. [DOI] [PubMed] [Google Scholar]
- (46).Yang HQ, Li X, Yang WM, Feng SM, and Ma JJ (2012) Neuroprotective effects of new Protein Kinase C activator TPPB against Aβ25-35 induced neurotoxicity in PC12 cells. Neurochem. Res 37, 2213–2221. [DOI] [PubMed] [Google Scholar]
- (47).Yi P, Schrott L, Castor TP, and Alexander JS (2012) Bryostatin-1 vs. TPPB: Dose-dependent APP processing and PKC-α, -δ, and -ε isoform activation in SH-SY5Y neuronal cells. J. Mol. Neurosci 48, 234–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Blumberg PM, Jaken S, König B, Sharkey NA, Leach KL, Jeng AY, and Yeh E (1984) Mechanism of action of the phorbol ester tumor promoters: Specific receptors for lipophilic ligands. Biochem. Pharmacol 33, 933–940. [DOI] [PubMed] [Google Scholar]
- (49).Nakagawa Y, Yanagita RC, Hamada N, Murakami A, Takahashi H, Saito N, Nagai H, and Irie K (2009) A simple analogue of tumor-promoting Aplysiatoxin is an antineoplastic agent rather than a tumor promoter: Development of a synthetically accessible Protein Kinase C activator with Bryostatin-like activity. J. Am. Chem. Soc, 131, 7573–7579. [DOI] [PubMed] [Google Scholar]
- (50).Miana GA, Riaz M, Shahzad-Ul-Hussan S, Paracha RZ, and Paracha UZ (2015) Prostratin: An overview. Mini-Rev. Med. Chem, 15, 1122–1130. [DOI] [PubMed] [Google Scholar]
- (51).Dijkgraaf GP, Alicke B, Weinmann L, Januario T, West K, Modrusan Z, Burdick D, Goldsmith R, Robarge K, Sutherlin D, Scales SJ, Gould SE, Yauch RL, and de Sauvage FJ (2011) Small molecule inhibition of GDC-0449 refractory Smoothened mutants and downstream mechanisms of drug resistance. Cancer Res. 71, 435–444. [DOI] [PubMed] [Google Scholar]
- (52).Yauch RL, Dijkgraaf GJP, Alicke B, Januario T, Ahn CP, Holcomb T, Pujara K, Stinson J, Callahan CA, Tang T, Bazan JF, Kan Z, Seshagiri S, Hann CL, Gould SE, Low JA, Rudin CM, and de Sauvage FJ (2009) Smoothened mutation confers resistance to a Hedgehog pathway inhibitor in medulloblastoma. Science 326, 572–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Ardecky R, Magnuson GK, Zou J, Ganji SR, Brown B, Ngo T, Lee J, Zeng FY, Sun Q, Stonich D, Salaniwal S, Sakata T, Rack PG, Casabar JKT, Mangravita-Novo A, Smith JH, Sergienko E, Chung TDY, Pinkerton AB, Pass I, and Chen JK (2012) Small-molecule antagonists of Gli function. [Updated 2014 May 13]. Probe Reports from the NIH Molecular Libraries Program [internet], National Center for Biotechnology Information (US), Bethesda, MD. [PubMed] [Google Scholar]
- (54).Horn ME, Ondrus AE, Sakata-Kato T, Rack PG, and Chen JK (2020) Bicyclic imidazolium inhibitors of Gli transcription factor activity. ChemMedChem, DOI: 10.1002/cmdc.202000169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Auld DS, and Inglese J (2016) Interferences with Luciferase Reporter Enzymes [Updated 2018 Jul 1], in Assay Guidance Manual [internet] Sittampalam GS, Grossman A, and Brimacombe K, et al. , Eds.), Eli Lilly & Company and the National Center for Advancing Translational Sciences, Bethesda, MD. [PubMed] [Google Scholar]
- (56).Hyman JM, Firestone AJ, Heine VM, Zhao Y, Ocasio CA, Han K, Sun M, Rack PG, Sinha S, Wu JJ, Solow-Cordero DE, Jiang J, Rowitch DH, and Chen JK (2009) Small-molecule inhibitors reveal multiple strategies for Hedgehog pathway blockade. Proc. Natl. Acad. Sci. U. S. A 106, 14132–14137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Sebolt-Leopold JS, Dudley DT, Herrera R, Van Becelaere K, Wiland A, Gowan RC, Tecle H, Barrett SD, Bridges A, Przybranowski S, Leopold WR, and Saltiel AR (1999) Blockade of the MAP kinase pathway suppresses growth of colon tumors in vivo. Nat. Med, 5, 810–816. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.