

Abstract
Polycystic liver disease (PLD) is a group of genetic disorders characterized by progressive development of cholangiocyte-derived fluid-filled hepatic cysts. PLD is the most common manifestation of autosomal dominant and autosomal recessive polycystic kidney diseases and rarely occurs as autosomal dominant PLD. The mechanisms of PLD are a sequence of the primary (mutations in PLD-causative genes), secondary (initiation of cyst formation), and tertiary (progression of hepatic cystogenesis) interconnected molecular and cellular events in cholangiocytes. Nonsurgical, surgical, and limited pharmacological treatment options are currently available for clinical management of PLD. Substantial evidence suggests that pharmacological targeting of the signaling pathways and intracellular processes involved in the progression of hepatic cystogenesis is beneficial for PLD. Many of these targets have been evaluated in preclinical and clinical trials. In this review, we discuss the genetic, molecular, and cellular mechanisms of PLD and clinical and preclinical treatment strategies.
Keywords: polycystic liver disease, cholangiocytes, cystogenesis, genetics, mechanisms, therapy
INTRODUCTION
Polycystic liver disease (PLD) is a group of genetic disorders characterized by the progressive growth of cholangiocyte-derived fluid-filled hepatic cysts. PLD was first described in 1856 as a pathological condition associated with polycystic kidney disease (PKD), and the existence of PLD as an isolated disorder was suggested in 1925 (1). The genetic landscape of PLD consists of 12 disease-causative genes. Nevertheless, multiple cases of PLD are still genetically unresolved.
PLD is classified as mild, moderate, and severe (2). Most individuals with PLD are asymptomatic, but in a subpopulation of patients, hepatomegaly leads to impaired liver function and, rarely, to liver failure requiring liver transplantation. Existent therapies for PLD patients include conservative management and nonsurgical and surgical options; off-label use of somatostatin analogs is the only currently available pharmacological treatment (3–6).
In this review, we discuss the clinical features of PLD, pathogenetic mechanisms of hepatic cystogenesis, and clinical and preclinical therapeutic options for PLD.
PLD
As of today, the PLD family includes (a) PLD associated with autosomal dominant PKD (ADPKD) (prevalence 1:400), (b) PLD associated with autosomal recessive PKD (ARPKD) (prevalence 1:20,000), (c) isolated autosomal dominant PLD (ADPLD) (prevalence 1:100,000), and (d) PLD with undetected mutations (3, 7–10). The focus of the current review is on PLD with a known genetic background.
The most common form of PLD is associated with ADPKD, a life-threatening hereditary (typically adult-onset) disorder characterized by cyst formation and enlargement in the kidney and other organs. The occurrence and disease severity correlate with age and are sex dependent (3, 9, 10). Up to 58% of patients develop hepatic cysts between the ages of 15 and 24 years old, up to 85% between 25 and 34 years old, and up to 94% between 35 and 46 years old. The majority of symptomatic PLD patients (up to 94%) are women (11).
ARPKD is a fibrocystic disease of the kidney and liver, commonly with a childhood onset. Clinically apparent hepatic involvement occurs in ~45 % of ARPKD cases and is characterized by bile duct dysplasia due to ductal plate malformation, congenital hepatic fibrosis, portal hypertension, and hepatosplenomegaly (12, 13).
In contrast to PKD-associated PLD, ADPLD is characterized by the presence of liver cysts with no or few kidney cysts. ADPLD manifests between 40 and 60 years of age. The number and volume of cysts in ADPLD are typically greater than in ADPKD-associated PLD (14).
PLD is diagnosed by the presence of at least 10 hepatic cysts (4, 15). However, patients with a family medical history can be diagnosed by the presence of 4 cysts (16). Hepatic cysts have different sizes and shapes and are filled with cystic fluid (Figure 1). Liver function in most PLD patients remains normal. Independent of liver cyst burden, ~20% of patients have elevated γ-glutamyl transferase, alkaline phosphatase, and total bilirubin, and ~45% of patients have increased levels of CA-19–9, which positively correlates with liver volume and/or cyst infection (11, 17).
Figure 1.
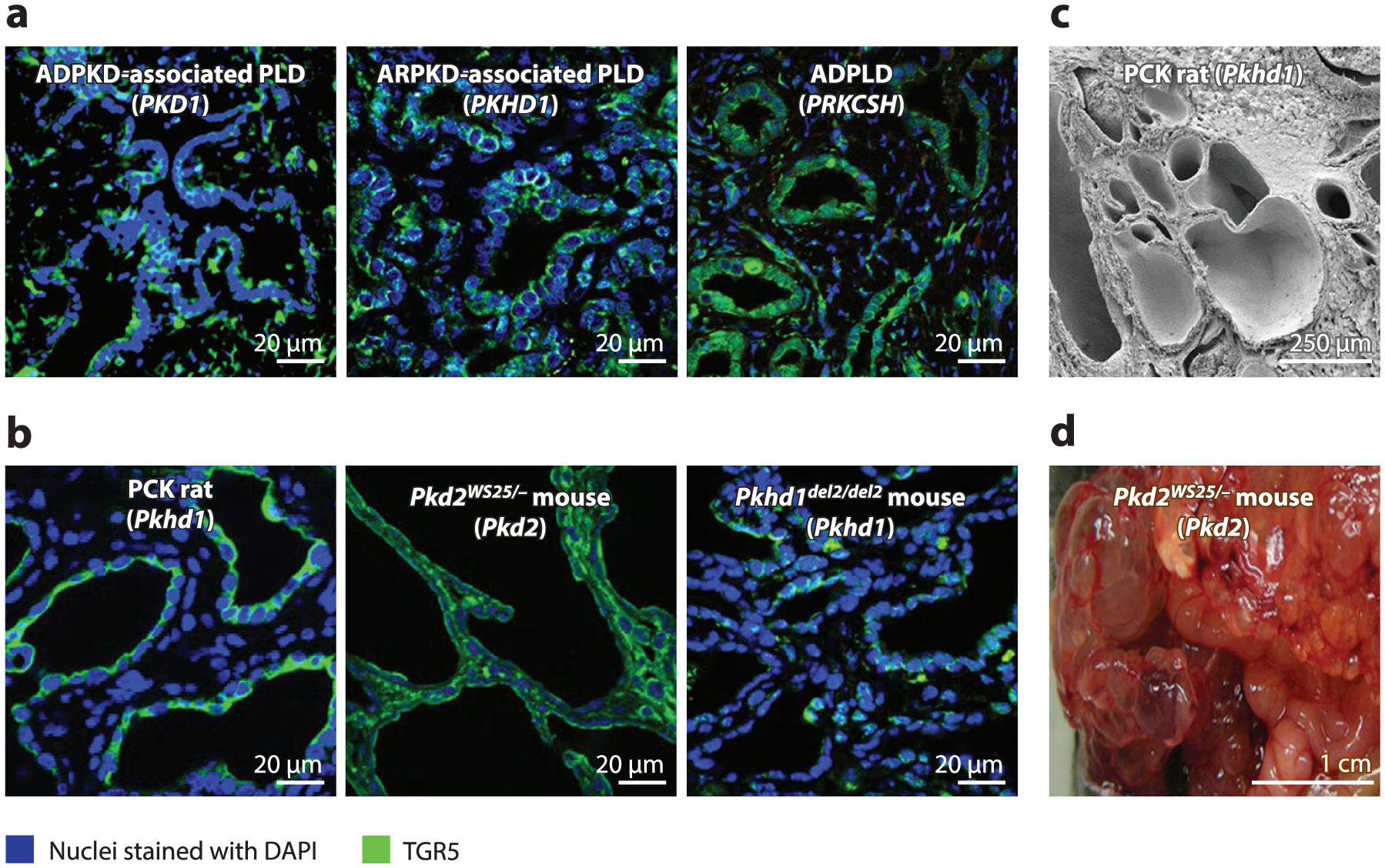
Hepatic cystogenesis in (a) patients and (b-d) animal models with different forms of PLD. Disease-causative genes are shown in parentheses. Despite genetic heterogeneity, mutations in PLD-causative genes result in similar end points—multiple hepatic cysts of different sizes and shapes. (a,b) Immunofluorescence confocal microscopy. (c) Scanning electron microscopy. (d) Gross appearance. Abbreviations: ADPKD, autosomal dominant polycystic kidney disease; ADPLD, autosomal dominant polycystic liver disease; ARPKD, autosomal recessive polycystic kidney disease; PLD, polycystic liver disease; TGR5, Takeda G protein-coupled receptor 5. Panel c adapted from Reference 45 with permission from Elsevier.
Progressive cyst growth correlates with enlargement of the liver, which increases in volume by up to 3.7% annually. In some patients, the liver can grow to up to 10 times the normal size, containing from 1 to 10 L of fluid. Both liver cysts and parenchyma contribute to hepatomegaly (2). Enlarged livers cause abdominal pain, nausea, vomiting, and early satiety, followed by weight loss, anorexia, shortness of breath, discomfort, and sleep apnea, affecting quality of life (2, 18–20).
MECHANISMS OF HEPATIC CYSTOGENESIS
We consider the mechanisms of hepatic cystogenesis as a sequence of the primary (mutations in PLD-causative genes), secondary (initiation of cyst formation), and tertiary (progression of hepatic cystogenesis) interconnected molecular and cellular events in cholangiocytes (Figure 2).
Figure 2.
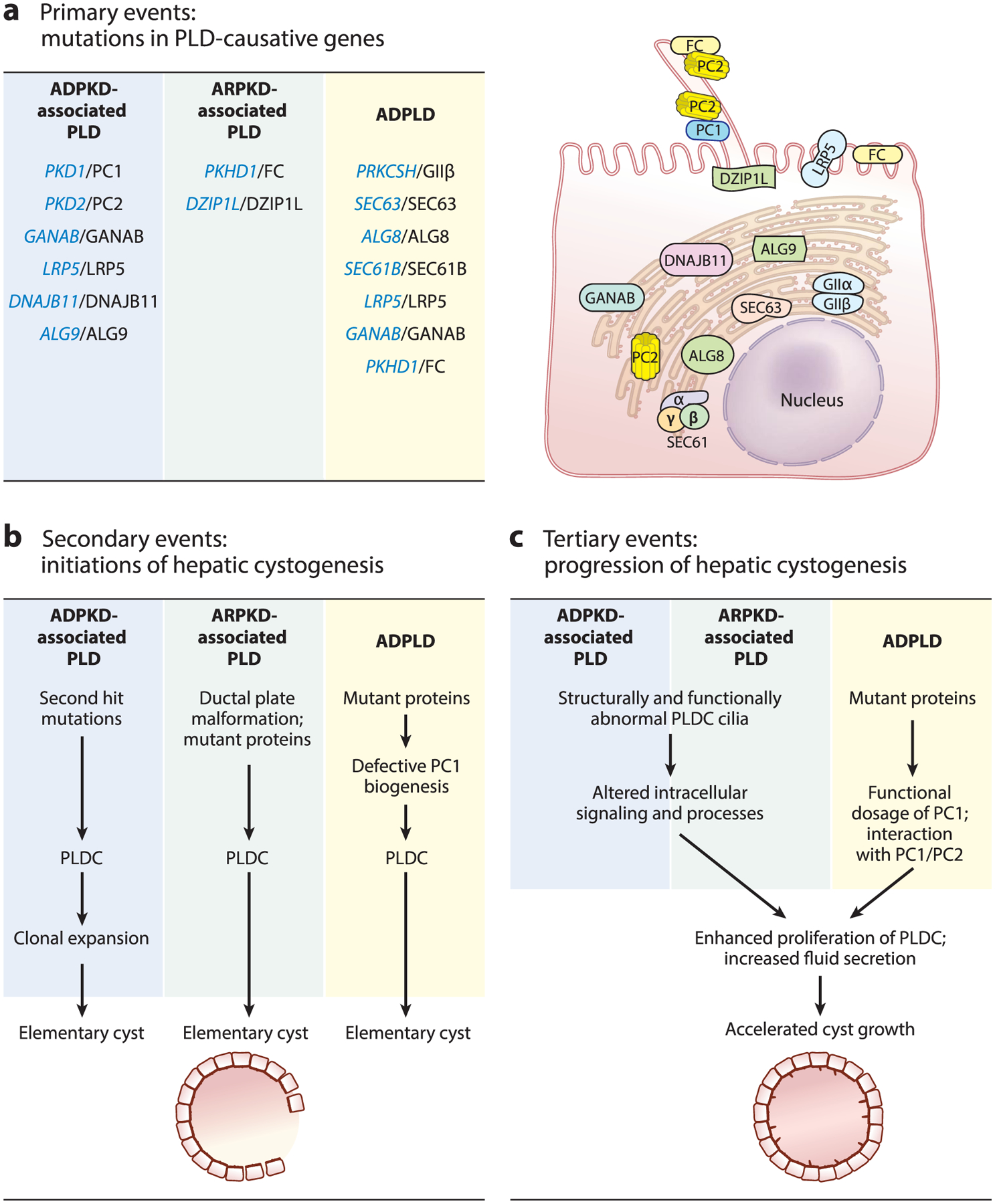
The mechanisms of hepatic cystogenesis: a sequence of the interconnected molecular and cellular (a) primary, (b) secondary, and (c) tertiary events in cholangiocytes. In panel a, PLD-associated genes are shown in blue and respective protein products are shown in black. Abbreviations: ADPKD, autosomal dominant polycystic kidney disease; ADPLD, autosomal dominant polycystic liver disease; ARPKD, autosomal recessive polycystic kidney disease; PLD, polycystic liver disease; PLDC, polycystic liver disease cholangiocyte.
PRIMARY MOLECULAR EVENTS: MUTATIONS IN PLD-CAUSATIVE GENES
Genetically, PLD is a monogenic disease, but allelically and phenotypically it is a heterogeneous disease (21). Mutations in six genes (i.e., PKD1, PKD2, GANAB, LRP5, DNAJB11, and ALG9) cause ADPKD-associated PLD. Mutations in two genes (i.e., PKHD1 and DZIP1L) result in ARPKD-associated PLD. ADPLD is caused by mutations in seven genes (i.e., PRKCSH, SEC63, LRP5, GANAB, ALG8, SEC61B, and PKHD1) (Figure 2a) (5, 9, 22–24). The number of PLD-causative genes and the observations that mutations in the same gene may cause both PKD-associated PLD and ADPLD emphasize the complexity of PLD genetics. It has been proposed that the genetic spectrum of PLD expands beyond the known 12 PLD-causative genes, and that mutations in more than one gene or activation/inhibition of gene modifiers might determine the severity of PLD (24).
Autosomal Dominant Polycystic Kidney Disease-Associated PLD: Genes and Proteins
The genetic nature of ADPKD was first suggested in 1925 and was accepted by 1957 (12). The PKD1 gene was identified in 1985; the second gene, PKD2, was reported in 1993 (25). To date, more than 1,650 mutations in PKD1 and 250 mutations in PKD2 cause approximately 80% and 15% of ADPKD cases, respectively, and 94% of ADPKD patients develop PLD (26).
PKD1 encodes the glycoprotein polycystin 1 (PC1) that functions as a G protein-coupled receptor (GPCR) (21, 27). The protein product of PKD2, membrane glycoprotein polycystin 2 (PC2), is a member of the transient receptor potential (TRP) family of ion channels (24). PC2 forms homotetramers and a heterotetramer with PC1 regulating [Ca2+]i (21, 27).
Mutations in GANAB, LRP5, DNAJB11, and ALG9 account for a small percentage of total ADPKD cases (24). As of today, 7% of ADPKD cases are still genetically unresolved.
GANAB encodes the catalytic α subunit of glucosidase II (GII), an endoplasmic reticulum (ER) protein responsible for N-linked glycosylation, a key quality-control process that regulates folding, maturation, and trafficking of membrane and secreted proteins. To date, several mutations of GANAB have been described (28–30).
The protein product of LRP5, a low-density lipoprotein receptor-related protein 5 (LRP5), is a part of the LRP5/LRP6/Frizzled coreceptor complex (10). LRP5, together with the Frizzled receptors, binds to the Wnt proteins, activating Wnt signaling (9). The LRP5 gene mutations are predicted to be loss-of-function mutations, although the exact role of LRP5 in hepatic cystogenesis remains unclear (3).
DNAJB11 encodes a soluble ER protein DNAJB11 (DNAJ/HSP40 homolog, subfamily B, member 11). DNAJB11 stabilizes the transfer of the translated peptides through the SEC61-SEC62-SEC63 channel to assure the correct protein trafficking and degradation. A dosage reduction of DNAJB11 is thought to cause cyst formation. To date, five different DNAJB11 pathogenic variants have been described (31).
Newly implicated in ADPKD and ADPKD-associated PLD, the ALG9 gene encodes ALG9, an ER protein involved in peptide glycosylation (32). The specific role of ALG9 in hepatic cystogenesis is unknown.
Mutations in PKD1, PKD2, and GANAB cause mild to severe PLD, while mutations in DNAJB11, LRP5, and ALG9 result in mild to moderate PLD (33). The severity of PLD does not directly correlate with the type of mutation (34).
Autosomal Recessive Polycystic Kidney Disease-Associated PLD: Genes and Proteins
ARPKD-associated PLD is caused by mutations in two genes, PKHD1 and DZIP1L, and is characterized by dysgenesis of the hepatic portal triad resulting from defective remodeling of the ductal plate, hyperplastic biliary ducts, and congenital hepatic fibrosis. In individuals surviving the neonatal period, portal hypertension and cholangitis are the major complications (24, 33, 35, 36).
PKHD1 encodes fibrocystin (FC), a transmembrane protein that functions as a membrane-bound receptor involved in regulation of proliferation and morphogenesis (37).
DZIP1L encodes DAZ interacting protein 1 like (DZIP1L), a soluble zinc-finger protein localized to the centrioles and at the distal end of the basal body (35). Dzip1l loss-of-function mice develop ductal plate malformations, a pathological characteristic of ARPKD-associated PLD (35).
Autosomal Dominant PLD: Genes and Proteins
Mutations in seven genes, PRKCSH, SEC63, LRP5, GANAB, ALG8, SEC61B, and PKHD1, have been implicated in ADPLD (8, 24, 28, 30, 38, 39). Despite such genetic heterogeneity, only 40% of cases are assigned to mutations in these genes; the rest of the cases are still unresolved. Mutations in PRKCSH, SEC63, and GANAB cause mild to severe PLD; mutations in ALG8, SEC61B, and LRP5 result in mild to moderate PLD; and mutations in PKHD1 result in congenital hepatic fibrosis and small liver cysts (33).
The protein product of PRKCSH, hepatocystin, is a noncatalytic β subunit of GII localized to the ER (40). Hepatocystin is involved in processing and quality control of newly synthesized glycoproteins (41). Mutations in PRKCSH result in abnormal hepatocystin, suggesting that ADPLD arises from a loss of function of this protein (10, 40).
SEC63 encodes an integral membrane protein of the ER, SEC63, which is a part of the multicomponent complex responsible for translocation of the integral membrane and secreted proteins. Mutations in PRKCSH and SEC63 together account for less than one-third of ADPLD cases (42, 43).
Mutations in the ALG8 and SEC61B genes have also been linked to ADPLD (10). ALG8 encodes alpha-1,3-glucosyltransferase (ALG8), an ER integral membrane protein. The SEC61B-encoded product is a component of the SEC63 protein complex in the ER. Both ALG8 and SEC61 control protein translocation and membrane insertion. Together, they account for ~4% of ADPLD cases (30).
Mutations in the LRP5 and PKHD1 genes (described above) may cause ADPLD and, respectively, ADPKD- and ARPKD-associated PLD. The detection rate of LRP5 and PKHD1 pathogenic variants in ADPLD is less than 1 % (9).
SECONDARY MOLECULAR AND CELLULAR EVENTS: INITIATION OF HEPATIC CYSTOGENESIS
Autosomal Dominant Polycystic Kidney Disease-Associated PLD
A heterozygous germline mutation in the PKD1 or PKD2 genes itself does not initiate hepatic cystogenesis. To initiate cyst formation, cholangiocytes must lose the wild-type allele after a second somatic mutation or a second-hit mutation at other loci. A cholangiocyte with a somatic mutation is then transformed into a hepatic cyst stem cell [i.e., a single PLD cholangiocyte (PLDC)] with aberrant expression and function of PC1 or PC2 and defective PC1- or PC2-mediated signaling (9). Clonal expansion of a PLDC initiates the formation of elementary hepatic cysts (9).
Thus, the transformation of healthy cholangiocytes into PLDCs with aberrant PC1 or PC2 expression and functions and the clonal expansion of PLDCs are the earliest molecular and cellular events that initiate hepatic cystogenesis (Figure 2b).
Autosomal Recessive Polycystic Kidney Disease-Associated PLD
The biliary cyst formation in ARPKD-associated PLD is primarily triggered by missense and truncating mutations in the PKHD1 gene and, in rare cases, by mutations in DZIP1L (21). The functions of proteins, FC, and DZIP1L encoded by these two genes remain obscure (24).
The initiation of hepatic cytogenesis in ARPKD-associated PLD is presumably linked to the embryological arrest of ductal plate development, which leads to the inability of biliary precursor cells to differentiate, defective maturation of primitive bile ducts, and/or abnormal bile duct enlargement (23).
FC is localized to cilia in healthy cholangiocytes but not in PLDCs, affecting ciliary morphology and sensory and signaling functions (44). DZIP1L normally is localized to the centrioles and the ciliary basal bodies maintaining the periciliary diffusion barrier at the ciliary transition zone. Mutated DZIP1L causes defects in the diffusion barriers, preventing delivery of sensory/signaling proteins to cilia (35). Thus, ductal plate malformation and ciliary abnormalities are presumably the earliest phenotypic manifestations of mutated PKHD1 and DZIP1L (Figure 2b).
Autosomal Dominant PLD
The protein products of ADPLD-causative genes, except for LRP5, reside in the ER and are involved in protein glycosylation, folding, quality control, and translocation. Correctly assembled proteins are transported out of the ER to their cellular location. Mutations in the PRKCSH, SEC63, ALG8, GANAB, and SEC61B genes compromise biogenesis and trafficking of many proteins, including PC1 (Figure 2b) (30). Mutations in GANAB may cause ADPKD-associated PLD and ADPLD, suggesting that hepatic cystogenesis in both diseases is likely driven by defects in PC1 maturation (22, 23). In contrast to ALG8, GANAB, and SEC61B, the pathogenic variants in PKHD1 contribute to ADPLD without affecting PC1 biogenesis, presumably via the aberrant PC1-FC interaction (22).
Missense variants in LRP5 coexist with PKHD1 variants, suggesting that interaction of these two genes may cause cyst growth in patients with mutated PKHD1 (22).
Thus, hepatic cystogenesis in ADPLD is initiated by the aberrant expression and functions of the ER proteins PRKCSH, SEC63, ALG8, SEC61B, and GANAB, which in many cases reduce the steady-state levels of PC1. The interaction of the protein products of the ADPLD-causative genes with PC1 supports the hypothesis that PC1 is a master regulator of the intersecting pathways in the initiation of hepatic cystogenesis (22, 23).
TERTIARY MOLECULAR AND CELLULAR EVENTS: PROGRESSION OF HEPATIC CYSTOGENESIS
The abnormalities in signaling functions of cholangiocyte primary cilia and altered cilia-dependent intracellular mechanisms, including enhanced proliferation and fluid secretion, are crucial for the progression of hepatic cystogenesis in ADPKD-associated PLD and ADPLD (Figure 2c).
ADPKD- and ARPKD-associated PLD belong to cholangiociliopathies, a group of liver diseases linked to the morphological and functional defects in cholangiocyte cilia (7, 45). In healthy cholangiocytes, PC1, PC2, and FC are localized to the primary cilium, a sensory organelle that extends from the apical plasma membrane into the bile duct lumen and detects, amplifies, and integrates diverse extracellular stimuli into the intracellular signaling and functional responses (46–48). According to a plausible hypothesis, in healthy cells, extracellular stimuli (e.g., fluid flow) activate the PC1-PC2 complexes in cilia, triggering a cilia-mediated [Ca2+]i response (48, 49). However, this hypothesis is now under revision because recent studies demonstrated that the PC1-PC2 complexes can induce PC2-dependent changes in the concentration of ciliary Ca2+ without affecting cytoplasmic Ca2+ (50).
In contrast to cilia in healthy cholangiocytes, malformed PLDC cilia do not express PC1, PC2, or FC, resulting in altered ciliary sensory and signaling functions and interrupted [Ca2+]i-cyclic adenosine monophosphate (cAMP) cross talk (44, 47, 48, 51). Aberrant expression and ciliary localization of PC1 decrease the levels of [Ca2+]i, subsequently activating [Ca2+]i-dependent cAMP signaling (52). Another potential mechanism of the cAMP increase in PLDC is overexpression and mislocalization of a cAMP-linked bile acid receptor, TGR5 (Takeda G protein–coupled receptor 5) (53). Activation of the ciliary-localized TGR5 by bile acids inhibits cAMP in healthy cholangiocytes (54). In contrast, in PLDC, TGR5 is overexpressed, localized predominantly at the apical plasma membrane, and, when activated, accelerates PLD progression (53, 54). In PLD, bile acids (i.e., TGR5 ligands) accumulate in the cystic fluid and can act as extracellular stimuli, regulating cAMP-dependent hepatic cyst growth (23, 54, 55).
In ARPKD-associated PLD, mutated FC and DZIP1L drive functional abnormalities in cilia and the downstream signaling pathways, accelerating cholangiocyte proliferation and hepatic cystogenesis (22, 23).
In ADPLD, a disrupted interaction of the protein products of ADPLD-causative genes with PC1 affects cholangiocyte sensory and signaling functions and ciliary-mediated progression of hepatic cystogenesis (23).
The primary, secondary, and tertiary molecular and cellular events that underlie hepatic cystogenesis are interconnected, but the mechanisms of their interconnection are largely unknown. We found that more than 30 biological pathways, including intracellular signaling, cell cycle regulation, proliferation, and secretion, are altered in PLDC (Figure 3) (56–58). Importantly, many of these key processes of hepatic cystogenesis can be pharmacologically targeted, creating new possibilities for therapeutic opportunities in PLD.
Figure 3.
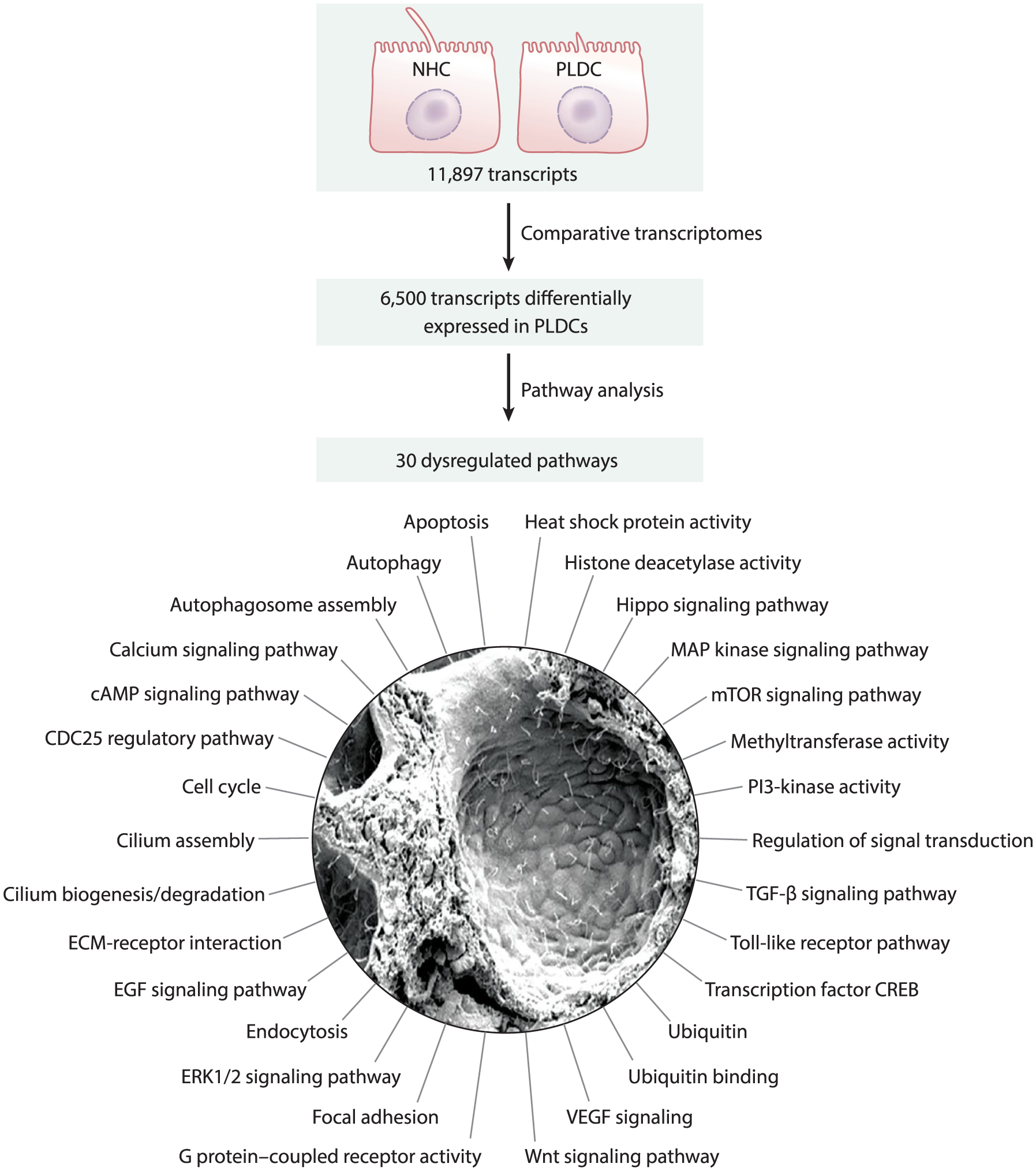
Pathways dysregulated in PLD. Transcriptome profiling of NHCs and PLDCs revealed that out of 11,897 transcripts present in both cell lines, 6,500 transcripts were differentially expressed in PLDCs (i.e., up- or downregulated). Functional annotation clustering of these transcripts by the Kyoto Encyclopedia of Genes and Genomes pathways indicates 30 altered pathways in PLDCs. Abbreviations: cAMP, cyclic adenosine monophosphate; CDC25, cell division cycle 25; CREB, cAMP-response element-binding protein; ECM, extracellular matrix; EGF, endothelial growth factor; ERK, extracellular signal-regulated kinase; MAP, mitogen-activated protein; mTOR mammalian target of rapamycin; NHC, normal human cholangiocyte; PI3, phosphoinositide 3; PLD, polycystic liver disease; PLDC, polycystic liver disease cholangiocyte; TGF, transforming growth factor; VEGF, vascular endothelial growth factor. Scanning electron microscope image adapted with permission from Reference 22.
THERAPEUTIC INTERVENTION
The treatment options for PLD depend on the severity of disease. Individuals without symptoms undergo conservative management (i.e., watchful waiting). Treatment of symptomatic patients includes nonsurgical and surgical options. Classification systems by Gigot and Schnelldorfer are commonly used to characterize the severity of PLD and to outline a potential treatment (Figure 4). In Gigot’s classification scheme, there are three types of PLD. Type I is defined by the presence of fewer than 10 hepatic cysts that are more than 10 cm in diameter; type II is defined by multiple cysts with large areas of noncystic parenchyma; and type III is defined by small- and medium-sized cysts with only a few areas of unaffected parenchyma (59). Schnelldorfer’s classification distinguishes four types of PLD. Type A is characterized by no or mild symptoms and cysts of any size. In type B, symptoms are moderate to severe; large and small cysts are present with multiple areas of noncystic parenchyma. Type C is characterized by severe symptoms; large and small cysts are present with reduced areas of noncystic parenchyma. Individuals with type D have severe symptoms; large and small cysts are present with significantly reduced areas of noncystic parenchyma (59, 60). Individuals with mild symptoms may experience back or flank pain. Moderate symptoms affect quality of life and include early satiety; nausea; bloating; gastroesophageal reflux; dyspnea; and abdomen, flank, or back pain. Individuals with severe PLD may experience liver displacement, stomach compression cause, early satiety, weight loss, and sarcopenia (61).
Figure 4.
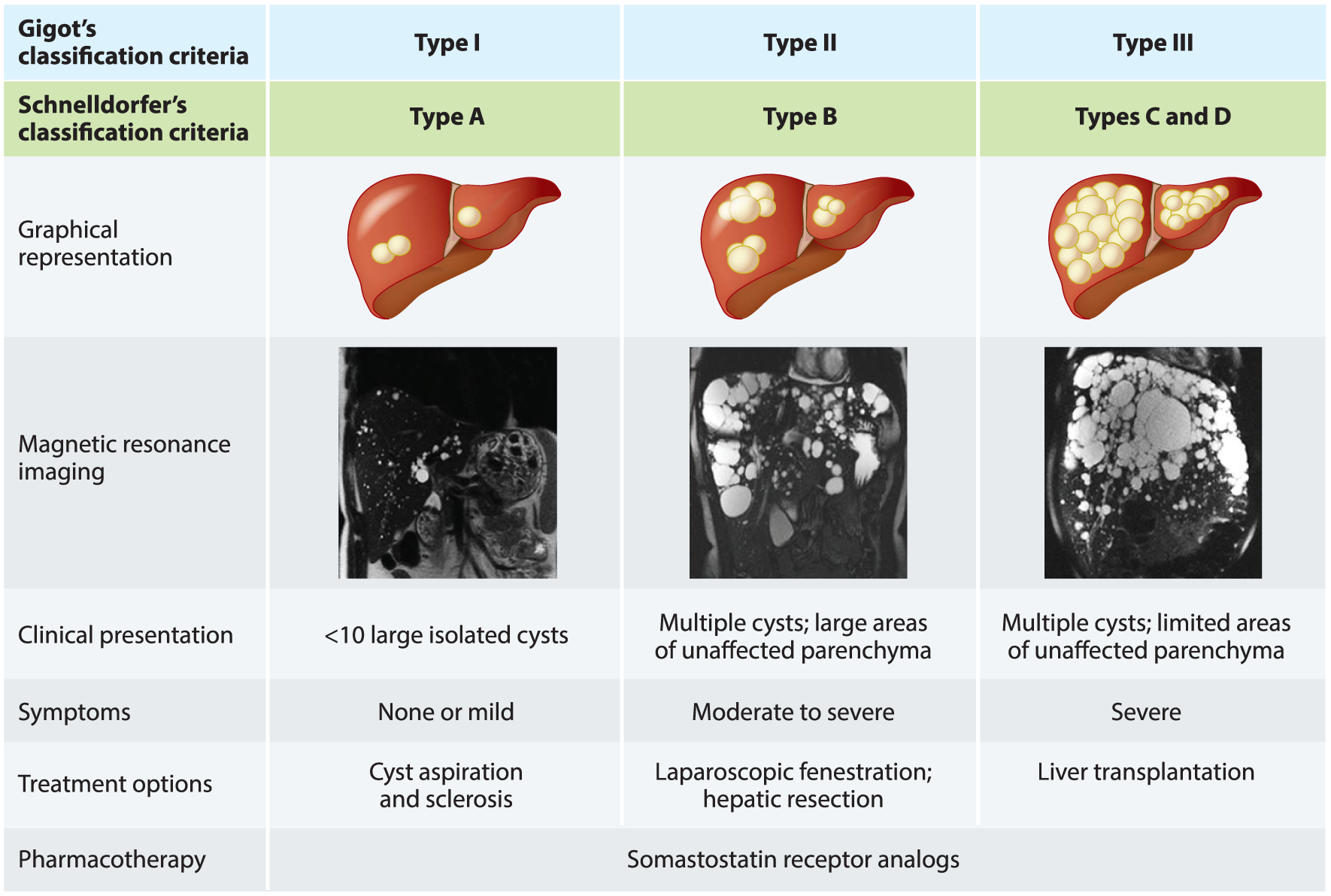
Classification criteria for polycystic liver disease severity, clinical presentation, symptoms, and treatment options. Gigot and Schnelldorfer classifications are based on the number and size of the cysts and the remaining liver parenchyma. Abdominal magnetic resonance imaging images provided by Dr. M. Hogan.
In patients with Gigot type I PLD, cyst aspiration and sclerosis are the recommended treatment. The success of the procedure correlates with the cyst size. Laparoscopic fenestration of hepatic cysts (a less common procedure since the results are often short lived) is used in patients with multiple cysts in Gigot types I and II or Schnelldorfer type B PLD (62, 63). In individuals with Gigot type II or Schnelldorfer type C, the treatment includes combined hepatic resection and fenestration. A liver transplantation is reserved for patients with impaired liver function or for whom a liver resection is not feasible (Gigot type III or Schnelldorfer type D) (60). PLD patients are frequently treated with multiple therapeutic options and are often treated more than once during the disease course (15). As of today, off-label use of somatostatin analogs is the only pharmacological option in symptomatic patients for whom surgical intervention is technically challenging or not justified (24, 64, 65).
Progress in the understanding of PLD pathogenic mechanisms has led to the identification of multiple molecular targets that have been explored in clinical and preclinical trials (Figure 5).
Figure 5.
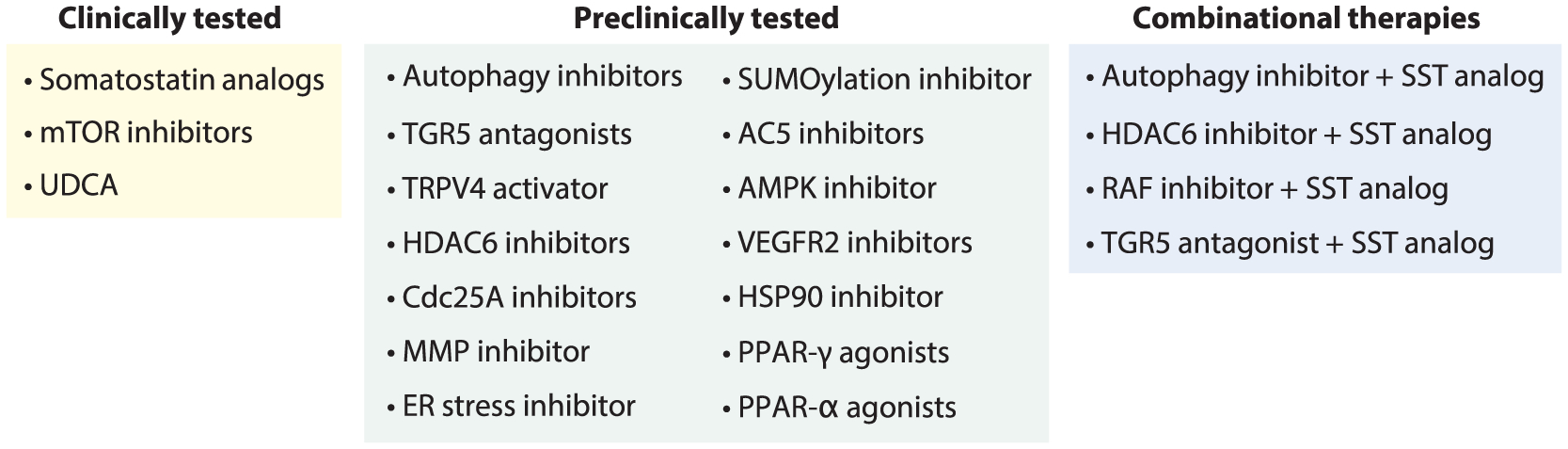
Therapeutic interventions in PLD. Abbreviations: AC5, adenylyl cyclase 5; AMPK, AMP-activated protein kinase; Cdc25A, cell division cycle 25A; ER, endoplasmic reticulum; HDAC, histone deacetylase; HSP, heat shock protein; MMP, matrix metalloproteinase; mTOR, mammalian target of rapamycin; PLD, polycystic liver disease; PPAR peroxisome proliferator-activated receptor; RAF, rapidly accelerated fibrosarcoma; SST, somatostatin; SUMO, small ubiquitin-like modifier; TGR5, Takeda G protein–coupled receptor 5; TRPV, transient receptor potential vanilloid; UDCA, ursodeoxycholic acid; VEGFR, vascular endothelial growth factor receptor.
CLINICALLY TESTED PHARMACOTHERAPIES
Somatostatin Analogs
Five Gαi protein–coupled somatostatin (SST) receptors (SSTR1 to SSTR5) are expressed in cholangiocytes (66, 67). Activation of the SST receptors in healthy cholangiocytes decreases cAMP, fluid secretion, and cell proliferation, the major cellular processes implicated in hepatic cystogenesis. Due to a short half-life of SST (~1–3 min), SST analogs with prolonged half-life [i.e., octreotide (~2 h), lanreotide (~2 h), and pasireotide (~12 h)] have been developed. Octreotide and lanreotide have a high affinity to SSTR2 and SSTR3 and a moderate affinity to SSTR5. Pasireotide binds with a high affinity to SSTR1, SSTR2, SSTR3, and SSTR5 (66, 67).
Our group was the first to demonstrate the promising role of SST analogs in PLD. We found that octreotide inhibits growth of hepatic cysts in PCK rats by decreasing cholangiocyte proliferation and cAMP levels (66). Comparative studies in PCK rats also demonstrated that pasireotide suppressed hepatic cystogenesis more effectively than octreotide (67).
A number of clinical trials evaluated the therapeutic effects of octreotide and lanreotide in PLD (19, 20, 68, 69). Collectively, these trials established that (a) the liver volume is decreased by 1.4% to 4.95% after 6–12 months of treatment; (b) the reduction in liver volume is maintained for 24 months posttherapy; (c) little or no further reduction in liver volume is noted upon continued treatment; (d) when the treatment is stopped, the liver volume begins to increase; (e) SST analogs are more effective in patients with the most severe PLD; (f) some patients do not respond to the treatment; (g) adverse events, such as abdominal pain and diarrhea, are well tolerated and quickly resolved; and (h) the quality of life is improved. Incidences of cholelithiasis, cholecystitis, and hepatic cyst infections have been reported in fewer than 5% of lanreotide-treated individuals (18, 19, 65, 70–72). As of today, off-label use of octreotide and lanreotide is the only available pharmacotherapy for individuals with PLD.
The effects of pasireotide have also been evaluated in patients with severe PLD in a randomized trial. Pasireotide reduced liver volume by ~3% but did not improve the quality of life, likely due to the high rates of diabetes and hyperglycemia (73). Another small trial showed that pasireotide administered 2 weeks before and 2 weeks after aspiration sclerotherapy did not affect cyst reduction (74).
Mammalian Target of Rapamycin Inhibitors
Mammalian target of rapamycin (mTOR) regulates multiple cellular processes including cell proliferation, metabolism, protein synthesis, and autophagy. In PLDC, mTOR is overexpressed (75). An mTOR inhibitor, rapamycin, attenuated hepatic cyst grown in Pkd2KO mice, an animal model of PLD (76). In PCK rats, sirolimus (formerly rapamycin) failed to reduce hepatic cystogenesis, whereas another mTOR inhibitor, everolimus, prevented cyst enlargement (77, 78).
Sirolimus and everolimus were also examined in clinical trials. In an individual with ADPKD-associated PLD, sirolimus decreased liver volume by ~12% (79). Comparative studies in PLD patients that assessed the reduction of the liver volume in response to octreotide alone or in combination with everolimus showed no additive effects of everolimus (80). Thus, the role of mTOR inhibitors in PLD treatment remains unclear.
Ursodeoxycholic Acid
Ursodeoxycholic acid (UDCA), an endogenous bile acid with choleretic and hepatoprotective properties, halted cyst growth in patients with enlarged polycystic livers (81). In a randomized clinical trial in individuals with severe ADPKD-associated PLD, UDCA delayed the growth of hepatic cysts after 24 weeks of treatment (82).
THERAPEUTIC TARGETS TESTED IN ANIMAL MODELS OF PLD
Autophagy
Autophagy is altered in PLDC, contributing to hepatic cyst growth. Molecular and pharmacologic intervention in autophagy reduced the proliferation of PLDC cholangiocytes in vitro and the growth of hepatic cysts in three-dimensional cultures. Hydroxychloroquine, an autophagy inhibitor, efficiently decreased hepatic cystogenesis in PCK rats (56).
TGR5
TGR5 is a cAMP-linked GPCR activated by bile acids. Our group was the first to report overexpression of TGR5 in PLDC and an elevated concentration of TGR5 ligands in the cystic fluid (54, 55). Activation of TGR5 in PLDC further increased cAMP and proliferation. In PCK rats, a TGR5 agonist, oleanolic acid, worsened hepatic cystogenesis. In contrast, genetic elimination of Tgr5 reduced hepatic cystogenesis in Tgr5−/− :Pkhd1del2/del2 mice (54). These data suggest to us that TGR5 inhibition is a promising approach in PLD treatment. Indeed, we showed that novel TGR5 antagonists developed by us effectively decreased cAMP levels, cholangiocyte proliferation, and cyst growth in in vitro and in vivo models of PLD (54).
Transient Receptor Potential Vanilloid 4
TRP vanilloid 4 (TRPV4), a calcium channel, is expressed in cilia of healthy cholangiocytes and functions as a chemosensor (83, 84). In PLDC, TRPV4 is overexpressed and mislocalized, being present intracellularly rather than in cilia. A specific TRPV4 activator, GSK1016790A, restored [Ca2+]i in PLDC, decreasing proliferation and cyst growth in vitro. However, only marginally reduced hepatic cystogenesis was observed in GSK1016790A-treated PCK rats (84).
Histone Deacetylase 6
Histone deacetylase 6 (HDAC6) is overexpressed in human and rodent PLDC. Several HDAC6 inhibitors (i.e., tubastatin A, tubacin, ACY1215, ACY738, and ACY241) and a pan-HDAC inhibitor, panobinostat, decreased hepatic cystogenesis in PCK rats by reducing cholangiocyte proliferation (85, 86).
Cell Cycle Protein CDC25A
Cell cycle–related proteins, including the master cell cycle regulator CDC25A, are overexpressed in PLDC (57). Genetic reduction of Cdc25a decreased cell proliferation and hepatic cystogenesis in Cdc25a−/−:Pkhd1del2/del2 mice (5, 51, 87). Two CDC25A inhibitors, menadione and PM-20, also suppressed hepatic cystogenesis in PCK rats by reducing Cdc25A activity (87).
Extracellular Matrix Remodeling and Matrix Metalloproteinases
Extracellular matrix (ECM) remodeling mediated by matrix metalloproteinases (MMPs) is essential for cyst expansion. Expression of MMP-3 is increased in PLDC. In PKC rats, the MMP-3 inhibitor marimastat decreased hepatic cystogenesis by reducing MMP hyperactivity (88). However, in clinical trials, marimastat showed musculoskeletal toxicity and thus does not appear to be a feasible therapy for PLD.
Endoplasmic Reticulum Stress
PLDCs are characterized by altered proteostasis (i.e., synthesis, folding, trafficking, and degradation of proteins), a markedly enlarged ER lumen, and hyperactivation of the proteasome. 4-phenyl butyric acid (4-PBA), an ER stress inhibitor, decreased hepatic cystogenesis in PCK rats by normalizing protein synthesis, folding, trafficking, and degradation and by reducing proteasome hyperactivity (89).
Posttranslational Protein Modification
Posttranslational modifications (including SUMOylation) are critical for proper protein functions. SUMOylation requires an enzymatic cascade consisting of heterodimer E1 activating enzyme (SAE1/UBA2), E2 conjugating enzyme (UBC9), and E3 ligase. In PCK rats, enhanced hepatic cystogenesis was associated with increased protein SUMOylation due to overexpression of SAE1, UBA2, and USB9. A UBC9-dependent SUMOylation inhibitor, S-adenosylmethionine, reduced proteosome activity, induced stress-related apoptosis, and attenuated cyst growth (90).
Adenylyl Cyclases
There are nine membrane-bound adenylyl cyclase (AC) isoforms and one cytosolic AC isoform. Cholangiocytes express at least seven ACs. One of the ACs, calcium-inhibitable AC5, has been implicated in PLD progression (91). The growth of biliary organoids formed by PC2-deficient cholangiocytes was reduced by two AC5 inhibitors, SQ22536 and NKY80. SQ22536 also suppressed cholangiocyte proliferation and attenuated hepatic cystogenesis in PC2-defective mice (92).
AMP-Activated Protein Kinase
The phosphorylation of AMP-activated protein kinase (AMPK), an upstream regulator of mTOR and CFTR, is reduced in PCK rats (93). Metformin, a biguanide drug, attenuated hepatic cystogenesis by increasing phosphorylation of AMPK, which subsequently suppressed phosphorylation of mTOR and ERK and expression of CFTR and AQP1 (93).
Vascular Endothelial Growth Factor Receptor 2
In Pkd2WS25/− and Pkd2KO mice, SU5416, an inhibitor of vascular endothelial growth factor receptor 2 (VEGFR2) and VEGFR1, decreased cholangiocyte proliferation and hepatic cystogenesis but did not affect these processes in Pkd1KO mice; this result suggests that underlying genetic background might determine a treatment outcome (94, 95).
Heat Shock Protein 90
Heat shock protein 90 (HSP90), a molecular chaperone, is required for stability of proteins involved in cell proliferation. In cholangiocytes of Pkd1−/− mice, inhibition of overexpressed HSP90 by a small molecule inhibitor, STA-2842, reduced the levels of two HSP90 client proteins, EGFR and ERK1/2, and inhibited hepatic cystogenesis (96).
Peroxisome Proliferator-Activated Receptors
Peroxisome proliferator-activated receptor gamma (PPAR-γ), a member of a family of nuclear hormone receptors, mediates inflammation, cell proliferation, and fibrosis. Two PPAR-γ agonists (a full agonist, pioglitazone, and a partial agonist, telmisartan) reduced hepatic cystogenesis in PCK rats, presumably by decreasing transforming growth factor β expression and ERK phosphorylation (97, 98).
The expression of PPAR-α, which promotes fatty acid β-oxidation and oxidative phosphorylation, is decreased in PLDC. In Pkd1RC/RC mice, fenofibrate, a PPAR-α agonist, reduced hepatic cystogenesis by increasing expression of PPAR-α (99).
COMBINATIONAL STUDIES
Considering that disturbances in multiple intracellular pathways underlie hepatic cystogenesis, a concurrent targeting of different pathways or synergistic targeting of different entities in a single pathway is an attractive concept. In PCK rats, a simultaneous administration of pasireotide and an autophagy inhibitor, hydroxychloroquine, attenuated hepatic cystogenesis more effectively than a single-drug treatment (56). Treatment of PCK rats with an HDAC6 inhibitor, ACY-1215, and pasireotide also reduced hepatic cystogenesis to a higher degree than each drug alone (86).
Cotreatment of PCK rats with octreotide and pasireotide showed that the effects of this drug combination on liver cystogenesis were comparable with those of pasireotide-only treatment (100). An important finding of this study is that the drug combination reduced glucagon concentration, since hyperglycemia caused by pasireotide is an adverse clinical effect (73). ATGR5 antagonist, SBI-115, and pasireotide together decreased cAMP levels, PLDC proliferation, and cyst growth in vitro more efficiently than each drug alone, demonstrating an additive beneficial effect on PLD (54).
CONCLUSION
Despite genetic diversity, PLD is characterized by development and progressive growth of hepatic cysts. Clinical management of PLD depends on disease severity and includes watchful waiting, nonsurgical and surgical interventions, and limited drug options. Significant advances in our understanding of the genetics and pathogenic mechanisms of PLD have triggered identification of novel therapeutic targets, which have been tested and shown promise in vitro and in vivo, in both preclinical studies and clinical trials.
ACKNOWLEDGMENTS
This work was supported by grants from the National Institutes of Health (DK24031), the Mayo Clinic, the Clinical and Optical Microscopy Cores of the Mayo Clinic Center for Cell Signaling in Gastroenterology (P30DK084567), and the Mayo Translational Polycystic Kidney Disease (PKD) Center (NIDDK P30DK090728); by a Mayo Translational PKD Center Pilot and Feasibility Award; and by the Eileen Creamer O’Neill Award from the PKD Foundation.
Footnotes
DISCLOSURE STATEMENT
T.V.M. and N.F.L. are inventors on US Patent 8,232,241 B2 for treating liver diseases.
LITERATURE CITED
- 1.Norio R 1983. Polycystic disease of liver: an entity of its own or not? Clin. Genet 23:78–79 [DOI] [PubMed] [Google Scholar]
- 2.Neijenhuis MK, Kievit W, Verheesen SM, D’Agnolo HM, Gevers TJ, Drenth JP. 2018. Impact of liver volume on polycystic liver disease-related symptoms and quality of life. United Eur. Gastroenterol. J 6:81–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barten TRM, Bernts LHP, Drenth JPH, Gevers TJG. 2020. New insights into targeting hepatic cystogenesis in autosomal dominant polycystic liver and kidney disease. Expert Opin. Ther. Targets 24:589–99 [DOI] [PubMed] [Google Scholar]
- 4.van Aerts RMM, Kolkman M, Kievit W, Gevers TJG, Nevens F, Drenth JPH. 2018. Drug holiday in patients with polycystic liver disease treated with somatostatin analogues. Therap. Adv. Gastroenterol 11. 10.1177/1756284818804784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Masyuk TV, Masyuk AI, LaRusso NF. 2017. Therapeutic targets in polycystic liver disease. Curr. Drug Targets 18:950–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Abu-Wasel B, Walsh C, Keough V, Molinari M. 2013. Pathophysiology, epidemiology, classification and treatment options for polycystic liver diseases. World J. Gastroenterol 19:5775–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Masyuk T, Masyuk A, LaRusso N. 2009. Cholangiociliopathies: genetics, molecular mechanisms and potential therapies. Curr. Opin. Gastroenterol 25:265–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cornec-Le Gall E, Torres VE, Harris PC. 2018. Genetic complexity of autosomal dominant polycystic kidney and liver diseases. J. Am. Soc. Nephrol 29:13–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.van de Laarschot LFM, Drenth JPH. 2018. Genetics and mechanisms of hepatic cystogenesis. Biochim. Biophys. Acta Mol. Basis Dis 1864:1491–97 [DOI] [PubMed] [Google Scholar]
- 10.Lee-Law PY, van de Laarschot LFM, Banales JM, Drenth JPH. 2019. Genetics of polycystic liver diseases. Curr. Opin. Gastroenterol 35:65–72 [DOI] [PubMed] [Google Scholar]
- 11.Gevers TJ, Drenth JP. 2013. Diagnosis and management of polycystic liver disease. Nat. Rev. Gastroenterol. Hepatol 10:101–8 [DOI] [PubMed] [Google Scholar]
- 12.Tahvanainen E,Tahvanainen P, Kaariainen H, Hockerstedt K 2005. Polycystic liver and kidney diseases. Ann. Med 37:546–55 [DOI] [PubMed] [Google Scholar]
- 13.Lasagni A, Cadamuro M, Morana G, Fabris L, Strazzabosco M. 2021. Fibrocystic liver disease: novel concepts and translational perspectives. Transl. Gastroenterol. Hepatol 6:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hoevenaren IA, Wester R, Schrier RW, McFann K, Doctor RB, et al. 2008. Polycystic liver: clinical characteristics of patients with isolated polycystic liver disease compared with patients with polycystic liver and autosomal dominant polycystic kidney disease. Liver Int. 28:264–70 [DOI] [PubMed] [Google Scholar]
- 15.Van Keimpema L, De Koning DB, Van Hoek B, Van Den Berg AP, Van Oijen MG, et al. 2011. Patients with isolated polycystic liver disease referred to liver centres: clinical characterization of 137 cases. Liver Int. 31:92–98 [DOI] [PubMed] [Google Scholar]
- 16.Qian Q 2010. Isolated polycystic liver disease. Adv. Chronic Kidney Dis 17:181–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Waanders E, van Keimpema L, Brouwer JT, van Oijen MG, Aerts R, et al. 2009. Carbohydrate antigen 19–9 is extremely elevated in polycystic liver disease. Liver Int. 29:1389–95 [DOI] [PubMed] [Google Scholar]
- 18.Hogan MC, Masyuk TV, Page L, Holmes DR 3rd, Li X, et al. 2012. Somatostatin analog therapy for severe polycystic liver disease: results after 2 years. Nephrol. Dial. Transplant 27:3532–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.van Keimpema L, Nevens F, Vanslembrouck R, van Oijen MG, Hoffmann AL, et al. 2009. Lanreotide reduces the volume of polycystic liver: a randomized, double-blind, placebo-controlled trial. Gastroenterology 137:1661–68.e2 [DOI] [PubMed] [Google Scholar]
- 20.Hogan MC, Masyuk TV, Page LJ, Kubly VJ, Bergstralh EJ, et al. 2010. Randomized clinical trial of long-acting somatostatin for autosomal dominant polycystic kidney and liver disease. J. Am. Soc. Nephrol 21:1052–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bergmann C, Guay-Woodford LM, Harris PC, Horie S, Peters DJM, Torres VE. 2018. Polycystic kidney disease. Nat. Rev. Dis. Primers 4:50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Masyuk TV, Masyuk AI, LaRusso NF. 2018. Polycystic liver disease: the interplay of genes causative for hepatic and renal cystogenesis. Hepatology 67:2462–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Perugorria MJ, Masyuk TV, Marin JJ, Marzioni M, Bujanda L, et al. 2014. Polycystic liver diseases: advanced insights into the molecular mechanisms. Nat. Rev. Gastroenterol. Hepatol 11:750–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Boerrigter MM, Bongers E, Lugtenberg D, Nevens F, Drenth JPH. 2021. Polycystic liver disease genes: practical considerations for genetic testing. Eur. J. Med. Genet 64:104160. [DOI] [PubMed] [Google Scholar]
- 25.Harris PC, Torres VE. 2002. Polycystic kidney disease, autosomal dominant. In GeneReviews, ed. Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, et al. Seattle, WA: Univ. Wash. [Google Scholar]
- 26.Mikolajczyk AE, Te HS, Chapman AB. 2017. Gastrointestinal manifestations of autosomal-dominant polycystic kidney disease. Clin. Gastroenterol. Hepatol 15:17–24 [DOI] [PubMed] [Google Scholar]
- 27.Sussman CR, Wang X, Chebib FT, Torres VE. 2020. Modulation of polycystic kidney disease by G-protein coupled receptors and cyclic AMP signaling. Cell. Signal 72:109649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Porath B, Gainullin VG, Cornec-Le Gall E, Dillinger EK, Heyer CM, et al. 2016. Mutations in GANAB, encoding the glucosidase IIα subunit, cause autosomal-dominant polycystic kidney and liver disease. Am. J. Hum. Genet 98:1193–207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Besse W, Choi J, Ahram D, Mane S, Sanna-Cherchi S, et al. 2018. A noncoding variant in GANAB explains isolated polycystic liver disease (PCLD) in a large family. Hum. Mutat 39:378–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Besse W, Dong K, Choi J, Punia S, Fedeles SV, et al. 2017. Isolated polycystic liver disease genes define effectors of polycystin-1 function. J. Clin. Investig 127:1772–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cornec-Le Gall E, Olson RJ, Besse W, Heyer CM, Gainullin VG, et al. 2018. Monoallelic mutations to DNAJB11 cause atypical autosomal-dominant polycystic kidney disease. Am. J. Hum. Genet 102:832–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Besse W, Chang AR, Luo JZ, Triffo WJ, Moore BS, et al. 2019. ALG9 mutation carriers develop kidney and liver cysts. J. Am. Soc. Nephrol 30:2091–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cornec-Le Gall E, Alam A, Perrone RD. 2019. Autosomal dominant polycystic kidney disease. Lancet 393:919–35 [DOI] [PubMed] [Google Scholar]
- 34.Chebib FT, Hogan MC, El-Zoghby ZM, Irazabal MV, Senum SR et al. 2017. Autosomal dominant polycystic kidney patients may be predisposed to various cardiomyopathies. Kidney Int. Rep 2:913–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lu H, Galeano MCR, Ott E, Kaeslin G, Kausalya PJ, et al. 2017. Mutations in DZIP1L, which encodes a ciliary-transition-zone protein, cause autosomal recessive polycystic kidney disease. Nat. Genet 49:1025–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Buscher R, Buscher AK, Weber S, Mohr J, Hegen B, et al. 2014. Clinical manifestations of autosomal recessive polycystic kidney disease (ARPKD): kidney-related and non-kidney-related phenotypes. Pediatr. Nephrol 29:1915–25 [DOI] [PubMed] [Google Scholar]
- 37.Onuchic LF, Furu L, Nagasawa Y, Hou X, Eggermann T, et al. 2002. PKHD1, the polycystic kidney and hepatic disease 1 gene, encodes a novel large protein containing multiple immunoglobulin-like plexin-transcription-factor domains and parallel beta-helix 1 repeats. Am. J. Hum. Genet 70:1305–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cnossen WR, te Morsche RH, Hoischen A Gilissen C, Chrispijn M, et al. 2014. Whole-exome sequencing reveals LRP5 mutations and canonical Wnt signaling associated with hepatic cystogenesis. PNAS 111:5343–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cnossen WR, te Morsche RH, Hoischen A, Gilissen C, Venselaar H, et al. 2016. LRP5 variants may contribute to ADPKD. Eur. J. Hum. Genet 24:237–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Drenth JP, te Morsche RH, Smink R Bonifacino JS, Jansen JB. 2003. Germline mutations in PRKCSH are associated with autosomal dominant polycystic liver disease. Nat. Genet 33:345–47 [DOI] [PubMed] [Google Scholar]
- 41.Drenth JP, Martina JA, te Morsche RH, Jansen JB, Bonifacino JS. 2004. Molecular characterization of hepatocystin, the protein that is defective in autosomal dominant polycystic liver disease. Gastroenterology 126:1819–27 [DOI] [PubMed] [Google Scholar]
- 42.Davila S, Furu L, Gharavi AG, Tian X, Onoe T, et al. 2004. Mutations in SEC63 cause autosomal dominant polycystic liver disease. Nat. Genet 36:575–77 [DOI] [PubMed] [Google Scholar]
- 43.Waanders E, te Morsche RH, de Man RA, Jansen JB, Drenth JP. 2006. Extensive mutational analysis of PRKCSH and SEC63 broadens the spectrum of polycystic liver disease. Hum. Mutat 27:830. [DOI] [PubMed] [Google Scholar]
- 44.Masyuk TV, Huang BQ, Masyuk AI, Ritman EL, Torres VE, et al. 2004. Biliary dysgenesis in the PCK rat, an orthologous model of autosomal recessive polycystic kidney disease. Am. J. Pathol 165:1719–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stroope A Radtke B, Huang B, Masyuk T, Torres V, et al. 2010. Hepato-renal pathology in Pkd2ws25/− mice, an animal model of autosomal dominant polycystic kidney disease. Am. J. Pathol 176(3):1282–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Masyuk AI, Gradilone SA, Banales JM, Huang BQ, Masyuk TV, et al. 2008. Cholangiocyte primary cilia are chemosensory organelles that detect biliary nucleotides via P2Y12 purinergic receptors. Am. J. Physiol. Gastrointest. Liver Physiol 295:G725–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Masyuk AI, Masyuk TV, LaRusso NF. 2008. Cholangiocyte primary cilia in liver health and disease. Dev. Dyn 237:2007–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Masyuk AI, Masyuk TV, Splinter PL, Huang BQ, Stroope AJ, LaRusso NF. 2006. Cholangiocyte cilia detect changes in luminal fluid flow and transmit them into intracellular Ca2+ and cAMP signaling. Gastroenterology 131:911–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nauli SM, Alenghat FJ, Luo Y, Williams E, Vassilev P, et al. 2003. Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat. Genet 33:129–37 [DOI] [PubMed] [Google Scholar]
- 50.Douguet D, Patel A, Honore E. 2019. Structure and function of polycystins: insights into polycystic kidney disease. Nat. Rev. Nephrol 15:412–22 [DOI] [PubMed] [Google Scholar]
- 51.Masyuk TV, Lee SO, Radtke BN, Stroope AJ, Huang B, et al. 2014. Centrosomal abnormalities characterize human and rodent cystic cholangiocytes and are associated with Cdc25A overexpression. Am. J. Pathol 184:110–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Calvet JP. 2015. The role of calcium and cyclic AMP in PKD. In Polycystic Kidney Disease, ed. Li X, pp. 169–96. Brisbane, Aust.: Codon; [PubMed] [Google Scholar]
- 53.Masyuk TV, Masyuk AI, LaRusso NF. 2015. TGR5 in the cholangiociliopathies. Dig. Dis 33:420–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Masyuk TV, Masyuk AI, Lorenzo Pisarello M, Howard BN, Huang BQ, et al. 2017. TGR5 contributes to hepatic cystogenesis in rodents with polycystic liver diseases through cyclic adenosine monophosphate/Gαs signaling. Hepatology 66:1197–218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Munoz-Garrido P, Marin JJ, Perugorria MJ, Urribarri AD, Erice O, et al. 2015. Ursodeoxycholic acid inhibits hepatic cystogenesis in experimental models of polycystic liver disease. J. Hepatol 63:952–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Masyuk AI, Masyuk TV, Lorenzo Pisarello MJ, Ding JF, Loarca L, et al. 2018. Cholangiocyte autophagy contributes to hepatic cystogenesis in polycystic liver disease and represents a potential therapeutic target. Hepatology 67:1088–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lee SO, Masyuk T, Splinter P, Banales JM, Masyuk A, et al. 2008. MicroRNA15a modulates expression of the cell-cycle regulator Cdc25A and affects hepatic cystogenesis in a rat model of polycystic kidney disease. J. Clin. Investig 118:3714–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Masyuk T, Masyuk A, LaRusso N. 2009. MicroRNAs in cholangiociliopathies. Cell Cycle 8:1324–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gigot JF, Jadoul P, Que F, Van Beers BE, Etienne J, et al. 1997. Adult polycystic liver disease: Is fenestration the most adequate operation for long-term management? Ann. Surg 225:286–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schnelldorfer T, Torres VE, Zakaria S, Rosen CB, Nagomey DM. 2009. Polycystic liver disease: a critical appraisal of hepatic resection, cyst fenestration, and liver transplantation. Ann. Surg 250:112–18 [DOI] [PubMed] [Google Scholar]
- 61.van Aerts RMM, van de Laarschot LFM, Banales JM, Drenth JPH. 2018. Clinical management of polycystic liver disease. J. Hepatol 68:827–37 [DOI] [PubMed] [Google Scholar]
- 62.Torres VE. 2007. Treatment of polycystic liver disease: One size does not fit all. Am. J. Kidney Dis 49:725–28 [DOI] [PubMed] [Google Scholar]
- 63.Drenth JP, Chrispijn M, Nagorney DM, Kamath PS, Torres VE. 2010. Medical and surgical treatment options for polycystic liver disease. Hepatology 52:2223–30 [DOI] [PubMed] [Google Scholar]
- 64.Takenaka T, Miura S, Kitajima M. 2020. The management of polycystic liver disease by tolvaptan. Clin. Mol. Hepatol 26:70–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Griffiths J, Mills MT, Ong AC. 2020. Long-acting somatostatin analogue treatments in autosomal dominant polycystic kidney disease and polycystic liver disease: a systematic review and meta-analysis. BMJ Open 10:e032620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Masyuk TV, Masyuk AI, Torres VE, Harris PC, Larusso NF. 2007. Octreotide inhibits hepatic cystogenesis in a rodent model of polycystic liver disease by reducing cholangiocyte adenosine 3′,5′-cyclic monophosphate. Gastroenterology 132:1104–16 [DOI] [PubMed] [Google Scholar]
- 67.Masyuk TV, Radtke BN, Stroope AJ, Banales JM, Gradilone SA, et al. 2013. Pasireotide is more effective than octreotide in reducing hepatorenal cystogenesis in rodents with polycystic kidney and liver diseases. Hepatology 58:409–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chrispijn M, Nevens F, Gevers TJ, Vanslembrouck R, van Oijen MG, et al. 2012. The long-term outcome of patients with polycystic liver disease treated with lanreotide. Aliment Pharmacol. Ther 35:266–74 [DOI] [PubMed] [Google Scholar]
- 69.Gevers TJ, Hol JC, Monshouwer R, Dekker HM, Wetzels JF, Drenth JP. 2015. Effect of lanreotide on polycystic liver and kidneys in autosomal dominant polycystic kidney disease: an observational trial. Liver Int. 35:1607–14 [DOI] [PubMed] [Google Scholar]
- 70.Hogan MC, Masyuk T, Bergstralh E, Li B, Kremers WK, et al. 2015. Efficacy of 4 years of octreotide long-acting release therapy in patients with severe polycystic liver disease. Mayo Clin. Proc 90:1030–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Neijenhuis MK, Gevers TJ, Nevens F, Hogan MC, Torres VE, et al. 2015. Somatostatin analogues improve health-related quality of life in polycystic liver disease: a pooled analysis of two randomised, placebo-controlled trials. Aliment Pharmacol. Ther 42:591–98 [DOI] [PubMed] [Google Scholar]
- 72.Pisani A, Sabbatini M, Imbriaco M, Riccio E, Rubis N, et al. 2016. Long-term effects of octreotide on liver volume in patients with polycystic kidney and liver disease. Clin. Gastroenterol. Hepatol 14:1022–30.e4 [DOI] [PubMed] [Google Scholar]
- 73.Hogan MC, Chamberlin JA, Vaughan LE, Waits AL, Banks C, et al. 2020. Pansomatostatin agonist pasireotide long-acting release for patients with autosomal dominant polycystic kidney or liver disease with severe liver involvement: a randomized clinical trial. Clin. J. Am. Soc. Nephrol 15:1267–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wijnands TFM, Gevers TJG, Lantinga MA, te Morsche RH, Schultze Kool LJ, Drenth JPH. 2018. Pasireotide does not improve efficacy of aspiration sclerotherapy in patients with large hepatic cysts, a randomized controlled trial. Eur. Radiol 28:2682–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ren XS, Sato Y, Harada K, Sasaki M, Furubo S, et al. 2014. Activation of the PI3K/mTOR pathway is involved in cystic proliferation of cholangiocytes of the PCK rat. PLOS ONE 9:e87660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Spirli C, Okolicsanyi S, Fiorotto R, Fabris L, Cadamuro M, et al. 2010. Mammalian target of rapamycin regulates vascular endothelial growth factor-dependent liver cyst growth in polycystin-2-defective mice. Hepatology 51:1778–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Temmerman F, Chen F, Libbrecht L, Vander Elst I, Windmolders P, et al. 2017. Everolimus halts hepatic cystogenesis in a rodent model of polycystic-liver-disease. World J. Gastroenterol 23:5499–507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Renken C, Fischer DC, Kundt G, Gretz N, Haffner D. 2011. Inhibition of mTOR with sirolimus does not attenuate progression of liver and kidney disease in PCK rats. Nephrol. Dial. Transplant 26:92–100 [DOI] [PubMed] [Google Scholar]
- 79.Qian Q, Du H, King BF, Kumar S, Dean PG, et al. 2008. Sirolimus reduces polycystic liver volume in ADPKD patients. J. Am. Soc. Nephrol 19:631–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Chrispijn M, Gevers TJ, Hol JC, Monshouwer R, Dekker HM, Drenth JP. 2013. Everolimus does not further reduce polycystic liver volume when added to long acting octreotide: results from a randomized controlled trial. J. Hepatol 59:153–59 [DOI] [PubMed] [Google Scholar]
- 81.Iijima T, Hoshino J, Suwabe T, Sumida K, Mise K, et al. 2016. Ursodeoxycholic acid for treatment of enlarged polycystic liver. Ther. Apher. Dial 20:73–78 [DOI] [PubMed] [Google Scholar]
- 82.D’Agnolo HM, Kievit W, Takkenberg RB, Riano I, Bujanda L, et al. 2016. Ursodeoxycholic acid in advanced polycystic liver disease: a phase 2 multicenter randomized controlled trial. J. Hepatol 65:601–7 [DOI] [PubMed] [Google Scholar]
- 83.Gradilone SA, Masyuk AI, Splinter PL, Banales JM, Huang BQ, et al. 2007. Cholangiocyte cilia express TRPV4 and detect changes in luminal tonicity inducing bicarbonate secretion. PNAS 104:19138–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Gradilone SA, Masyuk TV, Huang BQ, Banales JM, Lehmann GL, et al. 2010. Activation of Trpv4 reduces the hyperproliferative phenotype of cystic cholangiocytes from an animal model of ARPKD. Gastroenterology 139:304–14.e2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gradilone SA, Habringer S, Masyuk TV, Howard BN, Masyuk AI, Larusso NF. 2014. HDAC6 is overexpressed in cystic cholangiocytes and its inhibition reduces cystogenesis. Am. J. Pathol 184:600–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lorenzo Pisarello M, Masyuk TV, Gradilone SA, Masyuk AI, Ding JF, et al. 2018. Combination of a histone deacetylase 6 inhibitor and a somatostatin receptor agonist synergistically reduces hepatorenal cystogenesis in an animal model of polycystic liver disease. Am. J. Pathol 188:981–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Masyuk TV, Radtke BN, Stroope AJ, Banales JM, Masyuk AI, et al. 2012. Inhibition of Cdc25A suppresses hepato-renal cystogenesis in rodent models of polycystic kidney and liver disease. Gastroenterology 142:622–33.e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Urribarri AD, Munoz-Garrido P, Perugorria MJ, Erice O, Merino-Azpitarte M, et al. 2014. Inhibition of metalloprotease hyperactivity in cystic cholangiocytes halts the development of polycystic liver diseases. Gut 63:1658–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Santos-Laso A, Izquierdo-Sanchez L, Rodrigues PM, Huang BQ, Azkargorta M, et al. 2020. Proteostasis disturbances and endoplasmic reticulum stress contribute to polycystic liver disease: new therapeutic targets. Liver Int. 40:1670–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lee-Law PY, Olaizola P, Caballero-Camino FJ, Izquierdo-Sanchez L, Rodrigues PM, et al. 2021. Targeting UBC9-mediated protein hyper-SUMOylation in cystic cholangiocytes halts polycystic liver disease in experimental models. J. Hepatol 74:394–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Strazzabosco M, Fiorotto R,Melero S, Glaser S, Francis H, et al. 2009. Differentially expressed adenylyl cyclase isoforms mediate secretory functions in cholangiocyte subpopulation. Hepatology 50:244–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Spirli C, Mariotti V, Villani A, Fabris L, Fiorotto R, Strazzabosco M. 2017. Adenylyl cyclase 5 links changes in calcium homeostasis to cAMP-dependent cyst growth in polycystic liver disease. J. Hepatol 66:571–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Sato Y, Qiu J, Hirose T, Miura T, Sato Y, et al. 2021. Metformin slows liver cyst formation and fibrosis in experimental model of polycystic liver disease. Am. J. Physiol. Gastrointest. Liver Physiol 320(4):G464–73 [DOI] [PubMed] [Google Scholar]
- 94.Spirli C, Okolicsanyi S, Fiorotto R, Fabris L, Cadamuro M, et al. 2010. ERK1/2-dependent vascular endothelial growth factor signaling sustains cyst growth in polycystin-2 defective mice. Gastroenterology 138:360–71.e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Amura CR, Brodsky KS, Groff R, Gattone VH, Voelkel NF, Doctor RB. 2007. VEGF receptor inhibition blocks liver cyst growth in pkd2(WS25/–) mice. Am. J. Physiol. Cell Physiol 293:C419–28 [DOI] [PubMed] [Google Scholar]
- 96.Smithline ZB, Nikonova AS, Hensley HH, Cai KQ, Egleston BL, et al. 2014. Inhibiting heat shock protein 90 (HSP90) limits the formation of liver cysts induced by conditional deletion of Pkd1 in mice. PLOS ONE 9:e114403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Yoshihara D, Kurahashi H, Morita M, Kugita M, Hiki Y, et al. 2011. PPAR-γ agonist ameliorates kidney and liver disease in an orthologous rat model of human autosomal recessive polycystic kidney disease. Am. J. Physiol. Ren. Physiol 300:F465–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Yoshihara D, Kugita M, Sasaki M, Horie S, Nakanishi K, et al. 2013. Telmisartan ameliorates fibrocystic liver disease in an orthologous rat model of human autosomal recessive polycystic kidney disease. PLOS ONE 8:e81480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Lakhia R, Yheskel M, Flaten A, Quittner-Strom EB, Holland WL, Patel V. 2018. PPARα agonist fenofibrate enhances fatty acid β-oxidation and attenuates polycystic kidney and liver disease in mice. Am. J. Physiol. Ren. Physiol 314:F122–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kugita M, Nishii K, Yamaguchi T, Suzuki A, Yuzawa Y, et al. 2017. Beneficial effect of combined treatment with octreotide and pasireotide in PCK rats, an orthologous model of human autosomal recessive polycystic kidney disease. PLOS ONE 12:e0177934. [DOI] [PMC free article] [PubMed] [Google Scholar]