

Abstract
Fatty acid metabolism is essential for the biogenesis of cellular components and ATP production to sustain proliferation of cancer cells. Long chain fatty acyl-CoA synthetases (ACSLs), a group of rate-limiting enzymes in fatty acid metabolism, catalyze the bioconversion of exogenous or de novo synthesized fatty acids to their corresponding fatty acyl-CoAs. In this study, systematical analysis of ACSLs levels and the amount of fatty acyl-CoAs illustrated that ACSL1 were significantly associated with the levels of a broad spectrum of fatty acyl-CoAs, and were elevated in human prostate tumors. ACSL1 increased the biosynthesis of fatty acyl-CoAs including C16:0-, C18:0-, C18:1- and C18:2-CoA, triglycerides and lipid accumulation in cancer cells. Mechanistically, ACSL1 modulated mitochondrial respiration, β-oxidation and ATP production through regulation of CPT1 activity. Knockdown of ACSL1 inhibited the cell cycle, and suppressed the proliferation and migration of prostate cancer cells in vitro, and growth of prostate xenograft tumors in vivo. Our study implicates ACSL1 as playing an important role in prostate tumor progression, and provides a therapeutic strategy of targeting fatty acid metabolism for the treatment of prostate cancer.
Keywords: ACSL1, fatty acid metabolism, fatty acyl-CoAs, prostate cancer
INTRODUCTION
Prostate cancer (PCa) is notorious for dys-regulation of fatty acid metabolism (1, 2). Prostate cancer cells undergo metabolic reprogramming by up-regulation of the expression and activity of a variety of enzymes in de novo fatty acid synthesis (3). The dys-regulated enzymes include ATP citrate lyase, acetyl-CoA carboxylase α, fatty acid synthase, and stearoyl-CoA desaturase-1. These enzymes enhance the efficiency to convert acetyl-CoA derived from catabolic processes to long chain fatty acids (2, 3). The elevated amount of intracellular fatty acids further provides a variety of precursors for the biogenesis of cellular components in malignant cells (2).
The bioconversion of de novo synthesized or exogenous fatty acids into their corresponding acyl-CoAs is required for their participation in lipogenesis, β-oxidation, protein fatty acylation, and other biological processes (4). Our studies have demonstrated that biosynthesis of acyl-CoAs significantly influences acylation of oncogenic proteins including myristoylation of Src kinase and its oncogenic signaling (5). This process facilitates high-fat diet accelerated prostate tumor progression in vivo (6).
Long-chain acyl-CoA synthetases (ACSLs) are a group of enzymes that convert long-chain fatty acids (LCFA; 12–20 carbons) to fatty acyl-CoAs by esterification (7). The mammalian ACSL group contains five members including ACSL1, ACSL3, ACSL4, ACSL5, and ACSL6. The activity of ACSLs is essential for the proliferation of cancer cells and tumor progression. For example, inhibition of ACSLs by triacsinc C, a pan-inhibitor of ACSLs, induced apoptosis in cancer cells (8). Studies have indicated that an individual ACSL gene might be expressed in a tissue-specific manner or favor certain fatty acids as substrates (7, 9), however the biological significance of individual ACSL genes in prostate cancer progression is largely unknown.
In this study, we systematically measured expression levels of ACSL gene family members and the amount of fatty acyl-CoAs in multiple cell lines, and identified that expression levels of ACSL1 were significantly associated with a broad spectrum of fatty acyl-CoAs. We further demonstrated that ACSL1 expression levels were highly elevated in prostate tumors. Knockdown of ACSL1 inhibited prostate cancer cell proliferation and growth of xenograft tumors. Further analyses indicated that down-regulation of ACSL1 suppressed the biosynthesis of a variety of acyl-CoAs including C16:0-, C18:0-, C18:1- and C18:2-CoA, which resulted in a decrease of intracellular triglycerides and lipid accumulation, and a decrease in mitochondrial respiration, β-oxidation, ATP production, and cell cycle arrest. Our study reveals an important role of ACSL1-mediated fatty acid metabolism in the regulation of prostate tumor progression, and provides a potential therapeutic strategy for the treatment of prostate cancer.
RESULTS
Systematic analysis reveals that ACSL1 expression levels are associated with a broad spectrum of fatty acyl-CoAs levels.
Elevated fatty acid metabolism provides a variety of metabolites to support proliferation of tumorigenic cells (3). The de novo synthesized or exogenous fatty acids must be converted into acyl-CoAs to further participate in metabolic pathways (9). The bioconversion of fatty acids to acyl-CoAs is catalyzed by the ACSL family of genes including ACSL1, 3, 4, 5, and 6 (7). To identify which ACSL member is the most important player in contributing to fatty acyl-CoAs levels, the correlation of relative ACSLs mRNA levels and the amount of acyl-CoAs in multiple cell lines were analyzed. A multiple linear regression was employed to analyze the association between the amount of individual acyl-CoAs and expression levels of ACSL family genes. The amount of individual acyl-CoAs was treated as the response variable, and the expression levels of ACSL1, ACSL3, ACSL4, ACSL5, and ACSL6 were treated as covariates. The linear regression corresponding to 8 different acyl-CoAs were established. Expression levels of ACSL1 were significantly correlated with a wide spectrum of acyl-CoAs including 10:0-, 12:0-, 14:0-, 16:0-, 18:0-, 18:1-, 18:2- and 20:0-CoA (Fig. 1A–G). Additionally, the regression coefficients indicated that expression levels of ACSL1 contributed to the levels of 14:0-, 16:0-, 18:0-, 18:1-, and 18:2-CoAs to a greater degree than other fatty acyl-CoAs (Fig. 1G).
Figure 1. Correlation of ACSL1 expression with acyl-CoA levels and human prostate tumors.

(A-F) Systematic analysis of relative mRNA levels of ACSL family genes with the amount of fatty acyl-CoAs. Expression levels of ACSL1, 3, 4, 5, and 6 mRNA were examined in prostate cells including PNT2, LNCaP, 22Rv1, PC-3 and DU145, and hepatic cancer cells (HepG2 and Hep3B) by RT-PCR. To compare ACSLs mRNA levels among different cell lines, relative ACSLs mRNA levels (ACSLs/GAPDH) in a cell line were normalized to those in the PNT2 cells (a non-cancer prostate cell line). The relative mRNA level of ACSL1/GAPDH in PNT2 cells was set as 1. The absolute amount of C10:0-, C12:0-, C14:0-, C16:0-, C18:0-, C18:1-, C18:2-, C20:0-, and C20:4-CoA was analyzed by LC-MS/MS and normalized to total cellular protein (pmol/mg protein). Relative mRNA expression levels of each ACSL gene and individual acyl-CoA levels were analyzed by multiple linear regression. The expression levels of ACSL1 significantly correlated with C10:0-, C12:0-, C14:0-, C16:0-, C18:0-, C18:1-, C18:2-, and C20:0-CoAs (C10:0 and C14:0-CoAs regression figures not shown). The blue circles indicate the analyzed cell line and the solid lines are the regression lines. (G) The correlation of expression levels of ACSL 1, 3, 4, 5, 6 with individual fatty acyl-CoAs. ACSL1 levels were correlated with a broad spectrum of fatty acyl-CoAs. The amount of C14:0-CoA was correlated with expression levels of ACSL1, 3, 4, and 5, suggesting the redundancy of ACSL genes in catalysis of C14:0-CoA biosynthesis. ACSL4 and 5 only correlated with C14:0-CoA levels, and ACSL6 expression levels were negatively correlated with C10:0-, C12:0-, C16:0-, and C18:1-CoAs. The relative ACSL mRNA levels and the amount of fatty acyl-CoAs were measured from three independent experiments for each cell line. The correlation analysis of between them is based on the multivariate linear regression model (see Materials and Methods).
Additionally, the analysis shows that the amount of C14:0-CoA was correlated with expression levels of ACSL1, 3, 4, and 5 (Fig. 1G), suggesting the redundancy of ACSL isoforms in catalysis of C14:0-CoA biosynthesis. Finally, while some ACSL genes, such as ACSL4 and 5, were only correlated with C14:0-CoA levels, ACSL6 expression levels negatively correlated with C10:0-, C12:0-, C16:0-, and C18:1-CoAs (Fig. 1G). Collectively, the data suggest that while ACSL1 might regulate the biosynthesis of a broad spectrum of acyl-CoAs, other ACSL isoforms might have substrate preferences in acyl-CoA biosynthesis or negatively regulate the biosynthesis of certain acyl-CoAs.
Expression levels of ACSL1 are elevated in prostate tumors
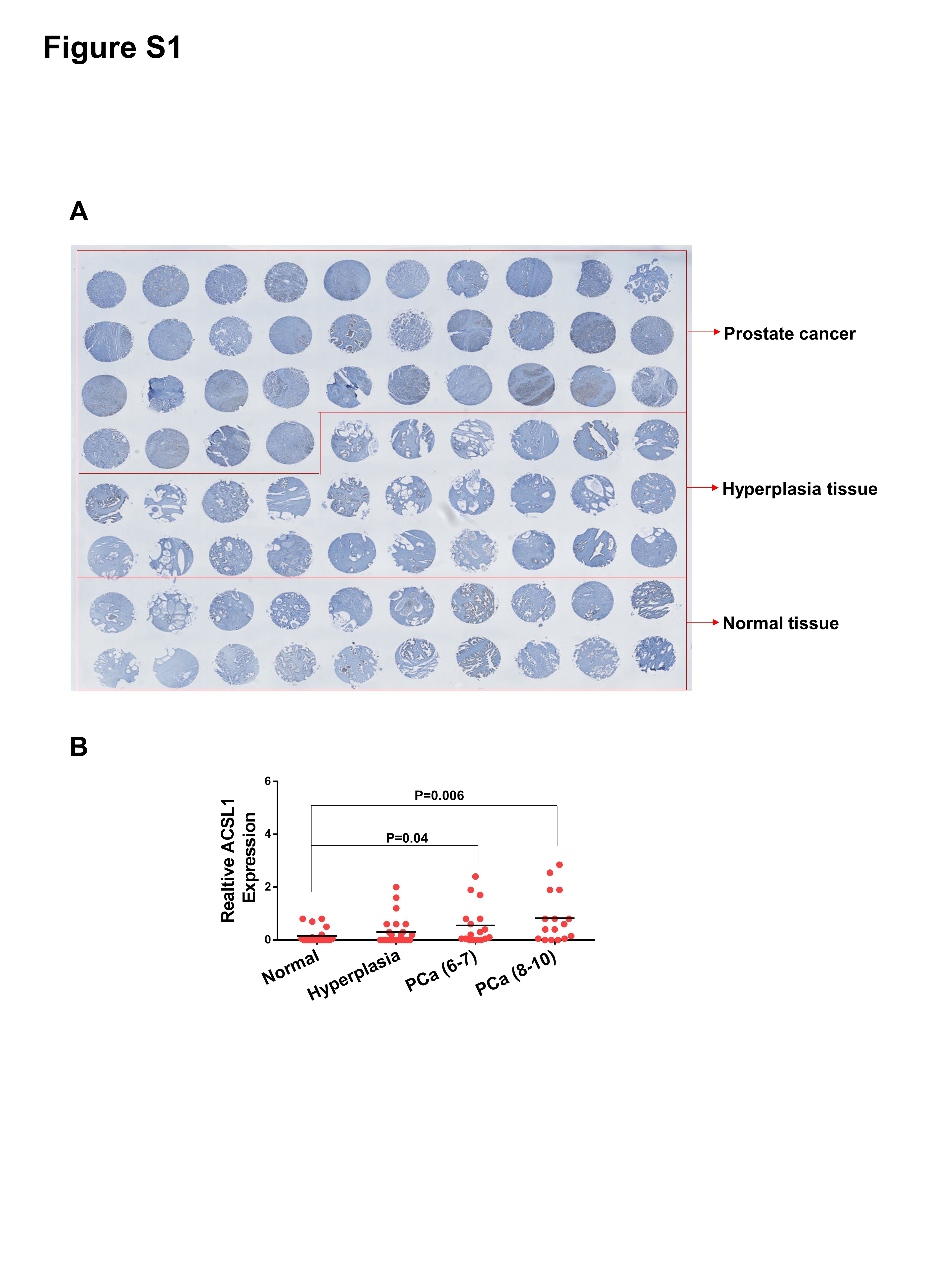
Lipid/fatty acid metabolism is highly dys-regulated in prostate cancer (2). Due to the important role of ACSL1 in regulating the biosynthesis of a broad spectrum of acyl-CoAs, we examined ACSL1 expression levels in a tissue array containing normal prostate, benign prostatic hyperplasia (BPH), and prostate cancer (PCa) with different Gleason scores by immunohistochemistry (IHC) (Fig. S1A). The expression levels of ACSL1 were significantly elevated in prostate tumors (Fig. 2A–D), especially those with high Gleason scores (higher than 7) (Fig. S1B). We also queried three independent microarray profiling datasets from Oncomine™ and Gene Expression Omnibus (GEO) for expression levels of ACSL1 mRNA. We found that ACSL1 levels were also significantly increased in PCa compared with normal prostate tissue (Fig. 2E–G). Collectively, the results indicate that elevated expression levels of ACSL1 are associated with high-grade prostate tumors, suggesting a potential oncogenic function.
Figure 2. ACSL1 expression levels were elevated in prostate tumors.

(A-D) ACSL1 expression levels in prostate tumor tissues. The tumor tissue array was purchased from US Biomax. ACSL1 expression levels were examined and analyzed by IHC. Representative images from normal tissue, hyperplasia, or prostate cancer (Gleason Score of 6–10) are presented. Scale bars: 100 μm. Expression levels of ACSL1 were quantified based on percentage of area and intensity of the staining (D). The tissue microarray contained 80 individual specimens including 34 cases of carcinoma, 26 cases of prostatic hyperplasia and 20 other cases classified as normal tissue. One-way ANOVA and Tukey post hoc test were performed. (E-G) ACSL1 mRNA levels in human prostate cancer tissue versus normal prostate tissue extracted from three public databases (GSE68907, GSE6956 and GSE6919) in Oncomine™ and Gene Expression Omnibus (GEO). GSE68907 contained 52 prostate tumors and 50 nontumor prostate samples; GSE6956 contained 69 prostate tumors and 20 nontumor prostate samples; and GSE6919 contained 90 prostate tumors and 18 nontumor prostate samples. The data were presented as relative change. Student’s t test was used for statistical analysis.
ACSL1 regulates the proliferation of prostate cancer cells
We further examined whether knockdown of ACSL1 inhibits the proliferation of prostate cancer cells. Prostate cancer cells including LNCaP, 22Rv1, PC-3 and DU145, or normal cells such as PNT2 and 293T cells were transduced with shRNA-control or shRNA-ACSL1 (Fig. S2A) by lentiviral infection. Knockdown of ACSL1 (Fig. S2B) significantly inhibited proliferation of 22Rv1, PC-3, DU145 and LNCaP (Fig. 3A–D and Fig. S3A–D), and migration of 22Rv1, PC-3 and DU145 (Fig. 3G–J and Fig. S3G–H). In contrast, ACSL1 knockdown had no effect on the proliferation of normal prostate cells (PNT2) or 293T cells (Fig. 3E–F and Supplementary Fig. S3E–F). Of note, LNCaP cells had the highest ACSL1 protein levels among the four prostate cancer cells tested (Fig. S2C), which is consistent with mRNA expression levels shown in Figure 1A–F. Knockdown of ACSL1 in LNCaP cells showed less inhibition of cell growth (Fig. 3D) in comparison with that in other cancer cells (Fig. 3A–C). This is likely due to the high level of residual ACSL1 since knockdown of ACSL1 in LNCaP cells was not as efficient as in other cell lines (Fig. S2B).
Figure 3. ACSL1 regulates proliferation, migration, and cell cycle of prostate cancer cells.

(A-F) Proliferation of prostate cancer cells (22Rv1, PC-3, DU145 and LNCaP) and normal cells (PNT2 and 293T) transduced with control (shCon) or two shRNA-ACSL1 by lentiviral infection was measured by the MTT assay. Data were compared to shCon, set as 1. Data are represented as mean ± SD (n=6). (G-J) Cell migration of 22Rv1, PC-3 and DU145 cells transduced with control or shRNA-ACSL1 examined by the Transwell migration assay. A representative image is shown (G-I), and the average number of migrated cells per field was calculated based on five different fields (J) (mean ± SD, n=6). (K-N) 22Rv1, PC-3, DU145 and PNT2 cells transduced with control or shRNA-ACSL1 were subjected to cell cycle analysis and the percentage of the cell population in G1, S, and G2/M phases was calculated (mean ± SD, n=3). (O) Expression levels of ACSL1 and cell cycle protein markers including Cyclin A2, B1, and E1 were examined by immunoblotting in PNT2, 22Rv1, PC-3, and DU145 transduced with control or shRNA-ACSL1 (two independent experiments). Scale bars: 100 μm. Student’s t test; *: p<0.05; **: p<0.01.
Cell cycle analysis was further performed to examine if ACSL1 regulates cell division of prostate cancer cells. Knockdown of ACSL1 arrested 22Rv1, PC-3, and DU 145 cancer cells at the G0/G1 phase with a decrease of cells entering S phase (Fig. 3K–M and Fig. S4A–C). In contrast, ACSL1 knockdown did not affect the cell cycle of normal prostate cells (PNT2) (Fig. 3N and Fig. S4D). We further examined the expression levels of cell cycle protein markers including cyclin A2, B1, and E1, elevation of which represent the entry into the G2, M, S phases, respectively. Expression levels of cyclin A2, B1, and E1 showed no significant change in PNT2 cells (normal prostate cell control) expressing shRNA-ACSL1 in comparison with the control shRNA. However, knockdown of ACSL1 significantly inhibited cyclin A2, B1, and E1 levels in 22Rv1 and PC-3 cells. Additionally, knockdown of ACSL1 also inhibited cyclin B1, a lesser extend in cyclin E1, and no change in cyclin A2 levels in DU145 cells (Fig. 3O). The results were consistent with the cell cycle analysis shown in Fig. 3K–N. Furthermore, knockdown of ACSL1 had no change in P21, cyclin D3, and slight elevation of p27 and CDK6 levels, but suppressed expression levels of cyclin D3 and CDK6, and increased expression levels of p21 in 22Rv1, PC-3, and DU145 cells, or increased p27 in 22Rv1 and PC-3 cells (Fig. S5). Collectively, the data suggest that ACSL1 facilitates proliferation of prostate cancer cells by regulation of the cell cycle, but has no significant effect on the cell cycle of normal prostate cells.
ACSL1 regulates biosynthesis of acyl-CoAs in prostate cancer cells
To mechanistically understand how ACSL1 regulates proliferation of prostate cancer cells, ACSL1 mediated fatty acid metabolism was further studied. Expression levels of ACSL1 were associated with intracellular levels of a broad spectrum of acyl-CoAs including 16:0-, 18:0-, 18:1- and 18:2-CoAs (Fig. 1G). Palmitic acid (PA) is a major dietary fatty acid and a final product of de novo fatty acid biosynthesis. We examined if ACSL1 regulates the biosynthesis of acyl-CoAs in the presence or absence of exogenous PA in prostate cancer cells. As expected, levels of C16:0-CoA were significantly elevated in the examined cells growing in the presence of exogenous palmitic acid (PA) (Fig. 4). Knockdown of ACSL1 significantly inhibited C16:0-, C18:0-, C18:1-, and C18:2-CoA levels in either all or some examined prostate cancer cells in the absence or presence of PA (Fig. 4), and to a lesser extent on C12:0- and C14:0-CoA levels (Fig. S6A–C).
Figure 4. ACSL1 regulates the biosynthesis of acyl-CoAs in prostate cancer cells.

(A-T) The amount of C16:0, C18:0, C18:1, and C18:2-CoA. 22Rv1, PC-3, DU145, PNT2, and 293T cells were transduced with shRNA-control (white bars) or shRNA-ACSL1 (black bars) by lentiviral infection. The transduced cells were grown in the medium with/without palmitic acid (PA) (400 μM). The levels of C16:0-CoA (A-E), C18:0-CoA (F-J), C18:1-CoA (K-O), and C18:2-CoA (P-T) were measured by LC-MS/MS. The amount of C12:0- and C14:0-CoAs are presented in Supplementary Figure 6. Data are represented as mean ± SD (n=3). Student’s t test; #, *: p<0.05; ##, **: p<0.01.
Similar to the response of exogenous PA in prostate cancer cells, an increase of PA elevated the amount of C16:0-CoA in normal cells (PNT2 and 293T) (Fig. 4D–E). However, in contrast to prostate cancer cells, knockdown of ACSL1 did not largely change the majority of fatty acyl-CoAs levels (Fig. 4 and Fig. S6D–E) in normal cells. The regulation of ACSL1 in the biosynthesis of acyl-CoAs in cancer cells, but not in normal cells, reinforces that ACSL1 is a potential target for the inhibition in prostate cancer progression.
ACSL1 regulates lipid biosynthesis in prostate cancer cells
Acyl-CoAs provide substrates for the biosynthesis of triglycerides and lipid accumulation through esterification of fatty acids (10). This anabolic process promotes proliferation of cancer cells (11). We further studied ACSL1 in the regulation of biosynthesis of triglycerides and lipogenesis in cancer cells. Knockdown of ACSL1 significantly reduced the endogenous levels of total triglycerides in 22Rv1, PC-3, and DU145 cells, but not total phospholipids except in DU145 cells (Fig. 5A–C). The addition of exogenous fatty acids significantly increased total triglyceride levels in PC-3 and DU145 cells. However, the elevated triglyceride levels were inhibited by knockdown of ACSL1 (Fig. 5A–C). Further analysis of the composition of the acyl chains in triglycerides indicated that the levels of C16:0, C18:0, C18:1ω9, and C18:2 acyl chains were significantly decreased in cancer cells expressing shRNA-ACSL1 (Fig. 5D–F), which was consistent with the role of ACSL1 in the biosynthesis of the acyl-CoAs shown in Figure 4.
Figure 5. Knockdown of ACSL1 inhibits lipid accumulation in prostate cancer cells.

22Rv1, PC-3, and DU145 cells transduced with shRNA-control or shRNA-ACSL1 were grown in the ATCC recommended medium with a mixture of oleic acid (OA, 400 μM) and palmitic acid (PA, 200 μM) for 48 h. (A-F) Total triglyceride (TG) and phospholipid (PL) (A-C) and acyl chain components of total triglyceride (D-F) were analyzed by gas chromatography. Data are represented as mean ± SD (n=3). One-way ANOVA and Tukey post hoc test; *: p<0.05; **: p<0.01. (G-I) The accumulation of cellular lipids was visualized by Oil Red O staining in 22Rv1 (G), PC-3 (H), and DU145 cells (I). A representative image of each group is shown.
Considering that C16:0- and C18-fatty acid chains are major components of triglycerides (Fig. 5D–F), we further examined if ACSL1 regulates lipogenesis in the presence of palmitic acid and oleic acid. Addition of fatty acids significantly induced lipid accumulation (Fig. 5G–I). However, this lipid accumulation was significantly inhibited by shRNA-ACSL1 in 22Rv1, PC-3, and DU145 (Fig. 5G–I), suggesting that ACSL1 regulates the esterification of exogenous fatty acids in lipogenesis. Collectively, the results indicate that ACSL1 regulates the anabolic process of exogenous fatty acid-induced triglyceride synthesis and lipid storage in prostate cancer cells.
ACSL1 regulates mitochondrial respiratory function and CPT1 activity
Palmitoyl-CoA (C16:0-CoA) is an important substrate for β-oxidation in mitochondria. Since ACSL1 regulates the biosynthesis of palmitoyl-CoA, we further examined if the expression levels of ACSL1 affect mitochondrial respiration and β-oxidation. Knockdown of ACSL1 significantly inhibited the basal oxygen consumption rate (OCR) with 52%, 56%, and 71% reduction in 22Rv1, PC-3 and DU145 (Fig. 6A–C), respectively. Additionally, oxidative phosphorylation (oligomycin-treated condition) and maximal respiration (FCCP-treated condition) were also significantly inhibited by shRNA-ACSL1 (Fig. 6A–C). Similarly, the extracellular acidification rate (ECAR), an indicator of active glycolysis, was also significantly suppressed by shRNA-ACSL1 (Fig. 6D–F).
Figure 6. Knockdown of ACSL1 inhibits mitochondrial respiration in prostate cancer cells.

(A-F) Oxygen consumption rate (OCR) (A-C) and extracellular acidification rate (ECAR) (D-F) were measured in 22Rv1, PC-3, and DU145 prostate cancer cells transduced with shRNA-control (shCon, blue circles) or shRNA-ACSL1 (red squares) using a Seahorse Extracellular Flux (XF) Analyzer after sequential injection of oligomycin (OM, 3 μM), FCCP (4 μM), and rotenone & antimycin (R/A, 1 μM). (G) Measurement of OCR when PC-3 cells were treated with palmitic acid or CPT1 inhibitor Etomoxir (Eto). (H-I) Basal respiration (H) and maximal respiration (I) were calculated based on the data from Panel G. Data are represented as mean ± SD (n=5). Student’s t test; *: p<0.05; **: p<0.01.
We further examined mitochondrial respiration with exogenous fatty acids. Oxygen consumption was significantly induced by the addition of palmitic acid (PA) in PC-3 cancer cells, and was inhibited by shRNA-ACSL1 (Fig. 6G). Knockdown of ACSL1 reduced endogenous and exogenous PA-induced oxygen consumption at basal and maximal respiration rates (Fig. 6H–I). Additionally, etomoxir (Eto), a carnitine palmitoyltransferase-1 (CPT-1) inhibitor, blocked fatty acid oxidation (FAO) in PC-3 cells and knockdown of ACSL1 further inhibited the process (Fig. 6H–I). Inhibition of β-oxidation was further confirmed by a reduction in ATP production in 22Rv1, PC-3, and DU145 cells expressing shRNA-ACSL1 (Fig. S7A). Taken together, the data indicate that ACSL1 is essential for the regulation of mitochondrial respiration and β-oxidation in prostate cancer cells.
ACSL1 is reported to be located at the mitochondrial outer membrane and interacts with carnitine palmitoyltransferase 1A (CPT1A), which is the rate-limiting enzyme responsible for translocating palmitoyl-CoA into the mitochondrial matrix for β-oxidation (12). We examined if suppression of ACSL1 inhibits CPT1 activity. The activity of CPT1 was determined based on the production of CoA-SH from carnitine and palmitoyl-CoA (Fig. S7B–D) (13). Knockdown of ACSL1 significantly inhibited CPT1 activity with a reduction of 30%, 56%, and 62% in 22Rv1, PC-3 and DU145, respectively (Fig. S7E–G). The data suggest that ACSL1 facilitates CPT1 activity in mitochondrial respiration.
Suppression of ACSL1 expression inhibits growth of prostate xenograft tumors
We further explored if suppression of ACSL1 inhibited growth of prostate xenograft tumors. 22Rv1, PC-3, and DU145 cells transduced with control vector or shRNA-ACSL1 were implanted into SCID mice subcutaneously (Fig. S8A–C). The size and weight of xenograft tumors derived from 22Rv1 (Fig. 7A–C), PC-3 (Fig. 7D–F), and DU145 cells (Fig. 7G–I) expressing shRNA-ACSL1were significantly reduced. The RFP fluorescence of xenograft tumors indicated successful transduction of the control or shRNA-ACSL1 (Fig. S8D) and knockdown of ACSL1 expression was confirmed by IHC staining (Fig. S8E). The expression levels of Ki67 and CD34 were inhibited in xenograft tumors expressing shRNA-ACSL1 (Fig. 7J–M), suggesting that ACSL1 regulates the proliferation of tumorigenic cells, and the formation of vasculature in the tumors. Collectively, the data indicate that ACSL1 is a potential target for the inhibition of prostate tumor proliferation and progression.
Figure 7. ACSL1 promotes growth of prostate xenograft tumors in vivo.

(A-I) 22Rv1, PC-3 and DU145 cells transduced with control (shCon) or shRNA-ACSL1 were implanted in SCID mice subcutaneously. Each animal carried a tumor expressing control or shRNA-ACSL1 in each flanking side (see Supplemental Figure S8). After 8 weeks, tumors were harvested and representative images are shown for shCon (top) and shACSL1 (bottom) in A, D, G. Volume and weight of 22Rv1 (B-C), PC-3 (E-F) and DU145 (H-I) xenograft tumors were determined. Data are represented as mean ± SD (n=6). Student’s t test; *: p<0.05; **: p<0.01. (J-M) Expression levels of Ki67 (J and L) and CD34 (K and M) were determined in xenograft tumors expressing shRNA-control (shCon) or shRNA-ACSL1 by IHC staining (scale bars: 100 μm), and were quantified based on IHC staining.
DISCUSSION
Our study has illustrated the important role of ACSL1 in prostate tumors. ACSL1 levels were upregulated in prostate tumors, and inhibition of ACSL1 significantly suppressed growth of prostate xenograft tumors in vivo and blocked proliferation and migration of prostate cancer cells in vitro. Genetically targeting ACSL1 suppressed the cell cycle by arresting prostate cancer cells in G1 phase and reducing the entry into G2/M and/or S phase. The cell cycle effects were reflected in the inhibition of cellular respiration as demonstrated by the reduced OCR and ATP production, and significant down-regulation of the biosynthesis fatty acyl-CoAs and lipid synthesis, suggesting that ACSL1 played a critical role for both the catabolism and anabolism of prostate cancer cells (Fig. 8). ACSL1 has been reported to be a prognostic biomarker or a predictor for survival in colon cancer. Elevated expression levels of ACSL1 together with expression of other genes such as ATP-binding cassette transporter A1, 1- acylglycerol-3-phosphate O-acyltransferase 1 and stearoyl-CoA desaturase could predict the survival rate in stage-II colon cancer patients (14). Additionally, a cohort of genes including ACSL1, ACSL4, and SCD is associated with migration and invasion of colorectal cancer cells and initiation of epithelial-mesenchymal transition (15). Our data emphasize that ACSL1 promotes cell proliferation and migration and is a potential therapeutic target for the treatment of prostate cancer.
Figure 8.

Graphic summary of the mechanisms underlying ACSL1-mediated tumor progression.
Our study has characterized the preference of ACSL1 in the biosynthesis of acyl-CoAs. ACSLs are essential for the activation of fatty acids, and further participation in fatty acid metabolism (Fig. 8). ACSLs facilitate the bioconversion of fatty acids to their corresponding acyl-CoAs (9). However, the catalytic preference of ACSL isoforms in the biosynthesis of acyl-CoAs is currently largely unknown. We show that expression levels of ACSL1 correlate with the amount of a broad spectrum of acyl-CoAs. Purified ACSL1 enzyme (previously named as ACS1) shows high enzymatic activity towards the bioconversion of saturated fatty acids including C10:0, C12:0, C14:0, C16:0, and C18:0, and unsaturated fatty acids including C18:1 and C18:2 (16). Some intracellular fatty acids such as C10/C12:0 are present at very low levels in cells, making them difficult to detect. We demonstrate that ACSL1 significantly regulates the production of 14:0-, 16:0-, 18:0-, 18:1- and 18:2-CoA in cancer cells. This result is supported by the reduction of C16:0, C18:0, C18:1 and C18:2-CoAs in liver cells under the challenge of a high-fat diet in ACSL1 knockout mice (17) or heart tissues (18).
We have also identified that certain ACSL isoforms, such as ACSL4, might have preferences for the biosynthesis of specific acyl-CoAs, while others appear to have functional redundancy in the biosynthesis of C14:0-CoA (ACSL1, ACSL3, ACSL4, and ACSL5). Although the catalytic preference of the individual ACSL members has not been fully delineated, it has become possible to profile their catalytic function with current technology (19). Given the essential role of acyl-CoAs in fatty acid metabolism, further delineation of the function of ACSL isoforms will be informative for developing ACSL isoform-specific inhibitors to target prostate cancer progression. It remains to be studied if other ACSL isoforms have pathological significance in prostate tumor progression. However, it has been shown that over-expression of ACSL4 promotes the proliferation of prostate cancer cells (20).
In addition to the regulation of acyl-CoA synthesis, ACSL1 exhibits a functional role in lipid biosynthesis in prostate cancer cells. Knockdown of ACSL1 inhibits triglyceride biosynthesis, specifically those containing C16:0, C18:0, C18:1, and C18:2 acyl chains, and lipogenesis under the induction of exogenous fatty acids. Lipid accumulation is considered a causative factor for the invasive ability of prostate cancer cells (21). Intracellular lipid contributes to invasiveness by releasing free fatty acid for ATP production in breast tumors (11), or to promote adaptation under stress conditions conferring a survival advantage for cancer cells (22). Therefore, suppression of lipid accumulation by targeting ACSL1 is a potential beneficial approach to inhibit invasive tumor growth.
Our study reveals that ACSL1 plays an essential role in ATP production via fatty acid catabolism through modulation of CPT1 activity in the mitochondria of prostate cancer cells. Loss of ACSL1 results in a reduction of CPT1 activity and a decrease of β-oxidation in mouse liver, heart and adipose tissues (17, 18). Interestingly, ACSL1 knockdown seems to only inhibit cell proliferation in cancer cells, but not in normal cells. It has been well-documented that cancer cells rely on elevated fatty acid metabolism evident by the elevated levels of enzymes in fatty acid de novo synthesis (2) and elevated fatty acid oxidation to supply ATP in prostate cancer (23). The expression of alpha-methylacyl-CoA racemase (AMACR) is a marker of dys-regulated fatty acid metabolism in prostate cancer (24). Therefore, targeting the “addicted” fatty acid metabolism pathway will lead to significant growth inhibition in prostate cancer. ACSL1 is primarily located at the mitochondrial outer membrane (25). CPT1 forms protein complexes with ACSL1 and the voltage-dependent anion channel (VDAC) allowing for transfer of activated fatty acids through the mitochondrial outer membrane for β-oxidation (12). It remains to be studied how the protein-protein interaction of ACSL1and CPT1 affect CPT1 function.
Tremendous effort has focused on targeting enzymes involved in de novo fatty acid synthesis such as FASN for inhibiting cancer progression (26). Onco-antigen 519 (OA-519) (fatty acid synthase) was strongly correlated with poor prognosis in breast and prostate cancer (27). Several inhibitors such as TVB-2640 have been developed and evaluated in clinical trials. These inhibitors have demonstrated that fatty acid metabolism is important for cancer progression. However, cells may adapt to suppression of de novo fatty acid synthesis by relying on exogenous fatty acid uptake or fatty acids from adipocytes (28, 29). ACSL family genes dictate the activation of fatty acids to participate in the metabolic process. Therefore, targeting ACSL family genes such as ACSL1, which facilitates the biosynthesis of a broad spectrum of acyl-CoAs, provides a potential therapeutic strategy for inhibiting prostate tumor progression.
Materials and Methods
Cell Culture
Human prostate cell lines (293T, PNT2, LNCaP, 22Rv1, PC-3 and DU145) and hepatic cell lines (HepG2 and Hep3B) were purchased from American Type Culture Collection (ATCC). Cells were maintained in a humidified incubator under 5% CO2 at 37 °C and grown in the recommended media supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin. Mycoplasma contamination was periodically examined. All cell lines were passaged for no more than 20 passages after resuscitation.
RNA Interference and Lentivirus Production
ACSL1 shRNA was generated using the following primers: ACSL1 shRNA 1, forward: 5’-TCCCTCCTTGGTGTATTTCTGTGATTTCAAGAGAATCATAGAAATACACCAAGGG-3’, reverse: 5’-AAAACCCTTGGTGTATTTCTATGATTCTCTTGAAATCACAGAAATACACC
AAGGA-3’. ACSL1 shRNA 2, forward: 5’- TCCCGCCTAGATGATACTTTGGTATTTCAAG
AGAATATCAAAGTATCATCTGGGC-3’, reverse: 5’- AAAAGCCCAGATGATACTTTGAT
ATTCTCTTGAAATACCAAAGTATCATCTAGGC-3’. Annealed primers were inserted into psiRNA-W[H1.4] vector at the BbsI site. The shRNAs with the H1 promoter were further reconstructed into FUCRW lentiviral vectors at the PacI site. The insertion in the new plasmid was confirmed by DNA sequencing. Lentivirus production and infection of prostate cell lines were performed as described previously (5). A single cell dilution was further performed to obtain a clonal population with stable gene knockdown. Lentivirus procedures followed the guidelines and regulations at the University of Georgia.
Extraction of ACSL1 Expression from the Oncomine or the Gene Expression Omnibus (GEO)
ACSL1 mRNA levels in human prostate cancer tissue versus normal prostate tissue were extracted from three public databases in the OncomineTM (GSE68907 and GSE6956) or the GEO (GSE6919). GSE68907 dataset includes 52 prostate tumors and 50 non-tumor prostate samples; GSE6956 dataset includes 69 prostate tumors and 20 normal prostate samples; and GSE6919 dataset includes 90 prostate tumors and 18 normal prostate samples. The data are presented as the fold change.
Cell proliferation, migration, and cell cycle analysis
The cell proliferation was assessed using the MTT assay. Cell migration assays were performed using the BD FluoroBlok Insert System (Becton-Dickinson, MA). Cell Cycle Analysis relied on Vybrant® DyeCycle™ Violet stain (Life Technologies, Eugene, OR) and analysis by a flow cytometer (CyAn ADP Analyzer). The details are described in Supplementary Materials and Methods.
Quantitative RT-PCR Analysis and Western blot
Total RNA was isolated from cell lines using the Qiagen RNeasy Mini Kit (Valencia, CA). For Western blots, cell lysates dissolved in RIPA buffer underwent standard electrophoresis and antibody detection. The details are described in the Supplementary Material and Methods. The following antibodies were used: ACSL1 (#4047), Cyclin A2 (#4656), Cyclin B1 (#4138), Cyclin E1(#4129), p21 (#2947), p27 (#3686), CDK6 (#3136), cyclin D3 (#2936), and CPT1A (#12252) from Cell Signaling (Boston, MA) or γ-tubulin (#T6557, Sigma-Aldrich).
In vivo Xenograft Animal Model
CB.17SCID/SCID (SCID) mice were purchased from Taconic (Hudson, NY). All animal procedures were approved by the Institutional Animal Care and Use Committee at the University of Georgia. All the experiments were consented by the approved protocols. Prostate cancer cell lines with ACSL1-shRNA or control vector were grown in recommended media. 22Rv1 cells (3×106), PC-3 cells (3×105) and DU145 cells (1×106) were mixed with 50 μL of collagen type I (pH 7.0) (BD Biosciences) to make grafts, and inoculated subcutaneously in both lateral flank sides of SCID mice (ACSL1 knockdown cells on the right flank and control on the left counterpart, n=6). The grafts were randomized or blinded to give a host SCID mouse (3-months old). The host mice were sacrificed, and xenograft tumors were harvested after an 8-week incubation. Tumor volume and weight were measured and then fixed in 10% formalin formaldehyde or stored at −80 °C. Tumor volume based on external caliper measurements was calculated by the modified ellipsoidal formula: Tumor volume = 1/2(length × width2).
Immunohistochemistry on mice xenograft tumors and human prostate tissue array
The above xenograft tumors were embedded in paraffin followed by tissue sectioning at a thickness of 4 μm. Immunohistochemical (IHC) was performed to measure expression levels of ACSL1 (1:100; Cell Signaling #4047), Ki67 (1:400; Novus Biologicals, #NB500–170B) and CD34 (1:2000; Abcam, # ab81289).
IHC staining of ACSL1 was also performed on human prostate tissue microarray slides purchased from US Biomax Inc. (catalog PR8011b, Rockwell, MD). The ACSL1 antibody was verified based on the IHC of PC-3 or DU145 xenograft tissues expressing shRNA-ACSL1 or control. The microarray contained 80 individual specimens including 34 cases of carcinoma, 26 cases of prostatic hyperplasia and 20 other cases classified as normal tissue. Pathology assessment and the ACSL1 staining were scored according to the area and intensity of the stains.
Measurement of Oxygen Consumption Rate and Extracellular Acidification Rate.
The effects of ACSL1 knockdown on mitochondrial respiratory function in prostate cancer cell lines were measured by detecting the oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) using the XF24 extracellular flux analyzer (Seahorse Biosciences, Billerica, MA). The details are described in the Supplementary Material and Methods.
Acyl-CoA Analysis
Long chain fatty acyl-CoAs were analyzed as described previously (19, 30). Briefly, cells were treated with/without 400 μM palmitic acid in serum-free media containing 2% fatty acid-free BSA for 24 h. Cells were incubated with 2 mL methanol including 15 μL pentadecanoyl CoA (10 μg/mL, internal standard) at −80 °C for 15 min after washing with PBS twice. The cell lysate was collected and centrifuged at 14,000 rpm at 4 °C for 10 min and the supernatant was transferred to a glass tube, mixed with 1 mL acetonitrile and evaporated in a vacuum concentrator at 55 °C for 2 h. The sample was reconstituted with 120 μL methanol, briefly vortexed, centrifuged, and 100 μL supernatant was transferred to an auto-sampler vial for LC-MS/MS analysis. The results were normalized to total cellular protein and are expressed as pmol/mg protein.
Oil Red O Staining and Lipids Analysis
Cells were treated with a mixture of oleic acid and palmitic acid. Lipid accumulation was stained by Oil Red O reagent, and visualized in a regular light microscope. For lipids measurements, lipids were extracted using Folch-Lees’s method (31). Triglyceride, phospholipid, and free fatty acids levels were analyzed using gas chromatography. The details are described in Supplementary Materials and Methods.
Statistical Analysis
The multiple linear regression model was used to study the association between fatty acyl-CoA levels and ACSL gene expression. The amount of fatty acyl-CoAs was treated as response variables and the expression levels of ACSL genes (ACSL1, ACSL3, ACSL4, ACSL5 and ACSL6) were treated as covariates. A regression coefficient was considered significant if its p-value was less than 0.05. All analyses were performed using the linear regression package ‘lm’ in statistical software R 3.1.2.
Additionally, Prism software 6 (Graph-Pad Prism Software, San Diego, CA) was used to carry out the statistical analysis and comparisons of relative expression levels of ACSLs in different cell lines. Variation of each group was estimated, and variance similarity was compared. The data are presented as mean ± SD, and statistical analysis including One-way ANOVA, Tukey post hoc test, and Student’s t test. “*”: p<0.05; “**”: p<0.01; “N.S.”: not significant.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
This work was supported by NIH (R01CA172495) and DOD (W81XWH-15-1-0507) to H. Cai.
Footnotes
Conflict of Interest
The authors disclose no potential conflicts of interest.
REFERENCES
- 1.Butler LM, Centenera MM, Swinnen JV. Androgen control of lipid metabolism in prostate cancer: novel insights and future applications. Endocr Relat Cancer. 2016;23(5):R219–27. [DOI] [PubMed] [Google Scholar]
- 2.Wu X, Daniels G, Lee P, Monaco ME. Lipid metabolism in prostate cancer. Am J Clin Exp Urol. 2014;2(2):111–20. [PMC free article] [PubMed] [Google Scholar]
- 3.Currie E, Schulze A, Zechner R, Walther TC, Farese RV. Cellular Fatty Acid Metabolism and Cancer. Cell Metab. 2013;18(2):153–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yan S, Yang XF, Liu HL, Fu N, Ouyang Y, Qing K. Long-chain acyl-CoA synthetase in fatty acid metabolism involved in liver and other diseases: an update. World J Gastroenterol. 2015;21(12):3492–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kim S, Alsaidan OA, Goodwin O, Li Q, Sulejmani E, Han Z, et al. Blocking Myristoylation of Src Inhibits Its Kinase Activity and Suppresses Prostate Cancer Progression. Cancer Res. 2017;77(24):6950–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kim S, Yang X, Li Q, Wu M, Costyn L, Beharry Z, et al. Myristoylation of Src kinase mediates Src-induced and high-fat diet-accelerated prostate tumor progression in mice. J Biol Chem. 2017;292(45):18422–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Soupene E, Kuypers FA. Mammalian long-chain acyl-CoA synthetases. Exp Biol Med (Maywood). 2008;233(5):507–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mashima T, Oh-hara T, Sato S, Mochizuki M, Sugimoto Y, Yamazaki K, et al. p53-defective tumors with a functional apoptosome-mediated pathway: a new therapeutic target. J Natl Cancer Inst. 2005;97(10):765–77. [DOI] [PubMed] [Google Scholar]
- 9.Grevengoed TJ, Klett EL, Coleman RA. Acyl-CoA metabolism and partitioning. Annual review of nutrition. 2014;34:1–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yen CL, Stone SJ, Koliwad S, Harris C, Farese RV Jr. Thematic review series: glycerolipids. DGAT enzymes and triacylglycerol biosynthesis. J Lipid Res. 2008;49(11):2283–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang YY, Attane C, Milhas D, Dirat B, Dauvillier S, Guerard A, et al. Mammary adipocytes stimulate breast cancer invasion through metabolic remodeling of tumor cells. Jci Insight. 2017;2(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee K, Kerner J, Hoppel CL. Mitochondrial Carnitine Palmitoyltransferase 1a (CPT1a) Is Part of an Outer Membrane Fatty Acid Transfer Complex. Journal of Biological Chemistry. 2011;286(29):25655–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bieber LL, Abraham T, Helmrath T. A rapid spectrophotometric assay for carnitine palmitoyltransferase. Anal Biochem. 1972;50(2):509–18. [DOI] [PubMed] [Google Scholar]
- 14.Vargas T, Moreno-Rubio J, Herranz J, Cejas P, Molina S, Gonzalez-Vallinas M, et al. ColoLipidGene: signature of lipid metabolism-related genes to predict prognosis in stage-II colon cancer patients. Oncotarget. 2015;6(9):7348–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sanchez-Martinez R, Cruz-Gil S, de Cedron MG, Alvarez-Fernandez M, Vargas T, Molina S, et al. A link between lipid metabolism and epithelial-mesenchymal transition provides a target for colon cancer therapy. Oncotarget. 2015;6(36):38719–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Iijima H, Fujino T, Minekura H, Suzuki H, Kang MJ, Yamamoto T. Biochemical studies of two rat acyl-CoA synthetases, ACS1 and ACS2. Eur J Biochem. 1996;242(2):186–90. [DOI] [PubMed] [Google Scholar]
- 17.Li LO, Ellis JM, Paich HA, Wang S, Gong N, Altshuller G, et al. Liver-specific loss of long chain acyl-CoA synthetase-1 decreases triacylglycerol synthesis and beta-oxidation and alters phospholipid fatty acid composition. J Biol Chem. 2009;284(41):27816–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ellis JM, Mentock SM, Depetrillo MA, Koves TR, Sen S, Watkins SM, et al. Mouse cardiac acyl coenzyme a synthetase 1 deficiency impairs Fatty Acid oxidation and induces cardiac hypertrophy. Mol Cell Biol. 2011;31(6):1252–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang X, Ma Y, Li N, Cai H, Bartlett MG. Development of a Method for the Determination of Acyl-CoA Compounds by Liquid Chromatography Mass Spectrometry to Probe the Metabolism of Fatty Acids. Anal Chem. 2017;89(1):813–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu X, Deng F, Li Y, Daniels G, Du X, Ren Q, et al. ACSL4 promotes prostate cancer growth, invasion and hormonal resistance. Oncotarget. 2015;6(42):44849–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yue S, Li J, Lee SY, Lee HJ, Shao T, Song B, et al. Cholesteryl ester accumulation induced by PTEN loss and PI3K/AKT activation underlies human prostate cancer aggressiveness. Cell Metab. 2014;19(3):393–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Koizume S, Miyagi Y. Lipid Droplets: A Key Cellular Organelle Associated with Cancer Cell Survival under Normoxia and Hypoxia. Int J Mol Sci. 2016;17(9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schlaepfer IR, Rider L, Rodrigues LU, Gijon MA, Pac CT, Romero L, et al. Lipid Catabolism via CPT1 as a Therapeutic Target for Prostate Cancer. Mol Cancer Ther. 2014;13(10):2361–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lloyd MD, Yevglevskis M, Lee GL, Wood PJ, Threadgill MD, Woodman TJ. alpha-Methylacyl-CoA racemase (AMACR): metabolic enzyme, drug metabolizer and cancer marker P504S. Progress in lipid research. 2013;52(2):220–30. [DOI] [PubMed] [Google Scholar]
- 25.Grevengoed TJ, Martin SA, Katunga L, Cooper DE, Anderson EJ, Murphy RC, et al. Acyl-CoA synthetase 1 deficiency alters cardiolipin species and impairs mitochondrial function. J Lipid Res. 2015;56(8):1572–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kuhajda FP, Jenner K, Wood FD, Hennigar RA, Jacobs LB, Dick JD, et al. Fatty acid synthesis: a potential selective target for antineoplastic therapy. Proc Natl Acad Sci U S A. 1994;91(14):6379–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Epstein JI, Carmichael M, Partin AW. Oa-519 (Fatty-Acid Synthase) as an Independent Predictor of Pathological Stage in Adenocarcinoma of the Prostate. Urology. 1995;45(1):81–6. [DOI] [PubMed] [Google Scholar]
- 28.Kuemmerle NB, Rysman E, Lombardo PS, Flanagan AJ, Lipe BC, Wells WA, et al. Lipoprotein lipase links dietary fat to solid tumor cell proliferation. Mol Cancer Ther. 2011;10(3):427–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gazi E, Gardner P, Lockyer NP, Hart CA, Brown MD, Clarke NW. Direct evidence of lipid translocation between adipocytes and prostate cancer cells with imaging FTIR microspectroscopy. J Lipid Res. 2007;48(8):1846–56. [DOI] [PubMed] [Google Scholar]
- 30.Kim S, Yang X, Li Q, Wu M, Costyn L, Beharry Z, et al. Myristoylation of Src kinase mediates Src induced and high fat diet accelerated prostate tumor progression in mice. J Biol Chem. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Folch J, Lees M, Sloane Stanley GH. A simple method for the isolation and purification of total lipides from animal tissues. J Biol Chem. 1957;226(1):497–509. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.