

Abstract
Lethal features of sepsis and acute respiratory distress syndrome (ARDS) relate to the health of small blood vessels. For example, infiltration of the alveoli with proteinaceous fluid is often driven by breach of the microvascular barrier. Spontaneous thrombus formation within inflamed microvessels exacerbates organ ischemia, and in its final stages, erupts into overt disseminated intravascular coagulation (DIC). A signaling axis in the vascular endothelium, the Angiopoietin-Tie2 pathway, may play a central role in the abrupt transition from microvascular integrity to pathological disruption. Tie2 signaling is highly active when vessels are in their quiescent state: stable junctions, anti-inflammatory, and anti-coagulant. During inflammation however, Tie2 signaling is rapidly toggled off as levels of its endogenous antagonist, Angiopoietin-2, rise by 1–2 orders of magnitude. When Tie2 signaling drops, the microvascular barrier collapses, endothelium becomes adhesive to leukocytes, and the vessel wall no longer prevents spontaneous coagulation. This review will summarize a large body of preclinical and clinical results that implicate the Tie2 pathway in the pathogenesis of sepsis and ARDS. Results from groups worldwide now identify Angiopoietins as dynamic circulating markers of the vasculature. Furthermore, Tie2 is emerging as a promising target to restore microvascular health in sepsis and ARDS. Translational pursuit of the Tie2 pathway may enable advances in the way critically ill patients are diagnosed, monitored, and treated.
4-8 keywords to direct and optimize search results: Angiopoietin, Tie2, sepsis, ARDS, vascular leakage, coagulation, VE-PTP
INTRODUCTION
Homeostasis of small blood vessels exerts a powerful influence on the disease course in critically ill patients. In systemic inflammation, dysregulated microvessels throughout the body leak their contents into the surrounding tissue, potentiate the penetration of inflammatory cells into major organs, promote the formation of in situ thrombi, and disrupt the vascular tone necessary to deliver oxygen and nutrients where they are needed. Vascular dysfunction arising from systemic inflammation may impact clinically meaningful adverse outcomes in the ICU. The morbidity, mortality, and cost from these pathophysiological processes is vast while clinical care in the ICU remains largely supportive rather than targeted at the underlying pathophysiological mechanisms [1–3]. Mechanisms that regulate the transition between microvascular homeostasis and dysregulation are therefore of substantial fundamental and translational interest.
The vascular endothelium receives signals from the circulating inflammatory milieu and responds to these signals to control tone, barrier function, adhesiveness to leukocytes, and coagulation. When inflammation arises locally, the transition from homeostasis to the activated state is likely adaptive: in response to local tissue injury, the breakdown of the microvascular barrier enables humoral and cellular effectors of immunity to access the parenchyma; local vasorelaxation increases the flux of such effectors into the target region; and thrombus formation limits the dissemination of pathogens from the portal of entry. Yet, when the very same sequence proceeds unchecked or unfolds simultaneously across multiple vascular beds, the results look like the septic patient: edematous soft tissues, inflamed and swollen major organs, vasoplegia, and DIC.
Molecular pathways within the vascular endothelium collaborate to facilitate homeostasis of blood vessels. Involved pathways include, but are not limited to, the Tie2-Angiopoietin axis [4–6]; vascular endothelial growth factor (VEGF) interactions with its receptors, VEGFR-2 and VEGFR-1 [7–9]; sphingosine-1 kinase and its receptors [10]; and mechanical regulators such as shear stress generated by the flow of blood [11, 12]. Among vascular signaling pathways, the Tie2 receptor is unique in that its phosphorylation provides an active signal to promote quiescence and counteract the “activated” state of endothelium [13–15]. Perhaps not surprisingly, diverse critical illness syndromes including sepsis, DIC, shock, ARDS, myocardial infarction, and acute kidney injury have all been linked to suppression of Tie2 signaling [4, 6, 16–21].
BIOLOGY OF THE ANGIOPOIETIN-TIE2 AXIS
Overview:
As a transmembrane tyrosine kinase, Tie2 is the central molecule in this axis. An orthologous protein called Tie1 is also expressed in endothelium, but does not directly bind Angiopoietins (Angpts) and remains an orphan receptor [22, 23]. A third transmembrane protein, vascular-endothelial protein tyrosine phosphatase (VE-PTP) is also expressed in the endothelium, where it closely associates with Tie2 to regulate its signaling.
In the quiescent state, Angiopoetin-1 (Angpt-1) binds to Tie2, an event that clusters individual Tie2 molecules into a cross-phosphorylating complex. Downstream of phosphorylated Tie2, the second messenger AKT is activated, preventing endothelial apoptosis and enhancing cellular quiescence [24]. During times of stress—e.g., inflammation signaled by cytokines such as TNFα—Angiopoietin-2 (Angpt-2) levels increase and competitively bind to the Tie2 receptor blocking Angpt-1 mediated phosphorylation [25, 26]. Because Angpt-2 is less efficient than Angpt-1 at clustering Tie2 monomers, the net effect of Angpt-2 induction in the context of inflammation is to suppress tonic Tie2 signaling. Loss of Tie2 signaling releases an important “brake” in the endothelium, enabling it rapidly shift its phenotype toward the activated state: weakened junctions [27, 28], expression of adhesion molecules for leukocytes including ICAM1 and VCAM1 [26], and enhancement of procoagulant proteins at the luminal surface [4, 29]. Each of these molecular-cellular events maps to major clinical manifestations in sepsis and ARDS. Weakened junctions lead to edema. Adhesion molecule induction promotes secondary inflammatory injury, and disruption of normal hemostatic mechanisms culminates in DIC. The subsequent sections summarize the molecular and cell biology of each major component in the Tie2 pathway.
Angiopoietin-1:
Angpt-1 is the canonical agonist ligand for the Tie2. Angpt-1 has a modular structure, starting at the amino terminus with a superclustering domain, then a coiled-coil domain, and finally, a fibrinogen-like domain. The N-terminal regions of Angpt-1 organize individual molecules into homotetramers and higher-order oligomers. The fibrinogen-like domain on the C-terminus is responsible for binding Tie2. Angpt-1 binds Tie2 very tightly at nanomolar affinity [25, 30]. The oligomeric status of Angpt-1 promotes the clustering of Tie2 monomers at the endothelial surface. The resulting molecular complex becomes an efficient signaling unit [31–33].
Angpt-1 knockout mice die in utero at ~E12.5 with a dilated, rudimentary vasculature that lacks branching and the support cells such as pericytes that are characteristic of a matured vasculature [34, 35]. The embryos exhibit edema and hemorrhage. Consistent with its role as an agonist, these features phenocopy global knockout of Tie2 [36]. Conversely, upregulation of Angpt-1 results in an increased quantity of branched vasculature that is no longer leaky [37–39].
Angpt-1 is expressed by platelets, mesenchymal cells, vascular smooth muscle cells, and pericytes [25, 34, 40, 41]. The last two cell types bathe the extracellular matrix surrounding endothelial cells in Angpt-1. Morevoer, Angpt-1 is naturally adhesive to the matrix [42, 43]. This provides a constant stimulus for Tie2 activation in the quiescent state. As will be discussed below, Tie2 activation plummets during inflammation with the induction of Angpt-2. However, in this latter context of endothelial “dyshomeostasis,” Angpt-1 derived platelet granules may play an adaptive role. Platelets store large quantities of Angpt-1 protein, releasing it from granules under the influence of inflammatory modulators such as thrombin. Degranulation of platelets could thereby help restore vascular homeostasis [4, 29, 40, 44, 45].
Angiopoietin-2 :
In contrast to the stabilizing actions of Angpt-1, Angpt-2 is a context-dependent antagonist of the Tie2 receptor despite significant primary sequence homology to Angpt-1 [25]. Indeed, overexpression of Angpt-2 results in an analogous phenotype to the Angpt-1 knockout model. Specifically, the vasculature is discontinuous and the endothelial cells themselves appear to have poor connection to the underlying extracellular matrix as noted by their spherical appearance [25]. Angpt-2 knockout mice remain relatively normal during the embryonic period, but exhibit poor vasculature remodeling in the post embryonic period and do not survive past two weeks of age secondary to lymphatic defects [25, 46].
Although Angpt-1 has a slightly higher binding affinity than Angpt-2 for the Tie2 receptor, when mixed together, Angpt-2 antagonizes the Tie2 receptor [15, 47, 48]. The basis for this signaling difference may be that Angpt-2 exists as a dimer rather than the higher-order oligomers of Angpt-1 [31–33, 49]. Therefore, at molar excess, the fibrinogen like domain of Angpt-2 may compete Angpt-1 off the receptor, leaving Tie2 molecules in a more dispersed—i.e., less phosphorylated—state on the endothelial surface.
Endothelial cells of the small vasculature express Angpt-2 [46, 50]. Pre-formed Angpt-2 protein is stored in the Weibel-Palade bodies of these cells, making Angpt-2 readily available for secretion after endothelial cells are stressed by triggers including hypoxia, TNFα, turbulent flow and thrombin [50–56]. Therefore, noxious stimuli trigger the endothelium to release pre-formed Angpt-2 protein. This Angpt-2 acts in a local, or paracrine, fashion to unseat Angpt-1, thereby deactivating Tie2 signaling. The switch from quiescence to disruption is fortified because activated Tie2 tonically inhibits the transcription of the ANGPT2 gene by keeping the transcription factor FoxO1 sequestered in the endothelial cytoplasm [47, 50, 57]. Therefore, when Tie2 phosphorylation is switched off by the rapid release of pre-formed Angpt-2 protein, that endothelial cell then commences to synthesize more Angpt-2 protein through FoxO1-mediated gene transcription.
Tie2:
Tie2 knockout is embryonically lethal at E10.5 due to a poorly developed heart and dilated leaky vessels that lack an investment of support cells [13, 36, 58]. (See Figures 1–2). These features are similar to the Angpt-1 knockout model [34, 35]. Tie2 is expressed in both active and quiescent endothelium and is necessary for adult vasculature. The quantity of receptor is downregulated in sprouting and inflamed vasculature [14, 59, 60]. Loss of Tie2 in vivo and in vitro results in a dysfunctional endothelium with impaired barrier function [4, 50, 61].
Figure 1:
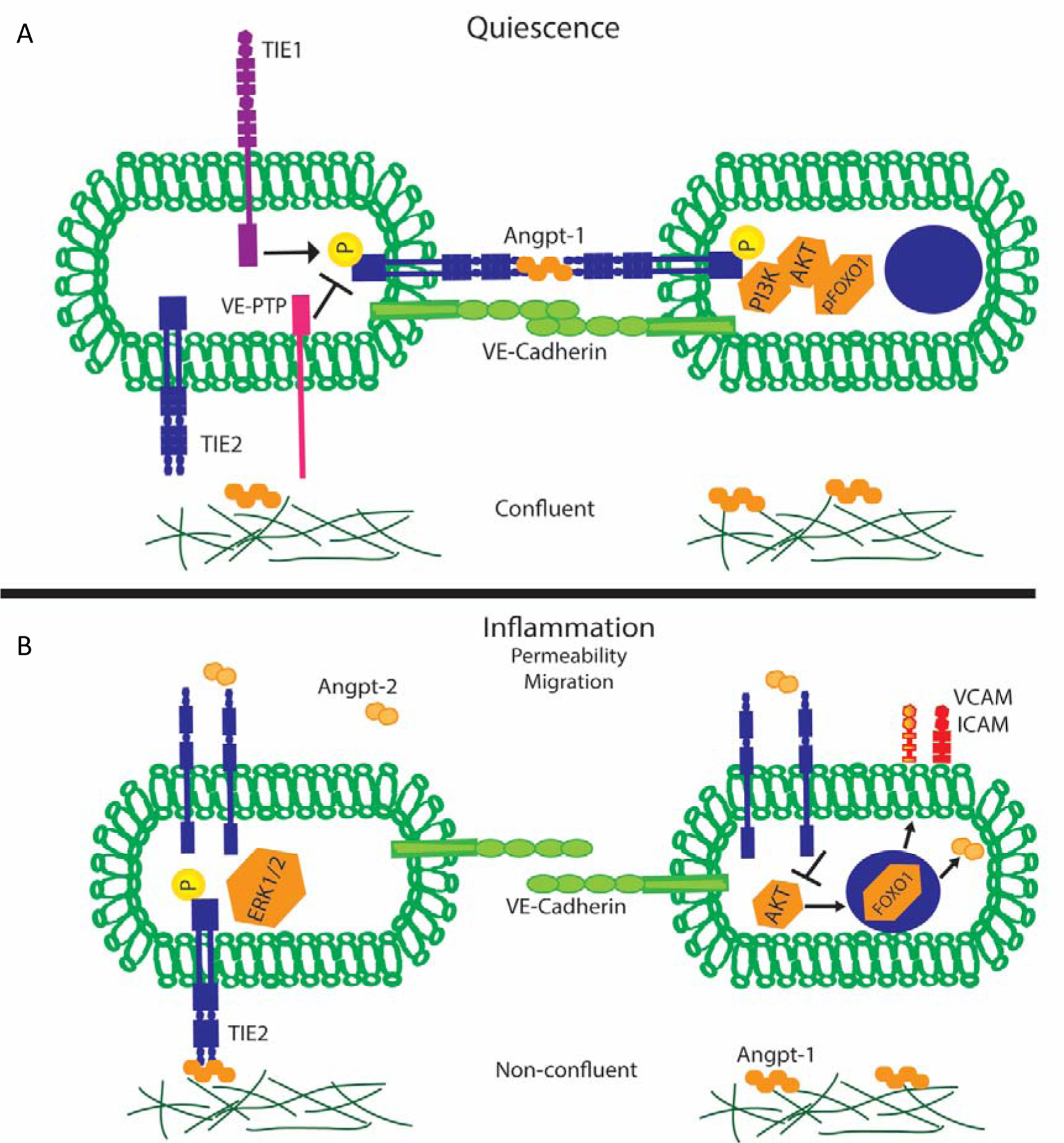
A) Endothelial cell Tie2 signaling during quiescence. Tie2 receptors cluster together and engage tetramers of Angpt-1 which results in activation of the receptor in a trans configuration. Furthermore, Tie1 binding further potentiates Tie2 activation. Downstream activation of Tie2 results in increased barrier function. B) Endothelial cell Tie2 signaling during inflammation. Tie2 receptor binds Angpt-2 dimers resulting in dephosphorylation and FOXO1 migration to the nucleus resulting in translation of inflammatory mediations and additional Angpt-2.
Figure 2:
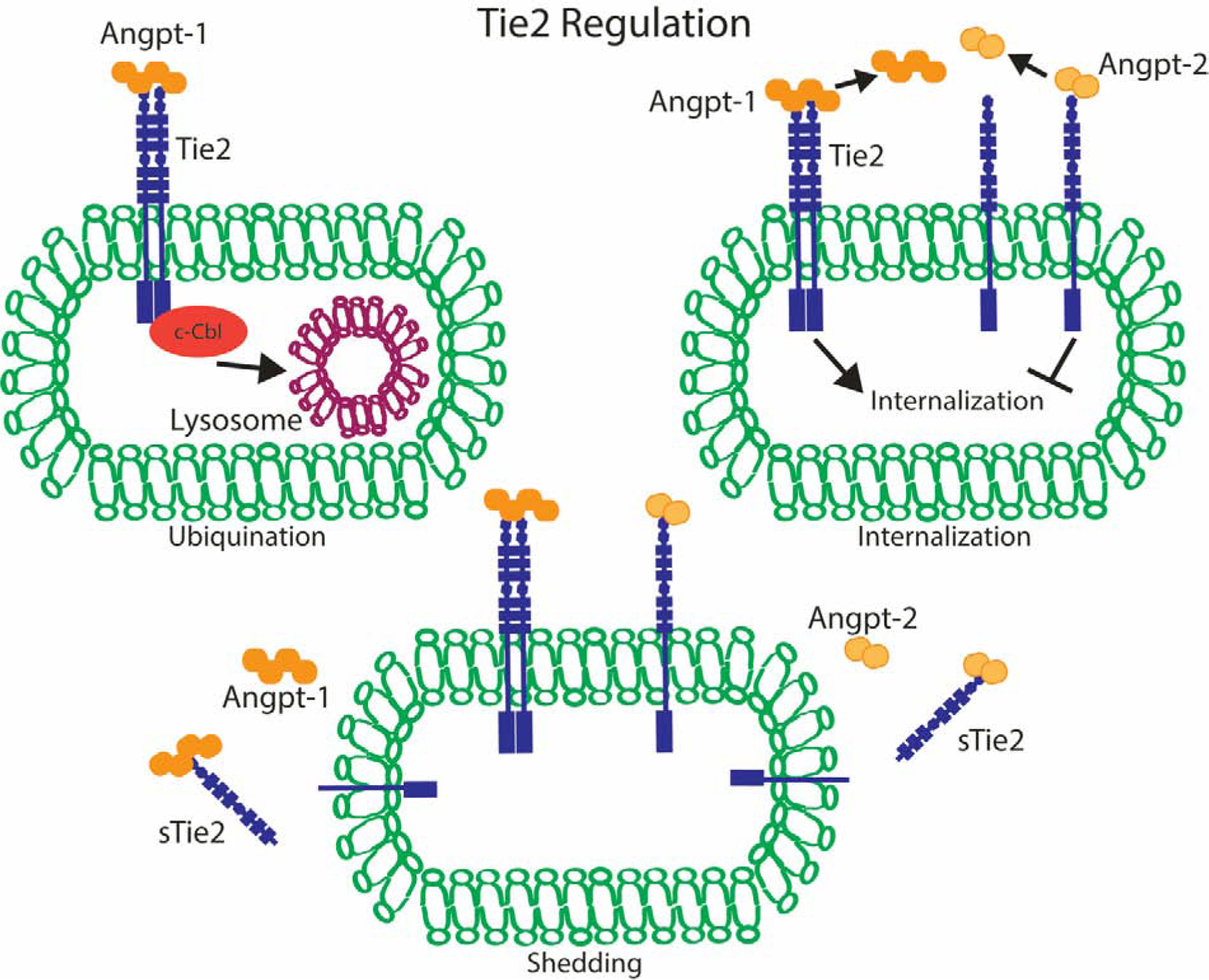
Tie2 regulation occurs through the above three mechanisms. Angpt-1 binding to Tie2 results in c-Cbl ubiquination and resulting trafficking to lysosomes for destruction. Angpt-1 binding to Tie2 results in internalization whereas Angpt-2 binding is only able to minimally internalize receptor. Both Angpt-1 and Angpt-2 are released from receptor after engagement. Finally, Angpt-2 and Angpt-1 engaged receptors are shed from endothelial cells by metalloproteinase resulting in decreased receptor pool available for receptor engagement.
In confluent or quiescent cells, Angpt-1 activates Tie2 at cell-cell borders in a trans configuration [62]. This signals PI3 kinase and Akt. Akt phosphorylates FoxO1 on key residues that inhibit its translocation to the nucleus and its binding of the ANGPT2 gene [50, 63–66]. Activated Akt also signals the molecular apparatus regulating the contractility of the endothelial cell and the essential junctional barrier protein VE-cadherin [6, 28, 67–69]. Expression of inflammatory adhesion molecules including ICAM1, VCAM1, and E-selectin is attenuated [70]. Finally, activated Akt suppresses key triggers of coagulation include phosphatidylserine and tissue factor [4]. The net output of these molecular events downstream of Tie2 activation is an endothelial layer that forms an effective barrier, suppresses the production of Angpt-2, inhibits leukocyte adhesion, and prevents the activation of coagulation.
Due to its critical function, Tie2 is regulated at multiple levels. Angpt-1 binding results in receptor ubiquitylation, which triggers receptor internalization and degradation [71]. Minimal receptor internalization occurs in the presence of Angpt-2. Tie2 releases both Angpt-1 and Anpt-2 after engagement, allowing for further activation of additional receptors [72]. Together, this indicates the system remains poised for recovery during critical insult as a quantity of Tie2 receptor remains available for phosphorylation during dysfunctional, Angpt-2 predominant states. Additional regulation of Tie2 is mediated by shedding of its extracellular domain. Matrix metalloproteinase cleavage facilitates both Angpt-1 and Angpt-2 induced shedding. Shed receptor binds additional ligand, preventing further receptor activation ultimately resulting in a smaller more directed pool of ligand and receptor [73, 74].
Tie1 and VE-PTP:
Further regulation of Tie2 receptor signaling occurs via transmembrane proteins that closely associate with Tie2. Although Tie1 does not directly bind Angpts, it is nonetheless critical for embryonic survival [75]. In in vitro and in vivo, Tie1 binding to Tie2 enhances the agonistic signaling of Angpt. Rapid cleavage of Tie1 occurs in response to inflammatory mediators such as TNFα and phorbol esters [22, 76].
VE-PTP enzymatically removes phosphates from proteins with which it associates. These include VEGFR-2, VE-cadherin, and Tie2 [77–79]. VE-PTP knockout mice die in utero of angiogenesis defects [80]. Inhibition of VE-PTP enhances Tie2 phosphorylation and strengthens the microvascular barrier against inflammatory triggers of permeability [81]. This barrier-enhancing effect of VE-PTP inhibition appears to require Tie2. Tie2 activation via VE-PTP inhibition is currently being explored in ocular conditions [82, 83].
PRE-CLINICAL EVIDENCE OF TIE2 REGULATING VESSELS DURING INFLAMMATION
Tie2 signaling appears to be a major determinant of the vascular response to a broad array of noxious stimuli. The earliest studies of the barrier-promoting effect of Tie2 stimulation demonstrated that Angpt-1 counteracts vascular leakage induced by unrelated triggers including VEGF, serotonin, and mustard oil [39, 84]. Since then, evidence from many laboratories testing diverse models of organ damage have coalesced around the central concept that Tie2 activation prevents vascular leakage, attenuates cellular inflammation, reduces spontaneous thrombus formation, and improves survival. As described next, this concept has been tested in preclinical models of sepsis, acute lung injury, and acute kidney injury. It has also been tested in experimental malaria, anthrax, and hemorrhagic fevers including Ebolavirus disease. The volume and diversity of the experimental settings convincingly propose that Tie2 signaling is a major determinant of outcomes across settings that acutely destabilize the vasculature.
Sepsis:
In the earliest report focusing on sepsis, Angpt-1 overexpression was shown to defend blood pressure, prevent leakage, attenuate inflammation, and improve survival in a model of endotoxic shock [85]. Angpt-1-mediated defense of vascular barrier function in the context of systemic endotoxin was thereafter shown to require p190RhoGap, a regulator of endothelial cell contraction [86], and p47phox, an upstream signal transducer [87]. Intravenous Angpt-1 administration improved multi-organ dysfunction and survival in a model of polymicrobial abdominal sepsis induced by cecal ligation and perforation (CLP) [88]. Acquired acute deficiency of Tie2 was found to be sufficient to weaken the vascular barrier in vivo [87]. Compared to wild-type littermates, loss of one Tie2 allele worsened vascular leakage and survival following either systemic endotoxin or CLP [87]. Tie1 expression must also be intact to counteract sepsis [22], and VE-PTP inhibition or genetic deletion has been shown to strengthen the vascular barrier against systemic inflammation [81].
Finally, Angpt-2 has been inhibited in several ways, all of which implicate this molecule as a mediator of adverse outcomes in experimental sepsis. First, partial genetic deletion of ANGPT2 was shown to protect CLP-induced mice from vascular leakage and to improve survival [89]. This result was verified by a second group, who also demonstrated protection against experimental sepsis with an Angpt-2 binding antibody [90]. The importance of the feed-forward loop that sustains Angpt-2 biosynthesis after the onset of inflammation was demonstrated in two studies that applied RNA interference to limit de novo production of Angpt-2 [66, 91]. Both reports demonstrated a positive impact on survival, suggesting that even a relatively late intervention—i.e., after pre-formed Angpt-2 protein has already been released—could still improve outcomes. The most recent work in this area has focused directly on the question of oligomerization by showing that a novel antibody that can “cluster” Angpt-2 also improves survival in two different models of sepsis [92]. Together, these studies propose that targeted manipulations to favor Tie2 signaling enhance organ function and survival whereas manipulations that weaken Tie2 signaling impair the organismic response to sepsis.
Lung injury/ARDS:
The Tie2 axis has been shown repeatedly to defend animals against disparate triggers of acute or chronic lung injury: this list includes intratracheal instillation of lipopolysaccharides [93]; parenteral lipopolysaccharides [81, 85, 86]; abdominal sepsis CLP [88, 91, 92, 94, 95]; hyperoxia in young animals [96]; the chemical warfare agent phosgene [97, 98]; and pulmonary hypertension following monocrotaline, serotonin or IL-6 [99, 100]. The salutary effect of Tie2 stimulation against unrelated noxious insults suggests a conserved role for Tie2 in the pathogenesis of lung injury.
Acute kidney injury:
The Tie2 axis has been tested in animal models of acute kidney injury (AKI) arising from sepsis. Angpt-2 deficiency was shown to protect against CLP-induced AKI [89]. Furthermore, intravenous Angpt-1 administration after the onset of CLP was shown to attenuate renal inflammation and expression of vascular adhesion molecules [88]. In models of acute injury that progress to fibrosis, administration of a recombinant Angpt-1 modified for better pharmacokinetic characteristics has been shown to ameliorate long-term outcomes [101, 102].
DIC:
Only two published studies have examined targeted manipulations of the Tie2 axis in the context of DIC. One study reported coagulation parameters in the CLP mouse model with and without treatment with the clustering antibody that converts Angpt-2 into a Tie2 agonist. That study demonstrated beneficial effects on sepsis-induced DIC with the use of the clustering antibody [92]. A second study examined the effects of partial Tie2 deletion and Angpt-1 addition in the early stages of endotoxemia or CLP [4]. Loss of one Tie2 allele was found to potentiate injury-induced fibrin deposition in vivo in the absence of inflammation. In the context of endotoxemia, the tendency of Tie2 heterozygotes to develop fibrin-rich clots was more severe than their wildtype littermates. Conversely, when Angpt-1 was administered in either the endotoxemia model or CLP, the tendency to clot formation was markedly attenuated.
Non-sepsis states of acute vascular destabilization:
Studies in models of acute vascular destabilization that do not involve sepsis collectively suggest that stimulation of Tie2 can defend or restore vascular quiescence in diverse contexts. These include hemorrhagic fevers (Puumala, hantavirus, Ebola, dengue), anthrax, malaria, influenza [95], chronic mycobacterial infection [50, 104], and systemic capillary leak syndrome (SCLS).
Hantavirus-infected endothelial cells are sensitized to triggers of permeability such as VEGF, an effect counteracted by Angpt-1 [105]. A study seeking to address causes of variation in the outcomes to Ebolavirus infection applied an experimental version of the virus to numerous outcrossed strains of mouse, then conduced genetics in the outliers. This study found that mice bearing hypomorphic alleles of TIE2 were more vulnerable to prolonged coagulation times, visceral hemorrhages and early death [106]. Certain TIE1 alleles were associated with increased weight loss, earlier death and increased overall mortality [106]. In a mouse model of dengue infection, treatment with Angpt-1 improved survival [107]. In a mouse model of malaria that develops cerebral edema, Angpt-1 was shown to restore vascular integrity, reduce edema, and improve survival [108].
Systemic administration of anthrax lethal toxin downregulated Tie2 signaling and increased vascular leakage in mice [103]. Partial deletion of Angpt-2 improved survival in this model. Partial loss of Angpt-2 attenuated survival, and administration of Angpt-1 by gene transfer improved survival in this model. Finally, in primates infected with an attenuated strain of Bacillus anthracis that nonetheless provokes vascular collapse, circulating Angpt-2 levels rose rapidly whereas Angpt-1 levels fell. The changes in circulating Angpts were proportional to the inoculum of bacteria [103].
CLINICAL EVIDENCE IMPLICATING THE TIE2 AXIS
Overview
Clinical studies of the Tie2 axis have primarily involved measurement of circulating levels of Angpt-1 and Angpt-2, although there are some reports on soluble levels of Tie2 and Tie1. Genetic polymorphisms influencing Angpt-2 and Tie2 expression have also been linked to risk of ARDS in the setting of both sepsis and non-sepsis.
Sepsis and ARDS
Early work in sepsis focused on the lung and showed that high circulating Angpt-2 was associated with impaired gas exchange, perhaps the result of vascular leak [109]. Subsequent studies in severe sepsis showed that Angpt-2 can increase dramatically in the circulation early in the course of disease and associated well with loss of vascular integrity in various organs [110, 111]. Angpt-2 levels even spike with infusion of low-dose endotoxin in heathy volunteers [112], leading many to propose that it could be a useful biomarker of infection. Furthermore, as Angpt-2 increases with systemic inflammation, Angpt-1 falls. Thus, the ratio of Angpt-2/Angpt-1 in the circulation may be a better indicator of Tie2 signaling impairment than either protein alone [111, 113]. However, “sterile inflammation” syndromes such as trauma or major surgery can also disrupt Tie2 signaling making it less specific for sepsis [114–116]. Finally, genetic polymorphisms linked to Angpt-2 over-secretion have shown an increased risk for ARDS [117].
Common variants reducing TIE2 gene expression itself have also been linked to the development of ARDS [95].
Sepsis and DIC
Disruption of the endothelial Tie2 axis also appears to be a sentinel event in septic DIC. We recently described proteomic analysis in septic DIC patients that revealed a network involving inflammation and coagulation with Angpt-2, occupying a central node [4]. Angpt-2 was strongly associated with traditional DIC markers including platelet counts, yet more accurately predicted mortality in 2 large independent cohorts (combined N = 1,077). Similarly, in endotoxemic mice, reduced Tie2 signaling preceded signs of overt DIC. Intravital imaging of the microvascular revealed excessive fibrin accumulation in a pattern reminiscent of Tie2 deficiency. Conversely, Tie2 activation normalized prothrombotic responses by inhibiting endothelial tissue factor and phosphatidylserine exposure. Importantly, Tie2 activation in this model had no adverse effects on bleeding. Given that interventions targeting Tie2 can normalize coagulation without increasing bleeding risk this approach could prove superior to current DIC therapies.
Cardiopulmonary bypass and acute kidney injury
One study measured serum Angpt-2 from 25 adult patients before and after cardiopulmonary bypass CPB, and compared it with indices of organ dysfunction, duration of mechanical ventilation, length of stay in the ICU, and hospital mortality. Angpt-2 levels steadily increased from 2.6 ± 2.4 ng/mL at 0 h up to 7.3 ± 4.6 ng/mL at 24 h following CPB (P < 0.001). The release of Angpt-2 correlated with the duration of CPB, aortic cross-clamp time, and post-CPB lactate levels. Changes in Angpt-2 during follow-up correlated with PaO2/FiO2 ratios, hemodynamics, fluid balance, and disease severity measures. Angpt-2 levels at 12 h predicted the durations of mechanical ventilation and ICU stay, and hospital mortality [118]. Another study of 21 patients who experienced a 50% or greater increase in serum creatinine after cardiac surgery compared circulating levels of Angpt-1, Angpt-2, and Tie2 to propensity matched controls. The investigators also measured kidney injury molecule-1 (KIM-1) and N-acetyl-beta-D-glucosaminidase (NAG) in the urine. Angpt-2 plasma levels increased over time in patients with AKI (from 4.2 to 11.6 ng/ml) but did so as well in control patients (from 3.0 to 6.7 ng/ml). Plasma levels of Tie2 and Angpt-1 also decreased in both AKI patients and controls. However, there was a positive correlation between plasma levels of Angpt-2 and urinary levels of NAG but not KIM-1 [21]. Although circulating Angpt-2 levels were predictive of AKI and renal failure in patients with acute pancreatitis the relationship may be non-specific [119]. Angpt-2 significantly correlated with acute phase proteins as well.
Other states of vascular destabilization
In patients with certain hemorrhagic fevers including hanta virus, dengue, and Crimean-Congo hemorrhagic fever the serum quantity of Angpt-2 increases in a similar fashion to that in septic patients [120–122]. In Clarkson disease (systemic capillary leak syndrome) patients have transient episodes of vascular leakage resulting in hemoconcentration and hypotension. Angpt-2 increases during disease flares and returns to baseline during remission [123]. Angpt-1 and Tie2 are both dysregulated in blood-outgrowth endothelial cells from affected humans suggesting a further role of this pathway in the systemic process [124]. Finally, P. falciparum cerebral malaria has also been associated with Angpt/Tie derangements. Once again as levels of Angpt-2 serum levels increased, the severity of falciparum malaria worsened [125].
CONCLUSIONS AND FUTURE DIRECTIONS
Vascular disruption underlies cardinal features of critical illness that culminate in death. A body of work spanning biochemistry, cell biology, rodent and primate models, human observational studies, and human genetics collectively propose that the Tie2 axis may be an important determinant of the vascular response to acute destabilizing stressors. While successful translation of preclinical observations to the bedside has been a major challenge in critical care medicine, the evidence emerging from laboratories around the world proposes several routes to clinical realization.
Genetic markers of the Tie2 axis and/or circulating Angpt measurements could one day inform clinicians regarding the susceptibility of a given patient to adverse vascular-related outcomes. In turn, such knowledge could be used to guide routine clinical care—e.g., fluid-conservative vs. - liberal resuscitation strategies or adjustment of ventilator settings to avoid secondary damage—or inform the selection of patients for trials of investigational agents that target the vasculature. Because circulating Angpt-2 levels indirectly report Tie2 signaling, sequential Angpt-2 measurements could be used to monitor the response to investigational therapies targeting Tie2. Such a surrogate marker of target engagement amenable to evaluation in a Phase 1b setting can significantly impact clinical development. As a circulating protein, Angpt-2 is amenable to neutralization with an antibody. A clinical-stage Angpt-2 neutralizing antibody has already been developed for cancer testing [126]. Conversely, Angpt-1 could be delivered as a modified recombinant protein or by gene transfer or even cell-based therapy that is already being tested in ARDS [127]. VE-PTP inhibition is also being tested in patients with retinal edema [83, 128]. Therefore, multiple “shovel-ready” approaches exist to test this promising hypothesis in patients.
Translational pursuit of the Tie2 axis could result in new ways to diagnose, stratify, and treat ICU patients. Any such advance would constitute a breakthrough for the many patients affected by sepsis, ARDS, and related conditions.
Table 1:
Factors modulating TIE2 signaling
| Quiescence | Inflammation/Critical Illness |
|---|---|
| Angpt-1 (tetramers) | Angpt-2 (dimers) |
| TIE1 | VE-PTP |
| Integrins | Cytokines |
| Confluence | Sub-confluence |
KEY POINTS
Cardinal clinical features of sepsis such as vascular leakage and coagulation relate to microvascular health. A vascular signaling pathway anchored by Tie2 actively promotes quiescence.
During sepsis, the endogenous Tie2 antagonist protein called Angiopoietin-2 is induced, leading to rapid reduction in Tie2 signaling. This renders microvessels leaky, proinflammatory, and prothrombotic.
Experimental models of sepsis, acute lung injury, and other acute stressors collectively demonstrate that restoration of Tie2 signaling exerts a protective effect and enhances survival.
ICU studies consistently demonstrate early and progressive elevation of circulating Angiopoietin-2 that is associated with worsening oxygenation, increased risk of DIC and other adverse outcomes.
Human genetic studies suggest that common variants in ANGPT2 and TIE2 genes may be linked to the risk of acute lung injury.
SYNOPSIS
Lethal features of sepsis and acute respiratory distress syndrome (ARDS) relate to the health of small blood vessels. For example, alveolar infiltration with proteinaceous fluid is often driven by breach of the microvascular barrier. Spontaneous thrombus formation within inflamed microvessels exacerbates organ ischemia, and in its final stages, erupts into overt disseminated intravascular coagulation (DIC). A vascular endothelial signaling axis, the Angiopoietin-Tie2 pathway, may mediate the abrupt transition from microvascular integrity to pathological disruption. Tonically activated Tie2 promotes vascular quiescence. During inflammation, however, Tie2 signaling is rapidly toggled off as levels of its endogenous antagonist, Angiopoietin-2, rise. When Tie2 signaling drops, the microvascular barrier collapses, endothelium becomes adhesive to leukocytes, and the vessel wall no longer prevents spontaneous coagulation. This review will summarize a large body of preclinical and clinical results that implicate the Tie2 pathway as a promising target to restore microvascular health in sepsis and ARDS.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DISCLOSURE STATEMENT
Disclose any relationship with a commercial company that has a direct financial interest in subject matter or materials discussed in article or with a company making a competing product. If nothing to disclose, please state “The Authors have nothing to disclose.”
S.M.P. is a member of the Scientific Advisory Board for Aerpio
REFERENCES
- 1.Cohen J, et al. Sepsis: a roadmap for future research. Lancet Infect Dis 2015;15:581–614. [DOI] [PubMed] [Google Scholar]
- 2.Iwashyna TJ, et al. Long-term cognitive impairment and functional disability among survivors of severe sepsis. JAMA 2010;304:1787–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liu V, et al. Hospital deaths in patients with sepsis from 2 independent cohorts. JAMA 2014;312:90–2. [DOI] [PubMed] [Google Scholar]
- 4.Higgins SJ, et al. Tie2 protects the vasculature against thrombus formation in systemic inflammation. J Clin Invest 2018;128:1471–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thamm K, et al. Molecular Regulation of Acute Tie2 Suppression in Sepsis. Critical care medicine 2018;46:e928–e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Parikh SM, et al. Excess Circulating Angiopoietin-2 May Contribute to Pulmonary Vascular Leak in Sepsis in Humans. PLOS Medicine 2006;3:e46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.van der Flier M, et al. PLASMA VASCULAR ENDOTHELIAL GROWTH FACTOR IN SEVERE SEPSIS. Shock 2005;23:35–8. [DOI] [PubMed] [Google Scholar]
- 8.Opal SM and van der Poll T. Endothelial barrier dysfunction in septic shock. Journal of internal medicine 2015;277:277–93. [DOI] [PubMed] [Google Scholar]
- 9.Olsson AK, et al. VEGF receptor signalling - in control of vascular function. Nature reviews Molecular cell biology 2006;7:359–71. [DOI] [PubMed] [Google Scholar]
- 10.Li X, et al. Basal and angiopoietin-1–mediated endothelial permeability is regulated by sphingosine kinase-1. Blood 2008;111:3489–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Backer DD, et al. Microvascular Blood Flow Is Altered in Patients with Sepsis. American Journal of Respiratory and Critical Care Medicine 2002;166:98–104. [DOI] [PubMed] [Google Scholar]
- 12.Heo K-S, Fujiwara K and Abe J.-i.. Disturbed-Flow-Mediated Vascular Reactive Oxygen Species Induce Endothelial Dysfunction. Circulation Journal 2011;75:2722–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sato TN, et al. Distinct roles of the receptor tyrosine kinases Tie-1 and Tie-2 in blood vessel formation. Nature 1995;376:70–4. [DOI] [PubMed] [Google Scholar]
- 14.Wong AL, et al. Tie2 expression and phosphorylation in angiogenic and quiescent adult tissues. Circ Res 1997;81:567–74. [DOI] [PubMed] [Google Scholar]
- 15.Yuan HT, et al. Angiopoietin 2 is a partial agonist/antagonist of Tie2 signaling in the endothelium. Mol Cell Biol 2009;29:2011–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee KW, Lip GYH and Blann AD. Plasma Angiopoietin-1, Angiopoietin-2, Angiopoietin Receptor Tie-2, and Vascular Endothelial Growth Factor Levels in Acute Coronary Syndromes. Circulation 2004;110:2355–60. [DOI] [PubMed] [Google Scholar]
- 17.Davis JS, et al. Angiopoietin-2 is increased in sepsis and inversely associated with nitric oxide-dependent microvascular reactivity. Critical Care 2010;14:R89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.David S, et al. Angiopoietin-2 may contribute to multiple organ dysfunction and death in sepsis*. Critical care medicine 2012;40:3034–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu K-L, et al. Elevated plasma thrombomodulin and angiopoietin-2 predict the development of acute kidney injury in patients with acute myocardial infarction. Critical Care 2014;18:R100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kümpers P, et al. Angiopoietin-2 in patients requiring renal replacement therapy in the ICU: relation to acute kidney injury, multiple organ dysfunction syndrome and outcome. Intensive Care Medicine 2010;36:462–70. [DOI] [PubMed] [Google Scholar]
- 21.Jongman RM, et al. Angiopoietin/Tie2 Dysbalance Is Associated with Acute Kidney Injury after Cardiac Surgery Assisted by Cardiopulmonary Bypass. PLoS One 2015;10:e0136205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Korhonen EA, et al. Tie1 controls angiopoietin function in vascular remodeling and inflammation. J Clin Invest 2016;126:3495–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Savant S, et al. The Orphan Receptor Tie1 Controls Angiogenesis and Vascular Remodeling by Differentially Regulating Tie2 in Tip and Stalk Cells. Cell Reports 2015;12:1761–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Papapetropoulos A, et al. Angiopoietin-1 Inhibits Endothelial Cell Apoptosis via the Akt/Survivin Pathway. Journal of Biological Chemistry 2000;275:9102–5. [DOI] [PubMed] [Google Scholar]
- 25.Maisonpierre PC, et al. Angiopoietin-2, a Natural Antagonist for Tie2 That Disrupts in vivo Angiogenesis. Science 1997;277:55–60. [DOI] [PubMed] [Google Scholar]
- 26.Fiedler U, et al. Angiopoietin-2 sensitizes endothelial cells to TNF-α and has a crucial role in the induction of inflammation. Nature Medicine 2006;12:235–9. [DOI] [PubMed] [Google Scholar]
- 27.Frye M, et al. Interfering with VE-PTP stabilizes endothelial junctions in vivo via Tie-2 in the absence of VE-cadherin. The Journal of Experimental Medicine 2015;212:2267–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gamble JR, et al. Angiopoietin-1 Is an Antipermeability and Anti-Inflammatory Agent In Vitro and Targets Cell Junctions. Circulation Research 2000;87:603–7. [DOI] [PubMed] [Google Scholar]
- 29.Daly C, et al. Angiopoietins bind thrombomodulin and inhibit its function as a thrombin cofactor. Scientific Reports 2018;8:505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Suri C, et al. Requisite role of angiopoietin-1, a ligand for the TIE2 receptor, during embryonic angiogenesis. Cell 1996;87:1171–80. [DOI] [PubMed] [Google Scholar]
- 31.Davis S, et al. Angiopoietins have distinct modular domains essential for receptor binding, dimerization and superclustering. Nature Structural Biology 2003;10:38–44. [DOI] [PubMed] [Google Scholar]
- 32.Kim K-T, et al. Oligomerization and Multimerization Are Critical for Angiopoietin-1 to Bind and Phosphorylate Tie2. Journal of Biological Chemistry 2005;280:20126–31. [DOI] [PubMed] [Google Scholar]
- 33.Procopio WN, et al. Angiopoietin-1 and −2 Coiled Coil Domains Mediate Distinct Homo-oligomerization Patterns, but Fibrinogen-like Domains Mediate Ligand Activity. Journal of Biological Chemistry 1999;274:30196–201. [DOI] [PubMed] [Google Scholar]
- 34.Davis S, et al. Isolation of Angiopoietin-1, a Ligand for the TIE2 Receptor, by Secretion-Trap Expression Cloning. Cell 1996;87:1161–9. [DOI] [PubMed] [Google Scholar]
- 35.Suri C, et al. Requisite Role of Angiopoietin-1, a Ligand for the TIE2 Receptor, during Embryonic Angiogenesis. Cell 1996;87:1171–80. [DOI] [PubMed] [Google Scholar]
- 36.Dumont DJ, et al. Dominant-negative and targeted null mutations in the endothelial receptor tyrosine kinase, tek, reveal a critical role in vasculogenesis of the embryo. Genes Dev 1994;8:1897–909. [DOI] [PubMed] [Google Scholar]
- 37.Thurston G, et al. Angiopoietin-1 protects the adult vasculature against plasma leakage. Nature Medicine 2000;6:460–3. [DOI] [PubMed] [Google Scholar]
- 38.Suri C, et al. Increased Vascularization in Mice Overexpressing Angiopoietin-1. Science 1998;282:468–71. [DOI] [PubMed] [Google Scholar]
- 39.Thurston G, et al. Leakage-resistant blood vessels in mice transgenically overexpressing angiopoietin-1. Science 1999;286:2511–4. [DOI] [PubMed] [Google Scholar]
- 40.Huang Y-Q, Li J-J and Karpatkin S. Identification of a family of alternatively spliced mRNA species of angiopoietin-1. Blood 2000;95:1993–9. [PubMed] [Google Scholar]
- 41.Mammoto T, et al. Platelet rich plasma extract promotes angiogenesis through the angiopoietin1-Tie2 pathway. Microvascular Research 2013;89:15–24. [DOI] [PubMed] [Google Scholar]
- 42.Xu Y and Yu Q. Angiopoietin-1, Unlike Angiopoietin-2, Is Incorporated into the Extracellular Matrix via Its Linker Peptide Region. Journal of Biological Chemistry 2001;276:34990–8. [DOI] [PubMed] [Google Scholar]
- 43.Saharinen P, et al. Angiopoietins assemble distinct Tie2 signalling complexes in endothelial cell–cell and cell–matrix contacts. Nat Cell Biol 2008;10:527. [DOI] [PubMed] [Google Scholar]
- 44.Li JJ, et al. Thrombin induces the release of angiopoietin-1 from platelets. Thrombosis and haemostasis 2001;85:204–6. [PubMed] [Google Scholar]
- 45.Ho-Tin-Noé B, et al. Platelet Granule Secretion Continuously Prevents Intratumor Hemorrhage. Cancer Research 2008;68:6851–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gale NW, et al. Angiopoietin-2 Is Required for Postnatal Angiogenesis and Lymphatic Patterning, and Only the Latter Role Is Rescued by Angiopoietin-1. Developmental cell 2002;3:411–23. [DOI] [PubMed] [Google Scholar]
- 47.Daly C, et al. Angiopoietin-2 functions as an autocrine protective factor in stressed endothelial cells. Proceedings of the National Academy of Sciences 2006;103:15491–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kim I, et al. Angiopoietin-2 at high concentration can enhance endothelial cell survival through the phosphatidylinositol 3′-kinase/Akt signal transduction pathway. Oncogene 2000;19:4549–52. [DOI] [PubMed] [Google Scholar]
- 49.Davis S, et al. Angiopoietins have distinct modular domains essential for receptor binding, dimerization and superclustering. Nature structural biology 2003;10:38–44. [DOI] [PubMed] [Google Scholar]
- 50.Kim M, et al. Opposing actions of angiopoietin-2 on Tie2 signaling and FOXO1 activation. J Clin Invest 2016;126:3511–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fiedler U, et al. The Tie-2 ligand Angiopoietin-2 is stored in and rapidly released upon stimulation from endothelial cell Weibel-Palade bodies. Blood 2004;103:4150–6. [DOI] [PubMed] [Google Scholar]
- 52.Mandriota SJ and Pepper MS. Regulation of Angiopoietin-2 mRNA Levels in Bovine Microvascular Endothelial Cells by Cytokines and Hypoxia. Circulation Research 1998;83:852–9. [DOI] [PubMed] [Google Scholar]
- 53.Oh H, et al. Hypoxia and Vascular Endothelial Growth Factor Selectively Up-regulate Angiopoietin-2 in Bovine Microvascular Endothelial Cells. Journal of Biological Chemistry 1999;274:15732–9. [DOI] [PubMed] [Google Scholar]
- 54.Goettsch W, et al. Flow-dependent regulation of angiopoietin-2. Journal of Cellular Physiology 2008;214:491–503. [DOI] [PubMed] [Google Scholar]
- 55.Tressel SL, et al. Laminar Shear Inhibits Tubule Formation and Migration of Endothelial Cells by an Angiopoietin-2–Dependent Mechanism. Arteriosclerosis, Thrombosis, and Vascular Biology 2007;27:2150–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dixit M, et al. Shear stress-induced activation of the AMP-activated protein kinase regulates FoxO1a and angiopoietin-2 in endothelial cells. Cardiovascular Research 2007;77:160–8. [DOI] [PubMed] [Google Scholar]
- 57.Daly C, et al. Angiopoietin-1 modulates endothelial cell function and gene expression via the transcription factor FKHR (FOXO1). Genes & Development 2004;18:1060–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dumont DJ, et al. Dominant-negative and targeted null mutations in the endothelial receptor tyrosine kinase, tek, reveal a critical role in vasculogenesis of the embryo. Genes & Development 1994;8:1897–909. [DOI] [PubMed] [Google Scholar]
- 59.Puri MC, et al. Interaction of the TEK and TIE receptor tyrosine kinases during cardiovascular development. Development 1999;126:4569–80. [DOI] [PubMed] [Google Scholar]
- 60.Felcht M, et al. Angiopoietin-2 differentially regulates angiogenesis through TIE2 and integrin signaling. The Journal of Clinical Investigation 2012;122:1991–2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ghosh CC, et al. Gene control of tyrosine kinase TIE2 and vascular manifestations of infections. Proceedings of the National Academy of Sciences 2016;113:2472–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Saharinen P, et al. Angiopoietins assemble distinct Tie2 signalling complexes in endothelial cell-cell and cell-matrix contacts. Nat Cell Biol 2008;10:527–37. [DOI] [PubMed] [Google Scholar]
- 63.Daly C, et al. Angiopoietin-1 modulates endothelial cell function and gene expression via the transcription factor FKHR (FOXO1). Genes Dev 2004;18:1060–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Daly C, et al. Angiopoietin-2 functions as an autocrine protective factor in stressed endothelial cells. Proc Natl Acad Sci U S A 2006;103:15491–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Goettsch W, et al. Flow-dependent regulation of angiopoietin-2. J Cell Physiol 2008;214:491–503. [DOI] [PubMed] [Google Scholar]
- 66.Ghosh CC, et al. Drug Repurposing Screen Identifies Foxo1-Dependent Angiopoietin-2 Regulation in Sepsis. Crit Care Med 2015;43:e230–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mammoto T, et al. Angiopoietin-1 Requires p190 RhoGAP to Protect against Vascular Leakage in Vivo. Journal of Biological Chemistry 2007;282:23910–8. [DOI] [PubMed] [Google Scholar]
- 68.Cascone I, et al. Tie-2–dependent activation of RhoA and Rac1 participates in endothelial cell motility triggered by angiopoietin-1. Blood 2003;102:2482–90. [DOI] [PubMed] [Google Scholar]
- 69.Alfieri A, et al. Angiopoietin-1 regulates microvascular reactivity and protects the microcirculation during acute endothelial dysfunction: Role of eNOS and VE-cadherin. Pharmacological Research 2014;80:43–51. [DOI] [PubMed] [Google Scholar]
- 70.Kim I, et al. Angiopoietin-1 Reduces VEGF-Stimulated Leukocyte Adhesion to Endothelial Cells by Reducing ICAM-1, VCAM-1, and E-Selectin Expression. Circulation Research 2001;89:477–9. [DOI] [PubMed] [Google Scholar]
- 71.Wehrle C, Van Slyke P and Daniel J Dumont. Angiopoietin-1-induced ubiquitylation of Tie2 by c-Cbl is required for internalization and degradation. Biochem J 2009;423:375–80. [DOI] [PubMed] [Google Scholar]
- 72.Bogdanovic E, Nguyen VPKH and Dumont DJ. Activation of Tie2 by angiopoietin-1 and angiopoietin-2 results in their release and receptor internalization. Journal of Cell Science 2006;119:3551–60. [DOI] [PubMed] [Google Scholar]
- 73.Findley Clarence M, et al. VEGF Induces Tie2 Shedding via a Phosphoinositide 3-Kinase/Akt–Dependent Pathway to Modulate Tie2 Signaling. Arteriosclerosis, Thrombosis, and Vascular Biology 2007;27:2619–26. [DOI] [PubMed] [Google Scholar]
- 74.Reusch P, et al. Identification of a soluble form of the angiopoietin receptor TIE-2 released from endothelial cells and present in human blood. Angiogenesis 2001;4:123–31. [DOI] [PubMed] [Google Scholar]
- 75.Puri MC, et al. The receptor tyrosine kinase TIE is required for integrity and survival of vascular endothelial cells. The EMBO journal 1995;14:5884–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Marron MB, et al. Regulated Proteolytic Processing of Tie1 Modulates Ligand Responsiveness of the Receptor-tyrosine Kinase Tie2. Journal of Biological Chemistry 2007;282:30509–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Fachinger G, Deutsch U and Risau W. Functional interaction of vascular endothelial-protein-tyrosine phosphatase with the angiopoietin receptor Tie-2. Oncogene 1999;18:5948–53. [DOI] [PubMed] [Google Scholar]
- 78.Nawroth R, et al. VE-PTP and VE-cadherin ectodomains interact to facilitate regulation of phosphorylation and cell contacts. EMBO J 2002;21:4885–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hayashi M, et al. VE-PTP regulates VEGFR2 activity in stalk cells to establish endothelial cell polarity and lumen formation. Nature communications 2013;4:1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Dominguez MG, et al. Vascular endothelial tyrosine phosphatase (VE-PTP)-null mice undergo vasculogenesis but die embryonically because of defects in angiogenesis. Proc Natl Acad Sci U S A 2007;104:3243–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Frye M, et al. Interfering with VE-PTP stabilizes endothelial junctions in vivo via Tie-2 in the absence of VE-cadherin. J Exp Med 2015;212:2267–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Shen J, et al. Targeting VE-PTP activates TIE2 and stabilizes the ocular vasculature. J Clin Invest 2014;124:4564–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Campochiaro PA, et al. Treatment of diabetic macular edema with an inhibitor of vascular endothelial-protein tyrosine phosphatase that activates Tie2. Ophthalmology 2015;122:545–54. [DOI] [PubMed] [Google Scholar]
- 84.Thurston G, et al. Angiopoietin-1 protects the adult vasculature against plasma leakage. Nat Med 2000;6:460–3. [DOI] [PubMed] [Google Scholar]
- 85.Witzenbichler B, et al. Protective role of angiopoietin-1 in endotoxic shock. Circulation 2005;111:97–105. [DOI] [PubMed] [Google Scholar]
- 86.Mammoto T, et al. Angiopoietin-1 requires p190 RhoGAP to protect against vascular leakage in vivo. J Biol Chem 2007;282:23910–8. [DOI] [PubMed] [Google Scholar]
- 87.Ghosh CC, et al. Angiopoietin-1 requires oxidant signaling through p47phox to promote endothelial barrier defense. PLoS One 2015;10:e0119577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.David S, et al. Acute administration of recombinant Angiopoietin-1 ameliorates multiple-organ dysfunction syndrome and improves survival in murine sepsis. Cytokine 2011;55:251–9. [DOI] [PubMed] [Google Scholar]
- 89.David S, et al. Angiopoietin-2 may contribute to multiple organ dysfunction and death in sepsis*. Crit Care Med 2012;40:3034–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ziegler T, et al. Angiopoietin 2 mediates microvascular and hemodynamic alterations in sepsis. J Clin Invest 2013;123:3436–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Stiehl T, et al. Lung-targeted RNA interference against angiopoietin-2 ameliorates multiple organ dysfunction and death in sepsis. Crit Care Med 2014;42:e654–62. [DOI] [PubMed] [Google Scholar]
- 92.Han S, et al. Amelioration of sepsis by TIE2 activation-induced vascular protection. Sci Transl Med 2016;20:335–55. [DOI] [PubMed] [Google Scholar]
- 93.McCarter SD, et al. Cell-based angiopoietin-1 gene therapy for acute lung injury. Am J Respir Crit Care Med 2007;175:1014–26. [DOI] [PubMed] [Google Scholar]
- 94.Kumpers P, et al. The synthetic Tie2 agonist peptide vasculotide protects against vascular leakage and reduces mortality in murine abdominal sepsis. Crit Care 2011;15:R261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ghosh CC, et al. Gene control of tyrosine kinase TIE2 and vascular manifestations of infections. Proc Natl Acad Sci U S A 2016;113:2472–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Bhandari V, et al. Hyperoxia causes angiopoietin 2-mediated acute lung injury and necrotic cell death. Nat Med 2006;12:1286–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Shen J, et al. Adenovirus-delivered angiopoietin-1 treatment for phosgene-induced acute lung injury. Inhal Toxicol 2013;25:272–9. [DOI] [PubMed] [Google Scholar]
- 98.Shao Y, et al. Mesenchymal stem cells overexpressing Ang1 attenuates phosgene-induced acute lung injury in rats. Inhal Toxicol 2018, 10.1080/08958378.2018.1521483:1-8. [DOI] [PubMed] [Google Scholar]
- 99.Kugathasan L, et al. The angiopietin-1-Tie2 pathway prevents rather than promotes pulmonary arterial hypertension in transgenic mice. J Exp Med 2009;206:2221–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Zhao YD, et al. Protective role of angiopoietin-1 in experimental pulmonary hypertension. Circ Res 2003;92:984–91. [DOI] [PubMed] [Google Scholar]
- 101.Kim W, et al. COMP-angiopoietin-1 ameliorates renal fibrosis in a unilateral ureteral obstruction model. J Am Soc Nephrol 2006;17:2474–83. [DOI] [PubMed] [Google Scholar]
- 102.Lee S, et al. Protective effect of COMP-angiopoietin-1 on cyclosporine-induced renal injury in mice. Nephrol Dial Transplant 2008;23:2784–94. [DOI] [PubMed] [Google Scholar]
- 103.Ghosh CC, et al. Impaired function of the Tie-2 receptor contributes to vascular leakage and lethality in anthrax. Proc Natl Acad Sci U S A 2012, 1120755109 [pii] 10.1073/pnas.1120755109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Tabruyn SP, et al. Angiopoietin-2-driven vascular remodeling in airway inflammation. Am J Pathol 2010;177:3233–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Gavrilovskaya IN, et al. Hantaviruses direct endothelial cell permeability by sensitizing cells to the vascular permeability factor VEGF, while angiopoietin 1 and sphingosine 1-phosphate inhibit hantavirus-directed permeability. J Virol 2008;82:5797–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Rasmussen AL, et al. Host genetic diversity enables Ebola hemorrhagic fever pathogenesis and resistance. Science 2014;346:987–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Phanthanawiboon S, et al. Acute Systemic Infection with Dengue Virus Leads to Vascular Leakage and Death through Tumor Necrosis Factor-α and Tie2/Angiopoietin Signaling in Mice Lacking Type I and II Interferon Receptors. PloS one 2016;11:e0148564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Higgins SJ, et al. Dysregulation of angiopoietin-1 plays a mechanistic role in the pathogenesis of cerebral malaria. Science Translational Medicine 2016;8:358ra128–358ra128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Parikh SM, et al. Excess circulating angiopoietin-2 may contribute to pulmonary vascular leak in sepsis in humans. PLoS Med 2006;3:e46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Orfanos SE, et al. Angiopoietin-2 is increased in severe sepsis: correlation with inflammatory mediators. Crit Care Med 2007;35:199–206. [DOI] [PubMed] [Google Scholar]
- 111.Ong T, et al. Ratio of angiopoietin-2 to angiopoietin-1 as a predictor of mortality in acute lung injury patients. Crit Care Med 2010;38:1845–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kumpers P, et al. Time course of angiopoietin-2 release during experimental human endotoxemia and sepsis. Crit Care 2009;13:R64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Giuliano JS Jr., et al. Admission angiopoietin levels in children with septic shock. Shock 2007;28:650–4. [PMC free article] [PubMed] [Google Scholar]
- 114.Ganter MT, et al. Angiopoietin-2, marker and mediator of endothelial activation with prognostic significance early after trauma? Ann Surg 2008;247:320–6. [DOI] [PubMed] [Google Scholar]
- 115.Fremont RD, et al. Acute lung injury in patients with traumatic injuries: utility of a panel of biomarkers for diagnosis and pathogenesis. J Trauma 2010;68:1121–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Meyer NJ, et al. ANGPT2 Genetic Variant is Associated with Trauma-Associated Acute Lung Injury and Altered Plasma Angiopoietin-2 Isoform Ratio. Am J Respir Crit Care Med 2011, 201005–0701OC [pii] 10.1164/rccm.201005-0701OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Su L, et al. Genetic variants in the angiopoietin-2 gene are associated with increased risk of ARDS. Intensive Care Med 2009;35:1024–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Clajus C, et al. Angiopoietin-2 is a potential mediator of endothelial barrier dysfunction following cardiopulmonary bypass. Cytokine 2012;60:352–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Sporek M, et al. Angiopoietin-2 Is an Early Indicator of Acute Pancreatic-Renal Syndrome in Patients with Acute Pancreatitis. Mediators Inflamm 2016;2016:5780903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Nusshag C, et al. Deregulation of levels of angiopoietin-1 and angiopoietin-2 is associated with severe courses of hantavirus infection. Journal of Clinical Virology 2017;94:33–6. [DOI] [PubMed] [Google Scholar]
- 121.Sancakdar E, et al. Important of Angiopoietic System in Evaluation of Endothelial Damage in Children with Crimean-Congo Hemorrhagic Fever. Pediatr Infect Dis J 2015;34:e200–5. [DOI] [PubMed] [Google Scholar]
- 122.van de Weg CAM, et al. Serum angiopoietin-2 and soluble VEGF receptor 2 are surrogate markers for plasma leakage in patients with acute dengue virus infection. Journal of Clinical Virology 2014;60:328–35. [DOI] [PubMed] [Google Scholar]
- 123.Xie Z, et al. Vascular endothelial hyperpermeability induces the clinical symptoms of Clarkson disease (the systemic capillary leak syndrome). Blood 2012;119:4321–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Druey KM and Parikh SM. Idiopathic systemic capillary leak syndrome (Clarkson disease). Journal of Allergy and Clinical Immunology 2017;140:663–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Yeo TW, et al. Angiopoietin-2 is associated with decreased endothelial nitric oxide and poor clinical outcome in severe falciparum malaria. Proceedings of the National Academy of Sciences 2008;105:17097–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Papadopoulos KP, et al. A Phase I First-in-Human Study of Nesvacumab (REGN910), a Fully Human Anti-Angiopoietin-2 (Ang2) Monoclonal Antibody, in Patients with Advanced Solid Tumors. Clin Cancer Res 2016;22:1348–55. [DOI] [PubMed] [Google Scholar]
- 127.Matthay MA, et al. Treatment with allogeneic mesenchymal stromal cells for moderate to severe acute respiratory distress syndrome (START study): a randomised phase 2a safety trial. Lancet Respir Med 2019;7:154–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Campochiaro PA, et al. Enhanced Benefit in Diabetic Macular Edema from AKB-9778 Tie2 Activation Combined with Vascular Endothelial Growth Factor Suppression. Ophthalmology 2016;123:1722–30. [DOI] [PubMed] [Google Scholar]