

Abstract
The NF-κB family of transcriptional activators is responsible for the expression of numerous genes that control key functions such as cell development and survival. Subunit p50 has been studied extensively and is known to include 13 tyrosines, but the extent and pattern of tyrosine phosphorylation that accompanies p50 function has not been defined in the literature, especially at the level of selectivity of gene expression. In this study, phosphorylated tyrosine (pTyr) was site-selectively incorporated into the p50 subunit using an E. coli in vitro expression system containing a modified ribosome. In human T cells, the NF-κBs containing a pTyr at position 60 or 82 of p50 strongly increased the expression of CD40, which is a potential target for cancer or viral immunotherapy. Promoter DNA binding was studied for CD40 promoters, and verified two pTyr residues in NF-κB p50/p65 heterodimers that facilitated this process, and that support the possible importance of phosphorylation stoichiometry. This study defines a new approach for studying tyrosine residues whose phosphorylation alters protein binding to DNA promoters, and contributes to the facility of DNA expression.
Graphical Abstract
Phosphorylation of key tyrosines in the NF-κB p50 subunit enhances DNA promoter binding in vitro and subsequent protein translation in vivo.
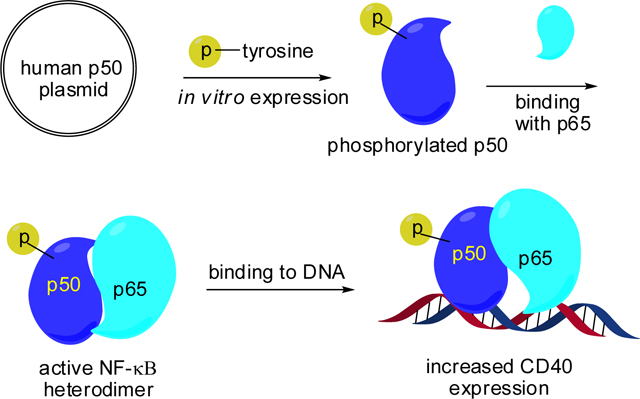
Nuclear factor-κB (NF-κB) is a family of transcriptional activators that modulate the expression of numerous genes in cells. Their key cellular functions include survival, development, proliferation and inflammation.1,2 The NF-κB family consists of at least 12 homo- and heterodimers that are formed by five subunits, namely p50, p52, RelA (p65), RelB and c-Rel.1,2 NF-κB plays key roles in the cell cycle, cell proliferation, and cell death via regulation of gene expression.3 Diseases such as neurodegeneration and cancer, as well as inflammatory diseases including atherosclerosis and autoimmunity, are associated with disorder of NF-κB expression.3,4
In the cytoplasm of mammalian cells, the NF-κB complex (e.g., p50/p65) is inhibited by binding to an inhibitory protein (IκB-α, IκB-β, or IκB-ε).5 Removal of the inhibitory protein enables the NF-κB complex to translocate into the nucleus and activate specific DNA sequences.6 It seems likely that the site-specific phosphorylation of NF-κB contributes to gene specific regulation via modulation of interactions with target genes or other factors.7
Numerous studies have explored NF-κB activities and function as regulated by phosphorylation of serine and threonine residues, which have been studied traditionally by either of two methods.7–10 One is the phosphorylation of specific serine or threonine residues using a protein kinase which can specifically phosphorylate individual Ser/Thr residues. A second method involves the site-directed replacement of Ser or Thr residues with alanine, which precludes their phosphorylation. This method generally demonstrates the importance of specific serine or threonine residues, but is inferential rather than direct. In this context, it is surprising that no site-specific tyrosine phosphorylation in NF-κB subunits has been reported to date, although the phosphorylation of p50 and p65 on tyrosine is known to modulate NF-κB transcriptional activity.11,12
The N-terminal domain of p50 contains the DNA binding region.13 In the X-ray crystal structure of a p50/p65-dsDNA complex (PBD 1VKX), four tyrosine residues (Y44, Y60, Y82 and Y90) located toward the N-terminus of p50 were selected for further study. Their distances to the bound DNA were measured, in the belief that phosphorylation close spatially to the DNA might well affect DNA binding by NF-κB. These distances ranged from 4.5 to 33.0 Å, with two (Tyr60, 4.5 Å; Tyr82, 13.3 Å) being relatively close to the DNA (Fig. 1A). To explore how the tyrosine phosphorylation of these residues affected the binding capacity of NF-κB to DNA, these were replaced systematically with phosphotyrosine. This was accomplished by altering each of these Tyr codons, one at a time, to TAG nonsense codons,14 readthrough of which was accomplished by suppression with phosphotyrosyl-tRNACUA (vide infra).15
Fig. 1.
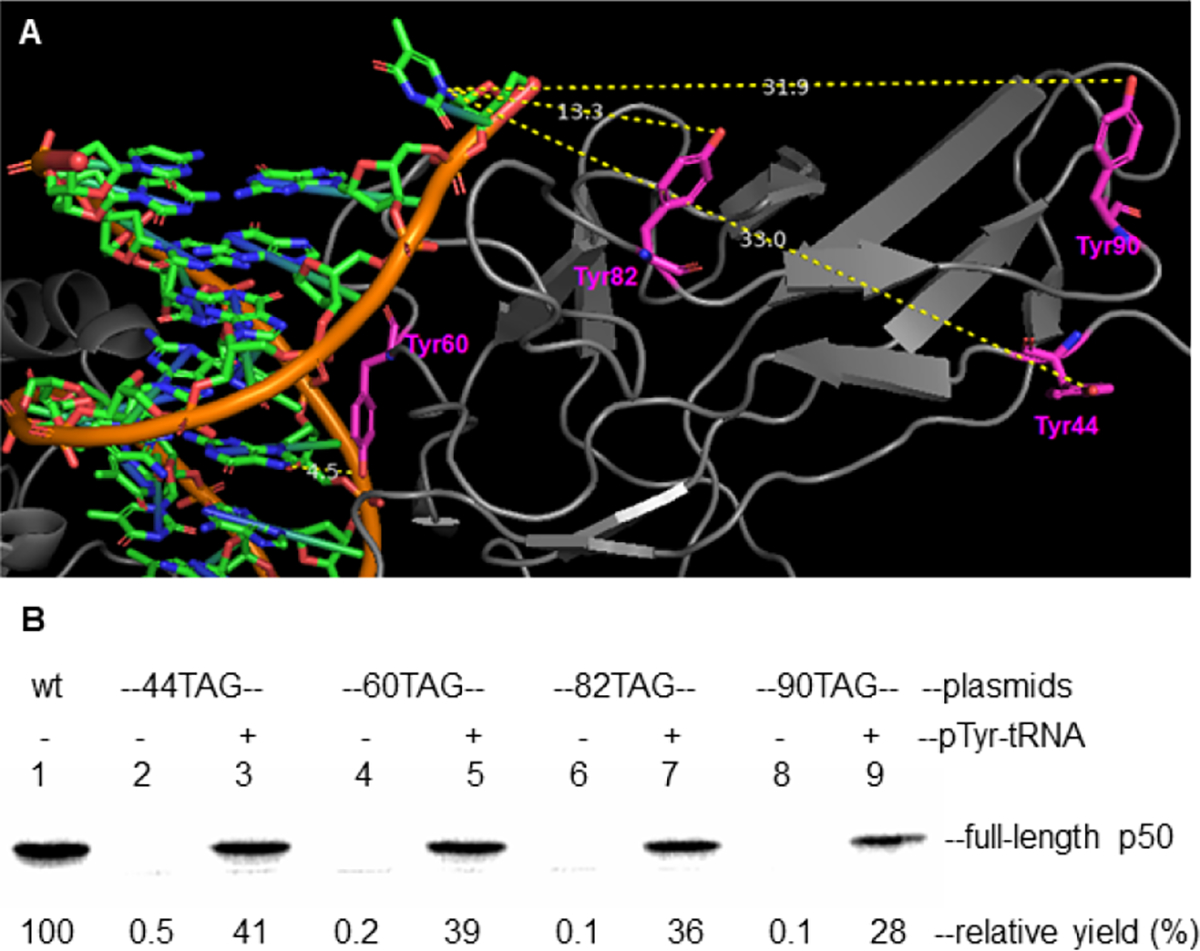
In vitro incorporation of NF-κB into p50 subunit. (A) X-ray crystal structure of NF-κB bound to a substrate duplex DNA (pdb 1VKX). Highlighted in the figure are the four tyrosine residues (Tyr44, 33.0 Å; Tyr60, 4.5 Å; Tyr82, 13.3 Å; Tyr90, 31.9 Å) closest to the DNA substrate. (B) Cell free expression of modified p50 subunits containing pTyr at positions 44, 60, 82 or 90, each synthesized by suppression of nonsense codon UAG in an E. coli protein biosynthesizing system. The samples were analyzed by 15% SDS-PAGE at 100 V for 2 h. Visualization was accomplished using the 35S-methionine incorporated into the proteins.
In a previous publication, we demonstrated that wild-type E. coli ribosomes could not incorporate phosphotyrosine into protein from a suppressor tRNACUA transcript to a significant extent, but could incorporate phosphotyrosine from a suppressor tRNA if the phosphate group was fully protected.15 Alternatively, we were able to select modified ribosomes capable of incorporating phosphotyrosine by the use of a phosphotyrosine derivative of puromycin.15,16 The latter technique was employed to introduce phosphotyrosine (pTyr) into position 42 of Iκ-Bα.
Following bacteriophage T4-mediated ligation of a suppressor tRNACUA-COH to phosphotyrosyl-pdCpA (Fig. S1, ESI), the full-length protected phosphotyrosyl-tRNACUA was obtained (Scheme 1).15 After removing the chemical protecting group,17 the phosphotyrosyl-tRNACUA was added into an in vitro protein biosynthesis reaction, which contained a modified ribosome.15 Plasmids containing a p50 gene having a TAG codon at positions 44, 60, 82 and 90, respectively, produced modified p50 proteins containing pTyr at the desired positions in suppression yields ranging from 28 – 41% (Fig. 1B and Scheme 1). No significant expression was observed in the absence of phosphotyrosyl-tRNACUA, and each of the modified p50 proteins (with or without 35S-methionine) was purified on a larger scale using a Strep-tactin column (Fig. S2 & S3, ESI).18,19 The wild-type and a modified p50 protein containing pTyr at position 60 was characterized using MS/MS analysis, which verified the presence of pTyr at position 60 (Fig. S4 & S5, ESI). Two additional full length modified p50 proteins containing one pTyr at different positions were analyzed by Q-TOF mass spectrometry. Protein 44pY-p50 gave a parent ion at m/z 42362.83, while 90pY-p50 exhibited m/z 42362.63 (both had calculated m/z 42354.29).
Scheme 1.
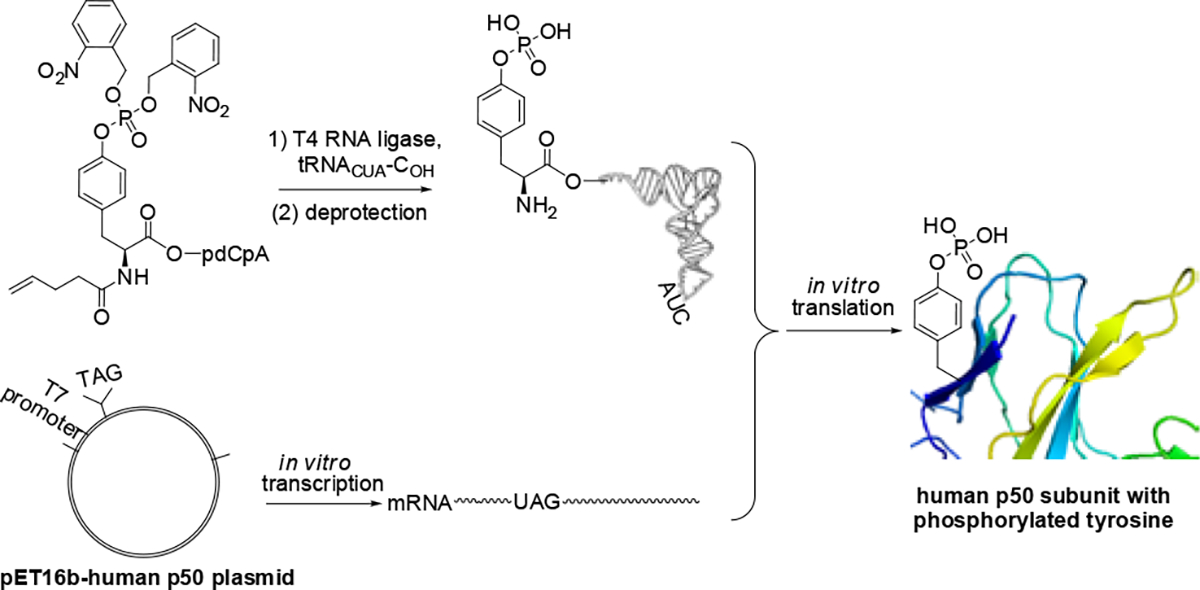
Strategy for the synthesis of human p50 protein phosphorylated on tyrosine.
By the same in vitro protein biosynthesis method, a wild-type plasmid containing a p65 gene was used to prepare and purify the His-tagged p65 subunit of NF-κB (Fig. S6 & S7, ESI). To determine whether a p50/p65 heteroduplex had formed, we prepared 35S-methionine labeled p50 and p65 subunits (Fig. S8, ESI) and demonstrated that their admixture resulted in a complex that migrated as expected for a p50/p65 heteroduplex. Additionally, heterodimers were formed between four different stoichiometrically phosphorylated p50s and wild-type p65.
Given that the transcription of >200 genes is regulated by NF-κB, it seems likely that only a small number of NF-κB heterodimers are involved in the activation of any single gene under physiological conditions. Accordingly, the focus of this study involved the possible involvement of the stoichiometry of phosphorylation of the p50 unit of NF-κB p50/p65 heterodimer in the mechanism by which selectivity of downstream gene expression is achieved. It was reported that 0.17 mM Triton-X100 can be used to deliver ferrocyanide into HeLa cells.20 In this study, it was found that 0.17 mM Triton-X100 could deliver the phosphorylated NF-κBs into Jurkat cells to induce protein overexpression (Fig. S9, ESI).
To the Triton-X100 pretreated Jurkat cells, 8 ng of phosphorylated NF-κBs (p50/p65, no 35S labeling) and 30 μCi of [35S]-L-methionine were added into RPMI-1640 cell culture medium. The in vivo expression mixture (10 μL total volume) was incubated at 37 °C for 1 h. The reaction mixture was treated with 5 μL of loading buffer (250 mM Tris-HCl, pH 6.8, containing 10% glycerol, 1% SDS, 0.01% bromophenol blue and 80 mM DTT) and heated at 90 °C for 2 min. The samples (15 μL each) were analyzed by 15% SDS-PAGE at 100 V for 2 h. As shown in Fig. 2A, one major protein band was overexpressed abundantly, Σ1.3 – 1.6-fold greater than wild-type NF-κB in the presence of phosphorylated NF-κBs (pTyr44, pTyr60 and pTyr82) and had a MW of about 40 kDa. To identify which genes were overexpressed, we used an RT2 Profiler™ PCR Array (Qiagen) to screen the over-transcription of 96 human NF-κB signaling pathway genes. These 96 genes were screened in the presence of wild-type and four mutant NF-κBs, each of the latter containing one stoichiometrically phosphorylated tyrosine at a different position (pTyr44, pTyr60, pTyr82 and pTyr90). These genes had different transcription levels regulated by each individual NF-κB. The transcription of the CD40 gene regulated by individual NF-κBs indicated increased transcription for three of the NF-κBs containing modified p50s (Fig. 2C). Western blot analysis of the band formed by this overexpressed protein was carried out with a goat monoclonal antibody to human CD40. This supported the conclusion that this band was due to CD40 (Fig. 2B). The molecular weight of CD40 can range from 32 – 39 kDa due to post-translational modifications.21 Interestingly, statistically significant overexpression of this protein was observed in the presence of NF-κB phosphorylated on p50 positions 44, 60, and 82. Among them, tyrosine phosphorylation at positions 60 and 82 generated greater overexpression of CD40.
Fig. 2.
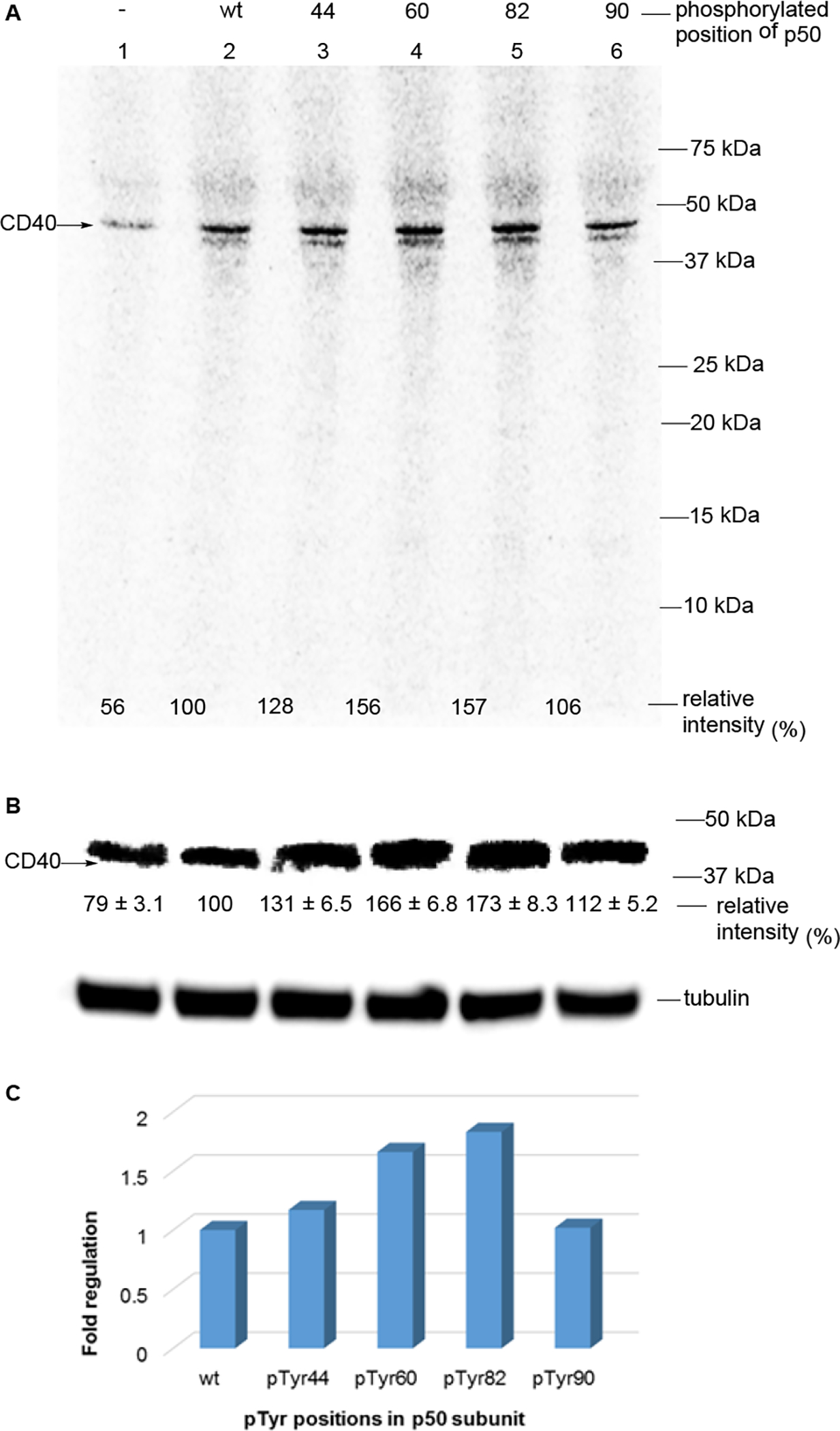
In vivo gene transcription and translation in activated Jurkat cells in the presence of NF-κB containing wild-type p65 and mutant p50 subunits. (A) The in vivo protein expression in activated Jurkat cells in the presence of [35S]-L-methionine. Lane 1, no external NF-κB was added; lane 2, wild-type NF-κB was added; lanes 3 – 6, modified NFκBs containing p50 subunits having pTyr at positions 44, 60, 82 or 90 were added, respectively. The samples were analyzed by 15% SDS-PAGE at 100 V for 2 h. (B) Western blot analysis was carried out with a goat monoclonal antibody to human CD40 to verify that the strongest band in panel A co-migrated with human CD40. The value of the desired band in the sample containing wild-type NF-κB was defined as 100%. The assays were repeated four times. The statistical significance was calculated using Student’s t-test. wt vs pTyr44-p50, p < 0.005; wt vs pTyr60-p50, p < 0.001; wt vs pTyr82-p50, p < 0.001. (C) The transcription of the CD40 gene in activated Jurkat cells in the presence of NF-κBs in the RT2 Profiler™ PCR Array assay. The mRNA levels of CD40 were compared to the reaction containing external wild-type NF-κB.
The cluster of differentiation 40 (CD40) is a member of the tumor necrosis factor receptor (TNFR) family, which is expressed in many kinds of cells, including T cells, B cells, monocytic cells and natural killer cells. 22–25 CD40 is a potential target for cancer immunotherapy, since it is essential in mediating a variety of inflammatory and immune responses.25–27 There are three NF-κB binding sites at positions upstream of the human CD40 gene.28
To explore how the tyrosine phosphorylation of NF-κBs increased the expression of CD40, we used all three CD40 DNA promoter sequences as substrates for enhanced binding by wild-type p50 and four stoichiometrically phosphorylated p50s, each used to form a heterodimer with wild-type p65. In the promoter DNA binding assay, phosphorylation of Tyr60 and Tyr82 in p50 enhanced the binding ability of the resulting p50/p65 dimer to promoter DNA1 (GGGAATTTCC) ~1.2-fold compared to wild-type p50/p65 dimer (Fig. S10, ESI). Comparatively, phosphorylation of Tyr44 and Tyr90 of the p50 subunit did not change their binding ability relative to the wild-type p50/p65 dimer. For promoter DNA2 (GGGAACTTCC), p50s modified at positions Tyr60 and Tyr82 also formed p50/p65 heterodimers that led to enhanced DNA binding. Finally, for binding to promoter DNA3 (GGGAAACTCC), the same two modified p50s (containing pTyr at positions 60 and 82) exhibited enhanced binding. Thus, CD40 DNA promoters were bound more extensively by a subset of the modified NF-κBs containing stoichiometrically phosphorylated p50s than wild-type NF-κB.
The p50 subunits containing phosphorylated Tyr44, Tyr60 and Tyr82 were studied in more detail for their concentration dependent behavior. NF-κB containing phosphorylated Tyr44 behaved in very similar fashion as wild type for all three DNA sequences, while the NF-κBs phosphorylated on Tyr60 or Tyr82 bound to the DNA more strongly at all tested concentrations (Fig. 3, Fig. S11 and Fig. S12, ESI). The aggregate data (for three different promoter regions of CD40 whose synthesis is regulated by NF-κB) suggests that phosphorylated residues Tyr60 and Tyr82 in the p50 subunit are important for facilitating DNA binding and protein expression.
Fig. 3.
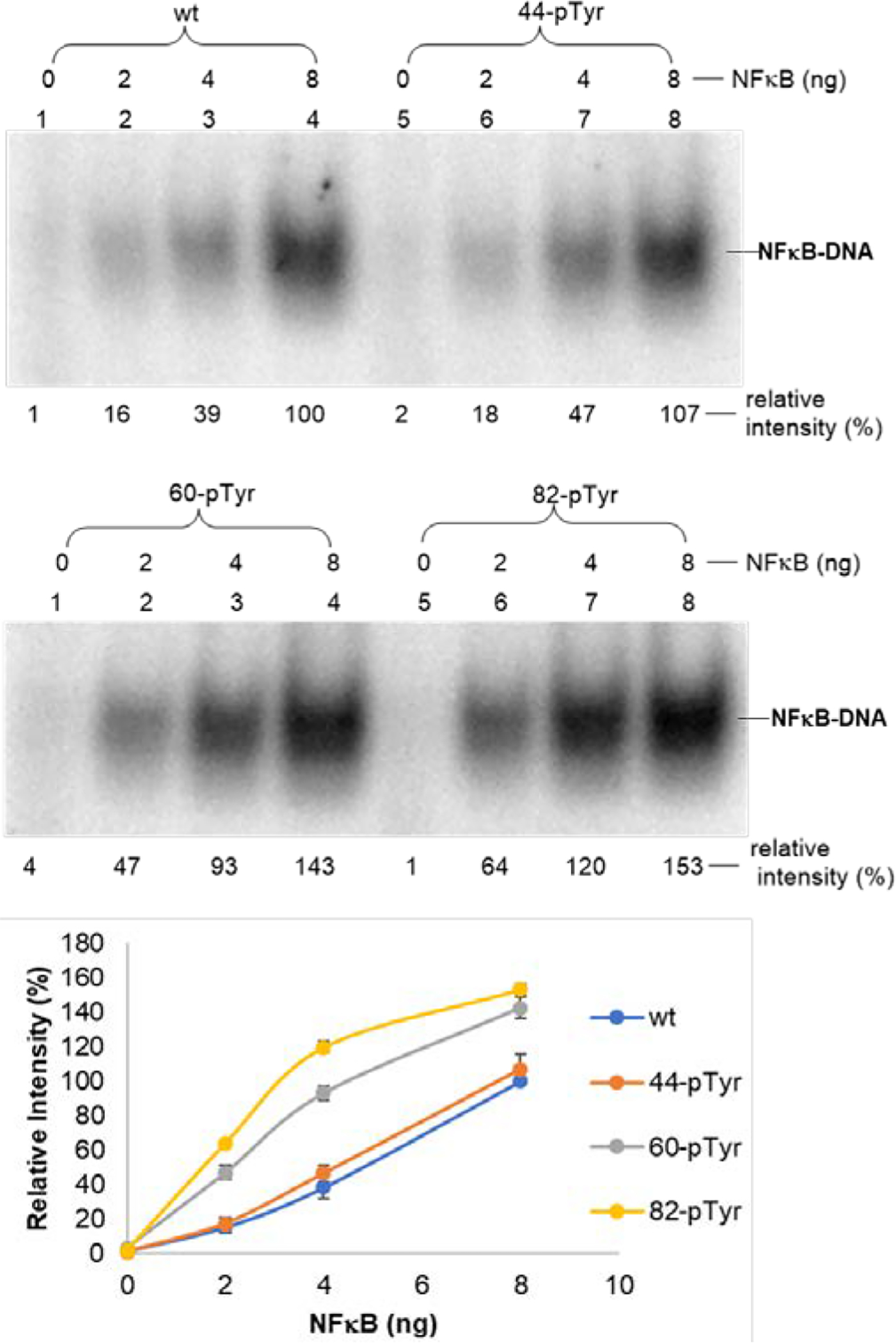
Concentration-dependent binding of wild-type NF-κB and three modified NF-κBs containing a single phosphorylated Tyr moiety to a CD40 promoter (DNA1) flanked by two tetranucleotides (CD40–1F, GCATGGGAATTTCCTACG). The underlined nucleotides are from the promoter sequence. The samples were analyzed by 5% native PAGE at 100 V for 1 h. The relative intensities of 32P-labeled complex were calculated with ImageQuant version 5.2 software from Molecular Dynamics based on the 32P signal. The band in the sample containing wild-type NF-κB was defined as 100%.
These findings support the proposal that modified NF-κBs phosphorylated at key p50 subunit tyrosines may enhance DNA promoter binding by NF-κB, as well as transcriptional activation and protein synthesis of a subset of cellular genes. While the present system lacks complete definition at a number of levels, including all of the amino acids whose phosphorylation is required for (selective) gene expression and protein synthesis, the ability to effect the selective phosphorylation of key amino acids, and to measure DNA binding and protein synthesis in specific assays, should facilitate a better understanding of the function of a key gene regulatory system. Plausibly, it may enable a subset of regulated genes to be expressed in a coordinated fashion experimentally.
In conclusion, we prepared proteins p50 and p65 in vitro, and also used modified bacterial ribosomes to incorporate pTyr stoichiometrically at one of four sites in p50 normally occupied by tyrosine. Purified p50 and p65 readily formed p50/p65 heterodimers that constitute one form of NF-κB. The modified p50s were introduced into activated Jurkat cells, resulting in enhanced transcription of the CD40 gene in the present of some modified p50 derivatives, as well as an increase in the expression of CD40 protein. A subset of the modified heterodimers was found to enhance DNA promoter binding in comparison with wild type, documenting selectivity of action by the modified NF-κBs and supporting the model of limited phosphorylation of wild-type NF-κB as a source of selective gene activation. Thus, in addition to the sites and order of phosphorylation of individual Tyr residues, phosphorylation stoichiometry may also regulate the selectivity, intensity and duration of a regulatory signal, and also constitute a useful experimental tool.
Supplementary Material
Acknowledgments
This study was supported by Research Grants R01GM103861 and R35GM104819, from the National Institute of General Medical Sciences, NIH.
Footnotes
Conflicts of interest
There are no conflicts to declare.
Notes and references
- (1).Liou H-C and Baltimore D, Curr. Opin. Cell Biol. 1993, 5, 477–487. [DOI] [PubMed] [Google Scholar]
- (2).Grimm S and Baeuerle PA, Biochem. J. 1993, 290, 297–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Baeuerle P and Henkel T, Annu. Rev. Immunol. 1994, 12, 141–179. [DOI] [PubMed] [Google Scholar]
- (4).Hayden MS and Ghosh S, Genes Dev. 2012, 26, 203–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Beg AA and Baldwin AS Jr., Genes Dev. 1993, 7, 2064–2070. [DOI] [PubMed] [Google Scholar]
- (6).Imbert V, Rupec RA, Livolsi A, et al. , Cell 1996, 86, 787–798. [DOI] [PubMed] [Google Scholar]
- (7).Christian F, Smith EL and Carmody RJ, Cells 2016, 5, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Hou S, Guan H and Ricciardi RP, J. Biol. Chem. 2003, 278, 45994–45998. [DOI] [PubMed] [Google Scholar]
- (9).Guan H, Hou S and Ricciardi RP, J. Biol. Chem. 2005, 280, 9957–9962. [DOI] [PubMed] [Google Scholar]
- (10).Smith EL, Somma D, Kerrigan D, McIntyre Z, Cole JJ, Liang KL, Kiely PA, Keeshan K and Carmody RJ Nucleic Acids Res. 2019, 47, 11151–11163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Pellegatta F, Bertelli AA, Staels B, Duhem C, Fulgenzi A and Ferrero ME, Am. J. Clin. Nutr. 2003, 77, 1220–1228. [DOI] [PubMed] [Google Scholar]
- (12).Kang JL, Jung HJ, Lee K and Kim HR, Toxicol. Sci. 2006, 90, 470–477. [DOI] [PubMed] [Google Scholar]
- (13).Chen FE, Huang DB, Chen YQ and Ghosh G, Nature 1998, 391, 410–413. [DOI] [PubMed] [Google Scholar]
- (14).Sawano A and Miyawaki A, Nucleic Acids Res. 2000, 28, E78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Chen S, Maini R, Bai X, Nangreave RC, Dedkova LM and Hecht SM, J. Am. Chem. Soc. 2017, 139, 14098–14108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Dedkova LM, Fahmi NE, Paul R, del Rosario M, Zhang L, Chen S, Feder G and Hecht SM, Biochemistry 2012, 51, 401–415. [DOI] [PubMed] [Google Scholar]
- (17).Lodder M, Golovine S and Hecht SM, J. Org. Chem. 1997, 62, 778–779. [DOI] [PubMed] [Google Scholar]
- (18).Bai X, Talukder P, Daskalova SM, Roy B, Chen S, Li Z, Dedkova LM and Hecht SM, J. Am. Chem. Soc. 2017, 139, 4611–4614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Chen S, Ji X, Gao M, Dedkova LM and Hecht SM, J. Am. Chem. Soc. 2019, 141, 5597–5601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Koley D and Bard AJ, Proc. Natl. Acad. Sci. USA. 2010, 107, 16783–16787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).van Kooten C and Banchereau J, J. Leukoc. Biol. 2000, 67, 2–17. [DOI] [PubMed] [Google Scholar]
- (22).Carbone A, Gioghini A, Gattei V, Aldinucci D, Degan M, De Paoli P, Zagonel V and Pinto A, Blood 1995, 85, 780–789. [PubMed] [Google Scholar]
- (23).Munroe ME and Bishop GA, J. Immunol. 2007, 178, 671–682. [DOI] [PubMed] [Google Scholar]
- (24).Carbone E, Ruggiero G, Terrazzano G, Palomba C, Manzo C, Fontana S, Spits H, Kärre K and Zappacosta S, J. Exp. Med. 1997, 185, 2053–2060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Zhang G, Lenardo MJ and Baltimore D, Cell 2017, 168, 37–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Elgueta R, Benson MJ, de Vries VC, Wasiuk A, Guo Y and Noelle RJ, Immunol Rev. 2009, 229, 152–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Bishop GA, Moore CR, Xie P, Stunz LL and Kraus ZJ, Adv. Exp. Med. Biol. 2007, 597, 131–151. [DOI] [PubMed] [Google Scholar]
- (28).Hinz M, Löser P, Mathas S, Krappmann D, Dörken B and Scheidereit C, Blood 2001, 97, 2798–2807. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.