

Abstract
Objective
Improved biomarkers of current disease activity and prediction of relapse are needed in antineutrophil cytoplasmic antibody–associated vasculitis (AAV). For clinical relevance, biomarkers must perform well longitudinally in patients on treatment and in patients with nonsevere flares.
Methods
Twenty‐two proteins were measured in 347 serum samples from 74 patients with AAV enrolled in a clinical trial. Samples were collected at Month 6 after remission induction, then every 3 months until Month 18, or at the time of flare. Associations of protein concentrations with concurrent disease activity and with future flare were analyzed using mixed‐effects models, Cox proportional hazards models, and conditional logistic regression.
Results
Forty‐two patients had flares during the 12‐month follow‐up period, and 32 remained in remission. Twenty‐two patients had severe flares. Six experimental markers (CXCL13, IL‐6, IL‐8, IL‐15, IL‐18BP, and matrix metalloproteinase‐3 [MMP‐3]) and ESR were associated with disease activity using all three methods (P < 0.05, with P < 0.01 in at least one method). A rise in IL‐8, IL‐15, or IL‐18BP was associated temporally with flare. Combining C‐reactive protein (CRP), IL‐18BP, neutrophil gelatinase‐associated lipocalin (NGAL), and sIL‐2Rα improved association with active AAV. CXCL13 and MMP‐3 were increased during treatment with prednisone, independent of disease activity. Marker concentrations during remission were not predictive of future flare.
Conclusion
Serum biomarkers of inflammation and tissue damage and repair have been previously shown to be strongly associated with severe active AAV were less strongly associated with active AAV in a longitudinal study that included mild flares and varying treatment. Markers rising contemporaneously with flare or with an improved association in combination merit further study.
INTRODUCTION
Antineutrophil cytoplasmic antibody (ANCA)‐associated vasculitis (AAV) encompasses two diseases, granulomatosis with polyangiitis (GPA) and microscopic polyangiitis (MPA), characterized by necrotizing vasculitis of small vessels in multiple organs and ANCA with specificity for either proteinase‐3 (PR3) or myeloperoxidase (MPO) (1). The course of the disease is highly variable after initial treatment, and it is often difficult to determine whether a disease flare is occurring or whether symptoms or abnormalities in diagnostic tests are attributable to infection, prior damage, medication toxicities, or other causes. Although ANCA titers and generic markers of inflammation (erythrocyte sedimentation rate [ESR] and C‐reactive protein [CRP]) are generally associated with predicting relapse and distinguishing active disease from remission (2, 3, 4, 5, 6, 7, 8), their ability to do so is not sufficient to determine the management of individual patients.
Twenty‐eight serum proteins were previously assessed for their ability to distinguish severe AAV from remission in 137 patients enrolled in the Rituximab in ANCA‐Associated Vasculitis (RAVE) clinical trial (9). These potential biomarkers were chosen from 108 proteins available on the assay platform to include 17 that had been tested previously in smaller studies and 11 additional proteins (limited by cost) to reflect different aspects of inflammation, injury, and repair. Twenty‐four of these 28 markers differed significantly between severe AAV and remission and also between active AAV and healthy controls. We found that serum levels of three markers, CXCL13 (also known as B cell attracting chemokine 1 [BCA‐1]), matrix metalloproteinase‐3 (MMP‐3), and tissue inhibitor of metalloproteinases‐1 (TIMP‐1), better distinguished severe AAV from remission than did the ESR or serum CRP levels, as measured by the areas under receiver operating characteristic curves (AUC‐ROC) or likelihood ratios.
The current study measured 22 of the 24 markers associated with severe active AAV from the original study in samples from 74 participants in the RAVE trial, collected longitudinally through Month 18 after enrollment. The goals were to: i) assess the ability of individual markers to distinguish active AAV from remission in a setting closer to clinical practice, with a wider range of disease activity and treatment and more measurements during remission; ii) preliminarily assess the ability of those markers in combination to distinguish active AAV from remission; and iii) determine whether marker concentrations or changes in levels during remission were associated with a future flare.
PATIENTS AND METHODS
Study cohort
The RAVE trial studied 197 patients with severe GPA or MPA, who were randomized to receive remission induction with a standardized regimen of glucocorticoids (GCs) plus either intravenous (IV) rituximab (RTX) 375 mg/m2 weekly for four treatments (RTX group) or oral cyclophosphamide (CYC) 2 mg/kg/day followed by azathioprine (AZA) 2 mg/kg/day (CYC/AZA group). Induction of remission was similar in the two groups (10). Relapse rates over the course of 18 months were also indistinguishable, even though B cell return was greater in the group that had received the single course of rituximab without additional maintenance therapy (11). Serum and plasma were collected at screening (at which point patients had severe active vasculitis, and many but not all had recently started high‐dose GCs); at Months 1, 2, 4, 6, 9, 12, 15, and 18; and additionally at the time of a flare. Patients who had severe flares were given the option of receiving RTX in an open‐label fashion. Patients with mild flares were usually treated with GCs alone without change in their baseline remission maintenance regimens (12). After Month 18, all patients were treated according to the judgment of their physicians. Data after Month 18 were not used in the current study.
Study design
The study included only patients in remission at Month 6. Remission was defined as Birmingham Vasculitis Activity Score for Wegener Granulomatosis (BVAS/WG) = 0 and active disease as BVAS/WG greater than 0. Delineation of disease activity as limited or severe was per definitions for BVAS/WG, in which some manifestations are automatically regarded as severe and others are usually considered nonsevere but may be classified as severe by the investigator (13). Flare was defined as an increase in BVAS/WG score from the previous visit. Data were reviewed to ensure that no patients with consecutive visits in remission had had flares between visits.
The size of the current study was limited to 74 patients by cost. Among participants in remission at Month 6, all patients in the trial who had severe flares after Month 6 were included (n = 22). Additional patients were selected on the basis of having been followed for at least 18 months with complete sample collections, in order to have representation among those who remained in remission continuously (n = 32, chosen randomly) or experienced one or more limited flares but no severe flares (n = 20, chosen randomly). Five of the patients who experienced severe flares had also experienced limited flares previously. In almost all cases, flare occurred on a baseline of remission (ie, from BVAS/WG = 0 to BVAS/WG > 0). In three cases, patients transitioned from limited flare to severe flare without an intervening remission.
Sera were assayed from all time points between screening and Month 18. We had previously analyzed concentrations of biomarkers at screening and Month 6 in 137 patients in RAVE (9, 14). The current study aimed to study the association of biomarkers with disease activity in a longitudinal set of samples obtained from the same trial and to determine the relationship between biomarker levels and the occurrence of relapse between Months 6 and 18. We did not analyze the time period between enrollment and Month 6 because disease activity and treatment were both in flux (eg, disease improvement, tapering of prednisone) and any severe flare that occurred during that time would be confounded by changes in treatment.
Biomarker assays
A custom microarray platform was used, as described previously (9, 15). Of the 28 markers in the previous study, 22 were included in this study, because they were strongly associated with severe AAV in the first study or because they were of interest in other forms of vasculitis to be studied separately.
Other variables
Demographic data were recorded at screening for enrollment in the clinical trial. Receipt of either RTX or CYC/AZA was used in some analyses. ESR and CRP were measured in the clinical labs of the participating sites at each visit. In the trial, B cell counts (cells per μl) were obtained from peripheral blood by flow cytometry, and serum concentrations of MPO‐ANCA and PR3‐ANCA concentrations were measured at a central location, as previously described (7). B cells were considered to be “depleted” if the count in blood was less than 10 cells/μl, “detectable” if between 10 and 69 cells/μl, and “reconstituted” if more than 69 cells/μl (16). For this study, the data used for MPO‐ANCA and PR3‐ANCA had been obtained using the Euroimmun assay. Current GC dose in mg (usually prednisone) was recorded at each visit, but for this study, GC use was modeled as a dichotomous variable (yes/no).
Statistical analysis
All markers were analyzed after natural‐log transformation. Distributions of granulocyte‐macrophage colony‐stimulating factor (GM‐CSF), IFNγ, IL‐6, and sIL‐2Rα remained highly skewed despite the transformation, and many samples had undetectable concentrations, so these markers were also analyzed as dichotomous variables in additional mixed models (not shown) and only as dichotomous variables in Cox models.
Distinguishing active vasculitis from remission
No single analytical technique can accomplish the goals of adjusting for repeated measures within‐patient, adjusting for the effects of treatment on marker levels, incorporating patients who remained in continuous remission as well as patients who had flares, and modeling the predictive ability of combinations of markers. Therefore, multiple analytical techniques with complementary strengths and weaknesses were used (Table 1, Supplementary Text): mixed‐effects models, Cox proportional hazards models, and stratified conditional logistic regression. Mixed models and conditional logistic regression were also applied to subgroups defined by treatment with RTX or CYC/AZA. Models that used data from multiple markers to predict disease activity were built using stratified conditional logistic regression starting with markers that were significantly associated with active disease individually (P < 0.05). The strength of association with active disease in these multivariable models was estimated by logistic regression, with the predictor variables being the markers' difference from that patient's mean level during remission.
Table 1.
Methods of analysis to test association of disease activity with biomarker concentration
| Regression method | Outcome variable 1 | Predictor variables | Within‐patient adjustment? | Includes continuous remission? | Caveats |
|---|---|---|---|---|---|
| Mixed‐effects | Biomarker | Activity ± treatment | Yes | No | One marker at a time |
| Cox (concentration) | Activity | Biomarker(s) ± covariates | No | Yes | Selective sampling |
| Cox (recent change) | Activity | Biomarker(s) ± covariates | No | Yes | Selective sampling |
| Conditional logistic regression | Activity | Biomarker(s) | Yes | No | |
| Logistic (change from mean) | Activity | Biomarker(s) | Yes | Yes | Repeated measures |
Biomarkers were modeled as continuous variables, and disease activity was modeled as a dichotomous variable (active or remission).
Predicting flare
Patients were classified into three groups based on disease course between Months 6 and 18: continuous remission, the occurrence of flare (any severity), or the occurrence of severe flare (a subset of the “any severity” group). Association of concentrations at Month 6 with time to flare was tested using Cox proportional hazards models. In a complementary and simpler method, concentrations at Month 6 were also compared between groups, independent of time, using Wilcoxon rank‐sum tests. To improve the precision of determining levels during remission, the means of all concentrations during remission (before severe flare) were determined for each patient, and means during remission were compared between groups. These approaches were also used in the subsets defined by RTX or CYC/AZA treatment.
Cox proportional hazard models were used to determine whether marker concentration during remission prior to flare differed from marker concentration during remission not immediately preceding flare. Time to flare (since randomization) was the outcome, with either marker concentration or change in marker concentration from the previous visit as the predictor. Covariates included age, sex, treatment group (RTX or CYC/AZA), new diagnosis versus relapsing disease at enrollment, and PR3‐ANCA versus MPO‐ANCA as a dichotomous variable.
Statistical analyses were performed using SAS version 9.1 or 9.3 (SAS Institute) or InStat (GraphPad). In light of the number of simultaneous tests, Cornfield's rule of thumb was followed and only regarded P values less than 0.01 as significant, but also report any result with P values less than 0.05. For research questions in which multiple methods were used (eg, association with concurrently active AAV), we regarded findings as significant if at least one method yielded P values less than 0.01 and the other methods yielded P values less than 0.05.
RESULTS
Characteristics of the cohort and summary of biomarker concentrations
Of the 74 patients with data in this study, 41 were female and 33 were male, with a median age of 51 years (interquartile range [IQR], 40‐63). Sixty‐eight were self‐described as white. Thirty‐five had new‐onset disease at trial entry, and 39 had relapsing disease. Fifty‐seven patients had GPA, 16 had MPA, and 1 could not be classified. Fifty‐four patients had previously tested positive for PR3‐ANCA and 20 for MPO‐ANCA. In the trial, 44 received RTX for induction (without re‐treatment or addition of another immunosuppressive drug), and 30 received CYC for induction and either were being transitioned to AZA for remission maintenance at the Month 6 visit or had already transitioned. Median serum creatinine was 1.1 mg/dl (IQR, 0.9‐1.7). All 74 patients were in remission at Month 6 (the first data point for this study). Sixty‐two patients were off prednisone and 12 were on prednisone at Month 6, either 3 to 10 mg (n = 8) or 15 to 40 mg (n = 4). In the 42 patients who had flares, the median time to first flare was 159 days after the Month 6 visit (IQR, 91‐215 days). In the 22 patients with severe flares, the median time to severe flare was 205 days (IQR, 86‐270). For patients who remained in remission through the Month 18 visit, the median follow‐up time after Month 6 was 362 days. Manifestations, total BVAS/WG score, and GC dosing at flare were diverse (Supplementary File).
Table 2 shows a summary of biomarker concentrations, separated by disease activity. Biomarker concentrations in individual patients at the time of flare are included in the Supplementary File.
Table 2.
Biomarker concentrations (medians and interquartile ranges) in antineutrophil cytoplasmic antibody–associated vasculitis during active disease and remission, with results from a previous study shown for comparison (9)
| AAV, current study, RAVE longitudinal | AAV, published study, RAVE screening visit and Month 6 | ||||
|---|---|---|---|---|---|
| Remission (287 samples from 74 patients) | Active (60 samples from 42 patients) | AAV active at screening visit (137 samples from 137 patients) | AAV remission Month 6 (137 from 137 patients) | Controls (n = 68) | |
| ACE | 170 (127;242) | 167 (106;233) | 105 (74;144) | 178 (130;252) | 97 (81;115) |
| BCA‐1 | 27 (16;50) | 41 (20;89) | 170 (74;489) | 32 (18;56) | 30 (20;45) |
| CRP | 5 (3;19) | 7 (3;32) | 12 (5;40) | 5 (3;12) | ND |
| ESR | 12 (5;24) | 16 (8;36) | 37 (16;60) | 14 (7;22) | ND |
| G‐CSF | 11 (3.9;28) | 18 (3.9;44) | 20 (8.0;46) | 11 (5.6;24) | 7.6 (4.9;13) |
| GM‐CSF | <1 (<1;2.6) | <1 (<1;4.3) | 28 (2.3;269) | 1.2 (<1;5.0) | 1.4 (<0.1;7.3) |
| IFNγ | <0.5 (<0.5;<0.5) | <0.5 (<0.5;<0.5) | <0.5 (<0.5;2.0) | <0.5 (<0.5;<0.5) | <0.5 (<0.5;<0.5) |
| IL‐6 | <0.5 (<0.5;1.8) | 1.3 (<0.5;5.0) | 2.1 (<0.5;20) | <0.5 (<0.5;0.8) | <0.5 (<0.5;<0.5) |
| IL‐8 | 9.8 (4.9;22) | 9.3 (2.1;56) | 20 (7.3;51) | 7.1 (3.6;15) | 3.0 (1.3;5.2) |
| IL‐15 | 6.8 (2.1;17) | 8.9 (1.8;22) | 22 (7.7;109) | 5.7 (2.6;14) | 2.9 (2.2;4.3) |
| IL‐18 | 40 (18;82) | 38 (22;99) | 57 (37;101) | 52 (31;86) | 36 (20;61) |
| IL‐18BP | 23 (7.9;57) | 37 (9.6;92) | 116 (22;768) | 15 (<6;55) | 14 (6.1;47) |
| IP‐10 | 11 (6.1;20) | 12 (5.5;24) | 11 (6.0;24) | 13 (7.7;25) | 3.3 (2.2;5.3) |
| MMP‐3 | 18 (10;31) | 27 (13;61) | 97 (47;148) | 16 (12;29) | 10 (7.0;16) |
| NGAL | 181 (107;294) | 190 (116;370) | 271 (176;399) | 172 (129;237) | 117 (92;150) |
| Osteopontin | 51 (29;89) | 52 (28;77) | 65 (39;101) | 54 (38;81) | 36 (30;42) |
| PAI‐1 | 2.2 (<1;4.4) | 2.9 (1.2;6.4) | 1.5 (<1;5.7) | 1.2 (<1;4.7) | 3.3 (1.1;7.2) |
| PDGF‐AB | 3.5 (1.3;6.1) | 2.9 (1.4;5.3) | 4.3 (1.6;6.6) | 3.3 (0.9;5.4) | 8.9 (5.8;12) |
| RANTES | 53 (30;111) | 51 (29;131) | 60 (33;107) | 52 (31;90) | 58 (28;91) |
| sICAM‐1 | 474 (300;887) | 507 (352;870) | 463 (307;933) | 537 (345;882) | 281 (226;337) |
| sIL‐2Rα | <2.5 (<2.5;7.1) | <2.5 (<2.5;13) | <2.5 (<2.5;153) | <2.5 (<2.5;<2.5) | <2.5 (<2.5;<2.5) |
| sIL‐6R | 22 (16;34) | 22 (16;35) | 27 (21;43) | 22 (15;33) | 16 (12;20) |
| sTNFRII | 2.1 (1.2;3.8) | 1.8 (1.1;3.0) | 2.7 (1.3;4.9) | 2.4 (1.4;5.8) | 0.5 (0.3;0.7) |
| TIMP‐1 | 183 (127;291) | 189 (136;366) | 477 (302;862) | 166 (125;233) | 117 (65;163) |
Units are mg/L (= mcg/ml) for CRP, mm/h for ESR, ng/ml for ACE, MMP‐3, NGAL, osteopontin, PAI‐1, PDGF‐AB, RANTES, sICAM‐1, sIL‐6R, sTNFRII, and TIMP‐1, and pg/ml for the remaining proteins, referring to the concentration in serum before dilution. Statistical analysis was not done to compare results, because patients in the current study are a nonrandom subset of the patients in the first study and because of repeated measures in the current study, differing in number among patients. With that caveat, marker levels were likely lower during active disease in the current study than in the first study, levels during remission were likely similar, and levels of many markers may have remained higher during remission (on treatment) than in controls.
Abbreviations: AAV, antineutrophil cytoplasmic antibody–associated vasculitis; CRP, C‐reactive protein; MMP‐3, matrix metalloproteinase‐3; RAVE, Rituximab in antineutrophil cytoplasmic antibody–associated Vasculitis trial; TIMP‐1, tissue inhibitor of metalloproteinases‐1.
Association of individual biomarker concentrations contemporaneous with relapse of vasculitis
Results of the three analytical approaches (mixed‐effects models, Cox proportional hazards models, and stratified conditional logistic regression) were largely in agreement. Six experimental markers (CXCL13/BCA‐1, IL‐6, IL‐8, IL‐15, IL‐18BP, and MMP‐3) and ESR were associated with disease activity at P values less than 0.01 with at least one method and at P values less than 0.05 with the other two methods (Table 3). Results for CRP and sIL‐2Rα were uncertain: P values less than 0.01 with mixed models and P values less than 0.05 with conditional logistic regression but P values more than 0.3 in Cox models. Markers in Cox models showed association with active vasculitis more commonly when modeled as a change between the value at relapse and the previous remission value than when modeled as absolute marker concentrations, reinforcing the importance of adjusting for differences among individuals' values at baseline (Table 3). This analysis, which was unique in allowing the inclusion of data from patients who remained in remission long term, identified IL‐8, IL‐15, and IL‐18BP as the most promising markers. Because marker concentrations were ln‐transformed, this analysis indicated that a 2.7‐fold increase in any of these markers was associated with a 1.4‐ to 1.8‐fold increase in odds of flare. The addition of demographic and clinical covariates (age, sex, treatment group, ANCA specificity, and new‐onset or relapsing disease) to Cox models did not change the association of markers with disease activity, and these clinical variables were not associated with disease activity (data not shown). There was also evidence of a broader tendency for markers to increase during active disease because at relapse the estimated β coefficients were greater than 0 or hazard ratios (HR) and odds ratios (ORs) greater than 1 in 20 to 23/24 experimental markers (Table 3). In separate analyses by treatment group, CXCL13/BCA were associated with the active disease only in patients treated with RTX, whereas neutrophil gelatinase‐associated lipocalin (NGAL) and osteopontin were associated with the active disease only in patients treated with CYC/AZA (Supplementary Table 1). With reduced numbers of patients and multiple tests, these results should be regarded as preliminary. Because disease manifestations were diverse, and the numbers of patients with a given manifestation were low (eg, seven patients with renal flare; Supplementary File), we did not attempt to analyze biomarker data relative to manifestations.
Table 3.
Association of biomarkers with active antineutrophil cytoplasmic antibody–associated vasculitis
| Marker 1 | Mixed models 2 | Mixed models, with treatment covariates | Cox, marker concentration | Cox, change in concentration | Conditional logistic regression | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| β | P | β | P | P (GC) | P (B) | HR | P | HR | P | OR | P | |
| ACE | −0.09 | 0.19 | −0.07 | 0.36 | 0.5 | 0.02 | 0.87 | 0.44 | 0.95 | 0.75 | 0.71 | 0.28 |
| CXCL13/BCA‐1 | 0.35 | 0.0006 | 0.18 | 0.10 | 0.0001 | 0.12 | 1.33 | 0.01 | 1.38 | 0.14 | 1.96 | 0.01 |
| CRP | 0.64 | <0.0001 | 0.63 | 0.0002 | 0.47 | 0.21 | 1.10 | 0.37 | 1.12 | 0.48 | 1.90 | 0.0005 |
| ESR | 0.46 | <0.0001 | 0.46 | <0.0001 | 0.41 | 0.10 | 1.66 | 0.003 | 1.28 | 0.29 | 3.01 | 0.0006 |
| G‐CSF | 0.20 | 0.13 | 0.12 | 0.42 | 0.06 | 0.31 | 1.15 | 0.11 | 1.23 | 0.16 | 1.45 | 0.13 |
| GM‐CSF | 0.14 | 0.28 | 0.11 | 0.46 | 0.44 | 0.95 | 1.81 | 0.06 | 0.86 | 0.69 | 1.19 | 0.35 |
| IFNγ | 0.02 | 0.79 | −0.15 | 0.11 | 0.01 | 0.001 | 3.66 | 0.06 | 3.58 | 0.18 | 1.11 | 0.76 |
| IL‐6 | 0.80 | <0.0001 | 0.57 | 0.001 | 0.65 | 0.0005 | 2.13 | 0.02 | 1.28 | 0.35 | 2.03 | 0.0001 |
| IL‐8 | 0.46 | 0.008 | 0.37 | 0.06 | 0.34 | 0.68 | 1.21 | 0.09 | 1.44 | 0.009 | 1.38 | 0.02 |
| IL‐15 | 0.24 | 0.02 | 0.18 | 0.11 | 0.13 | 0.01 | 1.09 | 0.29 | 1.85 | 0.0006 | 1.73 | 0.04 |
| IL‐18 | 0.001 | 0.99 | 0.07 | 0.61 | 0.32 | 0.02 | 1.16 | 0.17 | 1.57 | 0.04 | 0.94 | 0.83 |
| IL‐18BP | 0.33 | 0.0007 | 0.16 | 0.11 | 0.15 | 0.84 | 1.06 | 0.49 | 1.57 | 0.007 | 2.11 | 0.01 |
| CXCL10/IP‐10 | 0.11 | 0.17 | 0.14 | 0.14 | 0.15 | 0.99 | 1.11 | 0.44 | 1.70 | 0.02 | 1.47 | 0.21 |
| MMP‐3 | 0.33 | 0.005 | 0.13 | 0.28 | <0.0001 | 1.0 | 1.26 | 0.10 | 1.34 | 0.05 | 1.71 | 0.02 |
| NGAL | 0.19 | 0.05 | 0.20 | 0.06 | 0.09 | 0.96 | 1.17 | 0.42 | 1.29 | 0.21 | 1.98 | 0.03 |
| Osteopontin | 0.08 | 0.39 | 0.18 | 0.08 | 0.03 | 0.12 | 1.24 | 0.23 | 1.49 | 0.07 | 1.35 | 0.32 |
| PAI‐1 | 0.14 | 0.10 | 0.13 | 0.19 | 0.69 | 0.89 | 1.29 | 0.09 | 1.08 | 0.71 | 1.52 | 0.20 |
| PDGF‐AB | 0.08 | 0.41 | 0.06 | 0.56 | 0.38 | 0.57 | 1.09 | 0.53 | 1.10 | 0.65 | 1.32 | 0.40 |
| CCL5/RANTES | 0.02 | 0.77 | 0.08 | 0.34 | 0.76 | 0.30 | 1.19 | 0.33 | 1.23 | 0.36 | 1.14 | 0.74 |
| sICAM‐1 | 0.02 | 0.73 | 0.08 | 0.33 | 0.99 | 0.03 | 1.08 | 0.68 | 1.27 | 0.15 | 1.07 | 0.84 |
| sIL‐2Rα | 0.45 | 0.001 | 0.28 | 0.08 | 0.99 | 0.03 | 0.75 | 0.35 | 1.34 | 0.40 | 1.55 | 0.03 |
| sIL6R | 0.05 | 0.49 | 0.06 | 0.41 | 0.65 | 0.60 | 0.94 | 0.73 | 1.27 | 0.13 | 1.35 | 0.57 |
| sTNFR2 | 0.04 | 0.39 | 0.10 | 0.07 | 0.04 | 0.16 | 0.96 | 0.78 | 2.38 | 0.005 | 1.88 | 0.33 |
| TIMP‐1 | 0.06 | 0.36 | 0.07 | 0.32 | 0.18 | 0.36 | 1.35 | 0.13 | 1.30 | 0.19 | 1.50 | 0.48 |
All marker values were ln‐transformed for mixed models, conditional logistic regression, and most Cox models. Four markers (GM‐CSF, IFNγ, IL‐6, and sIL‐2Rα) were treated as dichotomous variables in the Cox model (see “Patients and Methods”).
In mixed models using ln‐transformed variables, the fold‐difference associated with active disease is 2.72 × β‐coefficient.
In mixed models, marker concentration is the dependent variable, and disease activity (active or remission), treatment with prednisone (yes/no), and B cell status (depleted, re‐detected, or reconstituted) are the independent variables. In Cox models and conditional logistic regression, disease activity (active or remission) is the dependent variable, and marker concentrations are the independent variables.
Abbreviations: Cox, Cox proportional hazards regression; CRP, C‐reactive protein; GC, glucocorticoid use at the time of sample collection (yes/no); B, B cells (depleted, detectable, or reconstituted), only in the rituximab‐treated group; HR, hazard ratio; MMP‐3, matrix metalloproteinase‐3; OR, odds ratio; TIMP‐1, tissue inhibitor of metalloproteinases‐1.
Restricting the analysis to severe flares (mild flares in the same individuals were excluded in this analysis) appeared to increase the strength of association (β coefficient, HR, and/or OR) between marker concentration and disease activity in most cases, although P values often increased, reflecting the decrease in the number of events from 186 to 73 (Supplementary Table 2).
Use of GCs was modeled as a dichotomous predictor variable in mixed models, because of the wide range of doses either during remission (range: 2‐40 mg oral prednisone; median: 8 mg) and at the time of visit for flare (range: 5‐80 mg and eight patients who received IV methylprednisolone 1,000 mg or more; Supplementary File), and uncertainty about whether high IV doses had been started before blood was drawn for biomarkers. Patients were using GCs at 37/60 visits during active disease (use not recorded for seven) and at 48/287 visits during remission (use not recorded for six), usually after minor flares. B cell levels (the best indicator of “current” rituximab treatment) were modeled as depleted (<10 cells/ul), re‐detected (10‐69 cells/ul), or reconstituted (>69 cells/ul). Concurrent use of GCs was strongly associated with increased concentrations of CXCL13/BCA‐1 and MMP‐3 (P ≤ 0.0001), and the association of these markers with active vasculitis was no longer significant after adjustment for treatment (Table 3). Concentrations of CXCL13/BCA‐1 and MMP3 were also elevated with GC use during remission (Figure 1).
Figure 1.
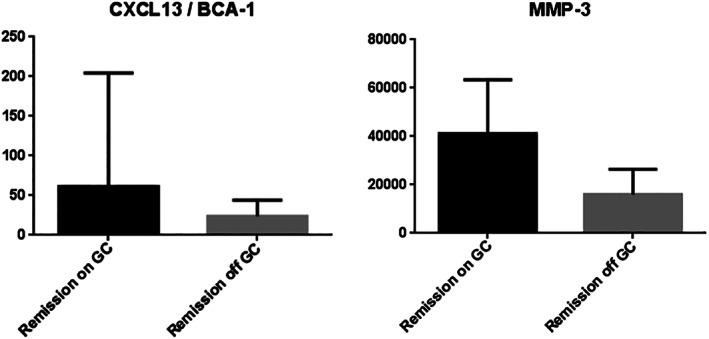
Concentrations of CXCL13/BCA‐1 and MMP‐3 (pg/ml) in patients with antineutrophil cytoplasmic antibody–associated vasculitis during remission, either on any dose of glucocorticoids (black bars) or off glucocorticoids (gray bars). P values less than 0.0001 for both proteins. GC, glucocorticoids; MMP‐3, matrix metalloproteinase‐3.
Conditional logistic regression and Cox models explored whether combinations of markers improved the strength of association with active disease. Because BCA‐1 and MMP‐3 were directly associated with current GC treatment, these markers were omitted from multivariable models. Using conditional logistic regression, multivariable models were built starting with markers that were individually significant (P < 0.05) and remained significant in combination with CRP, and then combinations of three to five markers were tested. As expected, CRP, ESR, and IL‐6 were highly correlated, so it was considered counterproductive to include more than one of them in a model. A combination of CRP, IL‐18BP, NGAL, and sIL‐2Rα was identified as a model in which each marker retained an association with active disease. The AUC‐ROC was estimated by logistic regression with marker variables modeled as differences from the individual patient's baseline (mean in remission) (17). Plots of these markers give a sense of the small differences in marker change between active disease and remission (Supplementary Figure 1). A combination of CRP, IL‐18BP, NGAL, and sIL‐2Rα had an AUC‐ROC of 0.72, indicating modest accuracy (72%) despite a P value less than 0.0001 for the multivariable model and a P value less than 0.05 for each individual marker. Subgroup analysis suggested better performance in patients treated with CYC/AZA (0.88) than in those treated with RTX (0.69). For comparison, the AUC‐ROC of CRP alone was 0.65 and was similar in the RTX (0.65) and CYC/AZA (0.64) groups. Combining marker concentrations using Cox models led to the loss of statistically significant association with active disease for most markers (data not shown), reinforcing the need for independent validation.
Predicting flare: marker concentrations during remission in patients who did or did not experience flares later
Among the 22 experimental proteins and the clinical markers CRP and ESR, values during remission specifically at Month 6 were associated with time to flare for CXCL13/BCA‐1 (P = 0.003), and possibly for GM‐CSF and plasminogen activator inhibitor‐1 (PAI‐1) (0.01 < P < 0.05) (Table 4). These associations were not strikingly different in the subgroups treated with RTX or CYC/AZA (data not shown). In the full cohort, a subtle overall tendency for markers to be elevated in patients destined to flare was evident in that 21/24 markers had HR greater than 1. The value of any experimental marker specifically at Month 6 was not convincingly associated with risk of flare by Month 18 (independent of time), because only soluble TNF‐receptor‐2 (sTNF‐R2) had a P value greater than 0.01 and less than 0.05 (Table 4). In contrast, levels of MPO‐ANCA at Month 6 were associated with risk of future flare (P = 0.01) despite being limited to only 20 patients, but levels of PR3‐ANCA were not (P = 0.49). Mean concentrations of CXCL13/BCA‐1 (P = 0.02 for any flare, P = 0.007 for severe flare) and sIL‐2Rα (P = 0.02 for any flare, P = 0.003 for severe flare) during all remission visits for a given patient were higher in patients who experienced flares of any severity (n = 32) or severe flares (n = 22) than in those who remained in remission through at least Month 18 (n = 42) (Table 4). Findings for CXCL13/BCA‐1 and sIL‐2Rα were similar in the subgroups treated with RTX or CYC/AZA, with P values greater than 0.05 in the context of small numbers of patients (data not shown).
Table 4.
Marker concentrations during remission in antineutrophil cytoplasmic antibody–associated vasculitis: association with future flare
| Cox Proportional Hazard Models, Time to Flare | Wilcoxon, Flare Versus No Flare by Month 18 | |||||
|---|---|---|---|---|---|---|
| Concentration in remission at Month 6 | Concentrations during remission, with time‐varying marker values | Concentration in remission at Month 6 | Mean concentration per patient during remission | |||
| HR | P | HR | P | P | P | |
| ACE | 0.83 | 0.28 | 0.89 | 0.56 | 0.96 | 0.75 |
| CXCL13/BCA‐1 | 1.32 | 0.003 | 1.22 | 0.08 | 0.26 | 0.02 |
| CRP | 1.02 | 0.84 | 1.05 | 0.65 | 0.18 | 0.32 |
| ESR | 1.41 | 0.01 | 1.50 | 0.02 | 0.26 | 0.77 |
| G‐CSF | 1.12 | 0.17 | 1.08 | 0.47 | 0.72 | 0.06 |
| GM‐CSF | 1.14 | 0.05 | 1.88 | 0.04 | 0.82 | 0.13 |
| IFNγ | 0.98 | 0.98 | 1.93 | 0.39 | 0.38 | 0.49 |
| IL‐6 | 1.06 | 0.43 | 1.52 | 0.17 | 0.70 | 0.54 |
| IL‐8 | 1.18 | 0.08 | 0.98 | 0.86 | 0.61 | 0.50 |
| IL‐15 | 1.07 | 0.40 | 0.97 | 0.77 | 0.91 | 0.93 |
| IL‐18 | 1.09 | 0.20 | 1.05 | 0.72 | 0.39 | 0.97 |
| IL‐18BP | 1.06 | 0.31 | 0.96 | 0.71 | 0.98 | 0.76 |
| CXCL10/IP‐10 | 1.10 | 0.38 | 0.94 | 0.70 | 0.24 | 0.64 |
| MMP‐3 | 1.22 | 0.11 | 0.99 | 0.93 | 0.76 | 0.32 |
| NGAL | 1.03 | 0.85 | 0.91 | 0.66 | 0.68 | 0.44 |
| Osteopontin | 1.22 | 0.15 | 0.96 | 0.82 | 0.19 | 0.56 |
| PAI‐1 | 1.32 | 0.03 | 1.26 | 0.16 | 0.31 | 0.79 |
| PDGF‐AB | 1.02 | 0.84 | 1.04 | 0.73 | 0.47 | 0.54 |
| CCL5/RANTES | 1.03 | 0.84 | 1.06 | 0.73 | 0.91 | 0.79 |
| sICAM‐1 | 1.11 | 0.51 | 0.89 | 0.47 | 0.60 | 0.69 |
| sIL‐2Rα | 1.10 | 0.09 | 0.53 | 0.07 | 0.18 | 0.02 |
| sIL6R | 1.02 | 0.94 | 0.80 | 0.11 | 0.99 | 0.93 |
| sTNFR2 | 0.92 | 0.48 | 0.82 | 0.19 | 0.03 | 0.05 |
| TIMP‐1 | 1.16 | 0.35 | 1.12 | 0.55 | 0.06 | 0.50 |
Abbreviations: CRP, C‐reactive protein; HR, hazard ratio; MMP‐3, matrix metalloproteinase‐3; TIMP‐1, tissue inhibitor of metalloproteinases‐1.
To determine whether a rise in marker level was detectable prior to flare, Cox analysis, with marker concentration as a time‐varying covariate, was restricted to visits during remission that were immediately preceded by other visits during remission. Groups were defined by whether the next visit was characterized as flare (n = 29) or ongoing remission (n = 122). No significant (P < 0.01) differences were found, and only 12/24 markers had an HR greater than 1 (Table 4).
CXCL13/BCA‐1, the only marker that was significantly associated with future flare in more than one analytical approach, showed higher concentrations in patients receiving GCs at the time of sample collection during remission (median 61 vs. 24 pg/ml in 48 and 239 samples, respectively; Figure 1), confounding determination of whether elevated levels during remission might be predictive of future flare.
DISCUSSION
Serum concentrations of CXCL13/BCA‐1, IL‐6, IL‐8, IL‐15, IL‐18BP, and MMP‐3, as well as the clinical marker ESR, were confirmed as being elevated during active vasculitis across a broad range of disease severity and over an 18‐month period. Results were usually consistent across multiple analytical techniques that adjust for varying patient baselines but have different strengths and limitations. Changes in IL‐8, IL‐15, and IL‐18BP contemporaneous with flare were particularly promising, with a 2.7‐fold increase in concentration versus the previous visit indicating a 1.4‐ to 1.8‐fold increase in odds of flare. A combination of CRP, IL‐18BP, NGAL, and sIL‐2Rα distinguished active disease from remission moderately well (estimated AUC‐ROC of 0.72) and would also be appropriate to test in an independent cohort. CXCL13/BCA‐1 and MMP‐3 were elevated in patients taking GCs at the time of sample collection, which could make their interpretation as biomarkers of active AAV difficult in patients on GCs.
The list of markers associated with active vasculitis was not enriched in markers that were recently reported as being more strongly associated with PR3‐ANCA or MPO‐ANCA in the same cohort (18). There was no indication that levels of the 22 experimental markers, nor the clinical markers CRP or ESR, would be useful during clinical remission for predicting future disease course, but a cohort with much less than 3 months between sample collections might be needed in order to detect relevant trends (19). In contrast, MPO‐ANCA during remission at Month 6 was associated with risk of future flare, which makes sense because it is a marker of underlying autoimmune disease rather than a marker of inflammation, injury, or repair.
This study had a number of strengths and weaknesses to consider. Strengths of this study include the standardized collection of clinical data by experts in the field, uniform collection and storage of samples, and the use of rigorously validated immunoassays. One weakness is the possibility that the clinical assessor was sometimes mistaken in distinguishing mild active AAV from remission, but misclassification should not be so common as to drown out a strong signal. In order for a biomarker to show promise to improve upon available methods of assessment, it should first show a good association with the available gold standard, which in this case was the clinician's assessment. Another weakness is the inevitable uncertainty about how to model variables with highly skewed distributions (biomarkers, treatment, and disease activity) and the lack of a single analytical method that would accomplish all of the study goals. It was reassuring that results tended to be similar with the use of different methods. Finally, in order to be most useful clinically, a prospective biomarker for active AAV should be tested in patients with bacterial or viral infections.
Although this study validated several markers as being associated with active AAV, the goal of finding generic markers of inflammation or ANCA titers that strongly reflect disease activity or predict future flare in AAV remains elusive. Additional hypothesis‐based studies and broader agnostic screens may both be useful in pursuing these goals. Regardless, attention to the direct effects of treatment on marker levels will be essential, either because of blunting of expected increases in inflammatory mediators (20, 21) or direct effects on production or metabolism independent of inflammation (22).
AUTHOR CONTRIBUTIONS
All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published. Dr. Monach had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Study conception and design
Monach, Warner, Tómasson, Johnson, Merkel.
Acquisition of data
Monach, Warner, Specks, Stone, Fervenza, Hoffman, Kallenberg, Langford, Seo, St. Clair, Spiera, Johnson, Merkel.
Analysis and interpretation of data
Monach, Lew, Merkel.
Supporting information
Disclosure Form
Table S1
Appendix S1: Supporting Information
This work was sponsored by the Vasculitis Clinical Research Consortium (VCRC). The VCRC is part of the Rare Diseases Clinical Research Network, an initiative of the Office of Rare Diseases Research, National Center for Advancing Translational Science (NCATS). The VCRC has received funding from NCATS, the National Institute of Arthritis and Musculoskeletal and Skin Diseases (U54 AR057319, RC1 AR058303, and P60 AR047785), and the National Center for Research Resources (U54 RR019497). The Rituximab in antineutrophil cytoplasmic antibodies (ANCA)‐Associated Vasculitis (RAVE) trial was performed with the support of the Immune Tolerance Network (NIH Contract #N01 AI15416), an international clinical research consortium supported by the National Institute of Allergy and Infectious Diseases. Genentech and Biogen Idec provided the study medications and partial funding. At the Mayo Clinic and Foundation, the trial was supported by a Clinical and Translational Science Award from the National Center for Research Resources (NCRR) (RR024150‐01); at Johns Hopkins University, by grants from the NCRR (RR025005) and career development awards (K24 AR049185 to Dr. Stone and K23 AR052820 to Dr. Seo); and at Boston University, by a Clinical and Translational Science Award (RR 025771), grants from the National Institutes of Health (M01 RR00533), and a career development award (K24 AR02224 to Dr. Merkel). Dr. Monach was also supported by an Arthritis Investigator Award from the Arthritis Foundation.
Author disclosures are available at https://onlinelibrary.wiley.com/action/downloadSupplement?doi=10.1002%2Facr2.11366&file=acr211366‐sup‐0001‐Disclosureform.pdf.
No potential conflicts of interest relevant to this article were reported.
REFERENCES
- 1. Weiner M, Segelmark M. The clinical presentation and therapy of diseases related to anti‐neutrophil cytoplasmic antibodies (ANCA). Autoimmunity Rev 2016;15:978–82. [DOI] [PubMed] [Google Scholar]
- 2. Tomasson G, Grayson PC, Mahr AD, Lavalley M, Merkel PA. Value of ANCA measurements during remission to predict a relapse of ANCA‐associated vasculitis–a meta‐analysis. Rheumatology (Oxford) 2012;51:100–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hind CR, Winearls CG, Lockwood CM, Rees AJ, Pepys MB. Objective monitoring of activity in Wegener's granulomatosis by measurement of serum C‐reactive protein concentration. Clinical Nephrol 1984;21:341–5. [PubMed] [Google Scholar]
- 4. Kälsch AI, Csernok E, Munch D, Birck R, Yard BA, Gross W, et al. Use of highly sensitive C‐reactive protein for followup of Wegener's granulomatosis. J Rheumatol 2010;37:2319–25. [DOI] [PubMed] [Google Scholar]
- 5. Sproson EL, Jones NS, Al‐Deiri M, Lanyon P. Lessons learnt in the management of Wegener's Granulomatosis: long‐term follow‐up of 60 patients. Rhinology 2007;45:63–7. [PubMed] [Google Scholar]
- 6. Finkielman JD, Merkel PA, Schroeder D, Hoffman GS, Spiera R, St Clair EW, et al. Antiproteinase 3 antineutrophil cytoplasmic antibodies and disease activity in Wegener granulomatosis. Ann Intern Med 2007;147:611–9. [DOI] [PubMed] [Google Scholar]
- 7. Fussner LA, Hummel AM, Schroeder DR, Silva F, Cartin‐Ceba R, Snyder MR, et al. Factors determining the clinical utility of serial measurements of antineutrophil cytoplasmic antibodies targeting proteinase 3. Arthritis Rheumatol 2016;68:1700–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Terrier B, Saadoun D, Sene D, Ghillani P, Amoura Z, Deray G, et al. Antimyeloperoxidase antibodies are a useful marker of disease activity in antineutrophil cytoplasmic antibody‐associated vasculitides. Ann Rheum Dis 2009;68:1564–71. [DOI] [PubMed] [Google Scholar]
- 9. Monach PA, Warner RL, Tomasson G, Specks U, Stone JH, Ding L, et al. Serum proteins reflecting inflammation, injury and repair as biomarkers of disease activity in ANCA‐associated vasculitis. Ann Rheum Dis 2013;72:1342–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Stone JH, Merkel PA, Spiera R, Seo P, Langford CA, Hoffman GS, et al. Rituximab versus cyclophosphamide for ANCA‐associated vasculitis. N Eng J Med 2010;363:221–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Specks U, Merkel PA, Seo P, Spiera R, Langford CA, Hoffman GS, et al. Efficacy of remission‐induction regimens for ANCA‐associated vasculitis. N Eng J Med 2013;369:417–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Miloslavsky EM, Specks U, Merkel PA, Seo P, Spiera R, Langford CA, et al. Outcomes of nonsevere relapses in antineutrophil cytoplasmic antibody‐associated vasculitis treated with glucocorticoids. Arthritis Rheumatol 2015;67:1629–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Stone JH, Hoffman GS, Merkel PA, Min YI, Uhlfelder ML, Hellmann DB, et al. A disease‐specific activity index for Wegener's granulomatosis: modification of the Birmingham Vasculitis Activity Score. International Network for the Study of the Systemic Vasculitides (INSSYS). Arthritis Rheum 2001;44:912–20. [DOI] [PubMed] [Google Scholar]
- 14. Monach PA, Tomasson G, Specks U, Stone JH, Cuthbertson D, Krischer J, et al. Circulating markers of vascular injury and angiogenesis in antineutrophil cytoplasmic antibody‐associated vasculitis. Arthritis Rheum 2011;63:3988–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Olle EW, Deogracias MP, Messamore JE, McClintock SD, Barron AG, Anderson TD, et al. Screening of serum samples for Wegener's granulomatosis patients using antibody microarrays. Proteomics Clin Appl 2007;1:1212–20. [DOI] [PubMed] [Google Scholar]
- 16. Berti A, Warner R, Johnson K, Cornec D, Schroeder DR, Kabat BF, et al. The association of serum interleukin‐6 levels with clinical outcomes in antineutrophil cytoplasmic antibody‐associated vasculitis. J Autoimmun 2019;105:102302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lieberthal JG, Cuthbertson D, Carette S, Hoffman GS, Khalidi NA, Koening CL, et al. Urinary biomarkers in relapsing antineutrophil cytoplasmic antibody‐associated vasculitis. J Rheumatol 2013;40:674–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Berti A, Warner R, Johnson K, Cornec D, Schroeder D, Kabat B, et al. Brief Report: Circulating Cytokine profiles and antineutrophil cytoplasmic antibody specificity in patients with antineutrophil cytoplasmic antibody‐associated vasculitis. Arthritis Rheumatol 2018;70:1114–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Orange DE, Yao V, Sawicka K, Fak J, Frank MO, Parveen S, et al. RNA Identification of PRIME cells predicting rheumatoid arthritis flares. N Eng J Med 2020;383:218–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Grayson PC, Monach PA, Pagnoux C, Cuthbertson D, Carette S, Hoffman GS, et al. Value of commonly measured laboratory tests as biomarkers of disease activity and predictors of relapse in eosinophilic granulomatosis with polyangiitis. Rheumatology (Oxford) 2015;54:1351–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dejaco C, Oppl B, Monach P, Cuthbertson D, Carette S, Hoffman G, et al. Serum biomarkers in patients with relapsing eosinophilic granulomatosis with polyangiitis (Churg‐Strauss). PloS One 2015;10:e0121737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Conklin LS, Merkel PA, Pachman LM, Parikh H, Tawalbeh S, Damsker JM, et al. Serum biomarkers of glucocorticoid response and safety in anti‐neutrophil cytoplasmic antibody‐associated vasculitis and juvenile dermatomyositis. Steroids 2018;140:159–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Disclosure Form
Table S1
Appendix S1: Supporting Information