

Abstract
Neural stem cells (NSCs) are tumor tropic and can be genetically modified to produce anti-cancer therapies locally in the brain. In a prior first-in-human study we demonstrated that a single dose of intracerebrally administered allogeneic NSCs, which were retrovirally transduced to express cytosine deaminase (CD), tracked to glioma sites and converted oral 5-fluorocytosine (5-FC) to 5-fluorouracil (5-FU). The next step in the clinical development of this NSC-based anti-cancer strategy was to assess the feasibility of administering multiple intracerebral doses of CD-expressing NSCs (CD-NSCs) in patients with recurrent high grade gliomas. CD-NSCs were given every 2 weeks using an indwelling brain catheter, followed each time by a 7-day course of oral 5-FC (and leucovorin in the final patient cohort). Fifteen evaluable patients received a median of 4 (range 2–10) intracerebral CD-NSC doses; doses were escalated from 50 x 106 to 150 x 106 CD-NSCs. Neuropharmacokinetic data confirmed that CD-NSCs continuously produced 5-FU in the brain during the course of 5-FC. There were no clinical signs of immunogenicity, and only three patients developed anti-NSC antibodies. Our results suggest intracerebral administration of serial doses of CD-NSCs is safe and feasible and identified a recommended dose for phase II testing of 150 x 106 CD-NSCs.
INTRODUCTION
Neural stem cells (NSCs) have an innate ability to track to sites of damage in the brain. Because of this inherent pathotropism and their pluripotency, there has been much interest in assessing NSCs for their regenerative potential to differentiate and replace damaged tissue in the central nervous system (CNS). For example, NSCs have been investigated as possible therapies for stroke (1), macular degeneration (2, 3), amyotrophic lateral sclerosis (4, 5), and spinal cord injury (6, 7). NSCs are also tumor tropic (8–10), and their intrinsic ability to migrate through brain parenchyma and efficiently localize to both the main tumor site and invasive foci provides an opportunity to achieve more effective, tumor selective therapy for brain tumor patients.
Glioblastoma, the most common malignant primary brain tumor in adults, remains incurable with a median overall survival of less than two years (11, 12). One major reason for the lack of success of current therapies is the highly invasive nature of glioblastoma, resulting in dissemination and formation of distant micro-tumor foci, which often “hide behind” an intact blood-brain barrier through which most chemotherapy agents cannot pass. Preclinical data have demonstrated that genetically modified NSCs can be used to deliver various anti-cancer agents, such as prodrug activating enzymes (13–17), interleukins (18), and tumor-specific oncolytic viruses (19, 20) to treat primary and metastatic brain tumors, in effect circumventing the blood-brain barrier by localizing therapeutic payloads to tumor sites in the brain.
We previously performed a first-in-human study in patients with recurrent high grade gliomas (21) of a clonal, genetically, and functionally stable NSC line (HB1.F3.CD21) that was retrovirally transduced to express cytosine deaminase (CD) (13, 14, 22–24). CD converts the prodrug 5-fluorocytosine (5-FC) to the cytotoxic agent 5-fluorouracil (5-FU), which kills surrounding tumor cells that are dividing. In this initial study, a single dose of CD-expressing NSCs (CD-NSCs) was administered intracerebrally to participants followed by a 7-day course of oral 5-FC every 6 h. Autopsy data documented CD-NSC migration from the site of injection to distant tumor foci, even when those foci were in the contralateral hemisphere. In addition, using intracerebral microdialysis (25), we showed that the CD produced by the NSCs converted 5-FC to 5-FU in the brain.
In contrast to applying NSCs for regenerative purposes, when used as drug delivery vehicles for treating cancer NSCs do not engraft and differentiate. Preclinical data in mice showed that NSCs remain in the brain for at least two weeks, with the majority eliminated by three weeks (14). Brain autopsies performed on two participants in our first-in-human study detected the presence of only single, non-dividing NSCs remaining approximately 1.5 and 2.5 months after intracerebral administration of 10 million NSCs. Therefore, when NSCs are used for the treatment of cancer, repeat administrations will likely be required to ensure that enough NSCs are present to continue generating the anti-cancer therapy. Based on these preclinical data, the initial clinical safety data for a single dose of intracerebrally administered CD-NSCs followed by oral 5-FC, as well as the proof-of-concept data regarding NSC migration to sites of tumor and NSC-mediated conversion of 5-FC into 5-FU from the first-in-human study, we next performed a phase I clinical trial to assess the feasibility and safety of administering multiple doses of CD-NSCs intracerebrally. We were able to give repeated doses of CD-NSCs to study patients and did not observe any CD-NSC related toxicities. The results of this phase I trial serve as a foundation for the design of future studies of genetically modified NSCs for the treatment of brain tumors.
PATIENTS AND METHODS
Study design
The primary objectives of the study were to assess the feasibility of serially administering CD-NSCs via a Rickham reservoir attached to an intracerebral catheter (Codman Holter Rickham Reservoir, [Integra LifeSciences, Princeton, NJ, USA]) and to determine the recommended dosing for phase II testing of CD-NSCs when given in combination with fixed doses of oral 5-FC. Secondary objectives included characterizing the relationship between intracerebral and systemic concentrations of 5-FC and 5-FU at the maximum feasible dose (MFD) level of CD-NSCs with the use of intracerebral microdialysis, assessing CD-NSC immunogenicity, and describing clinical activity.
A standard 3 + 3 dose escalation design (26) was used to evaluate 4 dose levels of study treatment (Table 1), in which three doses of CD-NSCs were assessed. The starting number of CD-NSCs was 50 x 106, which was the highest single dose assessed in the first-in-human study (21). To prevent cell aggregation, CD-NSCs were suspended at a highest density of 67 x 106 cells in 1 mL artificial CSF (Perfusion Fluid CNS, M Dialysis, North Chelmsford, MA, USA) and 2% human serum albumin. Furthermore, due to volume constraints of intracerebral administration the highest CD-NSC dose was limited to 150 x 106 cells in 2.25 mL. All study participants received the same dose of oral 5-FC (37.5 mg/kg every 6 h for 7 d), which is within the standard dosing range when 5-FC is used to treat fungal infections. Patients on dose level 4 also received leucovorin (25 mg every 6 h for 7 d), which potentiates the efficacy of 5-FU (27–29).
Table 1.
Dose escalation designa
| Dose Level | Number of CD-NSCs intracerebrally administered on day 1 of each cycleb | Oral 5-FC (mg/kg) taken every 6 hours on days 4–10 of each cycle | Oral leucovorin (mg) taken every 6 hours on days 4–10 of each cycle | Number of evaluable participants | DLTsc |
|---|---|---|---|---|---|
| 1 | 50 x 106 | 37.5 | --- | 3 | 0 |
| 2 | 100 x 106 | 37.5 | --- | 3 | 0 |
| 3 | 150 x 106 | 37.5 | --- | 3 | 0 |
| 4 | 150 x 106 | 37.5 | 25 | 6d | 1 |
This study implemented a standard 3+3 design for the treatment regimen of CD-NSCs with 5-FC (and concomitant leucovorin for dose level 4 participants).
The length of each cycle was 14 days.
DLT – dose limiting toxicity.
Seven participants started treatment on dose level 4, but one was unevaluable and had to be replaced after receiving the first dose of CD-NSCs due to the tip of the Rickham catheter migrating into the lateral ventricle.
The length of a treatment cycle was 14 d; the CD-NSCs were administered intracerebrally on day 1 of each cycle (Figure 1). The first intracerebral dose of CD-NSCs was manually injected during surgery. When tumor resection was performed, the CD-NSC dose was administered in divided 100–150 µL aliquots, as evenly spaced as possible throughout the resection cavity and slowly injected as the needle was withdrawn, to distribute the CD-NSCs within a 1–2.5 cm depth of the cavity wall. Then a Rickham catheter was placed in the resection cavity for subsequent intracerebral administrations of the CD-NSCs. If only a biopsy was performed, the Rickham catheter was placed in the residual tumor tissue through the biopsy track, and then up to 67 x 106 CD-NSCs in 1 mL were manually injected through the Rickham catheter over 10 min followed by a 0.3 mL artificial CSF flush. For biopsy patients treated on dose levels 2–4, the remainder of the CD-NSC dose was administered through the Rickham catheter a few hours after the surgery by slow infusion as described below.
Figure 1. Study schema.
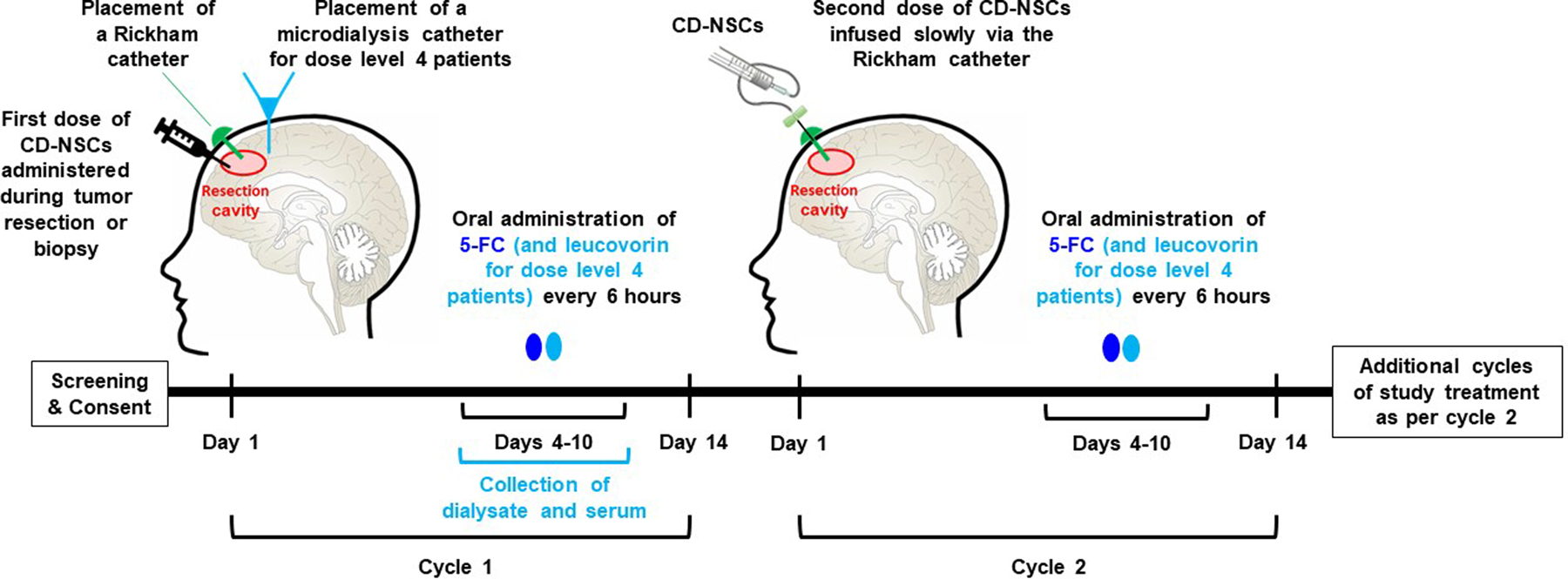
The cycle length was 14 d. On cycle 1 day 1, participants underwent surgery for either resection or biopsy of the tumor and manual injection of the first dose of CD-NSCs. A Rickham catheter (green) was placed in the resection cavity or residual tumor (if only a biopsy was performed) for subsequent intracerebral administrations of the CD-NSCs. For participants treated on dose level 4, a temporary intracerebral microdialysis catheter (light blue) was also placed during surgery to quantify CD-NSC-mediated conversion of 5-FC to 5-FU at the highest dose of CD-NSCs. Because NSCs cease dividing within 48 h (14), thus avoiding NSC destruction due to 5-FU cytotoxicity, and in order to give time for the NSCs to migrate to distant tumor foci, treatment with 5-FC did not begin until 3 d after surgery. The 5-FC was taken orally every 6 h for 7 d (dose level 4 patients also took leucovorin every 6 h for 7 d). For cycle 2 and subsequent cycles, CD-NSCs were slowly infused via the Rickham catheter. Administration of 5-FC and leucovorin was as per cycle 1. Brain MRIs were performed every 2 months to assess response.
Beginning with the second cycle, CD-NSCs were administered in the outpatient setting by slow infusion (0.5 mL/h) through the Rickham catheter followed by a 1 mL flush of artificial CSF. The total infusion time was 2.5 h to 5.5 h depending on the volume (dose level 1: 0.75 mL, dose level 2: 1.5 mL, dose level 3: 2.25 mL) of NSCs administered. Three days after each CD-NSC administration (to allow time for the NSCs to distribute among tumor foci) participants began taking the 7-day course of oral 5-FC (and leucovorin for participants on dose level 4). Possible development of anti-NSC antibodies after repeated exposure to the allogeneic NSCs was assessed using serial plasma samples from participants, as described below. Brain MRIs were obtained every 8 weeks to evaluate response to treatment. The clinical trial was approved by the City of Hope Institutional Review Board and conducted under an Investigational New Drug Application (IND# 14041). Each study participant gave written informed consent. The study was registered at ClinicalTrials.gov (NCT02015819).
Eligibility criteria
Patients 18 years or older who had radiographic findings consistent with recurrent, supratentorial high grade gliomas (defined as grade III or IV by the World Health Organization (30)) and needed tumor resection or biopsy were eligible to participate in this study. Study participants were required to have a Karnofsky performance status ≥ 70, received previous treatment with brain radiation and temozolomide, and recovered from any toxicities associated with their prior therapy. Other inclusion criteria were: no anticipated physical connection between the post-resection tumor cavity and the cerebral ventricles; adequate bone marrow function (defined as an absolute neutrophil count of ≥ 1,500 cells/mm3 and platelet count ≥ 100,000 cells/mm3); total bilirubin ≤ 2.0 mg/dL and aspartate aminotransferase ≤ 4 times the institutional upper limit of normal; serum creatinine within normal limits; a minimum of 2 weeks from the last dose of a targeted agent; and a minimum of 4 weeks from the last dose of a cytotoxic chemotherapy (6 weeks from treatment with a nitrosourea) or bevacizumab. There was no limit to the number of prior therapies a participant could have received for treatment of recurrent disease.
Exclusion criteria included the presence of anti-human leukocyte antigens (HLA) antibodies specific for class I or II HLA antigens expressed by the CD-NSCs, a chronic or active viral CNS infection, a coagulopathy or bleeding disorder, or a serious medical or psychiatric illness that might prevent the participant from completing study treatment. Patients were also excluded from study participation if they were currently receiving radiation or other chemotherapy, or if they were pregnant or breastfeeding.
Neural stem cells
The primary NSCs were obtained in accordance with the Guidelines of the Anatomical Pathology Department of Vancouver General Hospital (Vancouver, British Columbia, Canada). Permission to use fetal tissue was given by the Clinical Research Screening Committee Involving Human Subjects of the University of British Columbia (Vancouver, British Columbia, Canada). As previously described, the parental NSC line (HB1.F3) was generated by immortalizing dissociated NSCs using a retrovirus encoding gag-v-myc, which preserves the cells’ stem-like properties that are needed for migration (31). The NSCs were then retrovirally transduced a second time to express Escherichia coli cytosine deaminase (CD-NSCs), cloned, and characterized (14). A materials transfer agreement gave permission for use of the HB1.F3 NSCs and CD-NSCs at City of Hope.
A master cell bank (MCB) was manufactured under cGMP conditions in compliance with FDA requirements and was extensively characterized (21). Clinical cell banks of CD-NSCs were manufactured from the MCB under cGMP conditions and release tested prior to use in the clinical trial. All cryopreserved cell banks were placed on an annual stability testing program. Thaw qualification runs were performed on each bank prior to clinical use and included testing for cell counts, viability, identity (human nestin [NSC marker] and CD expression [transgene expression] assessed by flow cytometry), endotoxin, and sterility. For each clinical infusion, the dose level-appropriate number of frozen vials of CD-NSCs were thawed, rinsed, and resuspended in artificial CSF with 2% human serum albumin. These cell products were also tested for sterility prior to intracerebral administration.
Safety correlative studies
Assessment of possible NSC migration into the systemic circulation
Quantitative PCR (qPCR) for the NSC marker gag-v-myc was done using a Chromo 4 System for Real-Time PCR (BioRad, CA, USA) and TaqMan technology (Applied Biosystems, CA, USA). Briefly, custom primer/probe sets were ordered (ABI/ThermoFisher, CA, USA) that were specific for gag-v-myc (NSC marker; forward primer = 5’-CAGCCGCCGCGATG-3’; reverse primer = 5’-CGTAGTCGTAATCGTAGTTCTTGCT-3’; and probe = 5’-FAM-TCAGCGCCAGCCTC-NFQ-3’) and ApoB (housekeeping gene; forward primer = 5’-GGAGGAACTTTGCACTATGTTCATG-3’; reverse primer = 5’-GACTTTCGAATATACCTGGGACAGT-3’; and probe = 5’-FAM-CCGTCCCTACCTCCC-NFQ-3’). Genomic DNA was extracted from peripheral blood mononuclear cells obtained from blood samples obtained at enrollment; every 2 weeks just prior to administering doses 2, 3, and 4; and monthly while on study treatment. DNA (0.5–1 µg per sample) was added to a final reaction mixture that contained TaqMan Universal Master Mix II with UNG (ABI/ThermoFisher, CA, USA), nuclease free water, and primer/probe sets 900 nM/250 nM (final concentration). PCR conditions were: initial denaturation at 95°C for 10 min, followed by 40 cycles of denaturation and annealing/extension at 95°C for 15 s and 60°C for 60 s, respectively.
Assessment for the presence of replication-competent retrovirus (RCR)
DNA extracted from whole blood from study patients was assessed for RCR-specific sequences prior to surgery; at 3 months, 6 months, and 1 year after study treatment; and annually thereafter. Real-time PCR with 4070A envelope-specific primers (forward primer = 5’-AGGCGCTTAACCTCACCAA-3’; reverse primer = 5’-ACACTAAGCACAGCCAACATTCTT-3’; and probe = 5’FAM-TCCCGACAAGACCC-NFQ-3’) was used for this analysis. The same qPCR conditions were used as above for the assessment of possible NSC migration into the systemic circulation.
Intracerebral microdialysis
Intracerebral microdialysis is a sampling technique that continuously measures free drug concentrations in the brain extracellular fluid without disrupting normal tissue function. We and others have applied intracerebral microdialysis to define the neuropharmacokinetic profile of other chemotherapies/anti-cancer agents (25, 32–36). In vitro recovery experiments were performed to determine the fractional recoveries of 5-FC and 5-FU. The intracerebral microdialysis technique used in this study has been described previously (21).
During surgery at the beginning of study treatment, a temporary microdialysis catheter (70 Brain MD Catheter, membrane length 10 mm, membrane molecular weight cutoff 20 kDa, shaft length 100 mm; M Dialysis, North Chelmsford, MA, USA) was inserted within 5–15 mm of the resection cavity or biopsy site in participants on dose level 4 to collect dialysate from which to measure concentrations of 5-FC and 5-FU in the brain interstitium.
After surgery, the microdialysis catheter was perfused with artificial CSF at a flow rate of 1 µL/min, and dialysate was collected continuously. Beginning at the time participants took their first dose of oral 5-FC on cycle 1 day 4, the collecting microvial was replaced with a fresh vial every 60 min for 24 h and thereafter replaced every 3 h until the end of the 7-day course of 5-FC or until the microdialysis catheter stopped functioning. Dialysate-containing microvials were placed on dry ice until they were moved to an ultralow temperature freezer (≤ -70°C) for storage prior to analysis.
Blood samples were collected from participants before and after 5-FC reached steady-state to determine the plasma concentration-time profiles of 5-FC and 5-FU. These data were to be used to characterize the relationship between intracerebral and systemic concentrations of the two drugs. Blood samples were obtained prior to the first dose of 5-FC and then every 30 min for 3 h, with additional samples at 4 h and 6 h after the first dose of 5-FC was taken on days 4 and 5. On days 6, 7, and 8, blood samples were collected just before the morning dose and then 90 min later. The blood samples were centrifuged at 1500 x g for 10 min for harvesting plasma within 1 h after collection and then stored at ≤ -70°C until they were analyzed.
Evaluation of 5-FC and 5-FU concentrations in dialysate and plasma was performed by quantitative tandem mass spectrometry in the City of Hope Analytical Pharmacology Core Facility using an Agilent Technologies LC 1100 series system (Palo Alto, CA, USA) interfaced with a Micromass Quattro Ultima Triple Quadrupole Mass Spectrometer (Micromass, Inc., Milford, MA, USA). HPLC separation was achieved using a Synergi hydro-RP 4 m 150 mm x 2.0 mm analytical column (Phenomenex, Torrance, CA, USA). The auto-injector temperature was maintained at 5oC and the column temperature at 25oC. Water was purified using the Millipore Milli-Q system (Milford, MA, USA). An isocratic mobile phase of 0.1% formic acid (J.T. Baker, Phillipsburg, NJ, USA) in water was used to elute the analytes from the column at a flow rate of 0.2 mL/min. The total run time was 7 min. The electrospray ionization source of the mass spectrometer was operated in positive ion mode with a cone gas flow of 50 L/h and a desolvation gas flow of 750 L/h. The capillary voltage was set to 3.5 kV. The cone voltage was optimized at 45 V for 5-FC (Sigma, St. Louis, MO, USA) and 37 V for 5-FU (Sigma, St. Louis, MO, USA) and the internal standard (IS), 5-Fluorouracil-15N2, (C/D/N Isotopes, Inc., Quebec, Canada). The collision cell energy was 20 eV for 5-FC and 17 eV for 5-FU and the IS. The source temperature was 125oC and the desolvation temperature was 450oC. A solvent delay program was used from 0 to 2 min and from 6 to 7 min to minimize the mobile phase flow to the source. MassLynx version 4.1 software was used for data acquisition and processing.
Positive electrospray ionization of 5-FU, 5-FC, and the IS produced abundant protonated molecular ions. Fragmentation of these compounds was induced under collision-induced dissociation conditions and acidic mobile phase. The precursor → product ion combinations at m/z 131.1→114.12 for 5-FU, 130.1→113.25 for 5-FC, and 133.0→115.20 for the IS were used in multiple reaction monitoring (MRM) mode to determinate these compounds. The use of MRM provided sufficient specificity and sensitivity. MS/MS experimental conditions, such as collision energy and collision cell pressure, were optimized from continuous flow injection sample introduction of standard solutions. Under optimized assay conditions, the retention time was 3.14 min for 5-FC and 5.28 min for 5-FU and the IS, and the lower limits of quantitation were 6 nM and 0.25 nM, respectively.
Assessment of anti-NSC antibodies in serum
We also collected serial blood samples from participants to evaluate for possible development of humoral immune responses to the allogeneic NSCs. Blood was collected for isolation of serum at the time of participant enrollment; every 2 weeks during participants’ first two months on study (prior to starting cycles 1–4); and then monthly for the remainder of time the participant received study treatment. The presence of antibodies against the CD-NSCs and the parental NSC line were assessed using the same qualified flow cytometric method as in the first-in-human study. Aliquots of cultured NSCs (CD-NSCs and the parental line as a control) were incubated at 4°C for 20 min with cryopreserved patient serum samples that had been heat-treated to inactivate complement. After incubation, the NSCs were washed twice with 1 mL of FACS buffer (PBS with BSA [5 g/L] and sodium azide [0.06 g/L]). After each wash, NSCs were pelleted by centrifugation (1000 × g, 5 min). After the second wash and centrifugation, NSCs were resuspended in FACS buffer and analyzed using flow cytometry for bound antibodies detected using a fluorescein isothiocyanate (FITC)-conjugated AffiniPure F(ab’)2 fragment goat anti-human IgG, Fcγ (Jackson ImmunoResearch, PA, USA; Catalog No. 109–096-008). Binding of human antibodies to the NSCs was detected as a shift in the mean fluorescence intensity (MFI) compared to NSCs not treated with human serum. A significant shift was defined as a value outside the 95% confidence interval around the mean for 10 normal donors.
Statistical analysis
The recommended dose for phase II testing of intracerebrally administered CD-NSCs in combination with fixed doses of 5-FC and leucovorin was determined based on the maximum tolerated dose (MTD), or the MFD, and the full toxicity profile. The protocol definition for the MTD was the highest CD-NSC dose tested in which less than 33% of patients experienced a dose-limiting toxicity (DLT) attributable to the treatment regimen when at least 6 patients were treated at that dose and were evaluable for toxicity based on the rules of the 3+3 design. The MFD was defined as the highest dose studied when DLT was not the limiting factor for the chosen dose level. DLTs consisted of grade 3 or 4 non-hematologic (except grade 3 nausea/vomiting without maximal anti-emetic therapy and grade 3 fatigue) or grade 4 hematologic adverse events that were at least possibly related to the investigational agents. The DLT evaluation period was 28 d. During the DLT period, participants were treated with 2 intracerebral doses of CD-NSCs separated by 2 weeks and followed each time by a 7-day course of oral 5-FC (and leucovorin for dose level 4 participants only).
Rates and associated 90% confidence limits were estimated for the DLTs at the MTD/MFD and disease response. Toxicities were graded using the NCI Common Terminology Criteria and Adverse Events (CTCAE) version 4.0. Tables were created to summarize all toxicities and side effects by dose, course, organ, severity, and attribution. In evaluable study participants, Kaplan Meier methods were used to estimate median progression-free survival and overall survival. Statistical and graphical methods were used to describe correlative pharmacokinetic (PK) and immunologic data. Participant demographics were summarized using descriptive statistics.
The primary PK parameters of interest were the Cmax and AUC of 5-FC and 5-FU, measured both in the dialysate samples and in plasma. The 5-FC and 5-FU PK data were also compared to the PK data obtained from patients who underwent intracerebral microdialysis in the first-in-human study of CD-expressing NSCs (21) to determine if there was a dose effect at the MTD/MFD compared to lower doses of CD-NSCs. All summaries were exploratory, with the goal of developing further questions regarding the modulation of therapy or reasons for efficacy or lack of efficacy.
RESULTS
Patient enrollment and characteristics
Between October 2014 and May 2017, 16 patients with recurrent high grade gliomas were enrolled at City of Hope. A description of these participants is provided in Table 2. The majority of participants had recurrent glioblastoma (81%) and underwent tumor resection (75%). The median age of study participants was 57 (range 26–71).
Table 2.
Patient demographics
| Dose Level | Patient number | Age | Gender | Diagnosis | Type of surgery | Number of CD-NSC doses | Best response |
|---|---|---|---|---|---|---|---|
| 1 | 1 | 59 | M | Glioblastoma | Resection | 9 | Progressiona |
| 1 | 2 | 59 | F | Glioblastoma | Resection | 8 | Stable disease |
| 1 | 3 | 48 | F | Glioblastoma | Biopsy | 9 | Stable disease |
| 2 | 4 | 26 | M | Glioblastoma | Resection | 4 | Progression |
| 2 | 5 | 40 | M | Anaplastic pleomorphic xanthoastrocytoma | Resection | 4 | Progression |
| 2 | 6 | 64 | M | Glioblastoma | Biopsy | 3 | Progression |
| 3 | 7 | 65 | F | Glioblastoma | Resection | 2 | Progression |
| 3 | 8 | 57 | M | Glioblastoma | Resection | 2 | Progression |
| 3 | 9 | 54 | M | Glioblastoma | Resection | 4 | Progression |
| 4 | 10 | 51 | M | Glioblastoma | Resection | 3 | Progression |
| 4 | 11 | 71 | M | Glioblastoma | Resection | 3 | Progression |
| 4 | 12 | 58 | M | Glioblastoma | Biopsy | 4 | Progression |
| 4 | 13 | 58 | M | Glioblastoma | Resection | 1b | Not assessed |
| 4 | 14 | 56 | M | High grade glioma | Resection | 10 | Stable disease |
| 4 | 15 | 45 | M | Glioblastoma | Biopsy | 3 | Progression |
| 4 | 16 | 69 | M | Gliosarcoma | Resection | 2 | Not assessedc |
The changes on Patient 1’s first brain MRI after 8 weeks of study treatment were inconclusive, and since he was neurologically stable, he continued with study treatment. When the second brain MRI approximately 8 weeks later showed definite tumor progression, re-review of the first brain MRI determined that those initial changes were from tumor growing, and so best response for him was changed to progression.
Patient 13 received only one dose of CD-NSCs due to migration of the Rickham catheter tip into the lateral ventricle; therefore, the patient was taken off the study.
Patient 16 experienced a dose-limiting toxicity (grade 3 wound infection) 3 weeks after start of study treatment, requiring removal of the Rickham catheter. Since he did not continue study treatment and chose to enroll in a hospice program, no further imaging of his brain was performed.
Dose escalation
Of the 16 patients who started study treatment, fifteen were fully evaluable for the purpose of dose escalation. One participant on dose level 4 could not proceed with the second dose of CD-NSCs because the tip of his Rickham catheter subsequently penetrated into the adjacent lateral ventricle after resolution of brain shift during the post-operative period. Because this participant did not experience a DLT but could not receive all the planned treatment for the DLT evaluation period, he was replaced by another patient for dose escalation and response evaluation. However, pharmacokinetic and immunologic samples were still collected from him.
No DLTs occurred on dose levels 1–3. However, a participant treated on dose level 4 experienced a DLT consisting of a grade 3 wound infection due to Serratia liquefaciens and Candida parapsilosis that was possibly related to the Rickham catheter, 5-FC, and leucovorin. The MFD of intracerebrally administered CD-NSCs was determined to be 150 x 106 in combination with oral 5-FC (37.5 mg/kg) with or without leucovorin (25 mg) every 6 h for 7 d. The DLT rate at the MFD was 17% (90% confidence interval (CI), 1–58%).
Safety
Overall, study treatment was well tolerated by the participants. There were no grade 4 or 5 toxicities related to study treatment. Eleven participants [69% (90% CI, 45–87%)] experienced grade 2 or 3 toxicities attributed to study drug, where attribution was defined as possibly, probably, or definitely related to NSCs, 5-FC or leucovorin. Table 3 presents the maximum grade toxicities of each type and includes all attributed grade 3 toxicities and grade 2 toxicities that occurred in more than one participant. We did not observe any leakage from the Rickham reservoirs during the several hours-long infusions of the CD-NSCs.
Table 3.
Summary of toxicities related to study treatmenta
| Maximum Grade Toxicity | Grade 2 Number (%) |
Grade 3 Number (%) |
|---|---|---|
| Anemia | 2 (13) | 0 (0) |
| Lymphocyte count decreased | 0 (0) | 1 (6) |
| Fatigue | 4 (25) | 0 (0) |
| Wound infection | 0 (0) | 1 (6) |
| Brain abscess | 0 (0) | 1 (6) |
| Hypophosphatemia | 4 (25) | 0 (0) |
| Thromboembolic event | 0 (0) | 1 (6) |
This table includes grade 2 toxicities that occurred in more than one participant and all grade 3 toxicities. There were no grade 4 or 5 toxicities related to study treatment.
Our first-in-human study found no evidence of CD-NSC migration into the systemic circulation after a single administration (21). In the current study, we also did not detect CD-NSCs, as determined by qPCR using gag-v-myc primers (unique to the HB1.F3.CD21 NSC line), in any of the serial blood samples from participants after multiple doses of CD-NSCs were administered, including a participant who was treated for as long as 5 months (10 cycles).
Because replication-deficient retrovirus vectors were used to transduce the NSCs ex vivo with the gag-v-myc and CD genes, we assessed the clinical lots of CD-NSCs and participants’ blood samples for the possible development of replication-competent retrovirus, as required by the United States Food and Drug Administration. The CD-NSC clinical lots and all blood samples, including those obtained as far out as one year from the start of study treatment, tested negative for replication-competent retrovirus.
Feasibility
The median number of intracerebrally administered CD-NSC doses that evaluable study participants received was 4 (range 2–10). The Rickham catheters remained functional for the duration of study treatment in all participants, including in the participant who received 10 intracerebral doses of the CD-NSCs over 5 months. These results indicate that it is feasible to administer multiple doses of CD-NSCs using a Rickham catheter.
Measuring conversion of 5-FC to 5-FU by the CD-NSCs
Dialysate and plasma samples were collected from participants treated with the highest dose of CD-NSCs and 5-FC/leucovorin (dose level 4 participants, N=7) during their first 7-day course of 5-FC. Although only 6 patients were evaluable for DLTs, all 7 patients enrolled at this dose level had dialysate and plasma samples collected for pharmacokinetics. The concentrations of 5-FC and 5-FU in these samples were determined by quantitative tandem mass spectrometry. Prior in vitro recovery experiments showed fractional recoveries of >90% for 5-FC and 5-FU (21); therefore, the microdialysis data are presented as uncorrected values. The interstitial concentrations of both 5-FC and 5-FU increased rapidly in the brain after the first oral dose of 5-FC, reaching a steady-state within 8 h (Figure 2A). Average steady-state 5-FC and 5-FU concentrations in the brain were 213 ± 58 μmol/L and 26 ± 14 nmol/L, respectively. The microdialysis catheters remained functional for the full 7-day dosing interval of 5-FC in four participants. Analysis of dialysate and plasma samples revealed that the CD-NSCs continued to convert 5-FC to 5-FU throughout the entire 7 d of 5-FC administration (Figure 2B), confirming our initial proof-of-concept data (21).
Figure 2. Intracerebral microdialysis data: CD-NSC-mediated conversion of 5-FC to 5-FU.
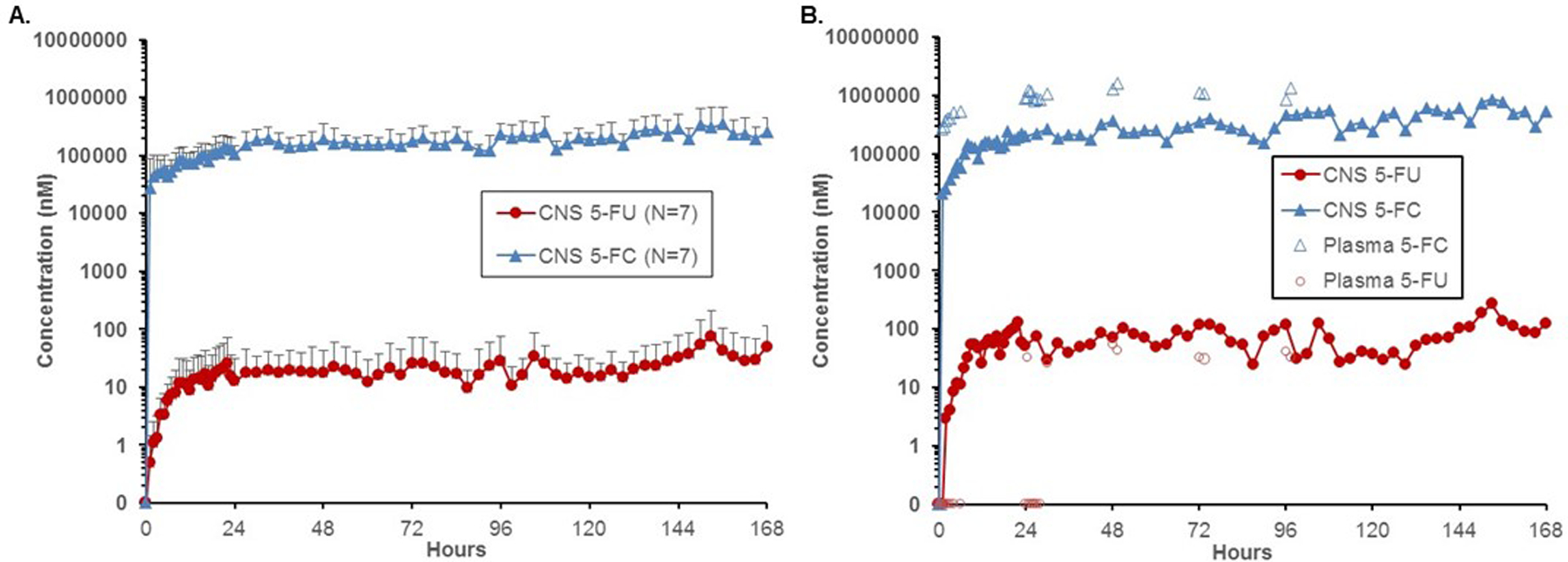
(A) Average brain interstitial 5-FC and 5-FU concentrations from patients on dose level 4 who were treated with an intracerebral CD-NSC dose of 150 x 106 and took oral 5-FC (37.5 mg/kg) and oral leucovorin (25 mg) every 6 h. Error bars represent standard deviations. (B) Representative example of intracerebral microdialysis and serum data from a patient, demonstrating the ability of the CD-NSCs to convert 5-FC to 5-FU in the brain throughout the 7-day course of oral 5-FC. In this patient, 5-FU was not detected in plasma until 24 h after the patient started taking oral 5-FC; nonetheless, 5-FU was present in the brain during the first 24 h, indicating that all of the initial 5-FU measured in the brain came from intracerebral NSC-mediated conversion of 5-FC. Although some of the later time points show that 5-FU concentrations in the brain were only slightly lower than plasma levels, knowing that only a small fraction of 5-FU crosses into the brain from plasma, these results indicate that most of the 5-FU measured in the brain was produced by the CD in the NSC converting 5-FC to 5-FU.
We also measured 5-FC and 5-FU plasma concentrations to assess the systemic exposure to the prodrug and to confirm that the measured intracerebral 5-FU concentrations were not just the result of circulating 5-FU produced from conversion of 5-FC by CD-expressing gut flora. Dose level 4 participants achieved an average steady-state plasma 5-FC concentration of 748 ± 218 μmol/L after taking 150 mg/kg/day of oral 5-FC. This concentration was similar to the results from our first-in-human study (21), as well as for patients taking 5-FC to treat cryptococcal meningitis (37). The mean ratio of average brain interstitial 5-FC concentrations to the average steady-state plasma levels was 29 ± 21%.
For all seven participants on dose level 4, plasma 5-FU levels were determined to be either low or undetectable. In nearly all instances where 5-FU was detected in the plasma, concentrations were below or at least slightly lower than the levels measured in the brain at the same time point (Figure 2B). Since the CNS penetration of 5-FU in a non-human primate model is between 10–20% of plasma levels (38), one would expect the brain 5-FU concentrations to be much lower than 5-FU levels in plasma if the only source of 5-FU in the brain was from 5-FU in plasma crossing the blood-brain barrier. So even though some intracerebral 5-FU data points were relatively similar to plasma 5-FU concentrations, we can confidently conclude that most of the 5-FU measured in the brain came from NSC-mediated production of 5-FU from 5-FC in the brain.
Humoral responses to CD-NSCs
Treating patients repeatedly with allogeneic NSCs raises concern about possible development of anti-NSC antibody responses, which could potentially decrease the efficacy of the NSCs. We assessed serial serum samples from 14 study participants for the presence of NSC-binding antibodies. Samples were collected at the time of enrollment and up to 5 months after start of treatment. The participants did not show any clinical signs of adverse immune responses to the NSCs, such as fever, rash, or unexplained cerebral edema. However, anti-NSC antibodies were detected in the serum of three of the 14 study participants (Figure 3) as compared with results obtained from normal donor sera (negative control) and sera from patients found to have antibodies against one of the HLA class I antigens on the CD-NSCs (positive control). Detectable antibody levels occurred after these three participants received their third dose of CD-NSCs. Although we were not able to determine from our assay if the anti-NSC antibodies were neutralizing ones, we found that the antibody responses were against both CD-NSCs and the parental HB1.F3 NSCs (data not shown) in all three patients, indicating that these responses were to the NSCs themselves and not the bacterially-derived cytosine deaminase.
Figure 3. Assessment of antibody responses to multiple doses of NSCs.
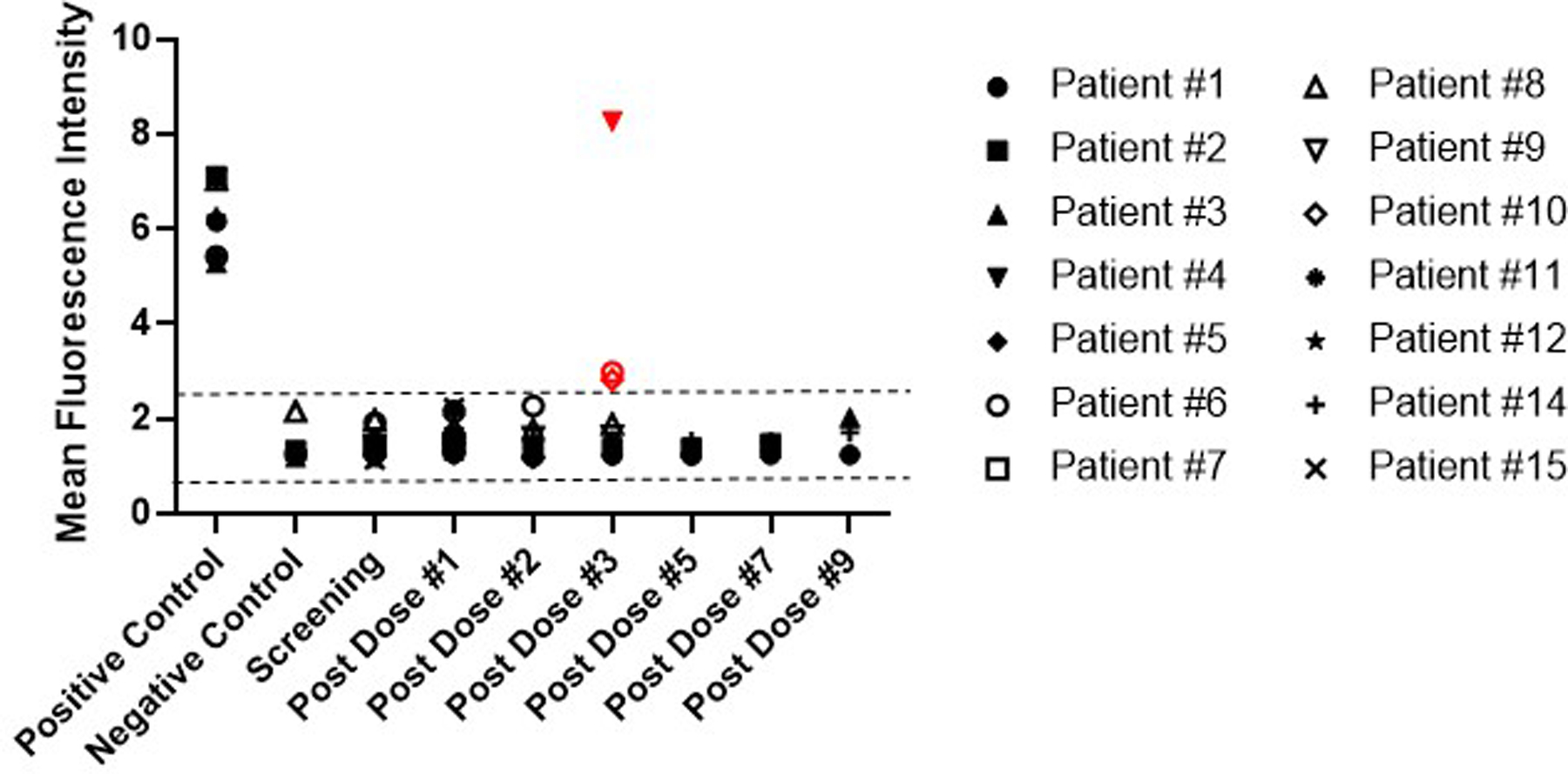
Patient serum samples were assessed by flow cytometry for the presence of anti-NSC antibodies at the time of enrollment, every 2 weeks (prior to administering the first four doses of CD-NSCs) for the first 2 months, and then monthly until the study treatment was stopped. The positive and negative controls (N=5 each) represent the results of 5 independent experiments and consisted of serum from subjects determined to have antibodies against the HLA-I antigens expressed on the CD-NSCs (positive control) and normal donors who did not express HLA-I antigens on the NSCs (negative control). All data points represent the mean of triplicate assays. The dashed lines indicate the range for a “negative” mean fluorescence intensity (MFI) determined from 10 normal donors, resulting in a range of 0.55 to 2.59 and representing a 95% confidence interval based on 2.3 SE around the population mean. The three positive antibody responses are indicated in red (Patients 4, 6, and 10). Patient 13 received only one dose of CD-NSCs and was not included in this analysis. Patient 16 was not included in this analysis either because we were unable to obtain a post second CD-NSC dose serum sample to analyze.
Of the three participants who showed antibody responses, Patient 4 had the highest measured antibody response. This patient had tapered off dexamethasone the day before receiving his third dose of CD-NSCs. Because we had a limited amount of sera for this patient, we were unable to repeat the HLA antibody assessment after the anti-NSC antibodies were detected. The other two participants (Patients 6 and 10) had anti-NSC antibody responses that were lower than that of Patient 4. At the time they exhibited antibody responses to the NSCs, both patients were found to have antibodies against HLA class I antigens known to be expressed on the NSCs (HLA class I A1, A31, and B7), despite being negative for these antibodies at screening. Similar to Patient 4, these other 2 participants had stopped taking dexamethasone for a period of time before the anti-NSC antibodies were detected. Patient 6, had stopped taking dexamethasone 12 d prior to receiving his second dose of CD-NSCs, but he then restarted dexamethasone (2 mg twice daily) 4 d before he was given his third dose of NSCs, and Patient 10 stopped taking dexamethasone 9 d before he received his third dose of CD-NSCs.
However, we do not believe generation of anti-NSC antibodies was directly related to dexamethasone because other study participants who also did not continuously take dexamethasone during study treatment did not develop these antibodies. For example, Patient 2, also tapered off dexamethasone 9 d before receiving her third dose of CD-NSCs and remained off dexamethasone for the rest of her study treatment, receiving a total of 6 doses of CD-NSCs during that time period without ever developing anti-NSC-antibodies. Similarly, Patient 5 stopped taking dexamethasone 4 d before receiving his second dose of CD-NSCs and remained off dexamethasone for his third and fourth doses of CD-NSCs without developing antibodies to the NSCs. In summary, of the 7 participants who took dexamethasone part of the time during their study treatment, 3 had detectable levels of antibodies against the CD-NSCs in their blood, while none of the 7 participants who took dexamethasone continuously developed antibodies to the CD-NSCs, even after receiving as many as 9 or 10 doses. Based on a one-sided Fisher’s exact test, the relationship between the development of antibodies and the use of dexamethasone was not statistically significant (P = 0.18) in this small post hoc analysis.
Clinical activity
For all study participants, the median progression-free survival was 1.8 months (95 % CI, 1.2–4.8) and median overall survival was 4.3 months (95% CI, 2.5–12.1). The best response was stable disease, which was seen in three participants and ranged from 4 to 5 months. One of these participants, Patient 14, did not actually experience tumor progression while receiving study treatment (Figure 4). He developed a brain abscess due to Propionibacterium acnei after five months of study therapy and was taken off treatment because his Rickham catheter had to be removed as part of the management of the brain abscess. At the time of study enrollment, he had a 6.5 cm by 3.7 cm aggressive left fronto-temporal lobe high grade glioma (O6-methylguanine-DNA methyl-transferase promoter unmethylated and isocitrate dehydrogenase wild type), which had not responded to prior treatment for recurrent disease that included lomustine, bevacizumab, and alternating electric fields (39). At the time of craniotomy to drain the brain abscess, biopsies of the surgical cavity showed no evidence of recurrent tumor. However, one month after stopping study treatment and completing a course of vancomycin, a follow up brain MRI documented tumor progression at the surgical site.
Figure 4. Serial brain MRIs from Patient 14 who had stable disease for 5 months.
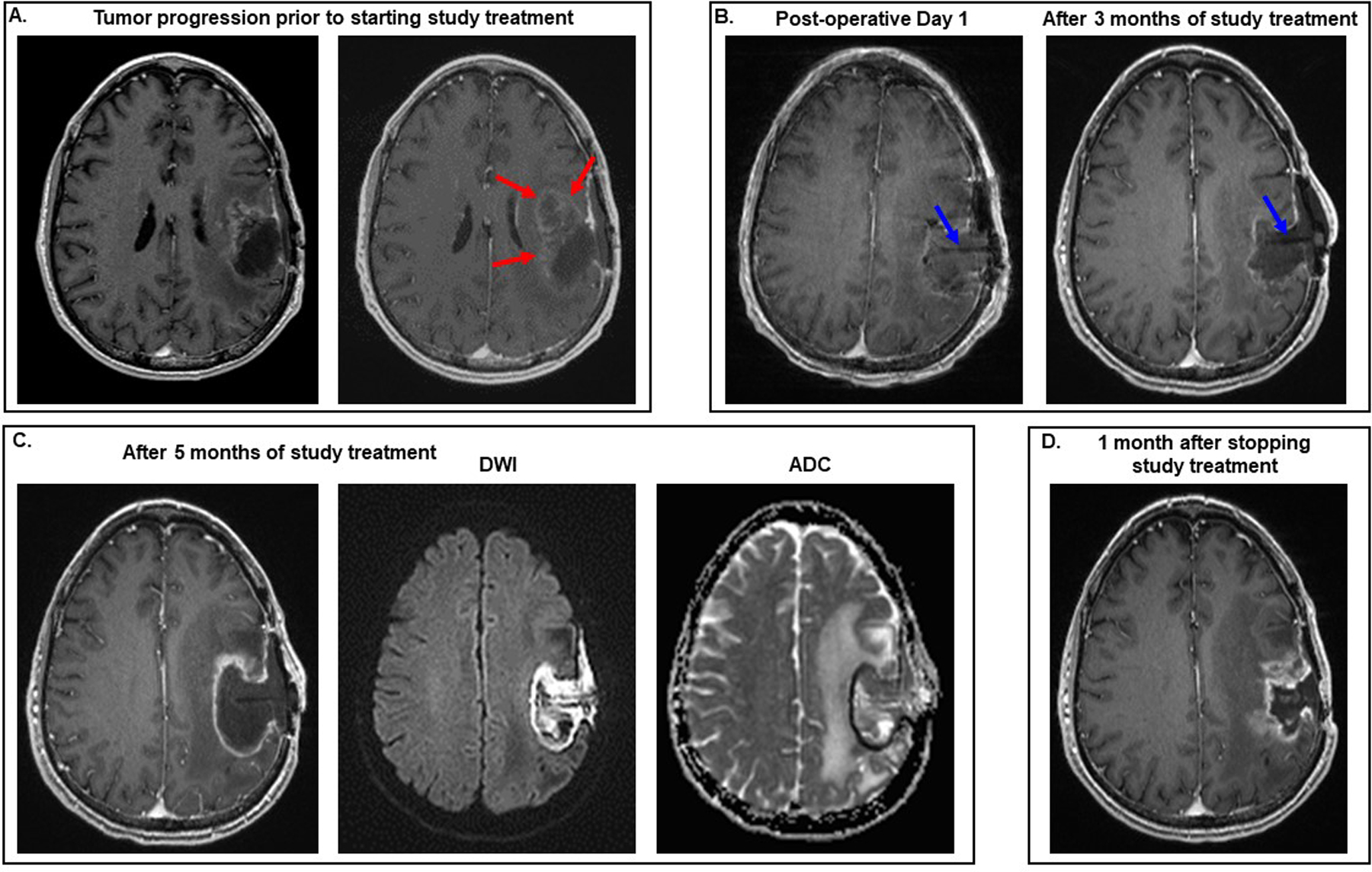
All images are T1 post-contrast except where indicated in the figure. (A) Prior to participating in the study, the patient was being treated with bevacizumab and alternating electric fields. The MRI on the right, which was performed 6 weeks later than the one on the left, documents that he developed tumor progression on this combination therapy, as evidenced by the increase in heterogeneous enhancement antero-medial to the surgical cavity (red arrows). (B) He underwent gross total resection of the recurrent tumor and started study therapy. No radiographic evidence of tumor growth was seen after 3 months of treatment with the CD-NSCs and 5-FU/leucovorin. Blue arrows point to the Rickham catheter seen within the surgical cavity. (C) His brain MRI performed approximately 5 months after the start of study treatment showed increase in the size of the surgical cavity and rim enhancement on the T1 post-contrast image. However, the rim and contents of the surgical cavity were bright on the diffusion weighted image (DWI) and dark on the apparent diffusion coefficient (ADC) image, indicating restricted diffusion. These imaging findings suggested that the changes in the surgical cavity and its contents on the T1 post-contrast image were due to the presence of a brain abscess rather than tumor progression. Aspiration of the Rickham reservoir revealed the presence of purulent fluid, and the patient then underwent surgery to drain the abscess. Biopsies of the surgical cavity obtained during this surgery showed no definite evidence of tumor regrowth. (D) MRI of his brain performed approximately 1 month after the surgery showed new enhancement at the surgical site consistent with tumor progression, which was then confirmed histopathologically by the presence of sheets of neoplastic cells with nuclear anaplasia associated with prominent microvasculature when another craniotomy was performed a week later.
DISCUSSION
NSCs are inherently tumor tropic, and when genetically modified they can be used to deliver anti-cancer agents for the treatment of brain tumors. This phase I study not only determined the recommended dose of CD-NSCs in combination with 5-FC and leucovorin for phase II testing, it demonstrated the feasibility of intracerebrally administering serial doses of NSCs. The Rickham catheters remained functional for up to 5 months.
Although our surgical site infection rate of 12.5% is higher than rates reported after a craniotomy for glioma resection (4–5%) (40, 41), this increased rate is not unexpected since the risk of infection is higher when an indwelling catheter is present. Comparing our study’s rate of infection to that of the more commonly used indwelling Ommaya reservoir/catheter, ours was still slightly higher than the infection rate of 8% reported in the largest study of Ommaya reservoir-related infections to date (40). The Rickham reservoir is similar to the Ommaya reservoir, except the Rickham’s diameter is smaller, and both reservoirs are positioned beneath the scalp. However, the catheter attached to the Ommaya reservoir is typically placed in the lateral ventricle to administer chemotherapy into the CSF via manual push over several minutes, whereas we inserted the Rickham reservoir’s catheter into the resection cavity or residual tumor and slowly infused the CD-NSCs over several hours. These differences in approach likely contributed to the difference in infection rate. Of note, neither the early nor the late surgical site infections that occurred in two of the study participants were life threatening, leading us to conclude that the somewhat increased risk of infection associated with our technique for intracerebral delivery of NSCs is acceptable in this patient population.
It is unknown how long CD-NSCs remain in the brain and are functional. Based on our preclinical data, autopsy results from the first-in-human study, and our intracerebral microdialysis data, we predict that the CD-NSCs persist for at least two weeks and then their numbers significantly decrease, thus necessitating serial administration of NSCs to sustain an anti-tumor effect. In this study, repeated treatment with the CD-NSCs and 5-FC with or without leucovorin was generally well tolerated. Participants received as many as 10 doses of intracerebrally administered CD-NSCs, and we did not detect any evidence of the NSCs migrating out of the brain into the systemic circulation. Moreover, testing for development of replication-competent retrovirus as far as one year out from the start of study treatment was negative.
Our microdialysis results validate the findings from our first-in-human study regarding the ability of the CD-NSCs to continuously convert 5-FC into 5-FU in the brain throughout the dosing period of oral 5-FC (21). These microdialysis data also corroborate our previous conclusion that diffusion of 5-FU from the peripheral circulation into the brain cannot account for the higher 5-FU concentrations measured in the brains of our study participants, confirming that the CD-NSCs were converting 5-FC to 5-FU locally in the brain.
We did not see any apparent NSC dose-dependent differences in intracerebral average steady-state 5-FU concentrations between patients treated with 150 x 106 CD-NSCs in the current study as compared to patients treated with 50 x 106 CD-NSCs in our first-in-human study, indicating that the ability of NSCs to distribute within tumor may be saturable. Furthermore, intracerebral concentrations of 5-FU in patients treated with the higher dose of CD-NSCs were still in the nM range, and below the accepted threshold for 5-FU cytotoxicity (typically in μM range). Because no increase in 5-FU concentrations in the brain was observed after administering the MFD of CD-NSCs, future directions could include possibly using a vector that yields a higher expression of CD or treating with 5-FC for longer than 7 d to assess the effect of continuous exposure from lower levels of 5-FU since the cytotoxic effect of 5-FU is time dependent (42, 43).
Administering multiple doses of allogeneic NSCs raises concern about possible development of immune responses that could neutralize the NSCs, thus limiting efficacy of the treatment. Although no clinical signs of immunogenicity were observed, a small number of participants had detectable levels of anti-NSCs antibodies in their blood after receiving a third dose of the NSCs. It remains unknown if anti-NSC antibodies were also present in the brain. The relationship between the development of anti-NSC antibodies in the blood and the use of dexamethasone was not found to be statistically significant; however, the fact that none of the participants who took dexamethasone continuously developed antibody responses could be hypothesis-driving. One potential strategy to investigate in future NSC-based studies would be to require all participants to take dexamethasone during the entire time that they receive study treatment to determine if doing so reduces or prevents the development of anti-NSC-antibodies.
In addition to determining the MFD of intracerebrally administered CD-NSCs, the results of this phase I study confirm proof-of-concept regarding the ability of NSCs to produce chemotherapy locally in the brain and establish the feasibility of administering serial doses of NSCs directly into the brain using a Rickham catheter. NSCs can be viewed as a platform technology for delivering a variety of anti-cancer therapies (19, 44–48) to sites of tumor. This NSC line, for example, has also been modified to express a conditionally replicating adenovirus (19, 20, 49). The rationale for combining NSCs with an oncolytic virus (NSC-OV) is that the NSCs can transport the oncolytic virus across normal brain to seed sites of tumor distant from the main mass while simultaneously protecting it from being destroyed by the immune system. Although a few participants in this phase I study of multiple doses of CD-NSCs developed anti-NSC antibodies after receiving three doses of NSCs, most patients did not. Furthermore, should patients develop anti-NSC antibodies, being able to effectively administer even three doses of an oncolytic virus before it is neutralized by the immune system would be an advance to the field of oncolytic virotherapy. A phase I study of a single dose of NSC-OV given intracerebrally to newly diagnosed high grade glioma patients in combination with standard focal brain radiation and temozolomide (NCT03072134) was recently completed at Northwestern University and City of Hope. Based on the documented safety and feasibility of intracerebrally administering multiple doses of NSCs reported here, a phase I study to assess intracerebral administration of serial doses of the NSC-OV in patients with high grade gliomas is being planned.
ACKNOWLEDGEMENTS
We thank Narine Arabyan, PhD, Keely Walker, PhD, and Alice Tuan, BS, for their editing and/or technical assistance with preparing this manuscript. We also thank Shiny Wu, BS, for her technical support in providing the PK data.
FUNDING
This research was supported by grants from the United States Food and Drug Administration R01 FD004816 (co-principal investigators: J. Portnow and K.S. Aboody), Phase One Foundation (PI: J. Portnow), The Rosalinde and Arthur Gilbert Foundation, and City of Hope. In addition, research reported in this publication included work performed in the Analytical Pharmacology and Biostatistics and Mathematical Modeling Cores supported by the National Cancer Institute of the National Institutes of Health under grant number P30CA033572. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
CONFLICT OF INTEREST
K.S. Aboody is a shareholder and board member of TheraBiologics, Inc.
REFERENCES
- 1.Kalladka D, Sinden J, Pollock K, Haig C, McLean J, Smith W, et al. Human neural stem cells in patients with chronic ischaemic stroke (PISCES): a phase 1, first-in-man study. Lancet 2016;388(10046):787–96. [DOI] [PubMed] [Google Scholar]
- 2.Hanus J, Zhao FK, Wang SS. Current therapeutic developments in atrophic age-related macular degeneration. Brit J Ophthalmol 2016;100(1):122–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Parameswaran S, Krishnakumar S. Pluripotent stem cells: A therapeutic source for age-related macular degeneration. Indian J Ophthalmol 2017;65(3):177–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mazzini L, Gelati M, Profico DC, Soraru G, Ferrari D, Copetti M, et al. Results from Phase I Clinical Trial with Intraspinal Injection of Neural Stem Cells in Amyotrophic Lateral Sclerosis: A Long-Term Outcome. Stem Cells Transl Med 2019;8(9):887–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Feldman EL, Boulis NM, Hur J, Johe K, Rutkove SB, Federici T, et al. Intraspinal neural stem cell transplantation in amyotrophic lateral sclerosis: Phase 1 trial outcomes. Ann Neurol 2014;75(3):363–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Curtis E, Martin JR, Gabel B, Sidhu N, Rzesiewicz TK, Mandeville R, et al. A First-in-Human, Phase I Study of Neural Stem Cell Transplantation for Chronic Spinal Cord Injury. Cell Stem Cell 2018;22(6):941–50 e6. [DOI] [PubMed] [Google Scholar]
- 7.Pereira IM, Marote A, Salgado AJ, Silva NA. Filling the Gap: Neural Stem Cells as A Promising Therapy for Spinal Cord Injury. Pharmaceuticals-Base 2019;12(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aboody KS, Brown A, Rainov NG, Bower KA, Liu S, Yang W, et al. Neural stem cells display extensive tropism for pathology in adult brain: evidence from intracranial gliomas. Proc Natl Acad Sci U S A 2000;97(23):12846–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kendall SE, Najbauer J, Johnston HF, Metz MZ, Li S, Bowers M, et al. Neural stem cell targeting of glioma is dependent on phosphoinositide 3-kinase signaling. Stem Cells 2008;26(6):1575–86. [DOI] [PubMed] [Google Scholar]
- 10.Zhang S, Xie R, Zhao T, Yang X, Han L, Ye F, et al. Neural stem cells preferentially migrate to glioma stem cells and reduce their stemness phenotypes. Int J Oncol 2014;45(5):1989–96. [DOI] [PubMed] [Google Scholar]
- 11.Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 2005;352(10):987–96. [DOI] [PubMed] [Google Scholar]
- 12.Stupp R, Taillibert S, Kanner A, Read W, Steinberg D, Lhermitte B, et al. Effect of Tumor-Treating Fields Plus Maintenance Temozolomide vs Maintenance Temozolomide Alone on Survival in Patients With Glioblastoma: A Randomized Clinical Trial. JAMA 2017;318(23):2306–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim SK, Kim SU, Park IH, Bang JH, Aboody KS, Wang KC, et al. Human neural stem cells target experimental intracranial medulloblastoma and deliver a therapeutic gene leading to tumor regression. Clin Cancer Res 2006;12(18):5550–6. [DOI] [PubMed] [Google Scholar]
- 14.Aboody KS, Najbauer J, Metz MZ, D’Apuzzo M, Gutova M, Annala AJ, et al. Neural stem cell-mediated enzyme/prodrug therapy for glioma: preclinical studies. Sci Transl Med 2013;5(184):184ra59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Teng J, Hejazi S, Badr CE, Tannous BA. Systemic Anticancer Neural Stem Cells in Combination with a Cardiac Glycoside for Glioblastoma Therapy. Stem Cells 2014;32(8):2021–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Seol HJ, Jin J, Seong DH, Joo KM, Kang W, Yang H, et al. Genetically engineered human neural stem cells with rabbit carboxyl esterase can target brain metastasis from breast cancer. Cancer Letters 2011;311(2):152–9. [DOI] [PubMed] [Google Scholar]
- 17.Hong SH, Lee HJ, An J, Lim I, Borlongan C, Aboody KS, et al. Human neural stem cells expressing carboxyl esterase target and inhibit tumor growth of lung cancer brain metastases. Cancer Gene Therapy 2013;20(12):678–82. [DOI] [PubMed] [Google Scholar]
- 18.Benedetti S, Pirola B, Pollo B, Magrassi L, Bruzzone MG, Rigamonti D, et al. Gene therapy of experimental brain tumors using neural progenitor cells. Nat Med 2000;6(4):447–50. [DOI] [PubMed] [Google Scholar]
- 19.Ahmed AU, Thaci B, Tobias AL, Auffinger B, Zhang L, Cheng Y, et al. A preclinical evaluation of neural stem cell-based cell carrier for targeted antiglioma oncolytic virotherapy. J Natl Cancer Inst 2013;105(13):968–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Thaci B, Ahmed AU, Ulasov IV, Tobias AL, Han Y, Aboody KS, et al. Pharmacokinetic study of neural stem cell-based cell carrier for oncolytic virotherapy: targeted delivery of the therapeutic payload in an orthotopic brain tumor model. Cancer Gene Therapy 2012;19(6):431–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Portnow J, Synold TW, Badie B, Tirughana R, Lacey SF, D’Apuzzo M, et al. Neural Stem Cell-Based Anticancer Gene Therapy: A First-in-Human Study in Recurrent High-Grade Glioma Patients. Clin Cancer Res 2017;23(12):2951–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim SU. Human neural stem cells genetically modified for brain repair in neurological disorders. Neuropathology 2004;24(3):159–71. [DOI] [PubMed] [Google Scholar]
- 23.Kim SU. Genetically engineered human neural stem cells for brain repair in neurological diseases. Brain Dev 2007;29(4):193–201. [DOI] [PubMed] [Google Scholar]
- 24.Kim SU, Lee HJ, Park IH, Chu K, Lee ST, Kim M, et al. Human neural stem cells for brain repair. Int J Stem Cells 2008;1(1):27–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Blakeley J, Portnow J. Microdialysis for assessing intratumoral drug disposition in brain cancers: a tool for rational drug development. Expert Opin Drug Met 2010;6(12):1477–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ratain MJ, Mick R, Schilsky RL, Siegler M. Statistical and ethical issues in the design and conduct of phase I and II clinical trials of new anticancer agents. J Natl Cancer Inst 1993;85(20):1637–43. [DOI] [PubMed] [Google Scholar]
- 27.Nadal JC, Van Groeningen CJ, Pinedo HM, Peters GJ. In vivo potentiation of 5-fluorouracil by leucovorin in murine colon carcinoma. Biomed Pharmacother 1988;42(6):387–93. [PubMed] [Google Scholar]
- 28.Borner MM, Castiglione M, Bacchi M, Weber W, Herrmann R, Fey MF, et al. The impact of adding low-dose leucovorin to monthly 5-fluorouracil in advanced colorectal carcinoma: results of a phase III trial. Swiss Group for Clinical Cancer Research (SAKK). Ann Oncol 1998;9(5):535–41. [DOI] [PubMed] [Google Scholar]
- 29.Modulation of fluorouracil by leucovorin in patients with advanced colorectal cancer: evidence in terms of response rate. Advanced Colorectal Cancer Meta-Analysis Project. J Clin Oncol 1992;10(6):896–903. [DOI] [PubMed] [Google Scholar]
- 30.Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK, et al. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta Neuropathol 2016;131(6):803–20. [DOI] [PubMed] [Google Scholar]
- 31.Kim SU, Nagai A, Nakagawa E, Choi HB, Bang JH, Lee HJ, et al. Production and characterization of immortal human neural stem cell line with multipotent differentiation property. Methods Mol Biol 2008;438:103–21. [DOI] [PubMed] [Google Scholar]
- 32.Bergenheim AT, Capala J, Roslin M, Henriksson R. Distribution of BPA and metabolic assessment in glioblastoma patients during BNCT treatment: a microdialysis study. J Neurooncol 2005;71(3):287–93. [DOI] [PubMed] [Google Scholar]
- 33.Blakeley JO, Olson J, Grossman SA, He X, Weingart J, Supko JG, et al. Effect of blood brain barrier permeability in recurrent high grade gliomas on the intratumoral pharmacokinetics of methotrexate: a microdialysis study. J Neurooncol 2009;91(1):51–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Portnow J, Badie B, Chen M, Liu A, Blanchard S, Synold TW. The neuropharmacokinetics of temozolomide in patients with resectable brain tumors: potential implications for the current approach to chemoradiation. Clin Cancer Res 2009;15(22):7092–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Portnow J, Badie B, Markel S, Liu A, D’Apuzzo M, Frankel P, et al. A neuropharmacokinetic assessment of bafetinib, a second generation dual BCR-Abl/Lyn tyrosine kinase inhibitor, in patients with recurrent high-grade gliomas. European Journal of Cancer 2013;49(7):1634–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Portnow J, Badie B, Liu X, Frankel P, Mi S, Chen M, et al. A pilot microdialysis study in brain tumor patients to assess changes in intracerebral cytokine levels after craniotomy and in response to treatment with a targeted anti-cancer agent. J Neurooncol 2014;118(1):169–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brouwer AE, van Kan HJ, Johnson E, Rajanuwong A, Teparrukkul P, Wuthiekanun V, et al. Oral versus intravenous flucytosine in patients with human immunodeficiency virus-associated cryptococcal meningitis. Antimicrob Agents Chemother 2007;51(3):1038–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kerr IG, Zimm S, Collins JM, O’Neill D, Poplack DG. Effect of intravenous dose and schedule on cerebrospinal fluid pharmacokinetics of 5-fluorouracil in the monkey. Cancer Res 1984;44(11):4929–32. [PubMed] [Google Scholar]
- 39.Stupp R, Wong ET, Kanner AA, Steinberg D, Engelhard H, Heidecke V, et al. NovoTTF-100A versus physician’s choice chemotherapy in recurrent glioblastoma: a randomised phase III trial of a novel treatment modality. Eur J Cancer 2012;48(14):2192–202. [DOI] [PubMed] [Google Scholar]
- 40.Chaichana KL, Kone L, Bettegowda C, Weingart JD, Olivi A, Lim M, et al. Risk of surgical site infection in 401 consecutive patients with glioblastoma with and without carmustine wafer implantation. Neurol Res 2015;37(8):717–26. [DOI] [PubMed] [Google Scholar]
- 41.Uzuka T, Takahashi H, Nakasu Y, Okuda T, Mitsuya K, Hayashi N, et al. Surgical Site Infection after Malignant Brain Tumor Resection: A Multicenter Study for Induction of a Basic Care Bundle. Neurol Med-Chir 2017;57(10):542–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Qin B, Tanaka R, Ariyama H, Shibata Y, Arita S, Kusaba H, et al. In-vitro differential metabolism and activity of 5-fluorouracil between short-term, high-dose and long-term, low-dose treatments in human squamous carcinoma cells. Anticancer Drugs 2006;17(4):439–43. [DOI] [PubMed] [Google Scholar]
- 43.Calabro-Jones PM, Byfield JE, Ward JF, Sharp TR. Time-dose relationships for 5-fluorouracil cytotoxicity against human epithelial cancer cells in vitro. Cancer Res 1982;42(11):4413–20. [PubMed] [Google Scholar]
- 44.Morshed RA, Gutova M, Juliano J, Barish ME, Hawkins-Daarud A, Oganesyan D, et al. Analysis of glioblastoma tumor coverage by oncolytic virus-loaded neural stem cells using MRI-based tracking and histological reconstruction. Cancer Gene Ther 2015;22(1):55–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bagci-Onder T, Wakimoto H, Anderegg M, Cameron C, Shah K. A dual PI3K/mTOR inhibitor, PI-103, cooperates with stem cell-delivered TRAIL in experimental glioma models. Cancer Res 2011;71(1):154–63. [DOI] [PubMed] [Google Scholar]
- 46.Hingtgen S, Ren X, Terwilliger E, Classon M, Weissleder R, Shah K. Targeting multiple pathways in gliomas with stem cell and viral delivered S-TRAIL and Temozolomide. Mol Cancer Ther 2008;7(11):3575–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Frank RT, Edmiston M, Kendall SE, Najbauer J, Cheung CW, Kassa T, et al. Neural Stem Cells as a Novel Platform for Tumor-Specific Delivery of Therapeutic Antibodies. Plos One 2009;4(12). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mooney R, Roma L, Zhao D, Van Haute D, Garcia E, Kim SU, et al. Neural stem cell-mediated intratumoral delivery of gold nanorods improves photothermal therapy. ACS Nano 2014;8(12):12450–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tobias AL, Thaci B, Auffinger B, Rincon E, Balyasnikova IV, Kim CK, et al. The Timing of Neural Stem Cell-Based Virotherapy Is Critical for Optimal Therapeutic Efficacy When Applied With Radiation and Chemotherapy for the Treatment of Glioblastoma. Stem Cell Transl Med 2013;2(9):655–66. [DOI] [PMC free article] [PubMed] [Google Scholar]