

Summary
Microtubules are essential to neuron shape and function. Acetylation of tubulin has the potential to directly tune the behaviour and function of microtubules in cells. Although proteomic studies have identified several acetylation sites in α-tubulin, the effects of acetylation at these sites remains largely unknown. This includes the highly conserved residue lysine 394 (K394), which is located at the αβ-tubulin dimer interface. Using a fly model, we show that α-tubulin K394 is acetylated in the nervous system and is an essential residue. We found that an acetylation-blocking mutation in endogenous α-tubulin, K394R, perturbs the synaptic morphogenesis of motoneurons and reduces microtubule stability. Intriguingly, the K394R mutation has opposite effects on the growth of two functionally and morphologically distinct motoneurons, revealing neuron-type-specific responses when microtubule stability is altered. Eliminating the deacetylase HDAC6 increases K394 acetylation, and the over-expression of HDAC6 reduces microtubule stability similar to the K394R mutant. Thus, our findings implicate α-tubulin K394 and its acetylation in the regulation of microtubule stability and suggest that HDAC6 regulates K394 acetylation during synaptic morphogenesis.
eTOC blurb
Neuronal microtubules are enriched in post-translational modifications, including acetylation. Saunders et al. reveal that a conserved α-tubulin lysine, K394, is acetylated in the fly nervous system, where it regulates neuronal morphogenesis by affecting microtubule stability. Loss- and gain-of-function studies indicate HDAC6 deacetylates K394.
Introduction
The microtubule cytoskeleton is integral to creating and maintaining neuronal morphology1–3. Microtubules are hollow filaments assembled from α- and β-tubulin heterodimers. The addition and removal of αβ-tubulin dimers from microtubule ends results in dynamic bouts of growth and shrinkage1,4. Microtubules that do not dramatically change in length at their tips are often referred to as stable. It is also possible for a microtubule to have a stable shaft, along which there is an infrequent exchange of dimers, but a dynamic end. Stable microtubules are typically long-lived, and some microtubule-associated proteins (MAPs), such as MAP1B, which is known as Futsch in Drosophila, promote microtubule stability by bundling microtubules together5. It is well-established that disrupting microtubules and microtubule-based activities alter neuronal structure and function.
In neurons and other cells, microtubule function is regulated by a combination of factors, including the intrinsic properties of tubulin, tubulin isotypes, proteins that interact with tubulin and microtubules, and post-translational modifications. Post-translational modifications, such as acetylation, have long been candidates to regulate microtubules in cells by affecting microtubule stability and dynamics, molecular-motor trafficking along microtubules, microtubule severing, and the binding of MAPs6. Although multiple sites of acetylation have been identified in tubulin, only a single site in α-tubulin, lysine 40 (K40), has been well characterised6. Acetylation of α-tubulin K40 frequently correlates with stable, long-lived microtubules in cells but does not itself regulate microtubule stability or dynamics. Rather, recent work has shown that K40 acetylation enables microtubules to withstand mechanical stresses, which may explain its correlation with long-lived microtubules in cells7,8. Although there is potential for microtubule function to be controlled by the acetylation of other sites in α-tubulin, this has not yet been explored.
Proteomic studies using tissue from mammals and flies have revealed that at least twelve conserved sites in α-tubulin in addition to K40 are acetylated in various organisms and tissues9–14. One site is consistently identified in all these studies: K394. In rats, α-tubulin K394 is acetylated in many different tissue types, ranging from brain, heart, kidney, lung, muscle, and skin, among others9. Additional studies have shown that K394 is acetylated in mouse brain and embryonic fibroblasts as well as several human cell lines11–15. Unlike K40, which is located in the microtubule lumen, K394 sits on the microtubule surface at the αβ-tubulin dimer interface, a region that likely regulates dimer stability and undergoes a conformational change as the dimer is added to the microtubule polymer (Figures 1A and 1B)4. Based on its position alone, it is possible that K394 could affect dimer stability and microtubule assembly, both of which could in turn affect microtubules and their function in cells. Indeed, studies carried out in HeLa cells showed that exogenously expressed K394R mutant α-tubulin, which cannot be acetylated, incorporates poorly into microtubules14. In CHO cells, K394 mutations were uncovered in a screen for α-tubulin mutations that would counteract the stabilising effects of taxol on microtubules, and these K394 mutations were postulated to make microtubules less stable16. Thus, K394 acetylation is a compelling candidate to regulate microtubules in cells.
Figure 1. Mutating conserved α-tubulin K394 to block acetylation disrupts synaptic morphogenesis.
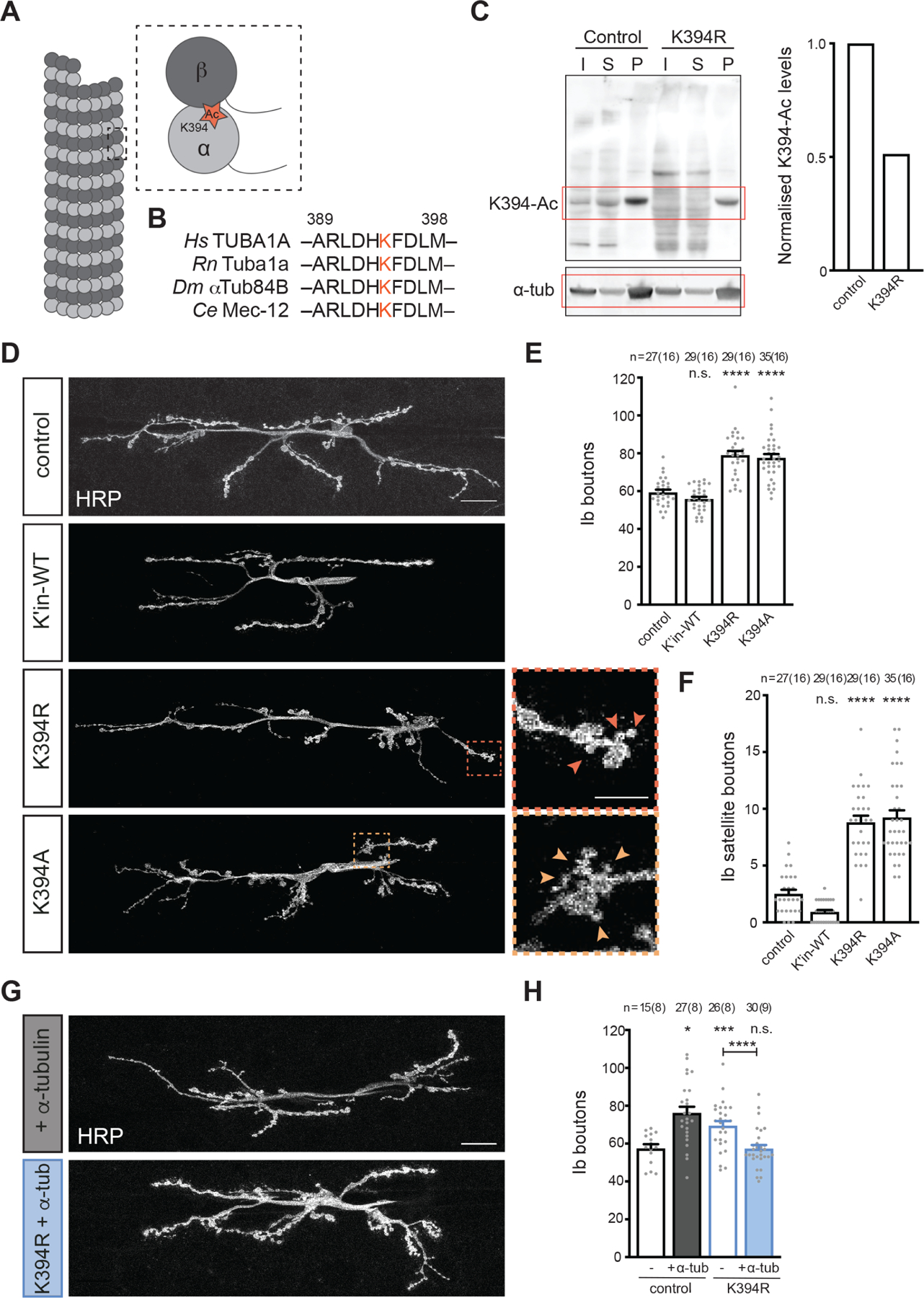
In this and subsequent figures, images are of synaptic terminals at m6/7 in 3rd instar larvae, and quantification is per m6/7, unless otherwise noted. All genotypes are homozygous for wild-type or mutant αTub84B alleles.
(A) Cartoon showing the position of K394 at the αβ-heterodimer interface.
(B) K394 (red) is conserved between humans, rats, flies and worms. This peptide sequence was used to create the anti-Ac-K394 antibody.
(C) Western blot analysis (left) and quantification (right) of K394 acetylation and total α-tubulin levels in control and K394R mutant fly head lysate. The anti-Ac-K394 signal in the tubulin pellet lane is normalised to total α-tubulin and represented as a fraction of the control values. α-tubulin migrates at ~50 kDa (red box). I=input, S=supernatant, P=pellet.
(D) Representative images of synaptic terminals in control and mutant animals. K394A mutants die as pupae, and K394R mutants are homozygous viable. Dashed-outline boxes: zoomed-in views to highlight ectopic satellite boutons (arrowheads). Scale bars: 25 μm and 10 μm (dashed-outline boxes). A neuronal membrane marker (HRP) illuminates the synaptic terminals.
(E and F) Quantification of total type Ib boutons (E) and satellite boutons (F) in control larvae and larvae in which wild-type (WT) or mutant alleles of tubulin were knocked into endogenous αTub84B. p-values: p=0.53, control v. wild-type αTub84B knock-in (K’in-WT); p<0.000001, control v. K394R; p<0.000001, control v. K394A (E). p-values: p=0.13, control v. K’in-WT; p<0.000001, control v. K394R; p<0.000001, control v. K394A (F). See also Figure S1 for quantification of bouton size in control and mutant animals.
(G and H) Neuronal over-expression of wild-type αTub84B (+α-tubulin) suppresses type Ib bouton overgrowth in K394R mutants. Representative images (G) and quantification (H). One copy each of OK6-Gal4 and UAS-αTub84B were used to over-express α-tubulin. p-values: p=0.023, control v. +α-tubulin; p=0.0001, control v. K394R; p=1.0, control v. K394R +α-tubulin; p=0.000002, K394R v. K394R +α-tubulin (H).
Quantification: One-way ANOVA with post-hoc Tukey. All data are mean ± SEM. n.s=non-significant; *p=0.01–0.05; ***p=0.001–0.0001; ****p<0.0001.
In this study, we asked whether α-tubulin K394 and its acetylation are important to neuronal morphogenesis using the fly neuromuscular junction (NMJ) as a model. We targeted both the K394 residue in endogenous α-tubulin and manipulated enzymes that might regulate K394 acetylation, including a putative K394 deacetylase, HDAC613. These complementary approaches are necessary to (a) determine the importance of K394 to tubulin and microtubule function and (b) interpret the effects of manipulating the modifying enzyme, particularly since most acetyltransferases and deacetylases, including HDAC6, have multiple protein targets17. Our studies of the fly NMJ show that K394 is an essential α-tubulin residue that is necessary for axon terminals to develop properly. Mutating K394 results in a dramatic reduction in microtubule stability as indicated by a decrease in microtubule loops, including those marked by Futsch, and an increase in sensitivity to the microtubule destabilising drug nocodazole. Our data indicate that this change in stability underlies the synaptic morphogenesis defects in the K394R mutants. Combined, our data implicate K394 and its acetylation in regulating microtubule stability during synaptic morphogenesis.
Results
Conserved α-tubulin K394 is acetylated in the nervous system and essential for normal synaptic morphogenesis
We selected the largely uncharacterised α-tubulin residue K394, whose acetylation is conserved across species9–14, as a candidate regulator of microtubule function in neurons. To investigate α-tubulin K394, we turned to Drosophila as an in vivo model. As in other organisms, there are multiple α-tubulin isotypes encoded in the fly genome. The four α-tubulin isotypes in Drosophila, which are named based on their cytological positions, are αTub84B, αTub84D, αTub85E and αTub67C. αTub84B is the essential, predominant, and ubiquitously expressed α-tubulin in flies18,19. αTub84D is almost identical to αTub84B, and it is also ubiquitously expressed; however, its expression is much lower, and αTub84D is not essential for survival20. αTub84B and αTub84D are the two principal α-tubulin proteins in the fly nervous system, with αTub84B predominating19. αTub84B is 97% identical to rat α-tubulin, and there are only five non-conservative amino acid differences, the majority of which are in the C-terminal tail. Conservation of the α-tubulin sequence and acetylation, combined with a powerful genetic toolbox for in vivo analysis, makes Drosophila an attractive model system for these studies.
We first set out to determine whether we could detect the acetylation of α-tubulin K394 in the fly nervous system. We were able to successfully raise a peptide antibody against acetylated K394. We pelleted microtubules from fly brain lysate and used the anti-acetylated-K394 antibody to probe a western blot (Figures 1B and 1C). The antibody recognised a prominent band that migrates at the expected size of α-tubulin in the lysate input, supernatant, and microtubule pellet. Next, to determine the specificity of the antibody for the acetylated residue, we probed lysate from the brains of flies expressing α-tubulin with a mutation, K394R, that blocks acetylation (the K394R mutation was knocked into the endogenous αTub84B gene, and lysate was obtained from flies homozygous for this mutation, see below). The antibody signal for acetylated K394 was reduced by at least half in the K394R mutant, indicating that the antibody recognises the acetylated residue (Figure 1C). The remaining signal may reflect antibody recognition of other α-tubulin isotypes acetylated at K394, and/or the antibody may retain some affinity for the K394R mutant α-tubulin. Consistent with published proteomic data showing that K394 is acetylated in the nervous system of rats and mice9,13, these western blot results indicate that α-tubulin K394 is acetylated in the Drosophila nervous system.
Mutagenesis is a powerful approach to analyse microtubule function in cells. Indeed, the function of α-tubulin K40 acetylation was primarily characterised using K40 mutations before the relevant modifying enzymes were identified decades later21–24. Thus, we leveraged K394 mutations to gain insight into the role that K394 plays in animals and neurons. To this end, we took advantage of a fly strain that we previously generated that enables us to readily create new alleles of endogenous α-tubulin20. We introduced two different point mutations into the endogenous αTub84B gene to block acetylation at K394: K394A and K394R. While the homozygous K394A mutation caused lethality during pupal stages of development, flies homozygous for the K394R mutation were viable. Alanine differs substantially from lysine in structure and charge, and the effects of a K-to-A mutation may not be due entirely to blocking acetylation. Unlike alanine, arginine is similar to lysine in structure and charge albeit cannot be acetylated; therefore, K-to-R mutations are frequently used to mimic the loss of acetylation. In contrast to K394, mutating K40 to alanine or arginine has no effect on survival20,23,25,26. The difference in survival between the K394A and K394R mutations suggests that arginine is likely a sufficient substitute for lysine in terms of structure and charge at this position and, thus, K394R is an advantageous mutation for characterising the loss of acetylation at this site.
We then asked whether α-tubulin K394 is important to neuronal morphogenesis. Disrupting microtubule function is frequently associated with defects in neuronal morphology, including the formation of boutons at the fly NMJ1,2. Boutons are swellings of the axon terminal that house synapses. Microtubules are present throughout the axon terminal and its boutons. A core of bundled microtubules extends along the length of the terminal, occasionally forming loop-like structures that correlate with stable microtubules27–30. Growing microtubules have been observed in both the main axon terminal branches and boutons, in which dynamic “pioneer” microtubules may promote bouton growth31. We used the type Ib (“big”) boutons as a model since they are a well-established system to investigate the role of the cytoskeleton in synaptic growth. We found that both the K394A and K394R mutations resulted in an increase of type Ib boutons and the formation of ectopic “satellite” boutons (Figures 1D–1F). While the number of type Ib boutons increased, the size of the boutons was equivalent between the controls and K394R mutants (Figure S1). Next, we tested whether we could revert the K394R mutant phenotypes by supplying wild-type α-tubulin in neurons. The neuronal over-expression of wild-type α-tubulin led to an increase in type Ib boutons in control animals but suppressed the synaptic overgrowth phenotype in K394R mutants (Figures 1G and 1H). This suggests that the K394R bouton overgrowth phenotype results from a disruption of the presynaptic (neuronal) microtubule cytoskeleton that can be rescued by wild-type α-tubulin. Altogether, our results indicate that α-tubulin K394, which is acetylated in the nervous system, plays a critical role in synaptic morphogenesis.
Stable microtubules are decreased at synaptic terminals in the K394R mutant
The morphology phenotype in the K394R mutant animals likely results from a perturbation in tubulin and/or microtubules (Figure 2A). First, we analysed α-tubulin levels in the K394R mutant. Our western blot analysis of larval brain lysate showed that α-tubulin levels were normal in K394R homozygous mutants (Figure 2B), suggesting that a change in the amount of α-tubulin was unlikely to cause the morphology defect. Next, we tested whether the K394R mutation might perturb synaptic morphogenesis by affecting microtubules. Using an antibody that recognises α-tubulin, we found that microtubules were still present in K394R homozygous mutant synaptic terminals, but that the number of microtubule loops, which are indicative of stable microtubules27–30, were reduced (Figure 2D). This suggests that microtubules still form in the K393R mutant but that the mutation may alter microtubule stability as indicated by the reduction in microtubule loops.
Figure 2. The acetylation-blocking K394R mutation does not affect tubulin levels or microtubule formation but decreases stable microtubules.
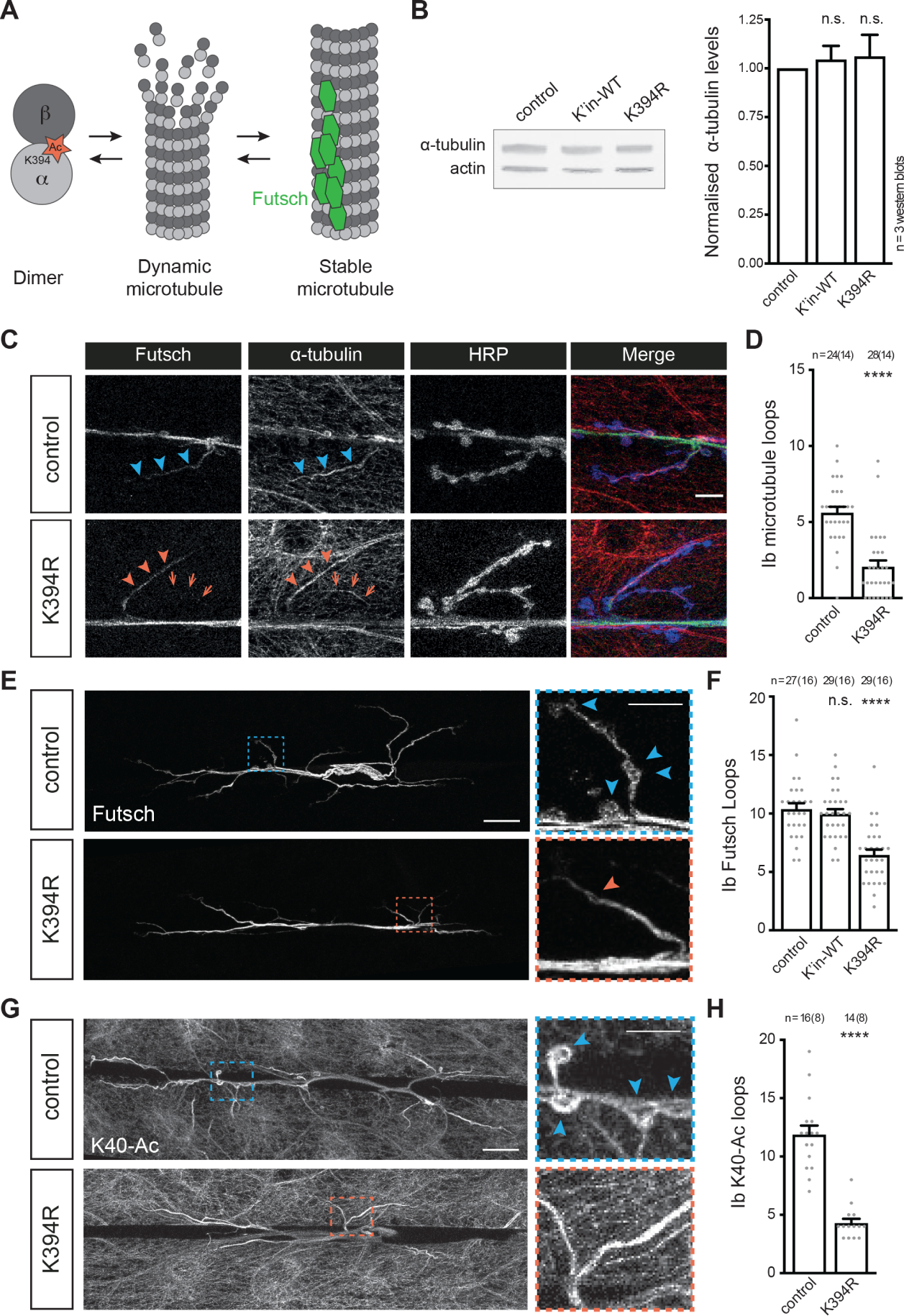
All genotypes are homozygous for wild-type or mutant αTub84B alleles.
(A) Schematic illustrating free tubulin dimer and two microtubule populations, dynamic and stable. Stable microtubules are bound by Futsch (green hexagons).
(B) Western blot analysis (top) and quantification (bottom) of α-tubulin levels (DM1α) in control, WT αTub84B knock-in, and K394R mutant lysate. DM1α signal is normalised to actin and represented as a fraction of the control values. p-values: p=0.98, control v. K’in-WT; p=0.95, control v. K394R.
(C and D) Representative images (C) of control and K394R synaptic terminals stained for α-tubulin (DM1α), Futsch, and a neuronal membrane marker (HRP), and the quantification (D) of microtubule loops in type Ib boutons. Arrowheads: Futsch labelled microtubule bundle; arrows: a microtubule branch lacking Futsch in the K394R mutant (C). p-value: p<0.000001, control v. K394R (D). (E and F) Representative images (E) of synaptic terminals stained for Futsch, which marks stable microtubules, and the quantification (F) of Futsch-positive microtubule loops (“Futsch loops”) in type Ib boutons in indicated genotypes. Dashed-outline boxes: zoomed-in views to highlight Futsch loops (arrowheads), which are decreased in K394R mutants. Scale bars: 25 μm and 10 μm (dashed-outline boxes). p-values: p=0.91, control v. K’in-WT; p<0.000001, control v. K394R (F). See also Figure S2.
(G and H) Representative images (G) of control and K394R synaptic terminals stained for α-tubulin acetylated at K40 (6–11B-1), and the quantification (H) of acetylated-K40-positive microtubule loops (arrowheads) in type Ib boutons. Dashed-outline boxes: zoomed-in views to highlight microtubule loops (arrowheads). Scale bars: 25 μm and 10 μm (dashed-outline boxes). p-value: p<0.00001, control v. K394R (H).
Scale bar: 10 μm.
Quantification: One-way ANOVA with post-hoc Tukey (B and F) and Student’s unpaired t-test (D and H). All data are mean ± SEM. n.s=non-significant; ****p<0.0001.
We further assessed microtubule stability in control and K394R homozygous mutants by staining for the microtubule-associated protein Futsch, which is homologous to the mammalian microtubule-associated protein (MAP) MAP1B3,27,32. Futsch colocalises with stable microtubules and the distinctive loops that they form within boutons5,33. The K394R mutation resulted in fewer Futsch-positive loops (Figures 2E and 2F), which indicates a decrease in stable microtubules. This decrease in Futsch loops was rescued by the expression of wild-type α-tubulin in neurons, but not in muscles, indicating that the effects of the K394R mutation on microtubules was cell autonomous (Figure S2). We also assessed α-tubulin K40 acetylation, which, as previously mentioned, correlates with stable microtubules in cells6. While anti-acetylated-K40 signal was still present in the K394R homozygous mutants, the number of acetylated-K40 loops was reduced (Figures 2G and 2H). This mirrors the finding that Futsch loops are reduced and is consistent with the idea that the K394R mutation causes a decrease in microtubule stability. Altogether, these results indicate that the K394R mutation does not affect tubulin levels nor drastically decreases microtubule formation; rather, the α-tubulin K394R mutation reduces microtubule loops that are indicative of stable microtubules.
The K394R mutant displays enhanced sensitivity to nocodazole and a reduction of futsch
The reduction of stable microtubules, as indicated by fewer microtubule loops, including Futsch-positive loops, in the K394R mutant suggests that the mutation may affect microtubule stability. To test this possibility, we used two complementary pharmacological and genetic approaches. First, we treated control and K394R homozygous mutant larvae with the microtubule destabilising agent nocodazole. We used a low concentration of nocodazole that did not affect the numbers of Futsch loops or boutons in control larvae (Figures 3A and 3B). In contrast, this nocodazole treatment dramatically reduced the number of Futsch loops in the K394R homozygous mutants (Figures 3A and 3B). This suggests that the K394R mutation makes microtubules more sensitive to microtubule destabilisation. Next, we took advantage of a hypomorphic allele of futsch (futschN94) to test for a genetic interaction with the K394R mutation. As a heterozygote, futschN94 had no effect on the numbers of Futsch loops or boutons (Figures 3C and 3D). Similarly, one copy of the K394R mutation did not affect either the number of Futsch loops or bouton formation (Figures 3C and 3D). However, Futsch loops were reduced in animals heterozygous for both futschN94 and the K394R mutation (e.g., futschN94/+; K394R/+) (Figures 3C and 3D). This indicates a genetic interaction between futsch and the K394R mutation. These findings that the K394R mutation enhances sensitivity to both nocodazole and a reduction in futsch are consistent with the idea that microtubule stability is reduced in the K394R mutant.
Figure 3. The K394R mutation enhances sensitivity to nocodazole and decreased futsch.
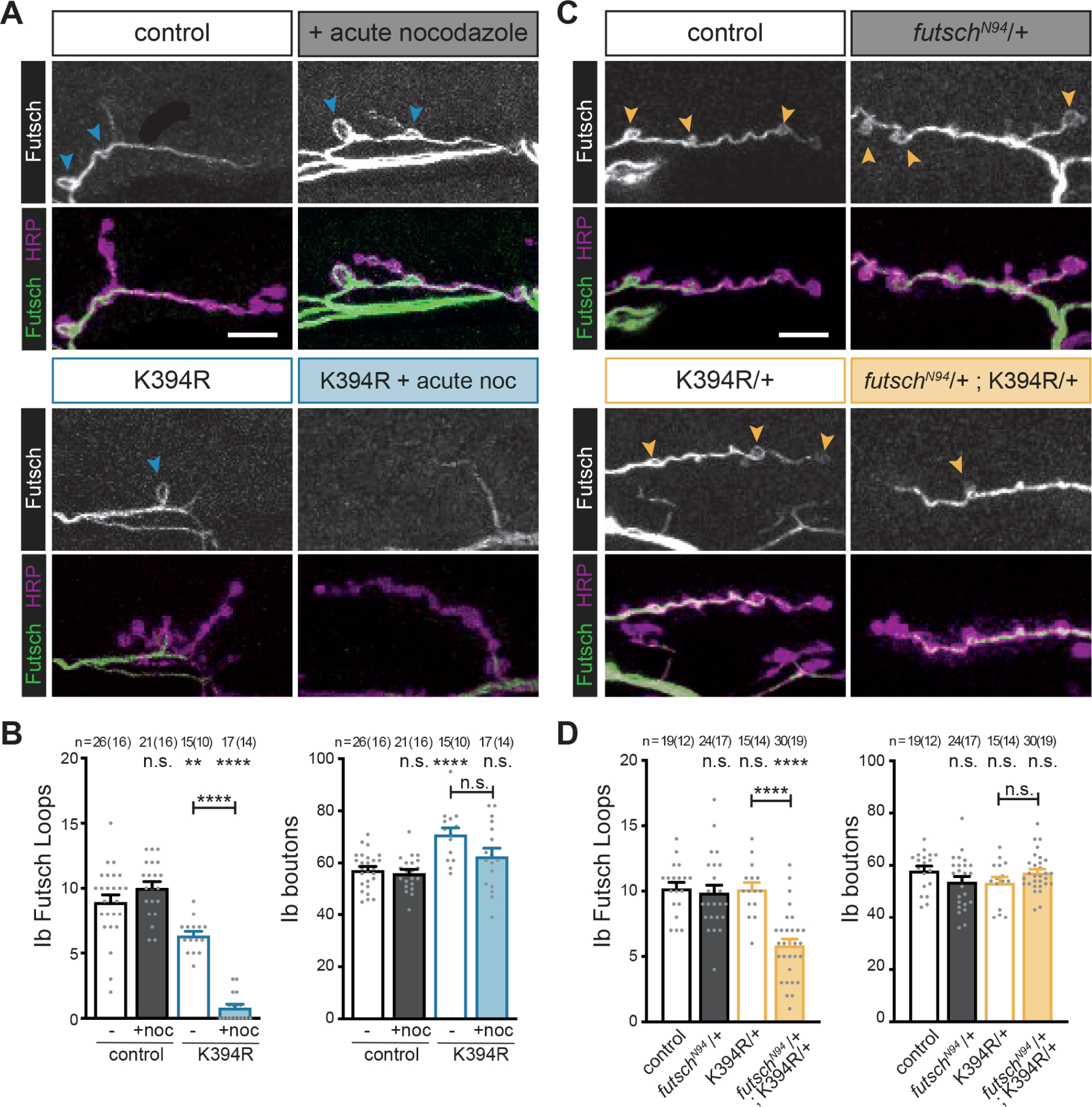
Larval fillets stained for stable microtubules (Futsch) and a neuronal membrane marker (HRP). Genotypes are homozygous for wild-type or mutant αTub84B alleles except where indicated. Arrowheads: Futsch loops in type Ib boutons. Futsch loops and type Ib boutons were quantified for genotype and treatment as indicated. Scale bar: 10 μm.
(A and B) Representative images (A) and quantification (B) of control and K394R mutant larvae treated with 30 μM nocodazole for 1 hour prior to fixation. p-values, Ib Futsch loops: p=0.30, control v. +nocodazole; p=0.0013, control v. K394R; p<0.000001, control v. K394R +nocodazole; p<0.000001, K394R v. K394R +nocodazole (B, left). p-values, Ib boutons: p=0.97, control v. +nocodazole; p<0.000001, control v. K394R; p=0.24, control v. K394R +nocodazole; p=0.052, K394R v. K394R +nocodazole (B, right).
(C and D) Representative images (C) and quantification (D) of control, futschN94/+, K394R/+, and futschN94/+ K394R/+ double mutant. futschN94 is a hypomorphic futsch allele. p-values, Ib Futsch loops: p=0.97, control v. futschN94/+; p=1.0, control v. K394R/+; p<0.000001, control v. futschN94/+; K394R/+; p=0.000003, K394R/+ v. futschN94/+ K394R/+ double mutant (D, left). p-values, Ib boutons: p=0.35, control v. futschN94/+; p=0.39, control v. K394R/+; p=0.99, control v. futschN94/+ K394R/+ double mutant; p=0.51, K394R/+ v. futschN94/+ K394R/+ double mutant (D, right).
Quantification: One-way ANOVA with post-hoc Tukey. All data are mean ± SEM. n.s= non-significant; **p=0.01–0.001; ****p<0.0001.
Taxol treatment and the over-expression of Futsch revert the K394R mutant phenotypes
Conditions that reduce microtubule stability, such as futsch loss-of-function mutants, are typically associated with a decrease in bouton numbers2,27,32. Our finding that the α-tubulin K394R mutation reduces the number of Futsch loops but increases bouton number raises the question of whether the decrease in stable microtubules in the mutant is causally related to the type Ib synaptic overgrowth phenotype. Again, we used a combination of pharmacological and genetic approaches to test whether enhancing microtubule stability would suppress the formation of additional type Ib boutons in the K394R mutant. First, we treated animals with the microtubule-stabilising drug taxol (also known as paclitaxel). An acute 30-minute treatment of larval fillets with taxol modestly increased the number of Futsch loops in control animals and was sufficient to rescue Futsch loop numbers to normal in the K394R homozygous mutant larvae (Figures 4A and 4B). This acute taxol treatment, however, did not suppress the formation of extra boutons in the K394R homozygous mutant. Since synaptic boutons develop over several days, it is possible that this 30-minute treatment was too short for taxol to exert an effect on synaptic morphogenesis. We next treated larvae by culturing them for 24 hours on taxol-containing food. The prolonged 24-hour treatment with taxol rescued microtubule stability and also fully suppressed the synaptic overgrowth phenotype in the K394R homozygous mutants, reducing the number of boutons in the K394R mutant to normal (Figures 4C and 4D). These taxol experiments indicate that a reduction in microtubule stability likely underlies the overgrowth of type Ib synaptic boutons in the K394R mutant.
Figure 4. K394R mutant phenotypes are reverted by promoting microtubule stability.
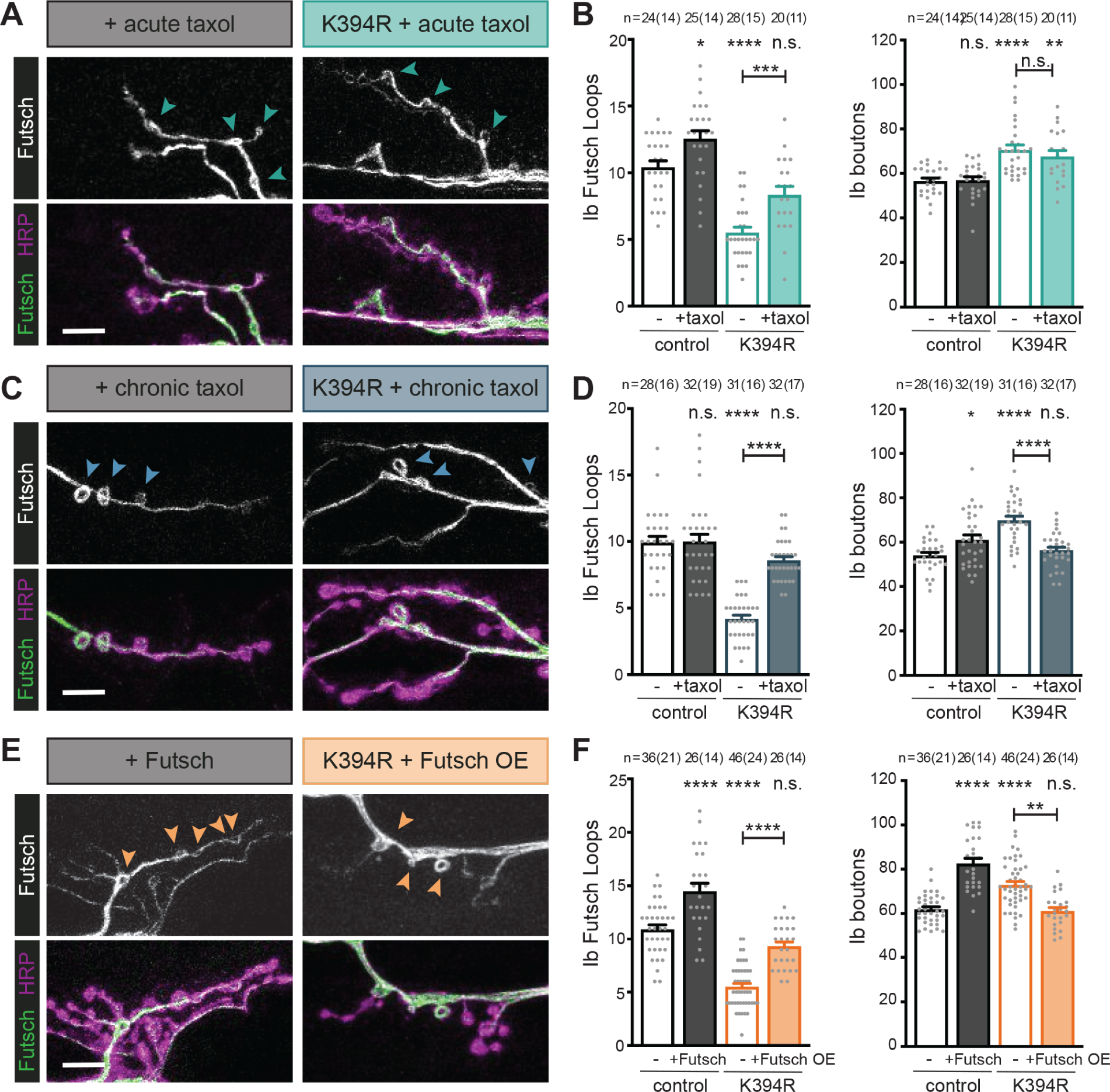
All genotypes are homozygous for wild-type or mutant αTub84B alleles. Larval fillets stained for stable microtubules (Futsch) and a neuronal membrane marker (HRP). Arrowheads: Futsch loops in type Ib boutons. Futsch loops and type Ib boutons were quantified for each experiment in the indicated genotype and treatment. Scale bar: 10 μm.
(A and B) Representative images (A) and quantification (B) of dissected control and K394R mutant larvae treated with 50 μM taxol for 30 minutes prior to fixation. p-values, Ib Futsch loops: p=0.023, control v. +taxol; p<0.000001, control v. K394R; p=0.056, control v. K394R +taxol; p=0.0018, K394R v. K394R +taxol (B, left). p-values, Ib boutons: p=0.99, control v. +taxol; p=0.000007, control v. K394R; p=0.0016, control v. K394R +taxol; p=0.72, K394R v. K394R +taxol (B, right).
(C and D) Representative images (C) and quantification (D) of control and K394R mutant larvae raised on food containing 10 μM taxol for 24 hours prior to dissection. p-values, Ib Futsch loops: p=1.0, control v. +taxol; p<0.000001, control v. K394R; p=0.12, control v. K394R +taxol; p<0.000001, K394R v. K394R +taxol (D, left). p-values, Ib boutons: p=0.027, control v. +taxol; p<0.000001, control v. K394R; p=0.77, control v. K394R +taxol; p=0.000001, K394R v. K394R +taxol (D, right).
(E and F) The neuronal over-expression of Futsch affects type Ib bouton growth in control and K394R mutants. One copy each of OK6-Gal4 and UAS-Futsch were used to over-express Futsch. Representative images (E) and quantification (F). p-values, Ib Futsch loops: p=0.000005, control v. +Futsch; p<0.000001, control v. K394R; p=0.12, control v. K394R +Futsch; p<0.000001, K394R v. K394R +Futsch (F, left). p-values, Ib boutons: p<0.000001, control v. +Futsch; p=0.000037, control v. K394R; p=0.99, control v. K394R +Futsch; p=0.0018, K394R v. K394R +Futsch (F, right).
Quantification: One-way ANOVA with post-hoc Tukey. All data are mean ± SEM. n.s= non-significant; *p=0.01–0.05; **p=0.01–0.001, ***p=0.001–0.0001; ****p<0.0001.
We took an additional genetic approach to test the model that the α-tubulin K394R mutation destabilises microtubules to alter synaptic morphogenesis. We over-expressed the microtubule-stabilising MAP Futsch in control and K394R homozygous mutant larvae. In control animals, elevating Futsch levels increased both the number of Futsch loops and type Ib boutons (Figures 4E and 4F), consistent with previous reports that increasing Futsch levels and microtubule stability stimulates type Ib bouton formation27,34,35. The over-expression of Futsch in K394R homozygous mutant neurons increased the number of Futsch loops to normal and also suppressed the formation of extra boutons (Figures 4E and 4F). Thus, elevating Futsch levels reverts the K394R mutant phenotypes. Combined with the results of the taxol experiments, these results indicate that the overgrowth of type Ib boutons in the K394R mutant is likely due to a decrease in microtubule stability.
Decrease in stable microtubules at axon terminals in K394R mutants is rescued by elevating tubulin cofactor levels
We next sought to gain insight into how the K394R mutation might affect microtubules. Previous studies have implicated K394 in microtubule polymerisation and have shown that K394R mutant α-tubulin is not incorporated into microtubules as efficiently as wild-type α-tubulin14,36,37. Based on these findings, we reasoned that the K394R mutation might disrupt microtubule polymerisation. To test this model, we analysed microtubule dynamics using EB1::GFP, which specifically binds to growing microtubule ends. To eliminate any effects on microtubule growth that might be caused by over-expressing EB1, we tagged endogenous EB1 with the split-GFP peptide sfGFP(11), which can be selectively visualised in neurons that express the complementary peptide sfGFP(1–10) (Figure 5A)38,39. Next, since it is difficult to quantitatively analyse microtubule dynamics at the larval NMJ, we turned to sensory neuron dendrites to analyse microtubule polymerisation. If the K394R mutation perturbs microtubule polymerisation, we predicted that EB1::GFP comet number and/or velocity would be reduced. Indeed, we observed significantly fewer EB1::GFP comets in the dendrites of K394R homozygous mutant sensory neurons, although there was no significant decrease in comet velocity (Figures 5B–5E). The reduction in EB1::GFP comet number in the K394R mutant neurons, combined with published reports14,36,37, is consistent with the idea that the K394R mutation may alter polymerisation. However, EB1::GFP comet velocity, which reflects the rate of microtubule polymerisation, was normal, raising the possibility that K394R has another (or different) effect on microtubules. A reduction in EB1::GFP comets might also indicate a reduction in microtubule number, an increase in microtubules that do not grow, and/or a decrease in microtubule nucleation40–42. Additional studies, ideally of microtubules reconstituted in vitro from wild-type and K394R mutant α-tubulin, will be needed to resolve the precise effect of the K394R mutation on microtubules.
Figure 5. Elevating tubulin chaperone levels reverts K394R mutant phenotypes.
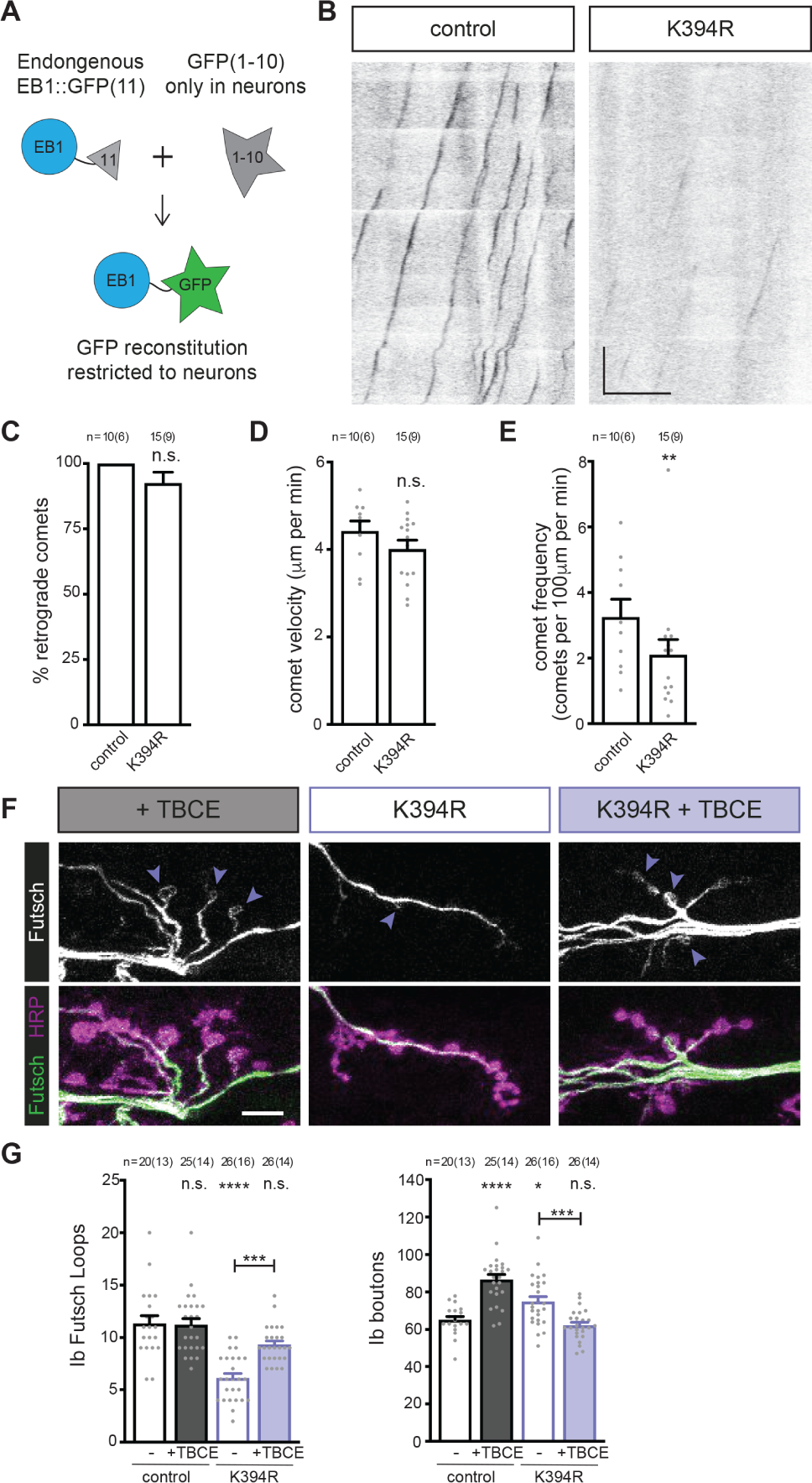
All genotypes are homozygous for wild-type or mutant αTub84B alleles.
(A) Cartoon showing the approach to visualise EB1::sfGFP specifically in neurons. Endogenous EB1 was tagged with the split-GFP peptide sfGFP(11), and pickpocket-Gal4 drove the expression of sfGFP(1–10) in sensory neurons to limit the reconstitution of sfGFP to neurons. (B) Representative kymographs of endogenous EB1 dynamics in control and K394R sensory neuron dendrites. Cell body is to the left. One copy each of EB1::sfGFP(11), pickpocket-Gal4, and UAS-sfGFP(1–10) were used. Scale bars: 10 μm (x-axis) and 30 seconds (y-axis). (C-E) Quantification of EB1::GFP comet trajectory (C), velocity (D), and frequency (E) in control and K394R mutants. p-values: p=0.16, control v. K394R (C); p=0.19, control v. K394R (D); p=0.0083, control v. K394R (E).
(F and G) Representative (F) images and quantification (G) of Futsch loops and type Ib boutons in controls and animals over-expressing TBCE, K394R mutants, and K394R mutants over-expressing TBCE. One copy each of OK6-Gal4 and UAS-TBCE were used to over-express TBCE. Larval fillets are stained for stable microtubules (Futsch) and a neuronal membrane marker (HRP). Arrowheads: Futsch loops. Scale bar: 10 μm. p-values, Ib Futsch loops: p=1.0, control v. +TBCE; p<0.000001, control v. K394R; p=0.057, control v. K394R +TBCE; p=0.00013, K394R v. K394R +TBCE (G, left). p-values, Ib boutons: p<0.000001, control v. +TBCE; p=0.024, control v. K394R; p=0.89, control v. K394R +TBCE; p=0.00045, K394R v. K394R +TBCE (G, right).
Quantification: Student’s unpaired t-test (C-E) and one-way ANOVA with post-hoc Tukey (G). All data are mean ± SEM. n.s=non-significant; *p=0.01–0.05, **p=0.01–0.001, ***p=0.001–0.0001, ****p<0.0001.
Microtubules grow as tubulin dimers are added to microtubule ends. The K394 residue is at the interface of the αβ-tubulin heterodimer. Based on its position, we theorised that K394 may contribute to the stability and/or conformation of the tubulin dimer, which may in turn affect microtubule behaviour or function. Tubulin dimer formation and homeostasis are regulated by a suite of tubulin cofactors, also known as chaperones, and α-tubulin is bound by the tubulin-specific chaperone E (TBCE)43. If the K394R mutation perturbs dimer assembly or reduces its stability, this may create a deleterious imbalance in the ratio of dimer to cofactor, e.g., the number of (unstable) dimers may increase relative to TBCE. TBCE and its partner tubulin cofactors likely exist at tightly controlled concentrations at synaptic terminals given that increasing or decreasing cofactor levels produces synaptic defects44,45. We tested the idea that the K394R mutation perturbs tubulin heterodimer formation or homeostasis by asking whether elevating TBCE expression would reverse the K394R mutant phenotypes. First, we found that higher TBCE levels resulted in significantly more boutons than controls41, albeit the number of Futsch loops was unaffected (Figures 5F and 5G). Increasing TBCE levels in the K394R homozygous mutant suppressed the formation of extra boutons and, significantly, also restored the formation of Futsch loops to wild-type levels (Figures 5F and 5G). Thus, the effects of the K394R mutation on microtubule stability can be reversed by increasing the amount of TBCE. While this experiment does not directly test the effects of the K394R mutation on tubulin dimer stability, these results are consistent with the notion that the K394R mutation may destabilise or otherwise alter the tubulin dimer.
α-tubulin K394 acetylation is regulated by HDAC6
We next wanted to identify the enzyme(s) that regulate the acetylation of K394. The enzyme α-tubulin acetyltransferase (αTAT, known as dαTAT in flies) has been shown to acetylate α-tubulin at K4023,24. However, the over-expression of dαTAT did not affect K394 acetylation (Figure S3), indicating that K394 is likely acetylated by another, unknown acetyltransferase. With regards to deacetylation, a previous study using mass spectrometry revealed that the deacetylase HDAC6 targets several tubulin residues, including both α-tubulin K40 and K39413. To determine whether HDAC6 indeed regulates the acetylation of α-tubulin at K394, we used our anti-Ac-K394 antibody to probe microtubules pelleted from the lysate of control and HDAC6-knock-out fly brains. If HDAC6 is responsible for deacetylating K394, K394 acetylation should increase when HDAC6 is knocked-out. When we pelleted microtubules from HDAC6 knock-out brain lysate, we indeed saw a dramatic increase in K394 acetylation (Figures 6A). We then over-expressed HDAC6 in neurons, which resulted in a decrease in K394 acetylation (Figure 6A). These data indicate that HDAC6 promotes the deacetylation of K394, either directly or indirectly.
Figure 6. α-tubulin K394 acetylation is regulated by HDAC6.
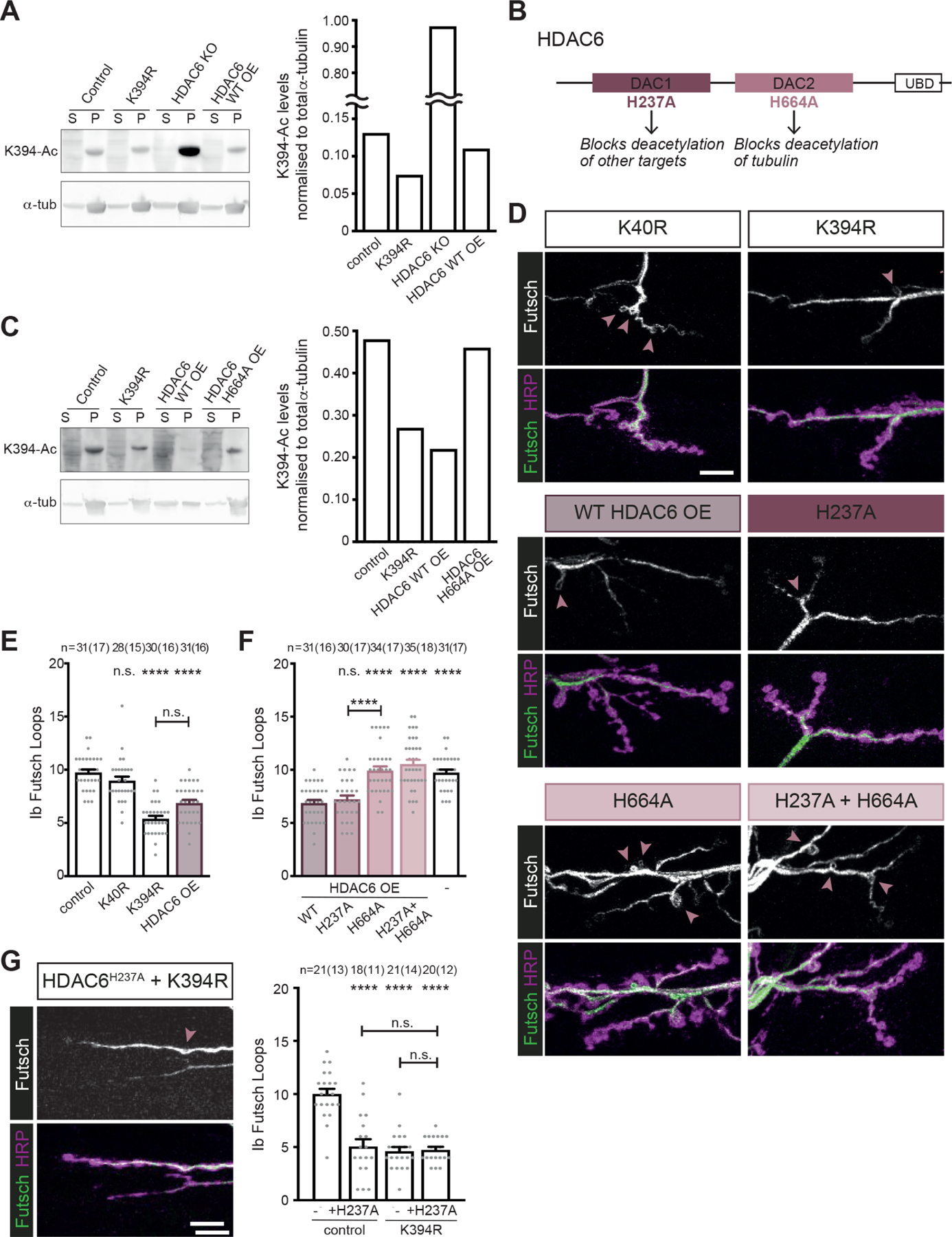
All genotypes are homozygous for wild-type or mutant αTub84B alleles and an HDAC6 knockout (KO) allele. elav-Gal4 (A and C) or OK6-Gal4 (D-G) were used to drive one copy of UAS-HDAC6 (wild-type or mutant as indicated). Larval fillets are stained for stable microtubules (Futsch) and a neuronal membrane marker (HRP) (D and G).
(A) Western blot analysis (left) and quantification (right) of K394 acetylation of microtubules pelleted from control, K394R, and HDAC6 KO, and HDAC6 over-expression (OE) fly heads. The anti-Ac-K394 signal in the tubulin pellet lane is normalised to total α-tubulin. S=supernatant, P=pellet. See also Figure S3 for analysis of K394 acetylation in control, K394R and dαTAT over-expression fly heads.
(B) Cartoon illustrating HDAC6 and its key domains, including two deacetylase domains (DAC1 and DAC2) and a ubiquitin-binding domain (UBD). The functional outcomes of point mutations in DAC1 and DAC2 are indicated.
(C) Western blot analysis (left) and quantification (right) of K394 acetylation of microtubules pelleted from control and K394R fly heads and the heads of flies over-expressing wild-type HDAC6 (WT OE) or mutant HDAC6 (H664A OE). The anti-Ac-K394 signal in the tubulin pellet lane is normalised to total α-tubulin. S=supernatant, P=pellet.
(D-F) The activity of DAC2 is necessary to reduce microtubule stability similar to the K394R mutants. Representative images (D) and quantification (E and F) of Futsch loops and type Ib boutons in control, K40R and K394R mutants, and animals over-expressing wild-type HDAC6 (HDAC6 OE) or mutant HDAC6 (H237A and/or H664A). All genotypes were included in the experiment, whose quantification is split between panels E and F. The quantification of the control and animals over-expressing wild-type HDAC6 are included in both E and F. p-values: p=0.74, control v. K40R; p<0.000001, control v. K394R; p<0.000001, control v. HDAC6 WT OE; p=0.062, K394R v. HDAC6 WT OE (E). p-values: p=0.99, HDAC6 WT OE v. HDAC6 H237A OE; p<0.000001, HDAC6 WT OE v. HDAC6 H664A OE; p<0.000001, HDAC6 WT OE v. HDAC6 H237A H664A OE; p<0.000001, HDAC6 WT OE v. control; p=0.000003, HDAC6 H237A OE v. HDAC6 H664A OE (F).
(G) Representative image (left) and quantification (right) of HDAC6 H237A overexpression in the K394R mutant. p-values: p<0.000001, control v. +HDAC6 H237A OE; p<0.000001, control v. K394R; p<0.000001, control v. K394R +HDAC6 H237A OE; p=0.997, K394R v. K394R +HDAC6 H237A OE; p=0.971, +HDAC6 H237A OE v. K394R +HDAC6 H237A OE.
Arrowheads: Futsch loops. Scale bar: 10 μm.
Quantification: One-way ANOVA with post-hoc Tukey. All data are mean ± SEM. n.s=non-significant; *p=0.01–0.05; ***p<0.0001.
HDAC6 is a multi-functional enzyme that has a role in diverse cellular activities: it targets multiple sites in α- and β-tubulin and sites in other proteins for deacetylation, and HDAC6 also functions via its ubiquitin-binding domain46. HDAC6 has two deacetylase domains (DAC1 and DAC2), only one of which, the second domain (DAC2), acts on tubulin (Figures 6B)47–49. To determine whether DAC2 plays a role in K394 deacetylation, we took advantage of a known mutation in DAC2 (H664A) that abrogates its deacetylation activity. While over-expression of wild-type HDAC6 reduces K394 acetylation, over-expression of the HDAC6 H664A mutant does not (Figure 6C). This indicates that HDAC6 relies on its DAC2 domain to deacetylate K394.
Next, we leveraged HDAC6 to ask whether K394 acetylation is important to synaptic microtubule stability. Since K394R is an acetylation-blocking mutation, we decided to over-express HDAC6, which should reduce acetylation, and compare the two. When we over-expressed wild-type HDAC6, we found that it reduced the number of Futsch loops similar to the acetylation-deficient K394R homozygous mutant (Figures 6D and 6E). We focused on the effects of HDAC6 on microtubule stability as HDAC6, acting via its DAC1 domain, has been implicated in disrupting synaptic morphogenesis by acetylating Bruchpilot, a scaffolding component of the active zone50. We over-expressed mutant HDAC6 enzymes that were defective in deacetylating non-tubulin targets (H237A in DAC1), tubulin (H664A in DAC2), or both (H237A, H664A double mutant). Over-expression of the mutant HDAC6 defective in deacetylating non-tubulin targets (H237A) reduced stable microtubules similar to the over-expression of wild-type HDAC6 (Figures 6E and 6F). In contrast, the over-expression of HDAC6 mutants that could not deacetylate tubulin (H664A and the H237A, H664 double mutant) had no effect on stable microtubules (Figures 6E and 6F). Thus, HDAC6 affects microtubule stability in type Ib boutons via the domain that deacetylates tubulin (DAC2).
We next asked whether HDAC6 might disrupt Futsch loops via an effect on K394 acetylation. If HDAC6 is exerting its effects via deacetylation of K394, then the effects of HDAC6 over-expression in the K394R homozygous mutant should be the same as the K394R homozygous mutant alone. In this experiment we took advantage of the HDAC6 H237A mutation, which prevents the deacetylation of non-tubulin targets, but leaves intact the DAC2 domain that deacetylates tubulin. Over-expression of HDAC6 H237A in the K394R homozygous mutant reduces Futsch loops similar to the K394R mutant alone (Figure 6G). This is consistent with the idea that HDAC6 exerts its effects on Futsch loops through the activity of its DAC2 domain, which deacetylates α-tubulin K394. In addition to K394, HDAC6 also deacetylates α-tubulin K40. Notably, mutating α-tubulin K40, another HDAC6 target, did not affect the number of Futsch loops, indicating that HDAC6 was unlikely to exert its effects on microtubule stability in type Ib boutons by modulating K40 acetylation (Figures 6D and 6E). Combined, our results suggest that acetylation of α-tubulin K394 is likely regulated by HDAC6, and deacetylation of K394 by HDAC6 is important to microtubule stability during the morphogenesis of type Ib synaptic terminals.
The K394R mutation and alterations in microtubule stability have neuron-type-specific effects on synaptic morphogenesis
We were struck by the observation that type Ib synaptic growth increased when microtubule stability was either reduced, as in the K394R mutation, or enhanced, as by the over-expression of Futsch in control animals. Type Ib synaptic terminals are known to be both physiologically and morphologically plastic51. For example, in response to enhanced activity, type Ib neurons generate additional boutons, the growth of which depends on the microtubule cytoskeleton52,53. We considered the possibility that the growth of the type Ib synaptic terminals in response to any alteration in microtubule stability may be characteristic of this particular neuron type. Another motoneuron type that innervates the same m6/7 muscle pair, type Is (“small”), is functionally and morphologically distinct from type Ib, and, unlike the type Ib neurons, type Is neurons do not form ectopic boutons in response to changes in activity (Figure 7A)51. Given these differences, we asked whether the type Is neurons would respond differently than type Ib to changes in microtubule stability.
Figure 7. Manipulation of microtubule stability has neuron-type-specific effects.
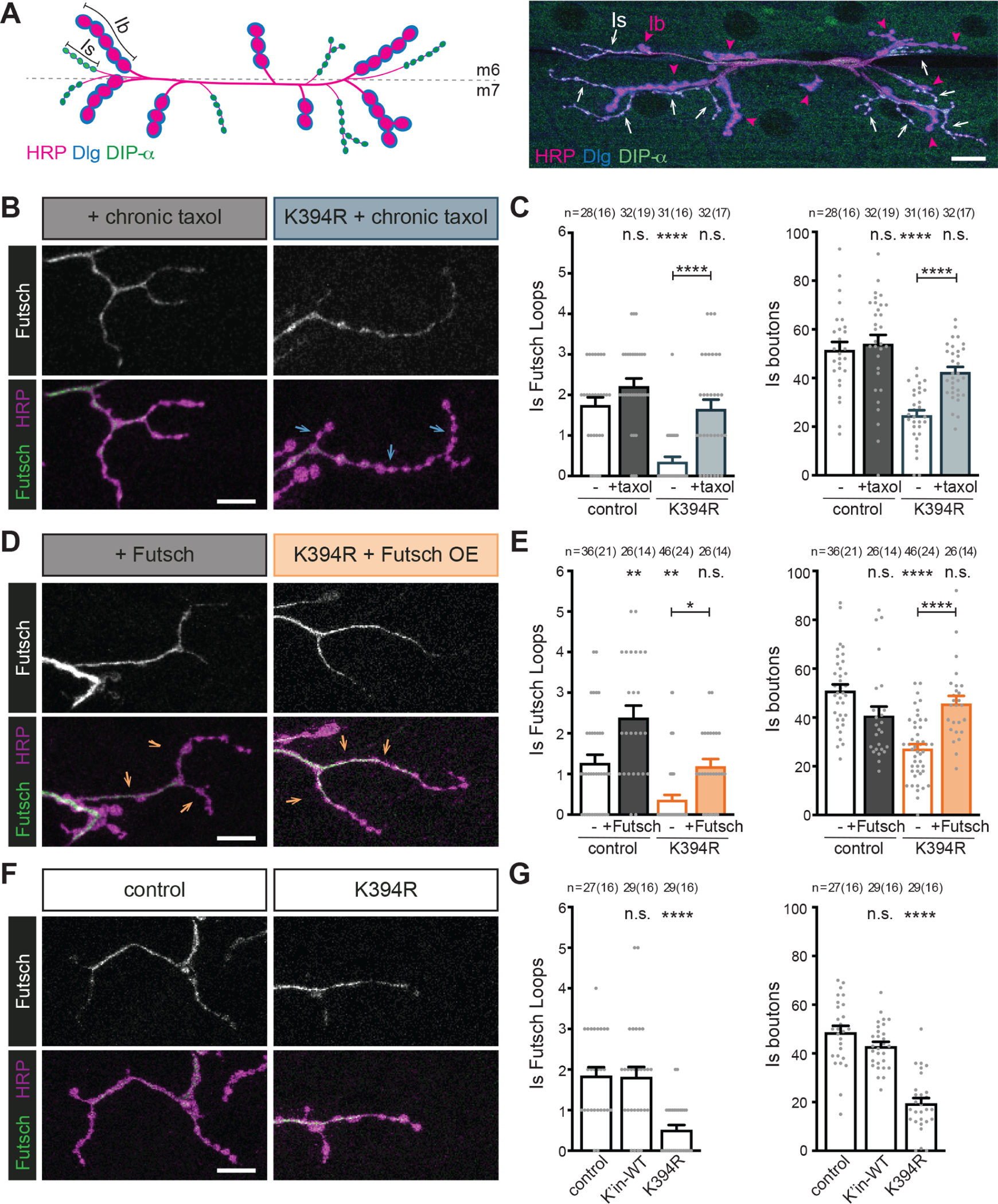
All genotypes are homozygous for wild-type or mutant αTub84B alleles. (A) Cartoon (left) and representative image (right) of type Ib and Is boutons that synapse on muscles 6 and 7 (m6 and m7) visualised with a neuronal membrane marker (HRP), Discs large 1 (Dlg), and DIP-α-GFP. The type Ib and Is boutons can be distinguished by differences in size (Ib are “big,” and Is are “small”) and molecular make-up: the type Ib boutons have high levels of Dlg and type Is neurons express DIP-α-GFP. Scale bar: 20 μm.
(B-G) Larval fillets stained for stable microtubules (Futsch) and a neuronal membrane marker (HRP). Arrows indicate branches of type Is boutons. Futsch loops and type Is boutons were quantified. Scale bar: 10 μm.
(B and C) Representative images (B) and quantification (C) of control and K394R mutant larvae raised on food containing 10 μM taxol for 24 hours prior to dissection. p-values, Is Futsch loops: p=0.3, control v. +taxol; p=0.000005, control v. K394R; p=0.96, control v. K394R +taxol; p=0.000012, K394R v. K394R +taxol (C, left). p-values, Is boutons: p=0.93, control v. +taxol; p<0.000001, control v. K394R; p=0.11, control v. K394R +taxol; p=0.000077, K394R v. K394R +taxol (C, right).
(D and E) Effects of Futsch over-expression on microtubule stability and the growth of type Is boutons. Representative images (D) and quantification (E) of control and neurons over-expressing Futsch (OK6-Gal4 and UAS-Futsch, hemizygous). p-values, Is Futsch loops: p=0.0018, control v. +Futsch; p=0.0036, control v. K394R; p=1.0, control v. K394R +Futsch; p=0.028, K394R v. K394R +Futsch (E, left). p-values, Is boutons: p=0.056, control v. +Futsch; p<0.000001, control v. K394R; p=0.64, control v. K394R +Futsch; p=0.000008, K394R v. K394R +Futsch (E, right).
(F and G) Representative images (F) and quantification (G) of control and K394R mutant. p-values, Is Futsch loops: p=1.0, control v. + K’in-WT; p=0.000017, control v. K394R (G, left). p-values, Is boutons: p=0.16, control v. K’in-WT; p<0.000001, control v. K394R (G, right).
Quantification: One-way ANOVA with post-hoc Tukey. All data are mean ± SEM. n.s=non-significant; ***p=0.01–0.001; ****p<0.0001.
We manipulated microtubule stability in several ways to test the response of type Is neurons. First, we asked whether enhancing microtubule stability in control animals would increase the number of type Is boutons. As we had done previously, we enhanced microtubule stability by raising control larvae on taxol-containing food or by over-expressing Futsch. Whereas both these manipulations increased type Ib bouton number (Figures 4C–4F), type Is bouton number was unchanged by either of these microtubule-stabilising treatments (Figures 7B–7E). Thus, unlike type Ib neurons, type Is neurons do not generate additional boutons when microtubule stability is enhanced. Next, we investigated the effects of the K394R mutation on type Is bouton growth. Consistent with its effects on the type Ib cytoskeleton, the K394R mutation decreased the number of Futsch loops in type Is synaptic terminals in homozygous mutant animals (Figures 7F and 7G). However, the K394R mutation resulted in significantly fewer type Is boutons than controls (Figures 7F and 7G). This decrease in type Is bouton number was rescued by treatment with taxol or by elevating Futsch levels (Figures 7B–7E), which indicated that type Is bouton loss was likely due to decreased microtubule stability. These results surprisingly reveal that the K394R mutation generates opposite synaptic growth phenotypes in two different neuron types even though the mutation has the same effect on stable microtubules. Combined with our other results, this indicates that the synaptic growth effects induced by altering microtubule stability may depend on the cellular context that varies between different neuron types.
Discussion
Microtubules are integral to neuronal structure and activity, and their function can be regulated by post-translational modifications such as acetylation. While the catalogue of tubulin acetylation sites has grown since tubulin was first shown to be acetylated over thirty-five years ago21, a central challenge remains: to understand how acetylation regulates microtubule function in neurons and other cell types. Here, we investigated a conserved acetylation site in α-tubulin, K394, that has been consistently identified in acetylome studies, yet remained uncharacterised. Leveraging an in vivo fly model system and mutagenesis of endogenous α-tubulin, we discovered that K394 is an essential residue that is acetylated in the nervous system and is required for microtubule stability and proper synaptic terminal development. Acetylation of K394 increases when the deacetylase HDAC6 is reduced and decreases when HDAC6 levels are elevated, indicating that HDAC6 regulates K394 acetylation. Consistent with HDAC6 functioning to deacetylate K394 during synaptic terminal development, we found that the over-expression of HDAC6 results in fewer Futsch loops similar to the acetylation-blocking K394R mutation and that this activity depends on its tubulin deacetylase domain. Together, our data suggest a model in which the α-tubulin K394 and its acetylation are important for normal microtubule function during synaptic terminal development.
Several pieces of data suggest that α-tubulin K394, and its acetylation, are likely to be important for microtubule stability during synaptic bouton development. We found that in the K394R mutant, and when HDAC6 is over-expressed, microtubule loops indicative of microtubule stability are reduced, and the K394R mutant has enhanced sensitivity to nocodazole and reduced futsch. The position of K394 in α-tubulin suggests that it has the potential to affect the intrinsic stability or dynamics of microtubules. For example, our EB1::GFP data suggest that the nucleation or polymerisation of microtubules may be disrupted. However, it is also possible that mutating K394 or altering its acetylation affects the binding of a protein(s) that regulates microtubules. For example, polyglutamylation of the α-tubulin C-terminal tail affects the affinity of microtubule severing enzymes for microtubules54–56. Another C-terminal tail modification, detyrosination, regulates microtubule dynamics by affecting the binding of CLIP-170, and subsequently EB1, to microtubule ends57–59. While our current work indicates that mutating α-tubulin K394 (and over-expressing HDAC6) reduces stable microtubules, additional studies will be needed to determine the mechanism.
Strikingly, the K394R mutant phenotype can be reversed by increasing the levels of the α-tubulin cofactor TBCE, which has been directly implicated in both the assembly and homeostasis of αβ-tubulin dimers43. TBCE functions as part of a cofactor complex that is postulated to regulate the interaction between α- and β-tubulin at the heterodimer interface, which is where K394 is located60,61. TBCE-containing cofactor complexes regulate the formation, dissociation, and degradation of tubulin heterodimers, and cofactors can also sequester tubulin monomers43,62. The over-expression of TBCE results in microtubule loss in some cell types, such as HeLa cells63, which likely reflects the sequestration of α-tubulin and/or dissociation of tubulin heterodimers by ectopic TBCE. In contrast, elevating TBCE levels has been reported to enhance microtubule stability at the fly neuromuscular junction44, which may result from TBCE’s positive role in tubulin biogenesis. These differing reports from different cell types and organisms suggest that the effects of TBCE over-expression may depend on the cellular context or relative increase in TBCE levels. In our experiments, the over-expression of TBCE induces the formation of extra boutons. Although we did not detect a significant change in the microtubule cytoskeleton as read-out by Futsch loops, the formation of extra boutons likely results from TBCE promoting tubulin heterodimer formation. The ability of TBCE over-expression to reverse K394R mutant phenotypes, coupled with the position of K394 at the αβ-tubulin heterodimer interface, leads us to hypothesise that the K394R mutation disrupts αβ-tubulin dimer formation, stability, and/or homeostasis. Notably, reducing TBCE levels at the fly neuromuscular junction also induces the formation of extra boutons similar to the α-tubulin K394R mutant44, which is consistent with the idea that bouton formation is sensitive to perturbations in αβ-tubulin dimer formation and stability. One possible model is that effects of the K394R mutation on the tubulin heterodimer in turn disrupts microtubules and microtubule stability. By extension, this suggests that K394 acetylation might similarly impact the microtubule cytoskeleton by affecting αβ-tubulin heterodimer formation and/or homeostasis. Interestingly, the acetylation of a β-tubulin residue (K252) that also resides at the αβ-tubulin heterodimer interface is proposed to regulate microtubule polymerisation by affecting dimer conformation64. Like β-tubulin K252 acetylation, it is possible that acetylation of α-tubulin K394 may impinge on the αβ-tubulin heterodimer to regulate microtubules and their stability.
Previous studies have suggested that changes in stable microtubules marked by Futsch loops and bouton numbers generally parallel each other (e.g., an increase in Futsch loops corresponds to an increase in boutons and vice versa). However, our results indicate that the relationship between stable microtubules and bouton growth is not direct. Bouton growth is regulated by multiple factors, including actin and the BMP signalling pathway, among others2,65. Our studies show that the K394R mutant significantly reduces Futsch loops but the number of type Ib boutons surprisingly increases. It is possible that the K394R mutation causes a change in the microtubule cytoskeleton that results in a compensatory increase in another pathway that promotes type Ib bouton development. Our preliminary analysis of some known bouton growth pathways did not uncover any obvious changes, however. Thus, the relationship between microtubule dynamics and stability and synaptic morphogenesis will require further investigation.
Our mutagenesis studies reveal that a reduction in stable microtubules in the K394R mutant correlates with striking neuron-type-specific morphogenesis defects. The type Ib and Is neurons have distinct roles in larval locomotion and respond differently to changes in neurotransmission51. For example, type Ib synaptic terminals can increase their number of boutons in response to changes in activity whereas type Is do not. Our work indicates that these two neuron types also respond differently to cytoskeletal changes: type Ib neurons overgrow when microtubule stability is reduced or increased. In contrast, the type Is neurons are sensitive to reduced microtubule stability, but enhanced microtubule stability does not induce ectopic synaptic growth. These data indicate that there is not a direct relationship between microtubule stability and bouton morphogenesis. Recent studies have uncovered some crosstalk between type Ib and Is when the activity (or presence) of one is disrupted, and it is possible that this may contribute to some of the morphological differences we observe66,67. Our findings raise the possibility that the microtubule cytoskeleton may be regulated in a neuron-type-specific manner in order to create the discrete morphologies that underlie the distinct functions of type Ib and Is neurons. It is likely that differences in the expression or activity of cytoskeletal regulators between these neuron types result in divergent phenotypes when microtubule stability is altered. Neuron-type-specific suites of cytoskeletal regulators, or possibly even α-tubulin isotypes, could contribute to the intrinsic differences between these two neuron types.
Elucidating the cellular effects of a microtubule modification includes identifying and manipulating the relevant modifying enzymes. The well-studied α-tubulin K40 is acetylated αTAT and deacetylated by HDAC6. Our experiments suggest that α-tubulin K394 is similarly deacetylated by HDAC6. However, this study and previous work by us and others suggests that αTAT is unlikely to target K394. Here, we found that increasing the amount of αTAT did not increase K394 acetylation. Previously others have shown that the mammalian α-tubulin acetyltransferase αTAT1 is found predominantly in the microtubule lumen where K40 is positioned but has minimal interaction with the microtubule surface where K394 is positioned, which is also inconsistent with K394 being an αTAT substrate68–70. Moreover, we and others have previously shown that the loss of αTAT in Drosophila does not phenocopy the K394R mutant: in dαTAT knock-out animals, microtubule dynamics are increased, and microtubule stability is unchanged at synaptic terminals19,53,71–73. Thus, additional studies will be needed to identify the relevant acetyltransferase for α-tubulin K394.
Our experiments indicate that HDAC6 regulates the deacetylation of α-tubulin K394 and microtubule stability during synaptic terminal development. Thus, HDAC6 may control microtubule stability in developing neurons by deacetylating K394. HDAC6, however, targets several sites in α-tubulin, including K4013. α-tubulin K40 acetylation has long been correlated with stable microtubules, and knocking-out HDAC6 increases K40 acetylation6. For example, the loss of HDAC6 in fly and mouse muscle cells makes microtubules more stable and resistant to either cold or nocodazole treatment74,75. While such examples of HDAC6’s effects on microtubule stability have been linked to K40 acetylation, our results suggest that some of these effects might also be due to HDAC6 regulating the acetylation of K394. We found that HDAC6 over-expression reduced microtubule stability in synaptic terminals similar to blocking acetylation at K394 but not K40 (K394R and K40R mutants, respectively). In addition, we observed that K40 is still acetylated in K394R mutants, which indicates that the effects of K394R are not likely due to a reduction in K40 acetylation. Thus, our K394 mutagenesis and HDAC6 experiments, combined with quantitative mass spectrometry results13, suggest that HDAC6 may regulate α-tubulin K394 acetylation to modulate microtubule stability in developing synaptic boutons. Our data suggest a model in which acetylation of α-tubulin K394 plays an important role in regulating microtubules to shape proper neuronal morphology.
STAR Methods
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Jill Wildonger (jwildonger@health.ucsd.edu).
Materials availability
All materials reported in this paper will be shared by the lead contact upon request.
Data and code availability
All data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyse the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Fly Husbandry and Strains
All stocks and crosses were maintained at 25°C under 12-hour light-dark cycles on standard cornmeal-molasses food unless otherwise stated. The following fly strains are from the Bloomington Drosophila Stock Center (BDSC): w1118 (stock # 6326), futschEP1419 (UAS-futsch, which was generated when a transposable element containing the UAS sequence inserted upstream of futsch32, stock # 10751), futschN94 (stock # 8805), OK6-Gal4 (stock # 64199), pickpocket-Gal4 (stock # 32079), elav-Gal4 (stock # 458), 24B-Gal4 (stock # 1767), UAS-TBCE (stock # 34536), UAS-dαTAT::GFP (J. Parrish, University of Washington)19, UAS-HDAC6WT (stock # 51181), UAS-HDAC6H664A (stock # 51184), UAS-HDAC6H237A (stock # 51183) and UAS-HDAC6H237A+H664A (stock # 51185). UAS-sfGFP(1–10) was created as previously described38,39. The following strains were created in this study (description of their creation is below): αTub84BK394A, αTub84BK394R, UAS-αTub84B, and EB1::sfGFP(11).
METHOD DETAILS
Drosophila strain creation
To create new αTub84B alleles, the pGE-attB-GMR integration plasmid containing the new allele was injected into αTub84BattP-KO embryos expressing the integrase PhiC31 (BestGene Inc.). The following mutations were introduced into the integration plasmid by Phusion high fidelity polymerase: K394R and K394A. αTub84BK’in-WT (in which wild-type αTub84B was knocked-into the locus) and αTub84BK40R were also generated using this method20. UAS-αTub84B was made by cloning αTub84B into the pIHEU-MCS plasmid (# 58375; Addgene) and was injected into P{CaryP}attP2 (BestGene Inc.).
CRISPR-Cas9-mediated genome editing was used to create EB1::sfGFP(11). A plasmid to express a guide RNA targeting the 3’-end of the EB1 coding sequence (pBSK-U63-EB1-gRNA) was injected with a repair template into nos-Cas9 embryos (BDSC stock # 78782) (BestGene Inc.). The repair template encoded a linker (GGSGG) plus seven tandem copies of sfGFP(11) separated by GGSGG linkers. To identify integration events, the repair template also included a 3xP3-dsRed cassette, which was flanked by the piggyBac inverted terminal repeat sequences. Candidate progeny expressing 3xP3-DsRed were crossed to a transposase-expressing strain to scarlessly excise 3xP3-dsRed cassette. The EB1::sfGFP(11) gene was then fully sequenced. EB1 guide RNA target sequence: 5’-TAATACTCCTCGTCCTCTGG-3’. Linker sequence: 5’-GGCGGATCCGGCGGA-3’.
sfGFP(11)x7: 5’-CGTGACCACATGGTCCTTCATGAGTATGTAAATGCTGCTGGGATTACAGGTGGCTCTGGAGGTAGAGATCATATGGTTCTCCACGAATACGTTAACGCCGCAGGCATCACTGGCGGTAGTGGAGGACGCGACCATATGGTACTACATGAATATGTCAATGCAGCCGGAATAACCGGAGGGTCCGGAGGCCGGGATCACATGGTGCTGCATGAGTATGTGAACGCGGCGGGTATAACTGGTGGGTCGGGCGGACGAGACCATATGGTGCTTCACGAATACGTAAACGCAGCTGGCATTACTGGCGGATCAGGTGGCAGGGATCACATGGTACTCCATGAGTACGTGAACGCTGCTGGAATCACAGGCGGTAGCGGCGGTCGGGACCATATGGTCCTGCACGAATATGTCAATGCTGCCGGTATCACC-3’
Imaging and Immunohistochemistry
All imaging was done on an SP5 confocal microscope (Leica Microsystems) using a 40 × 1.3 NA oil-immersion objective. Muscle pair 6/7 in abdominal segment A2 were imaged and analysed unless otherwise noted.
Wandering third instar larvae were dissected in PHEM buffer (80 mM PIPES pH 6.9, 25 mM HEPES pH 7.0, 7 mM MgCl2, 1 mM EGTA) and fixed in 4% PFA in 1X phosphate-buffered saline (PBS) with 3.2% sucrose for 30 minutes (mins), permeabilised in 1XPBS with 0.3% Triton-X100 for 20 mins, quenched in 50 mM NH4Cl for 10 mins, blocked in buffer containing 2.5% BSA (catalogue number A9647, Sigma), 0.25% FSG (catalogue number G7765, Sigma), 10 mM glycine, 50 mM NH4Cl, 0.05% Triton-X100 for at least 1 hour at room temperature. Fillets were then incubated in primary antibody in blocking buffer overnight at 4°C, washed in 1XPBS with 0.1% Triton-X100 at room temperature and incubated with secondary antibody in blocking buffer overnight at 4°C in the dark. After washing in 1XPBS with 0.1% Triton-X100, fillets were mounted in elvanol containing antifade (polyvinyl alcohol, Tris 8.5, glycerol and DABCO; DABCO, catalogue number 11247100, Fisher Scientific, Hampton, NH). The following antibodies were used on dissected larval fillets: mouse anti-Futsch 22C10 (1:50, Developmental Studies Hybridoma Bank, Iowa City, IA), rabbit anti-Futsch-LC (1:5,000, gift of R. Ordway, Penn State University)76, mouse-anti-Discs large (1:100, Developmental Studies Hybridoma Bank, Iowa City, IA), mouse anti-α-tubulin DM1α (1:500, Sigma-Aldrich), mouse anti-acetylated-K40 6–11B-1 (1:500, Sigma-Aldrich), goat anti-HRP conjugated Alexa Fluor 647 (1:3000, or 0.5 μg mL−1, Jackson ImmunoResearch, West Grove, PA), Dylight 550 anti-mouse (1:1000, or 0.5 μg mL−1, ThermoFisher, Waltham, MA), Dylight 488 anti-mouse (1:1000, or 0.5 μg mL−1, ThermoFisher, Waltham, MA).
Taxol and Nocodazole Treatment
For acute taxol treatment, wandering third instar larvae were dissected in PHEM buffer containing 50 μM taxol and incubated for 30 mins prior to fixation. For acute nocodazole treatment, larvae were dissected in PHEM buffer containing 30 μM nocodazole and incubated for 1 hour prior to fixation. For chronic taxol treatment, larvae were transferred onto cornmeal molasses food containing 10 μM taxol 24 hours before dissections. As a test, we also incubated larvae on 10 μM taxol-containing food for 12 hours, but at this time point the taxol treatment did not have a significant effect on either Futsch loops or bouton number in the K394R mutant larvae (Figure S4, compare 12-hour treatment to no taxol). Larvae were then dissected and fixed. Fixed tissue was subsequently stained following the methods above.
Quantification of Bouton Number and Loops
Type Ib and Is boutons and loops marked by DM1α (anti-α-tubulin), Futsch, and acetylated-K40 in these boutons were quantified at m6/7 of segment A2 in blinded images. Satellite boutons were defined as five or fewer smaller sized boutons coming from the main terminal branch of the nerve. Loops marked by DM1α (anti-α-tubulin), Futsch, and acetylated-K40 were defined as complete, unbroken loops of signal within a bouton.
EB1::GFP Imaging and Analysis
Wandering third instar larvae were immobilised in 50% glycerol in 1XPBS on a slide between two strips of vacuum grease. Dorsal class IV ddaC sensory neurons in abdominal segments 2–4 were imaged using a 40 × 1.3 NA oil-immersion objective. EB1 comet trajectories were visualised by reconstituting GFP fluorescence specifically in class IV neurons: pickpocket-Gal4 was used to drive the expression of UAS-sfGFP(1–10) in EB1::sfGFP(11) larvae. Videos were captured at a rate of 1.358 frames/second and a resolution of 1,024 × 512 pixels. Videos were stabilised using the FIJI Image Stabilizer plugin and kymographs generated in FIJI. Comet direction, velocity and frequency were then analysed in MetaMorph and data exported to Excel for analysis.
Anti-Acetylated-K394 Custom Antibody
To detect acetylated α-tubulin K394, the following peptide was used to generate polyclonal antibodies in rabbits: ARLDH(AcK)FDLMYAK (ThermoFisher Scientific). The antibodies were purified via negative selection against non-acetylated peptide ARLDHKFDLMYAK (ThermoFisher Scientific). The antibody did not produce any positive signal when incubated with fixed larval fillets.
α-tubulin Analysis and Microtubule Pelleting
To analyse α-tubulin levels, 10 larval fly brains were dissected in 1XPBS, then 30 μL of 1XSDS loading buffer was added, and the sample was boiled for 10 mins. Lysate from the equivalent of 2 brains (6 μL) was loaded per lane.
Microtubules were pelleted from the lysate of fly heads from controls (w1118), K394R flies, HDAC6 knock-out flies, elav-Gal4 UAS-HDAC6 (wild-type) flies, elav-Gal4 UAS-HDAC6 H664A flies, and elav-Gal4 UAS-dαTAT::GFP flies. Flies were flash frozen then vortexed for 1 min, and a sieve was used to separate heads from bodies. Heads were homogenised in 1 mL BRB-80 lysis buffer (80 mM PIPES, 1 mM MgCl2, 1 mM EGTA, 1 mM DTT, cOmplete EDTA-free protease inhibitor cocktail [Roche], 100 nM TSA, 1 mM sodium butyrate, 10 mM nicotinamide, 2 μM SAHA) per 1 gram of heads. To remove cell debris, samples were centrifuged at 20,000g at 4°C for 30–45 mins. Lysate was then further clarified by centrifuging at 209,000g for 14 mins at 4°C. 3 mM GTP and 20 μM taxol was then added to the lysate and microtubules allowed to polymerise for 30 mins at room temperature followed by incubation for 30 mins on ice. Samples were then centrifuged through a 15% sucrose cushion at 45,000g for 4 mins at 4°C to pellet microtubules. Pellets were resuspended in BRB-80 lysis buffer with 20 μM taxol.
Immunoblotting
Samples were run on an SDS-PAGE gel, and then proteins were transferred to low-fluorescence PVDF membrane (catalogue number GE10600022, GE Health and Life Sciences). Membranes were blocked in 5% milk in 1X Tris-buffered saline (TBS) with 0.1% Tween-20 (0.1% TBST) for 1 hour at room temperature and incubated with primary antibody in block overnight at 4°C. After washing in 0.1% TBST membranes were incubated with secondary antibodies for 2–4 hours at room temperature. Membranes were then imaged using either chemiluminescence (Super Signal West Femto ECL, catalogue number 34094, ThermoFisher Scientific) or fluorescence. The following antibodies were used for western blotting: mouse anti-α-tubulin DM1α (1:1000, Sigma-Aldrich), mouse anti-actin C4 (1:1000, Sigma-Aldrich), rabbit anti-Ac-K394 (1:50, this study), Cy5 anti-mouse (1:10000, Jackson ImmunoResearch, West Grove, PA), and HRP-conjugated anti-rabbit (1:10000, Bio-Rad Laboratories).
QUANTIFICATION AND STATISTICAL ANALYSIS
Data were blinded prior to analysis. Statistical analysis was performed in Excel and GraphPad Prism using a significance level of p < 0.05. Data were first analysed for normality using the Shapiro-Wilk test. Normally distributed data was then analysed for equal variance and significance using either an F-test and Student’s unpaired t-test (two samples) or one-way ANOVA with post-hoc Tukey (multiple samples). Data sets that were not normally distributed were analysed using Mann-Whitney U test (two samples) or Kruskal-Wallis test with post-hoc Dunn test for significance (multiple samples). Each graph reports both the exact n, which is the number of axon terminals (NMJs) that were analysed, and the number of larvae in the following format: # n (# larvae) above each bar for the different experimental genotypes or conditions. The statistical test used to evaluate the data for each experiment and the exact p-values are included in the figure legends.
Supplementary Material
Key resources table.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-Futsch 22C10 | Developmental Studies Hybridoma Bank | DSHB Cat# 22C10; RRID:AB_528403 |
| anti-Futsch-LC | Dr. Richard Ordway, Penn State University | NA |
| Anti-discs large 4F3 | Developmental Studies Hybridoma Bank | DSHB Cat# DLG1, RRID:AB_2314321 |
| anti-alpha tubulin DM1A | Sigma-Aldrich | Cat# T6199; RRID:AB_477583 |
| anti-acetylated tubulin 6-11B-1 | Sigma-Aldrich | Cat# T6793; RRID:AB_477585 |
| anti-HRP conjugated Alexa Fluor 647 | Jackson ImmunoResearch | Cat# 123-605-021; RRID:AB_2338967 |
| Dylight 550 anti-mouse | ThermoFisher | Cat# SA5-10167; RRID:AB_2556747 |
| Dylight 488 anti-mouse | ThermoFisher | Cat# SA5-10166; RRID:AB_2556746 |
| rabbit anti-Ac-K394 | This study | NA |
| anti-actin C4 | Millipore Sigma (Sigma Aldrich) | Millipore Cat# MAB1501, RRID:AB_2223041 |
| Cy5-AffiniPure Donkey Anti-Mouse IgG (H+L) | Jackson ImmunoResearch Labs | Cat# 715-175-151, RRID:AB_2340820 |
| HRP anti-rabbit | Bio-Rad | Cat# 170-6516, RRID:AB_11125547 |
| Chemicals, peptides, and recombinant proteins | ||
| BSA | Sigma-Aldrich | Cat#A9647 |
| FSG | Sigma-Aldrich | Cat#G7765 |
| DABCO | Fisher Scientific | Cat#11247100 |
| GTP | Thermo Scientific | Cat#AC226250500 |
| Paclitaxel (taxol) | Sigma-Aldrich | Cat#T7191 |
| nocodazole | Sigma-Aldrich | Cat#M1404 |
| Trichostatin A | Sigma-Aldrich | Cat#T8552 |
| Sodium butyrate | Thermo Scientific | Cat#AAA1107906 |
| Nicotinamide | Fisher Scientific | Cat#AC128271000 |
| SAHA | Fisher Scientific | Cat#46-5210 |
| cOmplete protease inhibitor cocktail | Millipore Sigma (Roche) | Cat#11836170001 |
| Experimental models: Organisms/strains | ||
| D. melanogaster: w[1118] | Bloomington Drosophila Stock Center (BDSC) | BL6326; RRID:BDSC_6326 |
| D. melanogaster: P{w[+mC]=EP}futsch[EP1419] w[1118] | BDSC | BL10751, RRID:BDSC_10751 |
| D. melanogaster: y[1] futsch[N94] | BDSC | BL8805, RRID:BDSC_8805 |
| D. melanogaster: P{w[+mW.hs]=GawB}OK6 | BDSC | BL64199, RRID:BDSC_64199 |
| D. melanogaster: w[*]; P{w[+mC]=ppk-GAL4.G}3 | BDSC | BL32079, RRID:BDSC_32079 |
| D. melanogaster: P{w[+mW.hs]=GawB}elav[C155] | BDSC | BL458, RRID:BDSC_458 |
| D. melanogaster: w[*]; P{w[+mW.hs]=GawB}how[24B] | BDSC | BL1767, RRID:BDSC_1767 |
| D. melanogaster: w[1118]; P{w[+mC]=UAS-tbce.J}3/TM6B, Tb[1] | BDSC | BL34536, RRID:BDSC_34536 |
| D. melanogaster: w[1118]; UAS-GFP-dTat-L | Dr. Jay Parrish, University of Washington | NA |
| D. melanogaster: y[1] w[*]; P{w[+mC]=UAS-HDAC6.D}3 | BDSC | BL51181, RRID:BDSC_51181 |
| D. melanogaster: y[1] w[*]; P{w[+mC]=UAS-HDAC6.H664A}2 | BDSC | BL51184, RRID:BDSC_51184 |
| D. melanogaster: y[1] w[*]; P{w[+mC]=UAS-HDAC6.H237A}2 | BDSC | BL51183, RRID:BDSC_51183 |
| D. melanogaster: y[1] w[*]; P{w[+mC]=UAS-HDAC6.H237A.H664A}2; MKRS/TM6B, Tb[1] | BDSC | BL51185, RRID:BDSC_51185 |
| D. melanogaster: y[1] sc[*] v[1] sev[21]; P{y[+t7.7] v[+t1.8]=nos-Cas9.R}attP2 | BDSC | 78782 |
| D. melanogaster: w[1118]; UAS-sfGFP(1–10) | [39] | NA |
| D. melanogaster: w[1118]; aTub84B[attP-KO] | [20] | NA |
| D. melanogaster: w[1118]; aTub84B[WT-K’in] | [20] | NA |
| D. melanogaster: w[1118]; aTub84B[K394R] | This study | NA |
| D. melanogaster: w[1118]; aTub84B[K394A] | This study | NA |
| D. melanogaster: w[1118]; aTub84B[K40R] | [20] | NA |
| D. melanogaster: w[1118]; UAS-aTub84B | This study | NA |
| D. melanogaster: w[1118]; EB1::sfGFP(11) | This study | NA |
| Oligonucleotides | ||
| Oligonucleotide primer sequences used in this study are listed in Table S1. | This study | NA |
| Recombinant DNA | ||
| pGE-attB-GMR | Drosophila Genomics Resource Center | 1295 |
| pGE-attB-GMR αTub84B K394R | This study | NA |
| pGE-attB-GMR αTub84B K394A | This study | NA |
| pIHEU-MCS | Addgene | 58375 |
| pIHEU-αTub84B | This study | NA |
| pBSK-U63-EB1-gRNA | This study | NA |
| Software and algorithms | ||
| ImageJ | National Institute of Health, USA | https://imagej.nih.gov/ij/RRID:SCR_003070 |
| Excel | Microsoft | https://products.office.com/en-gb/excel RRID:SCR_016137 |
| GraphPad PRISM | GraphPad Software | https://www.graphpad.com RRID:SCR_002798 |
| Leica LAS AF software | Leica LAS AF software | http://www.leica-microsystems.com/products/microscope-software/ RRID:SCR_016555 |
| Adobe PhotoShop | Adobe | http://www.adobe.com/products/photoshop.html RRID:SCR_014199 |
| Adobe Illustrator | Adobe |
http://www.adobe.com/products/illustrator.html RRID:SCR_010279 |
| Other | ||
| Super Signal West Femto ECL | ThermoFisher Scientific | 34094 |
| Low-fluorescence PVDF membrane | GE Health and Life Sciences | GE10600022 |
| Leica TCS SP5 microscope | Leica Microsystems | https://www.leica-microsystems.com RRID:SCR_008960 |
Highlights.
α-tubulin K394 is acetylated in the nervous system and is critical to neuronal growth.
An acetylation-blocking K39R mutation decreases microtubule stability.
The K394R mutation has neuron-type-specific effects on growth.
Loss of HDAC6 increases K394 acetylation, indicating that HDAC6 deacetylates K394.
Acknowledgements
We thank the Bloomington Drosophila Stock Center and Dr. Jay Parrish (University of Washington) for fly strains and Dr. Richard Ordway (Penn State University) and the Developmental Studies Hybridoma Bank for antibodies. We thank Helen Than and Madison Johnson for technical assistance. We thank members of the laboratories of Drs. Kate O’Connor-Giles laboratory (Brown University) and Melissa Gardner (University of Minnesota) and Dr. Erik Dent (University of Wisconsin-Madison) for helpful advice and input, and members of the Wildonger laboratory for thoughtful discussions and comments on the manuscript. This work was generously supported by the National Institutes of Health (NIH) grant R01NS116373 to J.W.
Footnotes
Declaration of Interests: The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Penazzi L, Bakota L, and Brandt R (2016). Microtubule Dynamics in Neuronal Development, Plasticity, and Neurodegeneration. International Review of Cell and Molecular Biology 321, 89–169. [DOI] [PubMed] [Google Scholar]
- 2.Bodaleo FJ, and Gonzalez-Billault C (2016). The Presynaptic Microtubule Cytoskeleton in Physiological and Pathological Conditions: Lessons from Drosophila Fragile X Syndrome and Hereditary Spastic Paraplegias. Front Mol Neurosci 9, 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Prokop A (2020). Cytoskeletal organization of axons in vertebrates and invertebrates. J Cell Biol 219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Knossow M, Campanacci V, Khodja LA, and Gigant B (2020). The Mechanism of Tubulin Assembly into Microtubules: Insights from Structural Studies. Iscience 23, 101511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Halpain S, and Dehmelt L (2006). The MAP1 family of microtubule-associated proteins. Genome Biol 7, 224–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Janke C, and Magiera MM (2020). The tubulin code and its role in controlling microtubule properties and functions. Nat Rev Mol Cell Bio 21, 307–326. [DOI] [PubMed] [Google Scholar]
- 7.Portran D, Schaedel L, Xu Z, Théry M, and Nachury MV (2017). Tubulin acetylation protects long-lived microtubules against mechanical ageing. Nat Cell Biol 19, 391–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xu Z, Schaedel L, Portran D, Aguilar A, Gaillard J, Marinkovich MP, Théry M, and Nachury MV (2017). Microtubules acquire resistance from mechanical breakage through intralumenal acetylation. Science 356, 328–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lundby A, Lage K, Weinert BT, Bekker-Jensen DB, Secher A, Skovgaard T, Kelstrup CD, Dmytriyev A, Choudhary C, Lundby C, et al. (2012). Proteomic Analysis of Lysine Acetylation Sites in Rat Tissues Reveals Organ Specificity and Subcellular Patterns. Cell Reports 2, 419–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Weinert BT, Wagner SA, Horn H, Henriksen P, Liu WR, Olsen JV, Jensen LJ, and Choudhary C (2011). Proteome-Wide Mapping of the Drosophila Acetylome Demonstrates a High Degree of Conservation of Lysine Acetylation. Sci Signal 4, ra48–ra48. [DOI] [PubMed] [Google Scholar]
- 11.Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, Olsen JV, and Mann M (2009). Lysine Acetylation Targets Protein Complexes and Co-Regulates Major Cellular Functions. Science 325, 834–840. [DOI] [PubMed] [Google Scholar]
- 12.Hansen BK, Gupta R, Baldus L, Lyon D, Narita T, Lammers M, Choudhary C, and Weinert BT (2019). Analysis of human acetylation stoichiometry defines mechanistic constraints on protein regulation. Nat Commun 10, 1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu N, Xiong Y, Li S, Ren Y, He Q, Gao S, Zhou J, and Shui W (2015). New HDAC6-mediated deacetylation sites of tubulin in the mouse brain identified by quantitative mass spectrometry. Sci Rep-uk 5, 16869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu N, Xiong Y, Ren Y, Zhang L, He X, Wang X, Liu M, Li D, Shui W, and Zhou J (2015). Proteomic Profiling and Functional Characterization of Multiple Post-Translational Modifications of Tubulin. J Proteome Res 14, 3292–3304. [DOI] [PubMed] [Google Scholar]
- 15.Weinert BT, Narita T, Satpathy S, Srinivasan B, Hansen BK, Schölz C, Hamilton WB, Zucconi BE, Wang WW, Liu WR, et al. (2018). Time-Resolved Analysis Reveals Rapid Dynamics and Broad Scope of the CBP/p300 Acetylome. Cell 174, 231–244.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yin S, Zeng C, Hari M, and Cabral F (2013). Paclitaxel resistance by random mutagenesis of α-tubulin. Cytoskeleton 70, 849–862. [DOI] [PubMed] [Google Scholar]
- 17.Narita T, Weinert BT, and Choudhary C (2019). Functions and mechanisms of non-histone protein acetylation. Nat Rev Mol Cell Bio 20, 156–174. [DOI] [PubMed] [Google Scholar]
- 18.Raff EC (1984). Genetics of microtubule systems. J Cell Biology 99, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yan C, Wang F, Peng Y, Williams CR, Jenkins B, Wildonger J, Kim H-J, Perr JB, Vaughan JC, Kern ME, et al. (2018). Microtubule Acetylation Is Required for Mechanosensation in Drosophila. Cell Reports 25, 1051–1065.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jenkins BV, Saunders HAJ, Record HL, Johnson-Schlitz DM, and Wildonger J (2017). Effects of mutating α-tubulin lysine 40 on sensory dendrite development. J Cell Sci 130, 4120–4131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.L’Hernault SW, and Rosenbaum JL (1985). Chlamydomonas .alpha.-tubulin is posttranslationally modified by acetylation on the .epsilon.-amino group of a lysine. Biochemistry-us 24, 473–478. [DOI] [PubMed] [Google Scholar]
- 22.Hubbert C, Guardiola A, Shao R, Kawaguchi Y, Ito A, Nixon A, Yoshida M, Wang X-F, and Yao T-P (2002). HDAC6 is a microtubule-associated deacetylase. Nature 417, 455–458. [DOI] [PubMed] [Google Scholar]
- 23.Akella JS, Wloga D, Kim J, Starostina NG, Lyons-Abbott S, Morrissette NS, Dougan ST, Kipreos ET, and Gaertig J (2010). MEC-17 is an α-tubulin acetyltransferase. Nature 467, 218–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shida T, Cueva JG, Xu Z, Goodman MB, and Nachury MV (2010). The major α-tubulin K40 acetyltransferase αTAT1 promotes rapid ciliogenesis and efficient mechanosensation. Proc Natl Acad Sci 107, 21517–21522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kalebic N, Sorrentino S, Perlas E, Bolasco G, Martinez C, and Heppenstall PA (2013). αTAT1 is the major α-tubulin acetyltransferase in mice. Nat Commun 4, 1962. [DOI] [PubMed] [Google Scholar]
- 26.Kim G-W, Li L, Gorbani M, You L, and Yang X-J (2013). Mice Lacking α-Tubulin Acetyltransferase 1 Are Viable but Display α-Tubulin Acetylation Deficiency and Dentate Gyrus Distortion. J Biol Chem 288, 20334–20350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Roos J, Hummel T, Ng N, Klämbt C, and Davis GW (2000). Drosophila Futsch Regulates Synaptic Microtubule Organization and Is Necessary for Synaptic Growth. Neuron 26, 371–382. [DOI] [PubMed] [Google Scholar]
- 28.Pennetta G, Hiesinger PR, Fabian-Fine R, Meinertzhagen IA, and Bellen HJ (2002). Drosophila VAP-33A Directs Bouton Formation at Neuromuscular Junctions in a Dosage-Dependent Manner. Neuron 35, 291–306. [DOI] [PubMed] [Google Scholar]
- 29.Tanaka EM, and Kirschner MW (1991). Microtubule behavior in the growth cones of living neurons during axon elongation. J Cell Biology 115, 345–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dent EW, Callaway JL, Szebenyi G, Baas PW, and Kalil K (1999). Reorganization and movement of microtubules in axonal growth cones and developing interstitial branches. J Neurosci Official J Soc Neurosci 19, 8894–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pawson C, Eaton BA, and Davis GW (2008). Formin-Dependent Synaptic Growth: Evidence That Dlar Signals via Diaphanous to Modulate Synaptic Actin and Dynamic Pioneer Microtubules. J Neurosci 28, 11111–11123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hummel T, Krukkert K, Roos J, Davis G, and Klämbt C (2000). Drosophila Futsch/22C10 Is a MAP1B-like Protein Required for Dendritic and Axonal Development. Neuron 26, 357–370. [DOI] [PubMed] [Google Scholar]
- 33.Ruiz-Canada C, Ashley J, Moeckel-Cole S, Drier E, Yin J, and Budnik V (2004). New synaptic bouton formation is disrupted by misregulation of microtubule stability in aPKC mutants. Neuron 42, 567–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shi Q, Lin YQ, Saliba A, Xie J, Neely GG, and Banerjee S (2019). Tubulin Polymerization Promoting Protein, Ringmaker, and MAP1B Homolog Futsch Coordinate Microtubule Organization and Synaptic Growth. Front Cell Neurosci 13, 192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nechipurenko IV, and Broihier HT (2012). FoxO limits microtubule stability and is itself negatively regulated by microtubule disruption. J Cell Biology 196, 345–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Szasz J, Yaffe MB, Elzinga M, Blank GS, and Sternlicht H (1986). Microtubule assembly is dependent on a cluster of basic residues in .alpha.-tubulin. Biochemistry-us 25, 4572–4582. [DOI] [PubMed] [Google Scholar]
- 37.Szasz J, Yaffe MB, and Sternlicht H (1993). Site-directed mutagenesis of alpha-tubulin. Reductive methylation studies of the Lys 394 region. Biophys J 64, 792–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kamiyama D, Sekine S, Barsi-Rhyne B, Hu J, Chen B, Gilbert LA, Ishikawa H, Leonetti MD, Marshall WF, Weissman JS, et al. (2016). Versatile protein tagging in cells with split fluorescent protein. Nat Commun 7, 11046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kelliher MT, Yue Y, Ng A, Kamiyama D, Huang B, Verhey KJ, and Wildonger J (2018). Autoinhibition of kinesin-1 is essential to the dendrite-specific localization of Golgi outposts. J Cell Biol 217, 2531–2547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hahn I, Voelzmann A, Parkin J, Fülle JB, Slater PG, Lowery LA, Sanchez-Soriano N, and Prokop A (2021). Tau, XMAP215/Msps and Eb1 co-operate interdependently to regulate microtubule polymerisation and bundle formation in axons. Plos Genet 17, e1009647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Consolati T, Locke J, Roostalu J, Chen ZA, Gannon J, Asthana J, Lim WM, Martino F, Cvetkovic MA, Rappsilber J, et al. (2020). Microtubule Nucleation Properties of Single Human γTuRCs Explained by Their Cryo-EM Structure. Dev Cell 53, 603–617.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bu S, Yong WL, Lim BJW, Kondo S, and Yu F (2021). A systematic analysis of microtubule-destabilizing factors during dendrite pruning in Drosophila. Embo Rep 22, e52679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Al-Bassam J (2017). Revisiting the tubulin cofactors and Arl2 in the regulation of soluble αβ-tubulin pools and their effect on microtubule dynamics. Mol Biol Cell 28, 359–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jin S, Pan L, Liu Z, Wang Q, Xu Z, and Zhang YQ (2009). Drosophila Tubulin-specific chaperone E functions at neuromuscular synapses and is required for microtubule network formation. Development 136, 1571–1581. [DOI] [PubMed] [Google Scholar]
- 45.Okumura M, Sakuma C, Miura M, and Chihara T (2015). Linking Cell Surface Receptors to Microtubules: Tubulin Folding Cofactor D Mediates Dscam Functions during Neuronal Morphogenesis. J Neurosci 35, 1979–1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Valenzuela-Fernández A, Cabrero JR, Serrador JM, and Sánchez-Madrid F (2008). HDAC6: a key regulator of cytoskeleton, cell migration and cell–cell interactions. Trends Cell Biol 18, 291–297. [DOI] [PubMed] [Google Scholar]
- 47.Xiong Y, Zhao K, Wu J, Xu Z, Jin S, and Zhang YQ (2013). HDAC6 mutations rescue human tau-induced microtubule defects in Drosophila. Proc National Acad Sci 110, 4604–4609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Haggarty SJ, Koeller KM, Wong JC, Grozinger CM, and Schreiber SL (2003). Domain-selective small-molecule inhibitor of histone deacetylase 6 (HDAC6)-mediated tubulin deacetylation. Proc National Acad Sci 100, 4389–4394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kaluza D, Kroll J, Gesierich S, Yao T, Boon RA, Hergenreider E, Tjwa M, Rössig L, Seto E, Augustin HG, et al. (2011). Class IIb HDAC6 regulates endothelial cell migration and angiogenesis by deacetylation of cortactin. Embo J 30, 4142–4156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Miskiewicz K, Jose LE, Yeshaw WM, Valadas JS, Swerts J, Munck S, Feiguin F, Dermaut B, and Verstreken P (2014). HDAC6 Is a Bruchpilot Deacetylase that Facilitates Neurotransmitter Release. Cell Reports 8, 94–102. [DOI] [PubMed] [Google Scholar]
- 51.Aponte-Santiago NA, and Littleton JT (2020). Synaptic Properties and Plasticity Mechanisms of Invertebrate Tonic and Phasic Neurons. Front Physiol 11, 611982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.McLaughlin CN, Nechipurenko IV, Liu N, and Broihier HT (2016). A Toll receptor–FoxO pathway represses Pavarotti/MKLP1 to promote microtubule dynamics in motoneurons. J Cell Biology 214, 459–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Coombes CE, Saunders HAJ, Mannava AG, Johnson-Schlitz DM, Reid TA, Parmar S, McClellan M, Yan C, Rogers SL, Parrish JZ, et al. (2020). Non-enzymatic Activity of the α-Tubulin Acetyltransferase αTAT Limits Synaptic Bouton Growth in Neurons. Curr Biol 30, 610–623.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lacroix B, Dijk J. van, Gold ND, Guizetti J, Aldrian-Herrada, Rogowski K, Gerlich DW, and Janke C (2010). Tubulin polyglutamylation stimulates spastin-mediated microtubule severing. J Cell Biology 189, 945–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Valenstein ML, and Roll-Mecak A (2016). Graded Control of Microtubule Severing by Tubulin Glutamylation. Cell 164, 911–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shin SC, Im S-K, Jang E-H, Jin KS, Hur E-M, and Kim EE (2019). Structural and Molecular Basis for Katanin-Mediated Severing of Glutamylated Microtubules. Cell Reports 26, 1357–1367.e5. [DOI] [PubMed] [Google Scholar]
- 57.Chen J, Kholina E, Szyk A, Fedorov VA, Kovalenko I, Gudimchuk N, and Roll-Mecak A (2021). α-tubulin tail modifications regulate microtubule stability through selective effector recruitment, not changes in intrinsic polymer dynamics. Dev Cell 56, 2016–2028.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Peris L, Thery M, Fauré J, Saoudi Y, Lafanechère L, Chilton JK, Gordon-Weeks P, Galjart N, Bornens M, Wordeman L, et al. (2006). Tubulin tyrosination is a major factor affecting the recruitment of CAP-Gly proteins at microtubule plus ends. J Cell Biology 174, 839–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bieling P, Kandels-Lewis S, Telley IA, Dijk J. van, Janke C, and Surrey T (2008). CLIP-170 tracks growing microtubule ends by dynamically recognizing composite EB1/tubulin-binding sites. J Cell Biology 183, 1223–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nithianantham S, Le S, Seto E, Jia W, Leary J, Corbett KD, Moore JK, and Al-Bassam J (2015). Tubulin cofactors and Arl2 are cage-like chaperones that regulate the soluble αβ-tubulin pool for microtubule dynamics. Elife 4, e08811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Serna M, Carranza G, Martín-Benito J, Janowski R, Canals A, Coll M, Zabala JC, and Valpuesta JM (2015). The structure of the complex between α-tubulin, TBCE and TBCB reveals a tubulin dimer dissociation mechanism. J Cell Sci 128, 1824–1834. [DOI] [PubMed] [Google Scholar]
- 62.Lewis SA, Tian G, and Cowan NJ (1997). The α- and β-tubulin folding pathways. Trends Cell Biol 7, 479–484. [DOI] [PubMed] [Google Scholar]
- 63.Bhamidipati A, Lewis SA, and Cowan NJ (2000). Adp Ribosylation Factor-like Protein 2 (Arl2) Regulates the Interaction of Tubulin-Folding Cofactor D with Native Tubulin. J Cell Biology 149, 1087–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chu C-W, Hou F, Zhang J, Phu L, Loktev AV, Kirkpatrick DS, Jackson PK, Zhao Y, and Zou H (2011). A novel acetylation of β-tubulin by San modulates microtubule polymerization via down-regulating tubulin incorporation. Mol Biol Cell 22, 448–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Menon KP, Carrillo RA, and Zinn K (2013). Development and plasticity of the Drosophila larval neuromuscular junction. Wiley Interdiscip Rev Dev Biology 2, 647–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wang Y, Lobb-Rabe M, Ashley J, Anand V, and Carrillo RA (2021). Structural and Functional Synaptic Plasticity Induced by Convergent Synapse Loss in the Drosophila Neuromuscular Circuit. J Neurosci 41, 1401–1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Aponte-Santiago NA, Ormerod KG, Akbergenova Y, and Littleton JT (2020). Synaptic plasticity induced by differential manipulation of tonic and phasic motoneurons in Drosophila. J Neurosci 40, JN-RM-0925–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Coombes C, Yamamoto A, McClellan M, Reid TA, Plooster M, Luxton GWG, Alper J, Howard J, and Gardner MK (2016). Mechanism of microtubule lumen entry for the α-tubulin acetyltransferase enzyme αTAT1. Proc National Acad Sci 113, E7176–E7184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Howes SC, Alushin GM, Shida T, Nachury MV, and Nogales E (2014). Effects of tubulin acetylation and tubulin acetyltransferase binding on microtubule structure. Mol Biol Cell 25, 257–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Szyk A, Deaconescu AM, Spector J, Goodman B, Valenstein ML, Ziolkowska NE, Kormendi V, Grigorieff N, and Roll-Mecak A (2014). Molecular Basis for Age-Dependent Microtubule Acetylation by Tubulin Acetyltransferase. Cell 157, 1405–1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Topalidou I, Keller C, Kalebic N, Nguyen KCQ, Somhegyi H, Politi KA, Heppenstall P, Hall DH, and Chalfie M (2012). Genetically Separable Functions of the MEC-17 Tubulin Acetyltransferase Affect Microtubule Organization. Curr Biol 22, 1057–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Neumann B, and Hilliard MA (2014). Loss of MEC-17 Leads to Microtubule Instability and Axonal Degeneration. Cell Reports 6, 93–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wei D, Gao N, Li L, Zhu J-X, Diao L, Huang J, Han Q-J, Wang S, Xue H, Wang Q, et al. (2017). α-Tubulin Acetylation Restricts Axon Overbranching by Dampening Microtubule Plus-End Dynamics in Neurons. Cereb Cortex New York N Y 1991, 1–15. [DOI] [PubMed] [Google Scholar]
- 74.Mao C-X, Wen X, Jin S, and Zhang YQ (2017). Increased acetylation of microtubules rescues human tau-induced microtubule defects and neuromuscular junction abnormalities in Drosophila. Dis Model Mech 10, 1245–1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Osseni A, Ravel-Chapuis A, Thomas J-L, Gache V, Schaeffer L, and Jasmin BJ (2020). HDAC6 regulates microtubule stability and clustering of AChRs at neuromuscular junctions. J Cell Biol 219, e201901099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zou B, Yan H, Kawasaki F, and Ordway RW (2008). MAP1 structural organization in Drosophila: in vivo analysis of FUTSCH reveals heavy- and light-chain subunits generated by proteolytic processing at a conserved cleavage site. Biochem J 414, 63–71. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyse the data reported in this paper is available from the lead contact upon request.