

Abstract
Dynamic regulation has been proved efficient in controlling gene expression at transcriptional, translational, and post-translational level. However, the dynamic regulation at gene replication level has been rarely explored so far. In this study, we established dynamic regulation at gene copy level through engineering controllable plasmid replication to dynamically control the gene expression. Prototypic genetic circuits with different control logic were applied to enable diversified dynamic behaviors of gene copy. To explore the applicability of this strategy, the dynamic gene copy control was employed in regulating the biosynthesis of p-coumaric acid, which resulted in a up to 78% increase in p-coumaric acid titer to 1.69 g/L in shake flasks. These results indicated the great potential of applying dynamic gene copy control for engineering biosynthesis of valuable compounds in metabolic engineering.
Keywords: Dynamic gene copy regulation, Controllable plasmid replication, Dynamic pathway regulation, p-coumaric acid
Introduction
Dynamic pathway regulations have seen growing applications in engineering microbial production of natural products, biofuels, biopharmaceuticals, and bulk chemicals (Dahl et al., 2013; Doong et al., 2018; Farmer and Liao, 2000; Gupta et al., 2017; Xu et al., 2014; Yang et al., 2018; Zhang et al., 2012). Unlike traditional static regulations, dynamic regulation can adjust gene expression and direct carbon flux based on intracellular chemical concentrations or environmental signals (Liu et al., 2018; Shen et al., 2019; Tan and Prather, 2017). To enable efficient dynamic pathway regulations, engineering efforts have been devoted to developing regulation toolsets and strategies at multiple levels. The transcriptional factors (Chen et al., 2018; Dinh and Prather, 2019; Gupta et al., 2017; Kim et al., 2017; Mahr and Frunzke, 2016), CRISPR based gene activation or inhibition (Konermann et al., 2015; Larson et al., 2013; Lian et al., 2017; Perez-Pinera et al., 2013), and ligand-binding DNA aptamers (Deng et al., 2019; Wang et al., 2017a), were used to target the transcription of target genes and thus achieving dynamic regulations at transcriptional level. At translational level, small regulatory RNA (Na et al., 2013; Yoo et al., 2013), antisense RNA (Yang et al., 2015), and riboswitches (Barrick and Breaker, 2007; Muranaka et al., 2009), were the most commonly used tools and strategies due to the short RNA lengths and the ease of manipulation. Besides, post-translational level regulation of protein availabilities by timely regulating the expression of heterologous proteases or assisting factors in protein degradation were also reported (Brockman and Prather, 2015; Dinh et al., 2020; Gao et al., 2019). All these regulation strategies were proven to be efficient in dynamic pathway regulation for engineering microbial production.
Beyond the regulations on transcription, translation and post-translation, controlling the copy numbers of genes, however, was less investigated in dynamic pathway regulations. While fine-tuning gene copies by using various plasmids has been commonly applied in metabolic engineering (Ajikumar et al., 2010; Jones et al., 2000), this strategy was usually used to statically or permanently adjust the gene copy during the production. Dynamically modulating the gene copy using biosensors in microbial production has not been explored so far. Dynamic gene copy regulations, in addition to the transcriptional, translational, or post-translational control, provides a new level of control to address the metabolic burdens arising from inappropriate high copies of genes or low-productivities from insufficient pathway copies by controlling the process of gene/plasmid replication based on cellular status or environmental conditions. Also, dynamic regulation of the gene copy through controllable plasmid replication, with just a simple regulatory device, can enable simultaneous up- or down-regulation of the expression levels for all the genes in the same plasmid. Therefore, this strategy may also be useful for efficient regulation of complex pathways with multiple gene targets.
With the goal of developing dynamic control at the gene copy level, we harnessed the replication mechanism of ColE1 derived origins. Plasmids with ColE1 derived origins, such as pUC18/19 (pMB1 variant) (Yanisch-Perron et al., 1985), pZE12luc (ColE1) (Lutz and Bujard, 1997), pCS27 (p15A) (Shen and Liao, 2008), and pET and its derivatives (pBR322) (Balbás and Bolívar, 2004), are commonly used in synthetic biology and metabolic engineering. Upon understanding the mechanism, we successfully established controllable replication of ColE1 derived origins. Based on this, three prototypic genetic circuits with different dynamic behaviors were constructed to enable diversified dynamic regulation logics on gene copy. Finally, the dynamic gene copy control strategy was applied in p-coumaric acid biosynthesis and enhanced its microbial production, which demonstrated the effectiveness of dynamic gene copy regulation in real-world application. Overall, our research provides a broadly applicable approach to dynamically and efficiently control microbial synthesis at DNA level.
Results
Establishing controllable plasmid replication to inhibit gene copy
The copy number of ColE1-derived plasmids is mainly regulated by a pair of plasmid-encoded RNAs, namely the RNAI and RNAII (Cesareni et al., 1991; Del Solar et al., 1998; Selzer et al., 1983). The RNAII serves as a pre-primer and folds into a secondary structure which can stabilize the interaction between the immature RNA and the DNA of origin. This hybrid is then processed by RNase H, which cleaves the RNA strand and exposes the 3′ hydroxyl group. The DNA Polymerase I will then begin to synthesize the leading strand and initiate the replication (Fig. 1a). RNAI is a section of antisense RNAII which can bind to the 5′ end of RNAII, forming kissing complex with the help of Rop protein. This will prevent the folding of RNAII and destroy the formation of the DNA-RNA hybrid, abolishing the aforementioned cleavage and slowing down the plasmid replication (Cesareni et al., 1991; Del Solar et al., 1998) (Fig. 1a). All regulatory components (RNAI, RNAII, and Rop protein) are encoded in the plasmid (Camps, 2010; Cesareni et al., 1991; Cesareni et al., 1982).
Figure 1. Validation the controllable inhibition on plasmid replication by overexpressing RNAI.
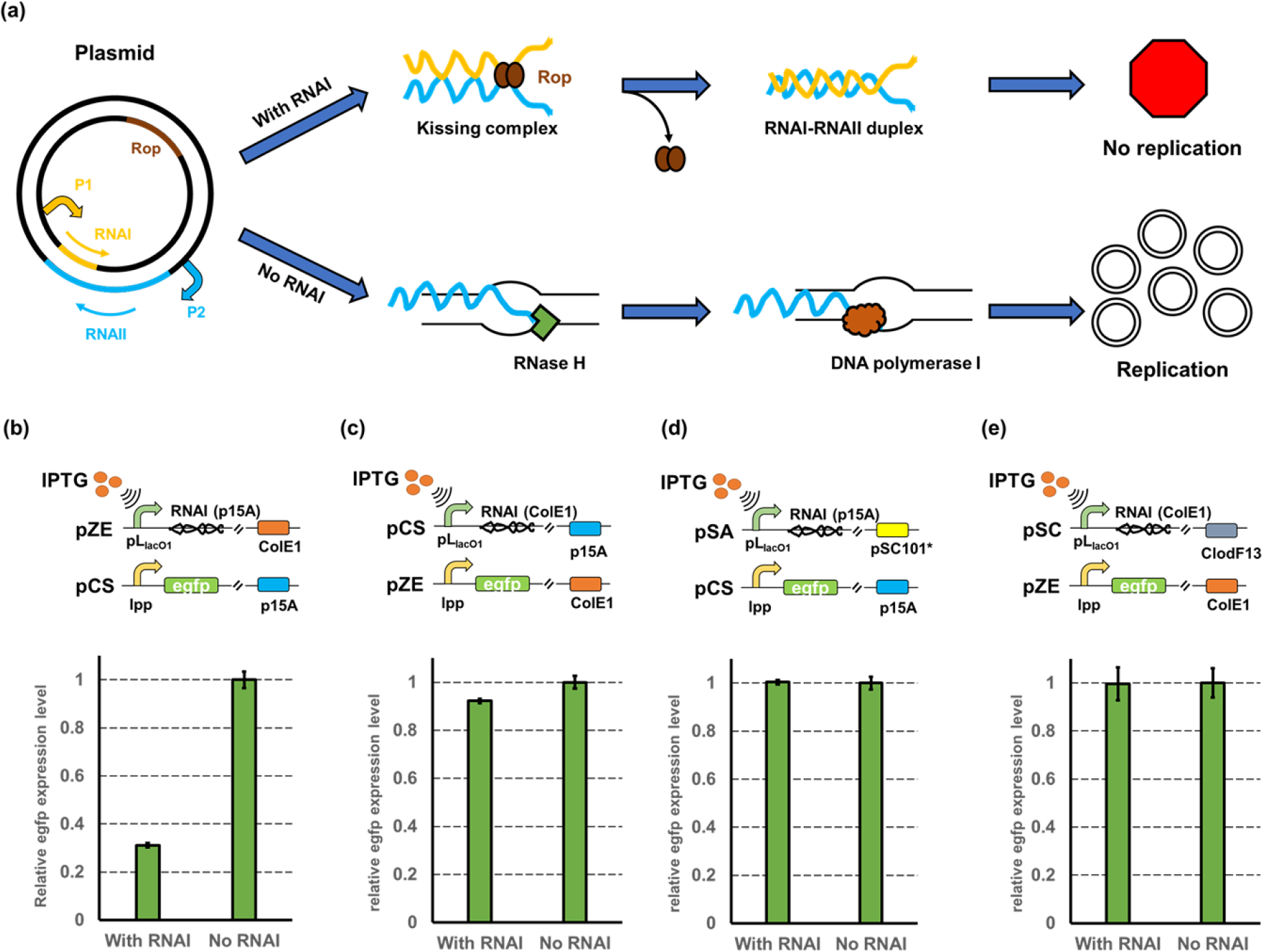
(a) Mechanism of how antisense RNAs regulate the replication of ColE1/p15A origin. (b) The inhibition efficiency on medium-copy plasmid pCS harboring the p15A origin. The RNAI of p15A was overexpressed in a high-copy plasmid pZE and the green fluorescence intensity was used for measuring the expression level of the reporter gene. (c) The inhibition efficiency on high-copy plasmid pZE harboring the ColE1 origin. The RNAI of ColE1 was overexpressed in a medium-copy plasmid pCS and the green fluorescence intensity was used for measuring the expression level of the reporter gene. (d) The inhibition efficiency on plasmid pCS when using a low-copy plasmid pSA to over-transcribe the RNAI (p15A). (e) The inhibition efficiency on plasmid pZE when using a high-copy plasmid pSC to over-transcribe the RNAI (ColE1). All data represent the mean of 3 biologically independent samples and error bars show standard deviation.
Based on this mechanism, we hypothesized that the copy number of the plasmid harboring ColE1 derived origins can be controlled by manipulating the relative RNAI/RNAII availability. To test this hypothesis, we first selected the p15A origin as the target. The p15A origin is a ColE1-derived origin with a medium copy number (15–20 copies per cell). A reporter gene encoding the enhanced green fluorescence protein (egfp) was placed in the pCS27 plasmid (harboring the p15A origin) under the control of a constitutive promoter lpp0.5 (Wang et al., 2017b). The fluorescence level normalized with the cell density (RFU/OD600) was employed as an observable evaluation of plasmid copy number (PCN). The RNAI (p15A) was constructed into a plasmid pZE12-luc under the control of an IPTG (isopropylthio-β-galactoside)-inducible promoter pLlacO1, resulting in pZE- RNAI (p15A). As expected, the over-transcribed RNAI (p15A) induced by IPTG resulted in a 68.87% decrease of the fluorescence level compared with the uninduced control (Fig. 1b). To further validate that the lowered fluorescence intensity was caused by a decreased copy number, the relative PCNs of pCS-lpp0.5-egfp with and without the over-transcription of RNAI (p15A) were measured by quantitative PCR (qPCR) (Lee et al., 2006). The result showed that a 71.63% reduction in relative PCN was observed when RNAI was over-transcribed (Supplementary Table S1). We noticed that the inhibition efficiency obtained by qPCR was slightly higher than what we observed using fluorescence assay. This was likely due to the lag of fluorescent protein degradation inside the cells after the decrease of PCN. These results demonstrated that the replication of p15A origin can be inhibited by increasing the RNAI availability.
With the success in regulating the replication of p15A origin, we then proceeded to test whether this strategy can be applied towards other ColE1 derived origins. Given that the p15A origin often provides a medium copy number in cells, we selected the ColE1 origin as our next target which gives a relatively higher copy number (70–100 copies per cell). The RNAI of the ColE1 origin was placed under the control of pLlacO1 in the pCS27 plasmid, resulting in pCS-RNAI (ColE1). The pZE-lpp0.5-egfp with the constitutively expressed egfp was used as the reporter plasmid. These two plasmids were co-transferred into E. coli BW25113 F’ to test the inhibition efficiency of RNAI on the replication of ColE1 origin. Surprisingly, the over-transcription of RNAI (ColE1) only resulted in a very slight decrease (less than 8%) in the fluorescence intensity (Fig. 1c), indicating an insufficient repression on the replication of the ColE1 origin. We hypothesized that this was due to an inadequate amount of RNAI (ColE1), as the plasmid replication was controlled by the relative balance of RNAI and RNAII. The successful manipulation of the replication of p15A origin was likely because the over-transcribed RNAI (p15A) posed evident alteration of the intracellular RNAI/RNAII ratio, while for the ColE1 origin, the over-transcribed RNAI (ColE1) did not result in significant increase in the ratio of RNAI/RNAII. To examine this hypothesis, two parallel experiments were designed: we increased the RNAI (ColE1) availability by moving it from the medium-copy plasmid pCS27 to a high-copy plasmid pSC74 (with a CloDF13 origin that gives 20–40 copies per cell) (Camps, 2010; Stuitje et al., 1979), resulting in pSC-RNAI (ColE1). Meanwhile, the RNAI (p15A) was also moved to the plasmid pSA74 (Huo et al., 2011) (with an origin of pSC101* that gives 3–5 copies in cells) to reduce its intracellular availability. The RNAI (p15A) transcribed from pSA only resulted in negligible inhibition on the replication of origin p15A (Fig. 1d), which was consistent with our expectation due to a decreased RNAI availability. However, the transcription of RNAI (ColE1) from the plasmid pSC-RNAI (ColE1) still did not result in any inhibition on the replication of ColE1 (Fig. 1e), indicating that the replication of pZE12 was insensitive to the increase of intracellular RNAI concentration, which was likely because the increased availability of RNAI (ColE1) did not result in significant change in the intracellular ratio of RNAI/RNAII. This suggested that further optimization of the copy number control strategy is required to manipulate the replication of the ColE1 origin. Our results indicated that the relative ratio of RNAI/RNAII was an important parameter for tuning the regulation efficiency on plasmid replication.
Modifying RNAI secondary structures for varied regulation efficiencies
The feasibility to inhibit the replication of p15A origin by over-transcribing RNAI was successfully demonstrated. However, this strategy was less successful when regulating the replication of the ColE1 origin which gives a relatively higher copy number. Therefore, we sought to investigate the factors related to the RNAI-RNAII interaction to enhance the inhibition efficiency. It is commonly recognized that the stability of RNA secondary structure plays an important role in RNA-RNA interactions (Cesareni et al., 1991; Guil and Esteller, 2015). Thus, we hypothesized that by optimizing the secondary structure of RNAI, the interaction between RNAI and RNAII can be enhanced and this will be beneficial for regulating the plasmid replication.
In our previous research, a double-stranded stem structure consisting of two inverted repeat DNA sequences (38 bp), namely the paired termini (PT) component, was developed to assist the inhibition of antisense RNA (Yang et al., 2015). We believe the long antisense stem in the PT component will stabilize the secondary structure of the RNAI and thus provide an enhanced inhibition efficiency on RNAII. Using the RNAI (p15A) as a proof-of-concept target, we simulated the secondary structures for both original RNAI (p15A) and RNAI (p15A) with the addition of PT component (PT_RNAI(p15A)) through a web-based modeling toolset named RNAfold (http://rna.tbi.univie.ac.at//cgi-bin/RNAWebSuite/RNAfold) (Fig. 2a). Based on the predicted outputs, the addition of PT component led to a drastically decreased Gibbs free energy ΔG0 of RNAI (p15A) from −42.29 to −129.21 kcal/mol (Fig. 2a). To experimentally test whether the improvement in RNAI stability by the addition of the PT component can enhance the regulation efficiency, the DNA sequence of PT_RNAI (p15A) was placed in the plasmid pZE12luc, forming pZE-PT_RNAI (p15A). The reporter plasmid pCS-lpp0.5-egfp and the pZE-PT_RNAI (p15A) were co-transferred into E. coli. The strain with the original RNAI (p15A) was used as a control. The normalized fluorescence intensities of induced and uninduced strains were tested and used to calculate the corresponding inhibition efficiencies. The transcription of PT_RNAI resulted in a 73.43% reduction in egfp expression, while the original RNAI (p15A) led to an inhibition efficiency of 68.58% (Fig. 2b). These results showed that the addition of the PT component on the RNAI slightly enhanced the regulation efficiency on plasmid replication. Observing this, we assumed that the addition of PT structure in RNAI (ColE1) might also improve the regulation effectiveness towards the ColE1 origin. Thus, the PT_RNAI (ColE1) was constructed and used to regulate the replication of ColE1 replication. This time, the introduction of PT_RNAI (ColE1) only enabled a 5% decrease in egfp expression level, indicating that the regulation efficiency of PT_RNAI (ColE1) was still insufficient to control the replication of ColE1 origin (Supplementary Fig. S1).
Figure 2. Optimizing the secondary structure of RNAI for enhanced inhibition efficiencies.
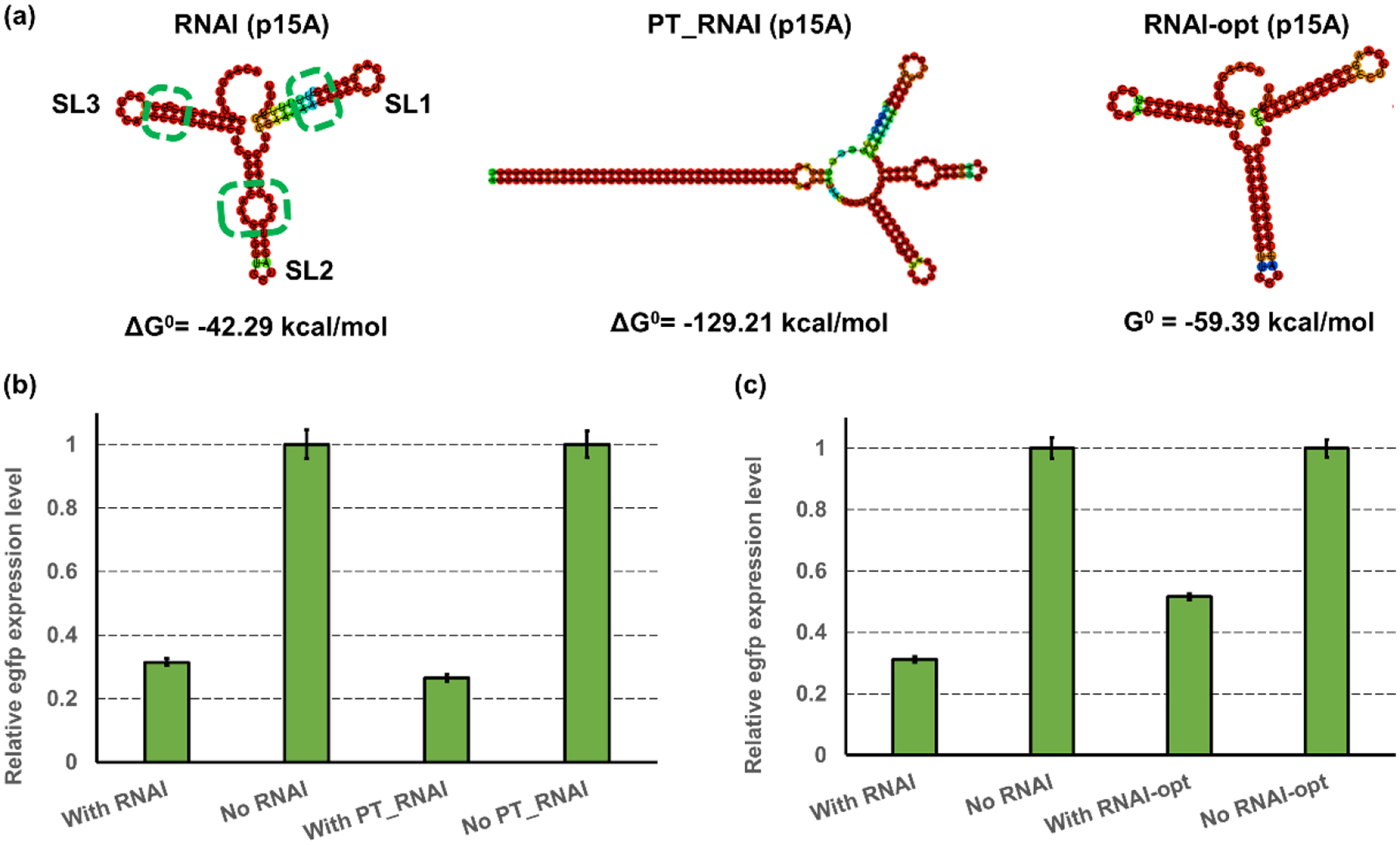
(a) The simulated secondary structures of wild-type RNAI (left), PT_RNAI (middle), and RNAI-opt (right). The structure predictions and their corresponding Gibbs free energy calculations were performed using a web-based prediction tool, RNAfold (http://rna.tbi.univie.ac.at//cgi-bin/RNAWebSuite/RNAfold). The green dashed circles label the imperfect match in each stem. (b) the inhibition efficiency on medium-copy plasmid pCS by RNAI (p15A) and PT_RNAI (p15A). The RNAI or PT_RNAI of p15A was overexpressed in a high-copy plasmid pZE and the normalized green fluorescence intensity was used for measuring the expression level of egfp. (c) the inhibition efficiency on medium-copy plasmid pCS by optimized RNAI (p15A) and RNAI-opt. The RNAI and RNAI-opt were overexpressed in a high-copy plasmid pZE and the normalized green fluorescence intensity was used for measuring the expression level of the egfp. All data represent the mean of 3 biologically independent samples and error bars show standard deviation.
After achieving enhanced stability of RNAI by the addition of an external structure, we next sought to remove the internal unstable factors in the RNAI to further enhance its stability. In the simulated structure of RNAI (p15A), there are three stem loops, SL1, SL2, and SL3 (Cesareni et al., 1991). There is an imperfect match in each of the stem loop located in the antisense paired stem (Fig. 2a). Specifically, an extra U is located in the stem of SL1, and an extra G is located in the stem of SL3. For SL2, the sequence C-AAA-G cannot pair perfectly with G-AGA-C, because “AAA” is not the reverse complementary counterpart of “AGA”. Thus, there is a bulgy ring in the stem of SL2 (Fig. 2a). We believe these mismatches may negatively affect the stability of RNAI, and the elimination of these unstable mismatches may improve the inhibition efficiency of RNAI due to an elevated stability. Therefore, we removed the extra U and G in SL1 and SL3, respectively, and the “C-AAA-G” sequence in the SL 2 was replaced by “C-TCT-G” to form a perfect antisense pair. The secondary structure of optimized RNAI was simulated in RNAfold and showed a decreased ΔG0 of −59.39 kcal/mol compared to the original RNAI (p15A) with −42.29 kcal/mol (Fig. 2a). To experimentally test the inhibition efficiency of the optimized RNAI (p15A), the plasmid pZE-RNAI-opt (p15A) using the optimized DNA sequence of RNAI (p15A) was constructed. This plasmid was co-transferred with the reporter plasmid pCS-lpp0.5-egfp. The strain with the original RNAI (p15A) was used as the control. Unexpectedly, while the unmodified RNAI (p15A) still maintained an inhibition efficiency of 68.81%, the transcription of optimized RNAI (p15A) showed a lower inhibition efficiency, with only 48.31% reduction in the egfp expression level (Fig. 2c). This decrease in inhibition efficiency was presumably because the optimized sequence of RNAI was no longer a perfect antisense counterpart of RNAII. The sequence modification introduced mismatches between RNAI and RNAII, which caused a decreased binding affinity between the RNAI and RNAII. These results demonstrated that although the stabilized secondary structure can enhance the inhibition efficiency, the perfect antisense match also plays a vital role in the RNAI-RNAII interaction. While the optimized RNAI (p15A) did not improve the regulation efficiency as we would expect, it can serve as a more relaxed control on the replication of p15A origin. The PT_RNAI, original RNAI, and RNAI-opt developed here provide varied inhibition efficiencies and may be useful in different application scenarios.
CRISPRi-mediated replication control on high-copy origin
While controllable replication inhibition can be established on the origins with medium or low copy number (e.g., p15A), efficient regulation of the high-copy origins, such as ColE1, remains challenging. As we shown before, even with the optimization of RNAI (i.e., addition of PT structure to RNAI(ColE1)), effective regulation on ColE1 replication was still difficult to achieve. We suspected that this was because the RNAI cannot efficiently neutralize RNAII due to a low ratio of RNAI/RNAII. Thus, a highly efficient regulatory tool that can control the RNAII transcription may be required to regulate the replication of high-copy origins. CRISPRi was demonstrated to be a very effective inhibition tool at the transcriptional level (Larson et al., 2013). We hypothesized that the replication of ColE1 origin can be efficiently controlled by introducing the CRISPRi to repress the transcription of RNAII, which can lead to decreased intracellular RNAII availability. Therefore, we adopted the CRISPRi into our strategy to test the feasibility of controlling the ColE1 replication. Two small guide RNAs (sgRNA) were designed to target different regions of the RNAII transcription cassette in the ColE1 origin. One is sgRNA-TIS with the target position located at 6–26 bp upstream of the transcription initiation site (TIS), namely the P2 promoter region. The other one is sgRNA-CDS with the target position located at 13–33 bp downstream of the TIS (Fig. 3a). These two sgRNAs were placed under the control of the IPTG-inducible promoter pLlacO1 and constructed into the plasmid pSA-dCas9 containing the deactivated Cas9 protein (D10A/H841A) from Streptococcus pyogenes (Wang et al., 2017c), forming pSA-dCas9-sgRNA (ColE1-TIS) and pSA-dCas9-sgRNA (ColE1-CDS), respectively.
Figure 3. CRISPRi-mediated controllable inhibition of replication.
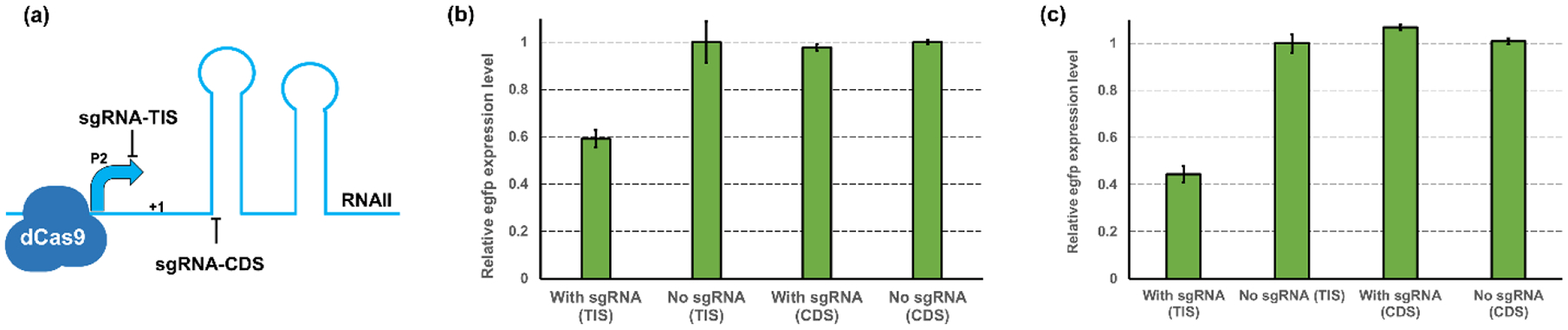
(a) Scheme of CRISPRi-mediated transcriptional repression on RNAII of ColE1. The sgRNA-TIS is targeting the promoter region (P2) of RNAII and the sgRNA-CDS is targeting the transcript sequence of RNAII. (b) The inhibition efficiency on high-copy plasmid pZE by sgRNA-TIS and sgRNA-CDS. These two sgRNA and the dCas9 protein were overexpressed in a low-copy plasmid pSA74. The normalized green fluorescence intensity was used for measuring the expression level of the reporter gene. (c) The inhibition efficiency on medium-copy plasmid pCS by sgRNA-TIS and sgRNA-CDS. These two sgRNA and the dCas9 protein were overexpressed in a low-copy plasmid pSA74. The normalized green fluorescence intensity was used for measuring the expression level of the reporter gene. All data represent the mean of 3 biologically independent samples and error bars show standard deviation.
A 40.87% decrease of egfp expression level was achieved when targeting the promoter region by sgRNA-TIS, while only a less than 3% inhibition can be observed when using sgRNA-CDS (Fig. 3b). The difference in the inhibition efficiency was likely because the binding of dCas9 protein guided by sgRNA-TIS caused steric hindrance and blocked the access of RNA polymerase to the promoter P2, but this hindrance was less prominent when the binding of dCas9 moved to the downstream sequence, and such decrease in regulation efficiency would no longer be able to cause obvious inhibition on plasmid replication. qPCR results also confirmed that the reduction in the egfp expression level was due to a decreased plasmid copy number, with a 53.98% decrease in relative plasmid concentration can be detected (Supplementary Table. S2). These results proved our hypothesis that inhibiting the transcription of RNAII can efficiently control the replication of ColE1 origin. We then proceed to see how well CRISPRi can perform in regulating the p15A origin, and a similar trend was observed when using CRISPRi to target the transcription of RNAII in p15A origin (Fig. 3c). The sgRNA-TIS for RNAII (p15A) caused a 55.56% reduction in egfp expression, while the sgRNA-CDS for RNAII (p15A) did not result in any obvious decrease in the normalized fluorescence intensity. We noticed that the regulation efficiency on plasmid pCS is slightly better than on pZE. This was likely because the copy number of pZE is higher than that of pCS, and this means a lower concentration of sgRNA is required to saturate the inhibition on the replication of plasmid pCS. However, the inhibition efficiency on p15A replication achieved by CRISPRi was lower than that by over-transcribing RNAI (p15A), which can maintain an inhibition efficiency of 68% (Fig. 1b). We suspect this was likely because the RNAI (p15A) was over-transcribed from a high-copy plasmid pZE12 (70–100 copies per cell), while the dCas9 and sgRNA were expressed from a low-copy plasmid pSA74 with pSC101* origin (3–5 copies per cell). Thus, the introduced dCas9 and sgRNA may not be adequate to efficiently repress the RNAII transcription, and this can lead to a decreased inhibition efficiency in comparison to RNAI (p15A).
Controllable gene copy enhancement by reducing RNAI availability
On validating the controllable inhibition on replication of ColE1 derived origins, we then sought to test whether the controllable activation is achievable by reducing the intracellular RNAI/RNAII ratio. We hypothesized that, if the intracellular RNAI/RNAII ratio can be reduced, either through lowering the RNAI concentration or repressing the transcription of RNAI, the plasmid replication would be enhanced, pushing the copy number to a higher level. To test this hypothesis, we designed the antisense RNAI (asRNAI, reverse complementary to RNAI) of p15A origin that would bind with RNAI and decrease the intracellular concentration of free RNAI. The coding sequence of asRNAI was placed under the control of pLlacO1 promoter, forming pZE-pLlacO1-asRNAI (p15A). This plasmid was used to target the replication of p15A origin using plasmid pCS-lpp0.5-egfp as the reporter (Fig. 4a). As expected, the introduction of asRNAI resulted in a 5.39-fold increase in the egfp expression level (Fig. 4b), which demonstrated that it was feasible to increase the copy number of p15A origin by decreasing the RNAI availability. Since the PT structure was demonstrated to be efficient in improving the regulation efficiency of RNAI, we also tested whether the introduction of the PT structure into the asRNAI would cause increased regulation effectiveness. While the PT_asRNAI still enabled a 5.08-fold increase in egfp expression level (Fig. 4b), The results suggested that both asRNAI and PT_asRNAI can achieve high activation efficiencies.
Figure 4. Controllable gene copy enhancement by reducing the RNAI availability.
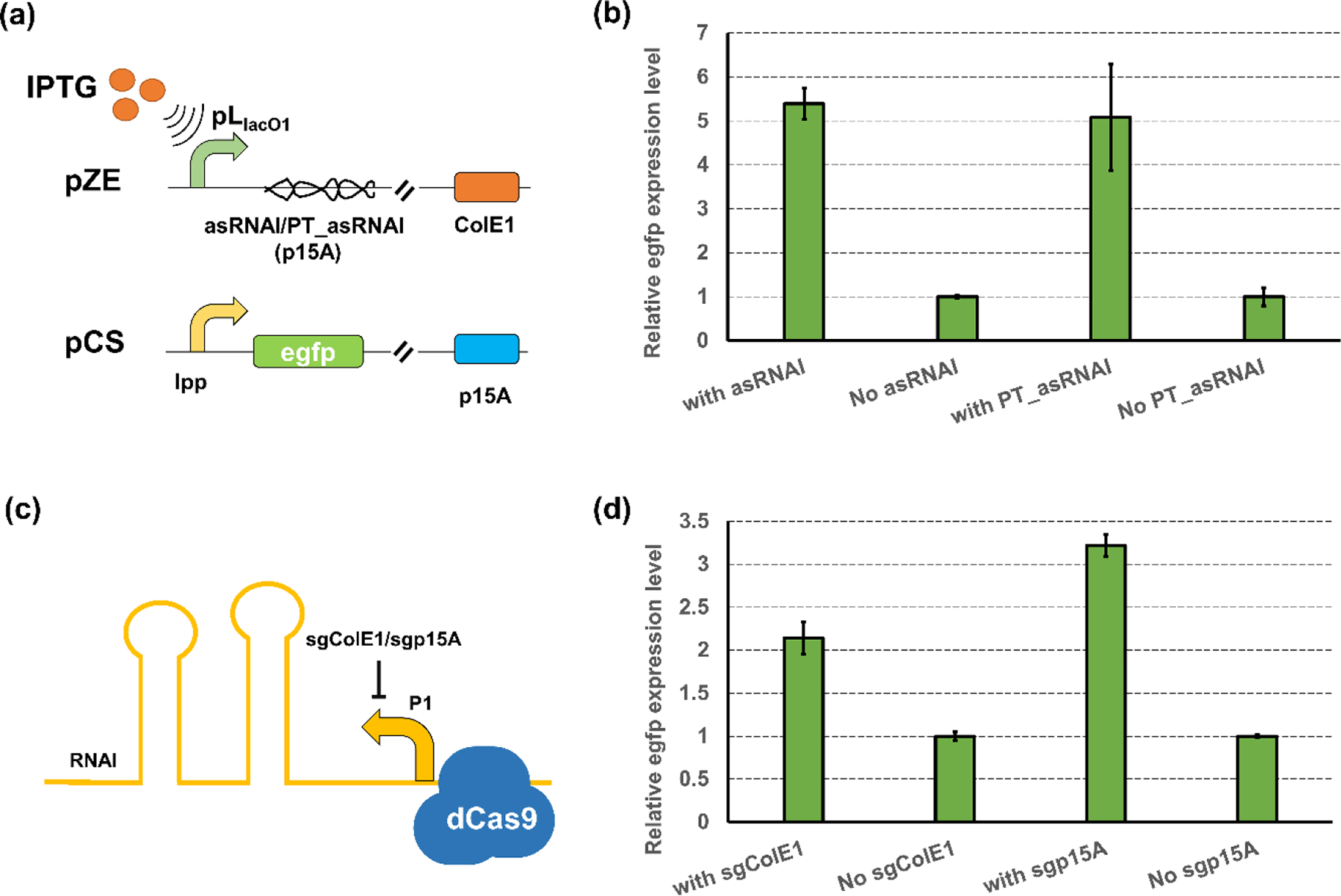
(a) Plasmid configuration of asRNAI- or PT_asRNAI-mediated repression on RNAI (p15A) availability (b) The activation efficiencies of asRNAI- or PT_asRNAI-mediated replication enhancement on p15A replication. (c) Scheme of CRISPRi-mediated transcriptional repression on RNAII of ColE1 (d). The activation efficiency when using sgColE1 or sgp15A to target RNAI transcription in ColE1 or p15A origin, respectively. All data represent the mean of 3 biologically independent samples and error bars show standard deviation.
After achieving the controllable activation on p15A replication, we next proceeded to investigate whether the replication of the high-copy ColE1 origin can be enhanced by repressing the transcription of RNAI. As the replication of ColE1 origin is insensitive to RNA-level control, the CRISPRi, which was demonstrated successful in inhibiting the ColE1 replication, was applied to target the transcription of RNAI (ColE1). Since the p15A origin can also be regulated by CRISPRi (Fig. 3c), we also include the p15A origin as a target in the test. Thus, two sgRNAs targeting the RNAI promoter region of ColE1 and p15A origin (Fig. 4c), namely the sgColE1 and sgp15A, respectively, were designed and constructed in the pSA-dCas9 plasmid containing the dCas9 protein expression cassette, forming pSA-dCas9-sgColE1 and pSA-dCas9-sgp15A. The strain harboring only the dCas9 protein expression cassette but without any sgRNA was used as a control. The introduction of CRISPRi on the RNAI (ColE1) transcription enabled a 2.14-fold increase in egfp expression over the control (Fig. 4d). Similarly, the inhibition on RNAI (p15A) transcription by CRISPRi also resulted in a 3.22-fold increase in the egfp expression level (Fig. 4d). The regulation on RNAI transcription led to a more prominent effect on plasmid pCS than on pZE, which was similar to what we observed when using CRISPRi to inhibit the transcription of RNAII. Also, the CRISPRi-mediated enhancement on p15A replication was less effective compared with what can be achieved by RNA-level control (3.22-fold vs 5.39-fold), which was consistent with what we observed in inhibition of the p15A replication (Fig. 1b,3c). Our results demonstrated that the decrease of RNAI/RNAII ratio, either by lowering the RNAI concentration or repressing the transcription of RNAI, would cause enhanced replication of ColE1 derived origins. The controllable activation, along with the controllable inhibition of the plasmid replication demonstrated in previous sections, can be used to enable diversified dynamic control of gene copy via regulating the plasmid copy number.
Exploration of simultaneous control of two plasmids
Applying the CRISPRi in dynamic gene copy control enabled the regulation on both the p15A and ColE1 origin. One advantage of CRISPRi is the ease of achieving multiplex regulation by designing different sgRNAs. As the two-plasmid configuration is usually used in metabolic engineering and synthetic biology research, we explored the potential of using sgRNAs to target the ColE1 and p15A origin at the same time for simultaneous regulation of both origins. As a demonstration, we selected the sgRNA-TIS (ColE1) and sgRNA-TIS (p15A), which target the RNAII transcription of ColE1 and p15A origin, respectively. The two sgRNA transcription cassettes were placed on plasmid pSA-dCas9, resulting in plasmid pSA-dCas9-Co_sgRNA. Both the pZE12 plasmid carrying the red fluorescence protein (RFP) and pCS27 plasmid carrying the egfp were used as the reporter plasmids. The induction of sgRNA transcriptions resulted in a 34.63% reduction of RFP expression level and a 47.49% in egfp expression level (Supplementary Fig. S2), demonstrating the capability of co-inhibiting two plasmids at the same time. To see whether the regulation on two plasmids is orthogonal, we individually targeted only one plasmid when both reporter plasmids were present. The inhibition on p15A origin successfully led to a 48.11% decrease in egfp expression level, but only slightly affected the red fluorescence level. The inhibition on ColE1 origin resulted in a 31.75% decrease in RFP expression level, and did not affect the egfp expression level (Supplementary Fig. S2). These results proved that the co-regulation of two origins is feasible, and the regulations on two plasmids are orthogonal.
Designing genetic circuits to enable diverse dynamic control of gene copy
With the validation of both controllable reduction and increase of gene copy through regulating the replication of origins, our next goal is to achieve diversified dynamic regulation of the gene copy using biosensor-enabled genetic circuits with different control logics. To this end, three different control logic were designed. The first control logic enables a dynamic behavior of direct “normal-to-low” control: the gene copy would be maintained at a normal level in the beginning but reduced when there is a signal input. As a result, the gene copy would be reduced to a lower level. The second control logic on the other hand, is direct enhancement of gene copy, forming a “normal-to-high” dynamic behavior: the gene copy would be maintained at the normal level when there is no signal input, but the replication would be enhanced to improve the gene copy when the input is present. The third control logic enables a more complex regulation: the inhibition on gene copy will be executed at the beginning, forcing a low-level of gene copy. When there is a signal input, the inhibition on gene copy would be released, and the gene copy would be restored to the normal level.
To apply the designed control logic, biosensor-enabled genetic circuits with the desired dynamic behaviors are employed. As a proof-of-concept demonstration, the optimized p-coumaric acid-responsive sensor system PadR-PpadC and its variants developed in our previous study (Jiang et al., 2021) were selected. PadR is a transcriptional repressor that can bind with PpadC (and its variant/hybrid promoters) to inhibit the promoter activity, and this inhibition would be released when p-coumaric acid is present (Jiang et al., 2021; Nguyen Thi Kim et al., 2011). To enable varied dynamic performance of the genetic circuits, we employed both the wild type (WT) PadR and a PadR (K64A) variant with increased responsiveness but less leaky activity from our previous study (Jiang et al., 2021) in demonstrating all three control logics. For each control logic, we designed two versions of genetic circuits, one for regulating the medium-copy p15A origin and the other is for high-copy ColE1 origin. When regulating the replication of medium-copy origin p15A, the PT_RNAI/asRNAI were used, while the CRISPRi system (dCas9 with sgRNAs) was applied when regulating the replication of high-copy origin ColE1. As we demonstrated in previous section, the control of p15A replication required a high concentration of RNA (PT_RNAI or asRNAI), while a small amount of sgRNA was sufficient to regulate the ColE1 replication when using CRISPRi. Thus, the strong promoter P9 that responds to p-coumaric acid (Jiang et al., 2021) was used to control the transcription of PT_RNAI or asRNAI, while the weaker promoter PpadC-FG (Jiang et al., 2021) was used for sgRNAs targeting the ColE1 replication.
For constructing the circuits with the first control logic (circuit 1), the PT_RNAI (p15A) and sgRNA-TIS (ColE1) were placed under the control of the p-coumaric acid responsive promoters P9 and PpadC-FG, respectively. The regulator gene padR was under the control of the constitutive promoter lpp0.2 (Wang et al., 2017b) for optimal sensor performance (Jiang et al., 2021) (Fig. 5a,c). As we designed, the promoter P9 and PpadC-FG can be repressed by PadR when no p-coumaric acid is present, but such repression will be released when p-coumaric acid is added. The P9 or PpadC-FG would then initiate the transcription of PT_RNAI (p15A) or sgRNA-TIS (ColE1), respectively, which would lead to inhibition on plasmid replication and reduce the gene copy. To test the circuit performance, gradient concentrations of p-coumaric acid (0, 100 mg/L, 500 mg/L) were fed into the media after 2h of inoculation. The dynamic performance of an unregulated control (without the PT_RNAI or sgRNA transcription operon) was also tested. As expected, with similar cell growth (Supplementary Fig. S3a,b), the addition of p-coumaric acid resulted in obvious reductions in egfp expression level when targeting either the p15A (Fig. 5b) or the ColE1 origin (Fig. 5d) (up to an inhibition efficiency of 37.45% for regulating p15A origin and 55.76% for ColE1 origin). Interestingly, we noticed that the dynamic performance of genetic circuits was largely dependent on the PadR regulator. For example, since the PadR (K64A) exhibits less leaking activity (Jiang et al., 2021), when no p-coumaric acid was added, the reduction in egfp expression level of the circuit harboring the PadR (K64A) variant (K64A-0, red line, 2.55% reduction at 12h) was less prominent compared with the circuit harboring the WT PadR (WT-0, orange line, 6.53% reduction at 12h) when regulating the p15A origin (Fig. 5b). This was especially obvious in regulating the ColE1 origin (17.79% reduction in WT-0 vs 3.08% reduction in K64A-0) (Fig. 5d). Also, due to a higher responsiveness of PadR (K64A) towards p-coumaric acid, the inhibition on gene copy was more prominent when using PadR (K64A), as both the highest inhibition efficiencies were achieved when the circuit was equipped with K64A and fed with 500 mg/L p-coumaric acid. (Fig. 5b,d). Prominent dose-dependent effect was observed in regulating ColE1 origin (Fig. 5d). The reduction in egfp expression level induced by 500 mg/L p-coumaric acid was higher than that of induced by 100 mg/L p-coumaric acid in both circuits harboring the WT PadR- (43.44% vs 54.97%) and PadR (K64A) (43.79% vs 55.76%). All these results showed that the performance of the biosensor was well-transited to the dynamic performance of the genetic circuits.
Figure 5. Constructing genetic circuits with different control logic to diversify the dynamic gene copy control strategy.
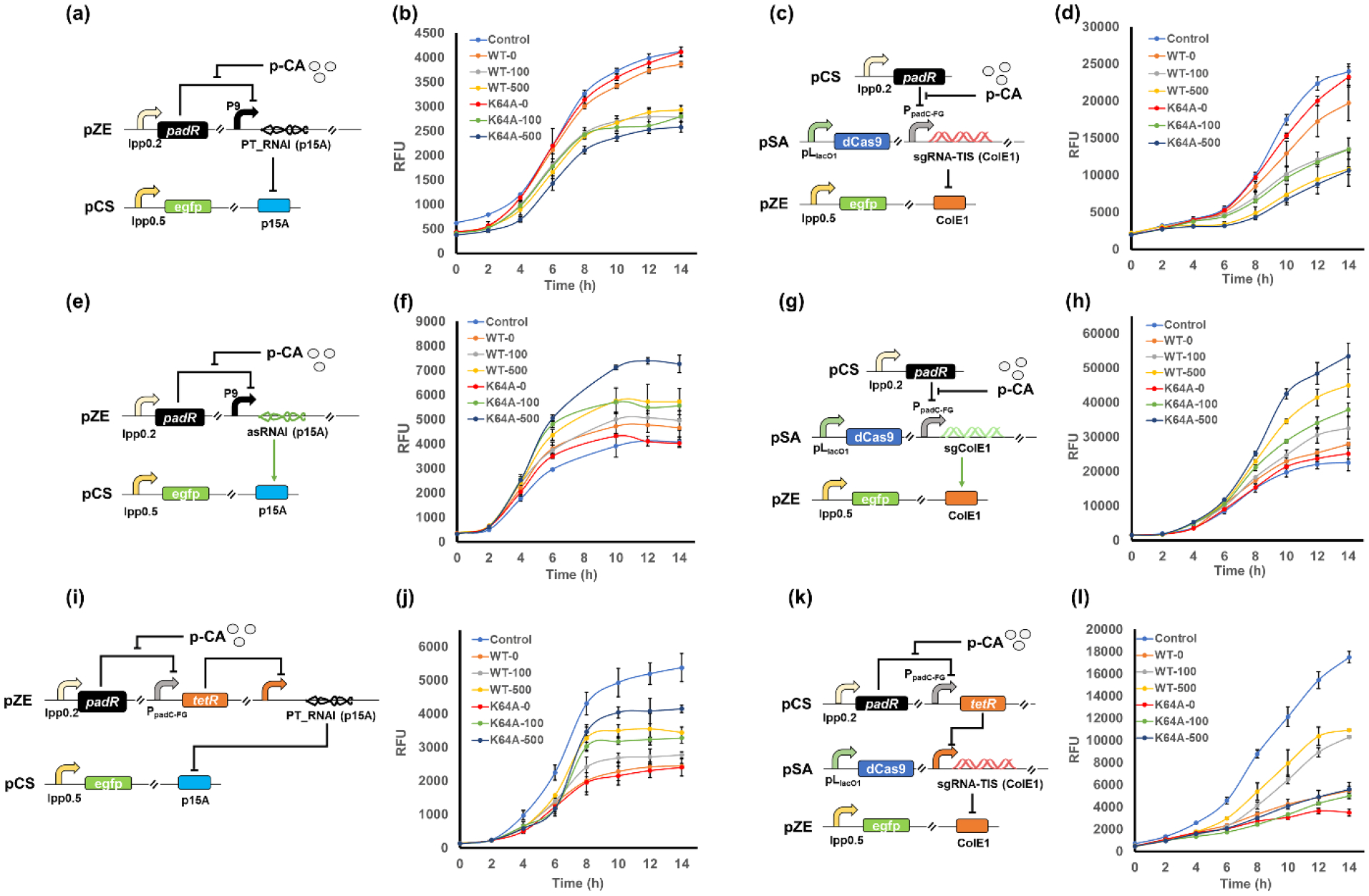
(a) Scheme of the dynamic genetic circuit 1 targeting p15A origin using PT_RNAI (p15A). (b) The time-profiled egfp expression level when applying circuit 1 to regulate the replication of p15A origin. WT, circuit harboring the wild type PadR. K64A, circuit harboring PadR (K64A) variant. The number behind the dash indicated the concentration (mg/L) of p-coumaric acid. (c) Scheme of the dynamic genetic circuit 1 targeting ColE1 origin using sgRNA-TIS (ColE1). (d) The time-profiled egfp expression level when applying circuit 1 to regulate the replication of ColE1 origin. WT, circuit harboring the wild type PadR. K64A, circuit harboring the PadR (K64A) variant. The number behind the dash indicated the concentration (mg/L) of p-coumaric acid. (e) Scheme of the dynamic genetic circuit 2 targeting p15A origin using asRNAI (p15A). (f) The time-profiled egfp expression level when applying circuit 2 to regulate the replication of p15A origin. (g) Scheme of the dynamic genetic circuit 2 targeting ColE1 origin using sgColE1. (h) The time-profiled egfp expression level when applying circuit 2 to regulate the replication of ColE1 origin. (i) Scheme of the dynamic genetic circuit 3 targeting p15A origin using PT_RNAI (p15A). (j) The time-profiled egfp expression level when applying circuit 3 to regulate the replication of p15A origin. (k) Scheme of the dynamic genetic circuit 3 targeting ColE1 origin using sgRNA-TIS (ColE1). (l). The time-profiled egfp expression level when applying circuit 3 to regulate the replication of ColE1 origin. RFU, relative fluorescence unit. All data represent the mean of 3 biologically independent samples and error bars show standard deviation.
The genetic circuits with the second regulation logic (circuit 2) were constructed with a similar configuration to the circuit 1, but the regulation tools that were under the control of p-coumaric acid-responsive promoters were changed to asRNAI (p15A) or sgColE1 (Fig. 5e,g). We also tested the dynamic performance of circuit 2 with both WT PadR and PadR (K64A), aiming to see whether the sensor dynamic properties would again, like what we observed in testing the first control logic, be transited to the dynamic performance of the circuit 2. Gradient concentrations of p-coumaric acid (0, 100 mg/L, 500 mg/L) were added to the media after 2h of inoculation. An unregulated strain (without the asRNAI or sgColE1 transcription cassettes) was also included in the test as a control. Both versions of the circuit 2 performed well as we expected. The addition of p-coumaric acid enhanced the plasmid replication and improved the egfp expression level (Fig. 5f,h), with an up to 1.78-fold and 2.37-fold increase can be observed at 14h for regulating p15A and ColE1 origin, respectively. Slight growth defects can be observed when applying circuit 2 in regulating the high-copy ColE1 origin (Supplementary Fig. S3c,d), which was likely because the further increased gene copy led to elevated burdens on cells. Dose-dependent effect was also observed in both regulations of p15A and ColE1 replication (Fig. 5f,h). Notably, the dynamic properties of the biosensor was again well transited to the dynamic performance of the circuit, as the circuit 2 harboring the PadR (K64A), compared to the circuit equipped with WT PadR, exhibited less leaky enhancement but higher activation efficiencies (Fig. 5f,h).
The control logic for the third type of genetic circuits (circuit 3) resembles the function of a genetic inverter, which adds an extra layer of regulation compared to the previous two strategies. To achieve this designed function, this circuit requires an additional regulator, for which we chose the well-studied and commonly used “repressor-promoter” combo, TetR-pLtetO1. The pLtetO1 is a constitutive promoter when the repressor TetR is not present, but its activity would be inhibited when TetR is expressed (Lutz and Bujard, 1997). Thus, the pLtetO1 promoter was used to control the transcription of PT_RNAI (p15A) or sgRNA-TIS (ColE1). The tetR gene was under the control of a weaker promoter PpadC-FG instead of hybrid promoter P9 due to the high sensitivity of the TetR-pLtetO1 system (a small amount of TetR protein is sufficient to fully repress the pLtetO1 promoter) (Lutz and Bujard, 1997). We first tested how this circuit can manipulate the gene copy via regulating the medium-copy p15A origin. An unregulated strain with only the sensor and the reporter plasmid (no PT_RNAI transcription cassette) was used as the positive control (Control). Gradient concentrations of p-coumaric acid (0, 100 mg/L, 500 mg/L) were added to the media after 2h of inoculation. The induction of tetR expression by p-coumaric acid enabled increased fluorescence intensities compared with the strain without any p-coumaric acid feeding (WT-0 and K64A-0), indicating the inverter function of circuit 3 was successfully established on regulating p15A origin (Fig. 5j). Dose-dependent effect was again observed, as the increased p-coumaric acid concentration resulted in a better recovery of egfp expression, with up to 77.17% activity can be recovered by the circuit harboring the PadR (K64A) induced by 500 mg/L p-coumaric acid (Fig. 5j). Similar to the previous two circuits, the sensor performance was also well-transited to the circuit, as the K64A variant can enable a better recovery of egfp expression (up to 77.17% egfp expression can be recovered) than that of WT PadR (up to 64.03%). We then tested whether this control logic could also work on regulating the high-copy ColE1 origin. For the circuit 3 controlled by WT PadR, the introduction of p-coumaric acid released the inhibition on ColE1 replication and resulted in increased egfp expression with dose-dependent effect (Fig. 5l). Up to 62.54% egfp expression level can be resumed by the genetic inverter. Notably, the strains with induced circuit 3 (WT-100 and WT-500) showed better cell growth (Supplementary Fig. S3f), indicating that the circuit 3 alleviated the cell burdens from excessive copy of egfp. However, unlike what we observed in regulating p15A replication (Fig.5j), the K64A variant did not result in better recovery of egfp expression this time. Instead, the circuit 3 harboring the PadR (K64A) can be barely activated by p-coumaric acid (Fig. 5l). We suspected that this was due to a stringent control of PpadC-FG by PadR (K64A), and this led to a stricter inhibition on TetR expression, which subsequently resulted in a higher activity of pLtetO1 and increased sgRNA concentration. As the regulation on ColE1 replication was sensitive to the sgRNA availability, the increased sgRNA concentration make it harder for the circuit 3 to eliminate the inhibition and restore the gene copy of egfp. Overall, the function of all three designed control logics have been demonstrated, and the developed genetic circuits with these dynamic behaviors would be beneficial for diversifying the dynamic regulation of gene copy in metabolic engineering.
Application of dynamic control of gene copy in p-coumaric acid biosynthesis
While we have validated and diversified the dynamic control of gene copy, the potential of this strategy in real-world applications remains unexploited. To test the applicability of dynamic control of gene copy, the biosynthesis of p-coumaric acid was selected as a proof-of-concept demonstration. p-coumaric acid is a high-value aromatic compound that serves as an important precursor for synthesizing many valuable natural products, such as naringenin, resveratrol, and apigenin (Siedler et al., 2014; Yang et al., 2015). Microbial production of p-coumaric acid is usually catalyzed by tyrosine ammonia lyase (TAL), which employs the L-tyrosine as the precursor. In our previous studies, we have established an efficient de novo p-coumaric acid biosynthesis pathway by employing four key enzymes from E. coli, TyrA*, PpsA, TktA, and AroG* (hereafter referred as TPTA, * indicated that the enzyme is feedback-resistant) along with the TAL from Rhodotorula glutinis (RgTAL) (Huang et al., 2013; Wang et al., 2017b) (Fig. 6a). Thus, the p-coumaric acid biosynthesis pathway containing the genes encoding the TPTA and RgTAL was assembled to the pCS27 plasmid, resulting in pCS-TPTA-RgTAL. As demonstrated in our previous study (Yang et al., 2018), the inhibition on ppc gene via antisense RNA would enhance the carbon flux towards shikimate pathway, which would be beneficial in improving the production of p-coumaric acid. Thus, the asppc (antisense RNA of ppc that inhibit the expression of genomic ppc) transcription cassette was also integrated with the p-coumaric acid pathway, forming pCS-TPTA-RgTAL-asppc. To direct more carbon flux to the p-coumaric acid biosynthesis and enhance the tyrosine accumulation, the competing pathways in E. coli BW25113 F’(Atsumi et al., 2008) were eliminated by knocking out pheA (for phenylalanine biosynthesis), pykAF (consuming the precursor phosphoenolpyruvate, PEP), and tyrR (encoding the regulator TyrR that conducts tyrosine-responsive feedback inhibition on tyrosine biosynthesis) (Fig. 6a). The final strain E. coli BW-PCA (BW25113 F’ ΔpheA ΔtyrR ΔpykA ΔpykF) was used for applying dynamic gene copy regulation in p-coumaric acid biosynthesis.
Figure 6. Implementing dynamic gene copy control in p-coumaric acid biosynthesis.
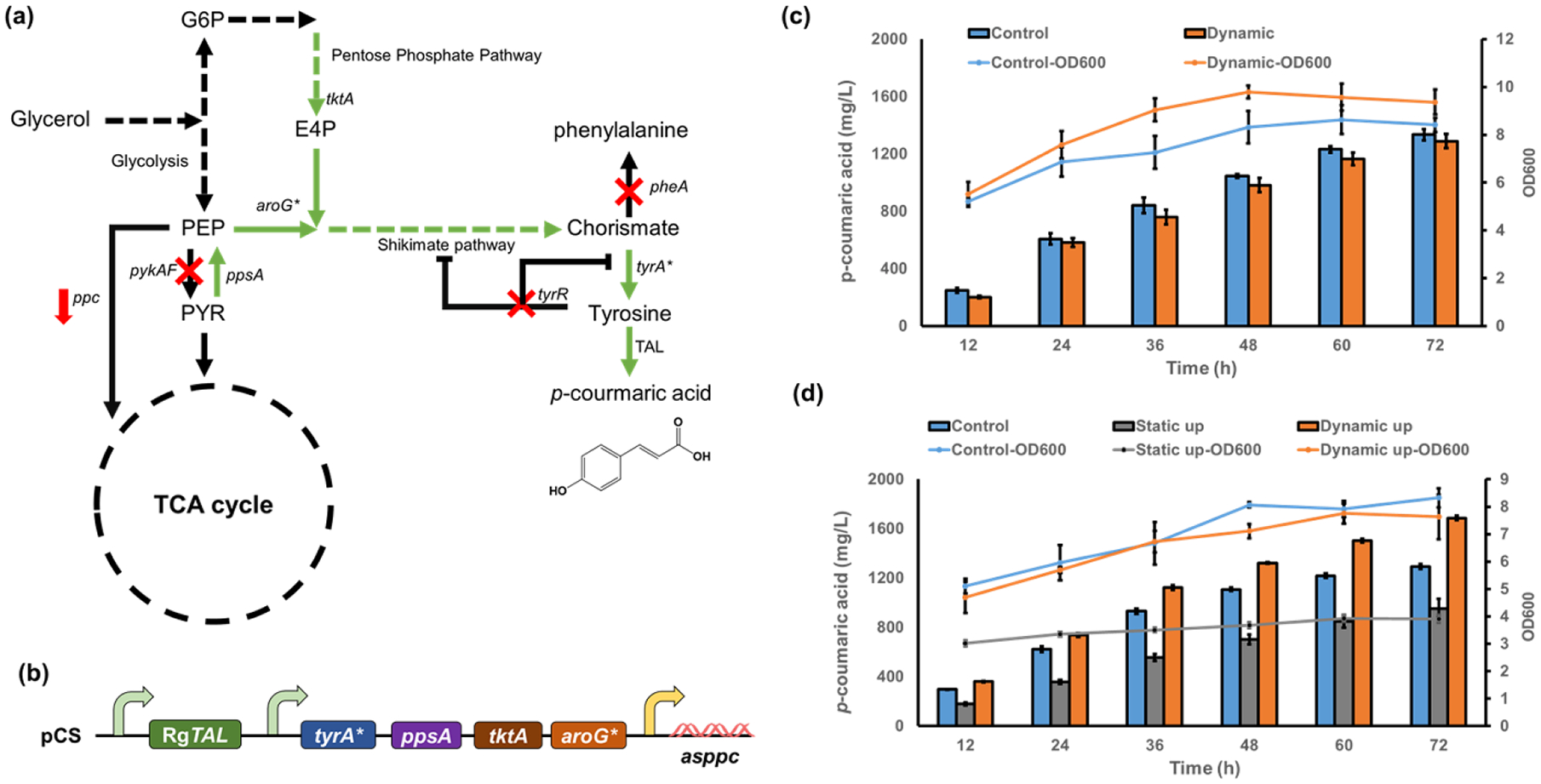
(a) The de novo biosynthetic pathway for p-coumaric acid in E. coli. (b) The plasmid harboring the p-coumaric acid biosynthesis pathway, which consist of three operons and a total of six target genes. (c) Growth curves and p-coumaric acid production profile by dynamic “low-to-normal” strategy (d) Growth curves and p-coumaric acid production profile by dynamic “normal-to-high” strategy. All data represent the mean of 3 biologically independent samples and error bars show standard deviation.
The biosynthesis of p-coumaric acid requires directing carbon flux from glycolysis to shikimate pathway (Fig. 6a). It will compete with central metabolism for PEP (phosphoenolpyruvate) and E4P (erythrose 4-phosphate), which may reduce the biomass generation and negatively affect the cell performance. To solve this problem, we first employed the dynamic “low-to-normal” control logic (Fig. 5i) to regulate the replication of all genes in the plasmid pCS-TPTA-RgTAL-asppc (with a p15A origin) (Fig. 6b). The copy numbers of the pathway genes would be kept at a low level when no p-coumaric acid is present. With cell growing and the accumulation of p-coumaric acid, the inhibition on gene replication would be released, and the pathway expression will be restored to a normal level, directing carbon flux towards the final product. We aimed to reduce the competition between cell growth and p-coumaric acid production using this design. To achieve the function, the circuit with the third control logic (Fig. 5i) was used to control the replication of all pathway genes in the plasmid. The circuit harboring the PadR (K64A) variant was used as this circuit exhibited a stricter inhibition and higher recovery efficiency of gene copy than the circuit with WT PadR (Fig. 5j). The strain without the dynamic gene copy regulation was used as the control. As expected, the cell growth was boosted by the dynamic gene copy regulation, with up to 24.52% increase in cell density at 36h and an 11.33% increase at 72h can be observed (Fig. 6c). However, the p-coumaric acid production did not further increased with the enhanced cell growth (Fig. 6c). A titer of 1.29 g/L p-coumaric acid can be accumulated using the dynamic gene copy control, while the unregulated control can generate a slightly higher titer of 1.33 g/L (Fig. 6c). We suspected this was due to the trade-offs between cell growth and production when using dynamic pathway control: when the pathway expression was at a low level, more carbon fluxes would be directed to biomass generation to improve the cell growth. However, the increased biomass generation, at the price of reduced pathway expression, reduced the carbon fluxes that can be used for microbial synthesis and led to a declined p-coumaric acid production.
As the better cell growth did not lead to increased production of p-coumaric acid, we hypothesized that the metabolic burdens arising from the pathway expression was still in an acceptable range. Next, the circuit with the “normal-to-high” control logic (circuit 2) was employed to dynamically improve the pathway gene copies. In this design, the pathway genes would maintain a normal copy number at the beginning of fermentation, but the gene replications would be dynamically enhanced with the accumulation of p-coumaric acid. Again, due to a better dynamic performance, the PadR (K64A)-enabled circuit was used to up-regulate the replication of p15A origin. The strain without dynamic gene copy regulation and the strain with static gene copy regulation (constitutively enhanced gene copy) were used as the control. As expected, the dynamic up-regulation of gene copies improved the production of p-coumaric acid, with the highest titer reaching 1.69 g/L in shake flasks (Fig. 6d). The unregulated strain accumulated 1.29 g/L p-coumaric acid, while the strain with static regulation exhibited growth defects and only produce a titer of 0.95 g/L p-coumaric acid (Fig. 6d). The dynamic regulation of gene copy resulted in 31.01% and 77.89% increase in p-coumaric acid titers compared to the unregulated control and static control, respectively (Fig. 6d). The plummeted titer by the static regulation confirmed that the excessive copies of pathway genes would negatively affect the cell growth and led to decreased production performance. The dynamic up-regulation, on the other hand, avoided the overwhelmed metabolic burden at the beginning compared to the static regulation, but gradually directed more carbon fluxes to p-coumaric acid synthesis when cells enter to post-log phase and stationary phase compared to the unregulated strain. Our results demonstrated the great potential of employing the dynamic gene copy control in real-world applications. Notably, our results also highlighted the importance of appropriately applying dynamic pathway control based on different regulation scenarios. When the metabolic burdens or competition between biomass generation and products accumulation did not cause significant defects on cell growth, applying dynamic down-regulation would reduce the carbon fluxes towards target compounds. This loss in compounds accumulation is unlikely to be made up by the better cell growth in this case. At this time, the “dynamic-up” regulation with gradually increased pathway expression would be more helpful in improving the biosynthesis, as demonstrated in our experiments. Overall, these results highlighted the great potential of applying the dynamic pathway control at gene replication level for engineering microbial cell factories.
Discussion
Dynamic regulation has attracted growing attentions in recent years as it provides a new solution to intelligently direct carbon flux, inhibit competing pathways that are essential to cell growth, or minimize accumulation of toxic intermediates, based on intracellular chemical concentrations and environmental signals. To enable efficient dynamic regulation, plenty of toolsets and strategies, as discussed in the introduction section, have been developed and engineered to improve the performance of microbial cell factories. While these strategies have been demonstrated efficient in regulating gene expression at transcriptional, translational, and post-translational level, the dynamic regulation at gene replication level, or gene copy level, to address the inherent burdens coming from unnecessary high copies of genes or low pathway efficiencies from insufficient gene replications, has been rarely explored.
Here, we successfully established dynamic control at gene replication level on plasmids with ColE1 derived origins using tunable biosensor and genetic circuits. As demonstrated in this study, by manipulating the transcription of RNAI and RNAII, the gene copy can either be dynamically increased or decreased in a tunable way. To diversify the control logic of regulating gene replication, genetic circuits equipped with biosensor were constructed and enabled different dynamic behaviors of gene replication. To investigate the industrial potential of this strategy, we applied dynamic gene copy control in p-coumaric acid biosynthesis, and enhanced the production of p-coumaric acid by up to 78% to a titer of 1.69 g/L in shake flasks. More importantly, the dynamic gene copy regulation developed in this study is tunable at multiple levels. By engineering the secondary structure of RNAI, using different regulatory tools (e.g., CRISPRi), and employing biosensor variants with different dynamic performance, the performance of the genetic circuits can be fine-tuned to meet different regulation requirements in various application scenarios. The p-coumaric acid titer was moderately enhanced by dynamic gene copy regulation. However, the production improvement by dynamic pathway regulation can be case-dependent, and it is also closely related to factors such as the dynamic properties of the biosensors, the pathway kinetics, and the cultivation conditions. Thus, the case of p-coumaric acid biosynthesis in this study is sufficient to demonstrate the applicability of the dynamic gene copy control in regulating chemical production.
Dynamic control of the gene copy expands the dynamic pathway regulation to gene replication level, which brings several advantages compared with current strategies of gene expression control at transcriptional, translational, and post-translational level. Taking the dynamic down-regulation as an example, the dynamic down-regulation of gene copy would repress the gene expression before it is replicated and save the cellular resources for replication, transcription, and translation. However, other level of regulations like transcriptional, translational, or post-translational control, repress the gene expression after the gene is replicated, transcribed, or even translated, which would cause a waste of cellular resources for the unnecessary gene expression. Besides, regulation at the replication level is generally more responsive compared with other level of controls that take place after the gene is replicated. An additional advantage of the dynamic gene copy control via controllable plasmid replication is the ease of achieving multiplex regulation. As demonstrated in the example of p-coumaric acid production, our approach enabled a simultaneous dynamic regulation of six targets in a plasmid using just one regulatory circuit. This feature can be especially useful when regulating complex pathways that involves multiple genes. Moreover, dynamic gene copy regulation can be combined with transcriptional, translational, or post-translational control to establish multi-layered dynamic regulation on multiple targets. Overall, the dynamic regulation of gene copy enabled a new level of dynamic control on gene expression and offered an additional layer of regulation on dynamic pathway control, which would be beneficial for engineering microbial production of valuable compounds in metabolic engineering.
However, the excessive use of plasmids is often not preferred by industrial biotechnology, mainly due to the low stability of the plasmid (Rugbjerg et al., 2018; Silva et al., 2012). Thus, developing stable plasmid regulation system will be necessary to improve the applicability of the dynamic gene copy control. There have been many recent engineering efforts at establishing strategies to stably maintain the plasmids (Kang et al., 2018; Zhang et al., 2019). Thus, future research can be conducted to combine such strategies and develop stable plasmid regulation systems for dynamic regulation of gene expression in metabolic engineering and synthetic biology.
Materials and Methods
Strains, plasmids and reagents
All plasmids and strains used in this study are listed in Supplementary Table S3. The E. coli strain XL1-Blue (Stratagene) was used for gene cloning and plasmid constructions. The E. coli strain BW25113 (F’) (Atsumi et al., 2008) was used for validation of controllable replication control of origins and dynamic performance test of genetic circuits. P1 transduction was used to sequentially knock out the pykA, pykF, tyrR, and pheA in E. coli strain BW25113 (F’), forming the strain BW-PCA that was used for p-coumaric acid biosynthesis. The high-fidelity Phusion DNA polymerase, restriction enzymes and Quick Ligase kit were purchased from NEB (New England Biolabs). Plasmid miniprep and DNA gel purification kits were purchased from Zymo Research (Irvine, CA, USA). Luria-Bertani (LB) medium was made from the LB powder purchased from Fisher Scientific and was used for plasmid propagation and cell inoculation. The M9Y medium containing 20 g/L glycerol, 5 g/L yeast extract, 6 g/L Na2HPO4, 0.5 g/L NaCl, 3 g/L KH2PO4, 1 g/L NH4Cl, 246.5 mg/L MgSO4·7H2O and 14.7 mg/L CaCl2·2H2O was used for production of p-coumaric acid. Antibiotics including ampicillin (100 mg/L), kanamycin (50 mg/L), or chloramphenicol (34 mg/L) were added to cultures as needed. The p-coumaric acid standard was purchased from MP Biomedical LLC.
DNA manipulation and genetic circuits construction
The plasmids constructed in this study were listed in Supplementary Table S3. The DNA sequences of the components used in this study were listed in Supplementary Table S4. The reporter plasmid pCS-lpp0.5-egfp was constructed in our previous study (Wang et al., 2017b), and the reporter plasmid pZE-lpp0.5-egfp was constructed by inserting the expression cassette of lpp0.5-egfp (containing lpp0.5 constitutive promoter, RBS, and egfp encoding gene) from pCS-lpp0.5-egfp into the plasmid pZE12luc using XhoI and XbaI. The pZE-lpp0.5-RFP was constructed by replacing the coding sequence of egfp in pZE-lpp0.5-egfp with the coding sequence of red fluorescence protein (RFP) using Acc65I and XbaI. The plasmid pZE12luc harboring the ColE1 origin was used as the template for amplifying the DNA sequence of RNAI (ColE1) by polymerase chain reaction (PCR). The fragment of RNAI (ColE1) was inserted into the multi-cloning site (MCS) of pCS27 using EcoRI and BamHI to form plasmid pCS-RNAI (ColE1). The plasmid pCS27 harboring the p15A origin was used as the template for amplifying the DNA sequence of RNAI (p15A) and asRNAI (p15A) by PCR. The fragments of RNAI (p15A) and asRNAI (p15A) were inserted into the multi-cloning site (MCS) of pZE12luc and pZE-PT (Yang et al., 2015) using EcoRI and XbaI, or Acc65I and BamHI to form the plasmid pZE-RNAI (p15A), pZE-PT_RNAI (p15A), pZE-asRNAI (p15A), pZE-PT_asRNAI (p15A) respectively. The RNAIopt (p15A) were generated by overlap extension PCR using RNAI (p15A) as the template. pZE-PT_RNAI (ColE1) was constructed by replacing the RNAI (p15A) in the pZE-PT_RNAI (p15A) using Acc65I and BamHI. pSC-PT_RNAI (ColE1) was constructed by inserting the PT_RNAI (ColE1) transcriptional cassette from pZE-PT_RNAI (ColE1) to plasmid pSC74 (Wang et al., 2020) using XhoI and XbaI. For the CRISPRi-based replication control of ColE1 origin, the sgRNA-TIS (ColE1) was designed to target the promoter region (from −7 to −27 bp) of RNAII, and the sgRNA-CDS (ColE1) was designed to target the transcript of RNAII (from +43 to +63 bp). Similarly, the sgRNA-TIS (p15A) was designed to target the promoter region (from −25 to −45 bp) of RNAII, and the sgRNA-CDS (p15A) was designed to target the transcript of RNAII (from +16 to +36 bp). The sgColE1 was designed to target the promoter region of RNAI in the ColE1 origin (from −3 to −23 bp) and the sgp15A was designed to target the promoter region of RNAI in the p15A origin (from +1to −20 bp). All DNA sequences of sgRNAs were listed in Supplementary Table S4. The DNA sequences of all designed sgRNAs were then constructed in pCS27 plasmid to form the pCS-sgRNA-TIS (ColE1), pCS-sgRNA-CDS (ColE1), pCS-sgRNA-TIS (p15A), pCS-sgRNA-CDS (p15A), pCS-sgColE1, and pCS-sgp15A. The transcriptional operons of the sgRNAs were constructed to plasmid pSA-dCas9 (Wang et al., 2017c) using AatII and XhoI, resulting in pSA-dCas9-sgRNA-TIS (ColE1), pSA-dCas9-sgRNA-CDS (ColE1), pSA-dCas9-sgRNA-TIS (p15A), pSA-dCas9-sgRNA-CDS (p15A), pSA-dCas9-sgColE1, and pSA-dCas9-sgp15A.
For genetic circuits construction, the plasmids pZE-lpp0.2-PadRwt and pZE-lpp0.2-PadR (K64A) were constructed by inserting the PadR expression cassettes from pCS-lpp0.2-PadRwt and pCS-lpp0.2-PadR (K64A) (Jiang et al., 2021) to pZE12luc using XhoI and AvrII. The PT_RNAI (p15A) and asRNAI (p15A) were constructed to pZE-P9-egfp using EcoRI and XbaI. The operons of P9-PT_RNAI (p15A) and P9-asRNAI (p15A) were constructed into pZE-lpp0.2-PadRwt and pZE-lpp0.2-PadR (K64A) to form pZE-lpp0.2-PadRwt-P9-PT_RNAI (p15A), pZE-lpp0.2-PadR (K64A)-P9-PT_RNAI (p15A), pZE-lpp0.2-PadRwt-P9-asRNAI (p15A), and pZE-lpp0.2-PadR (K64A)-P9-asRNAI (p15A). The tetR gene was placed under the control of promoter PpadC-FG. The expression operon of PpadC-FG-tetR was inserted to pZE-lpp0.2-PadRwt, pZE-lpp0.2-PadR (K64A), pCS-lpp0.2-PadRwt, and pCS-lpp0.2-PadR (K64A), resulting in plasmids pZE-lpp0.2-PadRwt-PpadC-FG-tetR, pZE-lpp0.2-PadR (K64A)-PpadC-FG-tetR, pCS-lpp0.2-PadRwt-PpadC-FG-tetR, and pCS-lpp0.2-PadR (K64A)-PpadC-FG-tetR. The pLtetO1-controlled transcriptional cassette of PT_RNAI (p15A) was then inserted to plasmids pZE-lpp0.2-PadRwt-PpadC-FG-tetR and pZE-lpp0.2-PadR (K64A)-PpadC-FG-tetR, forming pZE-lpp0.2-PadRwt-PpadC-FG-tetR-pLtetO1-PT_RNAI (p15A) and pZE-lpp0.2-PadR (K64A)-PpadC-FG-tetR- pLtetO1-PT_RNAI (p15A). For controlling ColE1 origin, the PpadC-FG-controlled transcriptional cassettes of sgRNA-TIS (ColE1) or sgColE1, and the pLtetO1-controlled transcriptional cassette of sgRNA-TIS (ColE1), were constructed to pSA-dCas9, resulting in plasmids pSA-dCas9-PpadC-FG-sgRNA-TIS (ColE1), pSA-dCas9-PpadC-FG-sgColE1, and pSA-dCas9-pLtetO1-sgRNA-TIS (ColE1). The plasmid for p-coumaric acid biosynthesis was constructed by inserting the RgTAL expression operon from pZE-RgTAL into plasmid pCS-TPTA, resulting in pCS-TPTA-RgTAL. The pLlacO1-controlled transcriptional cassette of asppc was constructed to pCS-TPTA-RgTAL to form the final plasmid pCS-TPTA-RgTAL-asppc.
Validation of controllable replication of ColE1 derived origins
The E. coli transformants were inoculated and cultivated for 12 h in 3.5 mL LB media at 37°C as seeds. 150 μl culture was transferred into test tubes with 3.5 mL fresh LB medium. IPTG (0.5 mM, final concentration) was used to induce the pLlacO1 promoter after 2 h of inoculation. The samples were collected at 14 h of inoculation. The cell densities (OD600, optical density at 600 nm) and the expression level of egfp (green fluorescence intensity) were measured using the plate reader Synergy HT purchased from BioTek. All values obtained from the plate reader misused the value of a blank control (pure water). For detecting the green fluorescence intensity, the emission and excitation parameter was set to 528/20 and 485/20, respectively.
Test dynamic performance of genetic circuits
The E. coli transformants were inoculated and cultivated for 12 h in 3.5mL LB media at 37°C as seeds. 150 μl culture was transferred into test tubes with 3.5 mL fresh LB medium. IPTG (0.5 mM, final concentration) was used to induce the pLlacO1 promoter and gradient concentrations of p-coumaric acid (0, 100 mg/L, 500 mg/L) were added into media after 2 h of inoculation. The samples were collected every 2 h for 14 h. The cell densities (OD600) and the expression level of egfp (green fluorescence intensity) were measured using the plate reader Synergy HT purchased from BioTek. All values obtained from the plate reader misused the value of a blank control (pure water). For detecting the green fluorescence intensity, the emission and excitation parameter was set to 528/20 and 485/20, respectively.
Relative plasmid copy number determination by quantitative PCR
The quantification of relative plasmid copy number was carried out by quantitative polymerase chain reaction (qPCR) (Skulj et al., 2008). Cultured samples were diluted to OD600=1.5 (corresponding to 1.5*106 cells/μL) and were centrifuged at 12,000 rpm for 2 min. The plasmid mixtures from every sample were purified using Zymo plasmid miniprep kit with 40 ul water for dissolving the final products. The plasmid mixtures were diluted 100 times (1:100) by water before used as templates for qPCR. The primer pairs (Supplementary Table S5) targeting the antibiotic markers (AmpR, KanR, and ChlR) were designed by inputting the corresponding sequences into Primer 3 (https://bioinfo.ut.ee/primer3-0.4.0/). The designed primers were then employed to specifically amplify the plasmids derived from pZE12 (AmpR), pCS27 (KanR), and pSA74 (ChlR), respectively. The sybr green supermix purchased from BIO-RAD was used for qPCR. The QuantStudio 3 Real-time PCR system (Thermo Fisher Scientific) was used for amplification and signal detection. The 2−ΔΔCt method was used to analyze the relative plasmid concentration after normalization with the corresponding controls.
p-coumaric acid biosynthesis and product analysis
The plasmid pCS-TPTA-RgTAL-asppc were used for p-coumaric acid biosynthesis. The plasmid harboring the circuit with the third control logic (pZE-lpp0.2-PadR (K64A)-PpadC-FG-tetR-P9-PT_RNAI (p15A)) or the plasmid harboring the circuit with the second control logic (pZE-lpp0.2-PadR (K64A)-P9-asRNAI (p15A)) were used to control the plasmid replication of pCS-TPTA-RgRAL-asppc. The transformants were inoculated in LB media for 12 h as seeds for fermentation. 1 mL seeds were then transferred into shake flasks with 20 mL fresh M9Y media and cultivated at 37°C. After 2.5 h of cultivation, IPTG was added to every culture with a final concentration of 0.5 mM to induce the p-coumaric acid pathway. The samples were collected every 12 h for 72 h and then subject to cell density measurement and HPLC analysis. The cell density (OD600) was measured using the V-1200 spectrophotometer (VWR) with the pure water as a blank. The concentrations of p-coumaric acid were quantitatively analyzed by Agilent HPLC 1260 Infinity II (1260 Infinity II Diode Array Detector WR) with a reverse-phase ZORBAX SB-C18 column. A methanol-water (containing 0.1% trifluoroacetic acid) gradient system at a flow rate of 1 mL/min was used. The analyzing method was set as follows: 5% methanol from 0–2 min, 5 to 80% methanol from 2–10 min, maintain 80% methanol from 10–14 min, 80 to 5% methanol from 14–18 min, and 5% methanol from 18–20 min. Standard curve was drawn using gradient concentrations of p-coumaric acid solution prepared from commercially available p-coumaric acid powder (MP Biomedical LLC). The retention time of p-coumaric acid is at around 8.89 min.
Supplementary Material
Highlights:
Established controllable plasmid replication to dynamically regulate the gene copy
Constructed prototypic genetic circuits with different control logic to diversify dynamic gene copy control
Applied dynamic gene copy control in the biosynthesis of p-coumaric acid and enhanced the final production by 78%.
Acknowledgement
This work was supported by the National Institute of General Medical Sciences of the National Institutes of Health under award number R35GM128620. We also acknowledge the support from the College of Engineering, The University of Georgia, Athens. We would like to thank Dr. Pengpeng Bi for their technical assistance on quantitative PCR.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest
The authors declare no competing interest.
Data availability
All relevant data are included in the article and/or its supplementary information files. The datasets and materials generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
References:
- Ajikumar PK, Xiao W-H, Tyo KE, Wang Y, Simeon F, Leonard E, Mucha O, Phon TH, Pfeifer B, Stephanopoulos G, 2010. Isoprenoid pathway optimization for Taxol precursor overproduction in Escherichia coli. Science. 330, 70–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atsumi S, Cann AF, Connor MR, Shen CR, Smith KM, Brynildsen MP, Chou KJY, Hanai T, Liao JC, 2008. Metabolic engineering of Escherichia coli for 1-butanol production. Metabolic Engineering. 10, 305–311. [DOI] [PubMed] [Google Scholar]
- Balbás P, Bolívar F, 2004. pBR322 and Protein Expression Systems in E. coli. Recombinant Gene Expression. 77–90. [DOI] [PubMed] [Google Scholar]
- Barrick JE, Breaker RR, 2007. The distributions, mechanisms, and structures of metabolite-binding riboswitches. Genome Biol. 8, R239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brockman IM, Prather KLJ, 2015. Dynamic knockdown of E. coli central metabolism for redirecting fluxes of primary metabolites. Metab Eng. 28, 104–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camps M, 2010. Modulation of ColE1-Like Plasmid Replication for Recombinant Gene Expression. Recent Patents on DNA & Gene Sequences. 4, 58–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cesareni G, Helmer-Citterich M, Castagnoli L, 1991. Control of ColE1 plasmid replication by antisense RNA. Trends in Genetics. 7, 230–235. [DOI] [PubMed] [Google Scholar]
- Cesareni G, Muesing MA, Polisky B, 1982. Control of ColE1 DNA replication: the rop gene product negatively affects transcription from the replication primer promoter. Proceedings of the National Academy of Sciences. 79, 6313–6317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Ho JML, Shis DL, Gupta C, Long J, Wagner DS, Ott W, Josic K, Bennett MR, 2018. Tuning the dynamic range of bacterial promoters regulated by ligand-inducible transcription factors. Nat Commun. 9, 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahl RH, Zhang F, Alonso-Gutierrez J, Baidoo E, Batth TS, Redding-Johanson AM, Petzold CJ, Mukhopadhyay A, Lee TS, Adams PD, Keasling JD, 2013. Engineering dynamic pathway regulation using stress-response promoters. Nat Biotechnol. 31, 1039–46. [DOI] [PubMed] [Google Scholar]
- Del Solar G, Giraldo R, Ruiz-Echevarría MJ, Espinosa M, Díaz-Orejas R, 1998. Replication and control of circular bacterial plasmids. Microbiol. Mol. Biol. Rev 62, 434–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng J, Chen C, Gu Y, Lv X, Liu Y, Li J, Ledesma-Amaro R, Du G, Liu L, 2019. Creating an in vivo bifunctional gene expression circuit through an aptamer-based regulatory mechanism for dynamic metabolic engineering in Bacillus subtilis. Metabolic Engineering. 55, 179–190. [DOI] [PubMed] [Google Scholar]
- Dinh CV, Chen X, Prather KLJ, 2020. Development of a Quorum-Sensing Based Circuit for Control of Coculture Population Composition in a Naringenin Production System. ACS Synthetic Biology. 9, 590–597. [DOI] [PubMed] [Google Scholar]
- Dinh CV, Prather KL, 2019. Development of an autonomous and bifunctional quorum-sensing circuit for metabolic flux control in engineered Escherichia coli. Proceedings of the National Academy of Sciences. 116, 25562–25568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doong SJ, Gupta A, Prather KLJ, 2018. Layered dynamic regulation for improving metabolic pathway productivity in Escherichia coli. Proc Natl Acad Sci U S A. 115, 2964–2969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farmer WR, Liao JC, 2000. Improving lycopene production in Escherichia coli by engineering metabolic control. Nature biotechnology. 18, 533–537. [DOI] [PubMed] [Google Scholar]
- Gao C, Hou J, Xu P, Guo L, Chen X, Hu G, Ye C, Edwards H, Chen J, Chen W, Liu L, 2019. Programmable biomolecular switches for rewiring flux in Escherichia coli. Nat Commun. 10, 3751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guil S, Esteller M, 2015. RNA-RNA interactions in gene regulation: the coding and noncoding players. Trends Biochem Sci. 40, 248–56. [DOI] [PubMed] [Google Scholar]
- Gupta A, Reizman IM, Reisch CR, Prather KL, 2017. Dynamic regulation of metabolic flux in engineered bacteria using a pathway-independent quorum-sensing circuit. Nat Biotechnol. 35, 273–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Q, Lin Y, Yan Y, 2013. Caffeic acid production enhancement by engineering a phenylalanine over-producing Escherichia coli strain. Biotechnol Bioeng. 110, 3188–96. [DOI] [PubMed] [Google Scholar]
- Huo Y-X, Cho KM, Rivera JGL, Monte E, Shen CR, Yan Y, Liao JC, 2011. Conversion of proteins into biofuels by engineering nitrogen flux. Nature Biotechnology. 29, 346–351. [DOI] [PubMed] [Google Scholar]
- Jiang T, Li C, Yan Y, 2021. Optimization of a p-Coumaric Acid Biosensor System for Versatile Dynamic Performance. ACS Synthetic Biology. 10, 132–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones KL, Kim S-W, Keasling J, 2000. Low-copy plasmids can perform as well as or better than high-copy plasmids for metabolic engineering of bacteria. Metabolic engineering. 2, 328–338. [DOI] [PubMed] [Google Scholar]
- Kang CW, Lim HG, Yang J, Noh MH, Seo SW, Jung GY, 2018. Synthetic auxotrophs for stable and tunable maintenance of plasmid copy number. Metabolic Engineering. 48, 121–128. [DOI] [PubMed] [Google Scholar]
- Kim EM, Woo HM, Tian T, Yilmaz S, Javidpour P, Keasling JD, Lee TS, 2017. Autonomous control of metabolic state by a quorum sensing (QS)-mediated regulator for bisabolene production in engineered E. coli. Metab Eng. 44, 325–336. [DOI] [PubMed] [Google Scholar]
- Konermann S, Brigham MD, Trevino AE, Joung J, Abudayyeh OO, Barcena C, Hsu PD, Habib N, Gootenberg JS, Nishimasu H, 2015. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature. 517, 583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson MH, Gilbert LA, Wang X, Lim WA, Weissman JS, Qi LS, 2013. CRISPR interference (CRISPRi) for sequence-specific control of gene expression. Nature Protocols. 8, 2180–2196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C, Kim J, Shin SG, Hwang S, 2006. Absolute and relative QPCR quantification of plasmid copy number in Escherichia coli. Journal of Biotechnology. 123, 273–280. [DOI] [PubMed] [Google Scholar]
- Lian J, HamediRad M, Hu S, Zhao H, 2017. Combinatorial metabolic engineering using an orthogonal tri-functional CRISPR system. Nat Commun. 8, 1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D, Mannan AA, Han Y, Oyarzun DA, Zhang F, 2018. Dynamic metabolic control: towards precision engineering of metabolism. J Ind Microbiol Biotechnol. 45, 535–543. [DOI] [PubMed] [Google Scholar]
- Lutz R, Bujard H, 1997. Independent and tight regulation of transcriptional units in escherichia coli via the LacR/O, the TetR/O and AraC/I1-I2 regulatory elements. Nucleic Acids Research. 25, 1203–1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahr R, Frunzke J, 2016. Transcription factor-based biosensors in biotechnology: current state and future prospects. Appl Microbiol Biotechnol. 100, 79–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muranaka N, Abe K, Yokobayashi Y, 2009. Mechanism-guided library design and dual genetic selection of synthetic OFF riboswitches. Chembiochem. 10, 2375–81. [DOI] [PubMed] [Google Scholar]
- Na D, Yoo SM, Chung H, Park H, Park JH, Lee SY, 2013. Metabolic engineering of Escherichia coli using synthetic small regulatory RNAs. Nature Biotechnology. 31, 170–174. [DOI] [PubMed] [Google Scholar]
- Nguyen Thi Kim C, Tran Ngoc P, Cavin J-F, 2011. Genetic and Biochemical Analysis of PadR-padC Promoter Interactions during the Phenolic Acid Stress Response in Bacillus subtilis 168. Journal of Bacteriology. 193, 4180–4191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Pinera P, Kocak DD, Vockley CM, Adler AF, Kabadi AM, Polstein LR, Thakore PI, Glass KA, Ousterout DG, Leong KW, 2013. RNA-guided gene activation by CRISPR-Cas9–based transcription factors. Nature methods. 10, 973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rugbjerg P, Myling-Petersen N, Porse A, Sarup-Lytzen K, Sommer MOA, 2018. Diverse genetic error modes constrain large-scale bio-based production. Nat Commun. 9, 787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selzer G, Som T, Itoh T, Tomizawa J. i., 1983. The origin of replication of plasmid p15A and comparative studies on the nucleotide sequences around the origin of related plasmids. Cell. 32, 119–129. [DOI] [PubMed] [Google Scholar]
- Shen CR, Liao JC, 2008. Metabolic engineering of Escherichia coli for 1-butanol and 1-propanol production via the keto-acid pathways. Metabolic engineering. 10, 312–320. [DOI] [PubMed] [Google Scholar]
- Shen X, Wang J, Li C, Yuan Q, Yan Y, 2019. Dynamic gene expression engineering as a tool in pathway engineering. Curr Opin Biotechnol. 59, 122–129. [DOI] [PubMed] [Google Scholar]
- Siedler S, Stahlhut SG, Malla S, Maury J, Neves AR, 2014. Novel biosensors based on flavonoid-responsive transcriptional regulators introduced into Escherichia coli. Metab Eng. 21, 2–8. [DOI] [PubMed] [Google Scholar]
- Silva F, Queiroz JA, Domingues FC, 2012. Evaluating metabolic stress and plasmid stability in plasmid DNA production by Escherichia coli. Biotechnol Adv. 30, 691–708. [DOI] [PubMed] [Google Scholar]
- Skulj M, Okrslar V, Jalen S, Jevsevar S, Slanc P, Strukelj B, Menart V, 2008. Improved determination of plasmid copy number using quantitative real-time PCR for monitoring fermentation processes. Microb Cell Fact. 7, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuitje AR, Veltkamp E, Nijkamp HJJ, Weijers PJ, 1979. Origin and direction of replication of the bacteriocinogenic plasmid Clo DF13. Nucleic Acids Research. 6, 71–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan SZ, Prather KL, 2017. Dynamic pathway regulation: recent advances and methods of construction. Curr Opin Chem Biol. 41, 28–35. [DOI] [PubMed] [Google Scholar]
- Wang J, Cui X, Yang L, Zhang Z, Lv L, Wang H, Zhao Z, Guan N, Dong L, Chen R, 2017a. A real-time control system of gene expression using ligand-bound nucleic acid aptamer for metabolic engineering. Metabolic Engineering. 42, 85–97. [DOI] [PubMed] [Google Scholar]
- Wang J, Li C, Zou Y, Yan Y, 2020. Bacterial synthesis of C3-C5 diols via extending amino acid catabolism. Proceedings of the National Academy of Sciences. 117, 19159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Mahajani M, Jackson SL, Yang Y, Chen M, Ferreira EM, Lin Y, Yan Y, 2017b. Engineering a bacterial platform for total biosynthesis of caffeic acid derived phenethyl esters and amides. Metab Eng. 44, 89–99. [DOI] [PubMed] [Google Scholar]
- Wang J, Wu Y, Sun X, Yuan Q, Yan Y, 2017c. De novo Biosynthesis of Glutarate via α-Keto Acid Carbon Chain Extension and Decarboxylation Pathway in Escherichia coli. ACS Synthetic Biology. acssynbio 7b00136-acssynbio.7b00136. [DOI] [PubMed] [Google Scholar]
- Xu P, Li L, Zhang F, Stephanopoulos G, Koffas M, 2014. Improving fatty acids production by engineering dynamic pathway regulation and metabolic control. Proc Natl Acad Sci U S A. 111, 11299–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Lin Y, Li L, Linhardt RJ, Yan Y, 2015. Regulating malonyl-CoA metabolism via synthetic antisense RNAs for enhanced biosynthesis of natural products. Metab Eng. 29, 217–226. [DOI] [PubMed] [Google Scholar]
- Yang Y, Lin Y, Wang J, Wu Y, Zhang R, Cheng M, Shen X, Wang J, Chen Z, Li C, 2018. Sensor-regulator and RNAi based bifunctional dynamic control network for engineered microbial synthesis. Nature communications. 9, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanisch-Perron C, Vieira J, Messing J, Chambers S, Prior S, Barstow D, Minton N, Gilbert W, Messing J, Messing J, 1985. Improved M13 phage cloning vectors and host strains: nucleotide. Gene. 33, 103–119. [DOI] [PubMed] [Google Scholar]
- Yoo SM, Na D, Lee SY, 2013. Design and use of synthetic regulatory small RNAs to control gene expression in Escherichia coli. Nature Protocols. 8, 1694–1707. [DOI] [PubMed] [Google Scholar]
- Zhang F, Carothers JM, Keasling JD, 2012. Design of a dynamic sensor-regulator system for production of chemicals and fuels derived from fatty acids. Nat Biotechnol. 30, 354–9. [DOI] [PubMed] [Google Scholar]
- Zhang R, Yang Y, Wang J, Lin Y, Yan Y, 2019. Synthetic symbiosis combining plasmid displacement enables rapid construction of phenotype-stable strains. Metabolic Engineering. 55, 85–91. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All relevant data are included in the article and/or its supplementary information files. The datasets and materials generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.