

Abstract
Programmed cell deaths are pathways involving cells playing an active role in their own destruction. Depending on the signaling system of the process, programmed cell death can be divided into two categories, pro-inflammatory and non-inflammatory. Pyroptosis is a pro-inflammatory form of programmed cell death. Upon cell death, a plethora of cytokines are released and trigger a cascade of responses from the neighboring cells. The pyroptosis process is a double-edged sword, could be both beneficial and detrimental in various inflammatory disorders and disease conditions. A physiological outcome of these responses is tissue damage, and sometimes death of the host. In this review, we focus on the inflammatory response triggered by pyroptosis, and resulting tissue damage in selected organs.
1. Introduction
Pyroptosis is a form of pro-inflammatory programmed cell death [1]. The term “pyroptosis” was first coined by Cookson and Brennan in 2001, to describe “the screaming, alarm-ringing pro-inflammatory death of a potentially dangerous cell in an organism” [2]. The term is constructed from the Greek roots “pyro” for fire or fever and “ptosis (to-sis)” for falling. Although discovered relatively recently, pyroptosis has been observed to occur in different types of cells ranging from the immune system, digestive system, central nervous system, reproductive system, to cardiovascular system. Pyroptosis is provoked by stimuli including damage-associated molecular patterns (DAMPs) such as stress signals, uric acid crystals, oxidized lipoproteins, or pathogen-associated molecular patterns (PAMPs) such as flagellin, type 3 secretion system (T3SS) structure proteins, lipopolysaccharide (LPS), and nucleic acids [3].
Pyroptosis has initially been defined as a process of “caspase 1-dependent programmed cell death” [3]. However, later other caspases have been reported to lead to pyroptosis as well. Intracellular LPS from Gram-negative bacteria activates caspase-11 (in mice) and caspase-4/5 (in human), leading to pyroptosis [4–9]. More recently, caspase-8 has been shown to be activated by a bacterial virulent effector YopJ and flagellin, and subsequently cleaves both gasdermin D (GSDMD) and gasdermin E (GSDME) in murine macrophages, resulting in pyroptosis [10, 11]. In cancer chemotherapy, caspase-3 can cleave GSDME to induce pyroptosis in certain GSDME-expressing cancer cells [12]. GSDME serves as a switch molecule in the transformation between apoptosis and pyroptosis in tumor cells [12–14]. GSDME is cleaved by caspase-3, an apoptotic caspase activated by intrinsic and extrinsic apoptotic pathways [14]. Following cleavage by caspase-3, the N-terminal domain of GSDME behaves very much like the GSDMD N-terminal domain, forming pores in plasma membrane and induce pyroptosis [14]. In tumor cells with low GSDME expression, the hypermethylation of its promoter suppressed GSDME production. During chemotherapy using the DNA methyltransferase inhibitor decitabine, the hypermethylation of the GSDME promotor was inhibited and GSDME expression level increased. This change of GSDME level promoted the occurrence of pyroptosis in tumor cells in a caspase-3-dependent manner [12]. Other chemotherapy drugs that induce caspase-3-mediated apoptosis in GSDME-negative cell lines were shown to induce pyroptosis in GSDME-positive cell lines [12]. Due to the involvement of multiple caspases, more recently pyroptosis is defined as “Gasdermin-Mediated Programmed Necrotic Cell Death”, as gasdermin activation and pore formation in the cell membrane are features shared by both canonical (Caspase-1 dependent pyroptosis) and non-canonical (depending on caspases other than caspase-1) pyroptosis [15]. In addition to the difference in triggering signals and caspases involved, pyroptotic cells undergo membrane blebbing, swelling, and flattening before cell lysis, different from the explosive rupture observed in necroptotic cells, or the shrinkage and withering of apoptotic cells [16].
2. Mechanism of pyroptosis
The mechanism of pyroptosis has been the topic of several excellent reviews [3, 15, 17, 18] (Figure 1). Mammalian cells have sensors and receptors on their surface to detect environmental cues. DAMPs and PMAPs activate pattern recognition receptors (PRRs) on the cell surface [19]. Upon stimulation by extracellular signals, a cascade of responses inside the cells are triggered, including the activation of caspases and transcriptional effectors such as nuclear factor-κB (NF-κB). Activated NF-κB promotes the expression of pro-inflammatory cytokines, which are activated by caspases and secreted to escalate immune response. Activated NF-κB also upregulates the expression of NLPR3, which is critical for the activation of the NLRP3 inflammasome. In parallel, certain bacteria such as Salmonella typhimurium, Shigella flexneri, Pseudomonas aeruginosa, Vibrio parahaemolyticus, Burkholderia pseudomallei, and Yersinia enterocolitica can directly deliver effector proteins into the cytoplasm of host cells through the T3SS [20, 21]. Another route of entry of pathogenic signal molecules to the cytoplasm is endocytosis [22]. Together with host-derived cytoplasmic signal molecules, they interact with intracellular receptor NAIPs (NLR family apoptosis inhibitory proteins) and NLRs (Nod-like receptors) to trigger the activation and assembly of multiple protein complexes called inflammasomes [23, 24].
Figure 1. Inflammasome activation and pyroptosis.
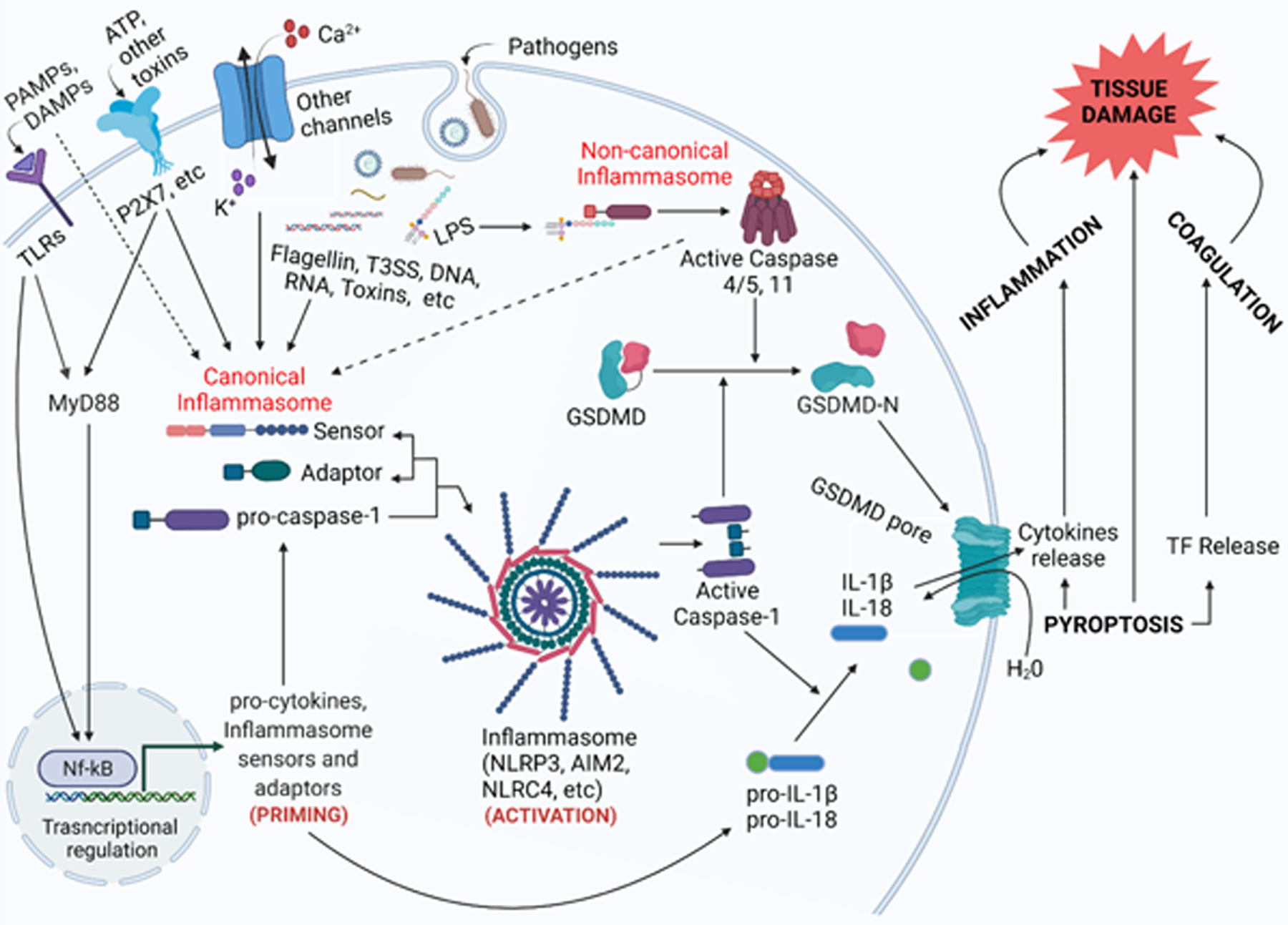
Various PAMPs and DAMPs are recognized by both membrane and cytoplasmic receptors, leading to a series of signaling cascades, expression of cytokines and inflammasome sensers and adaptors, and assembly of either canonical or non-canonical inflammasome. As a result, caspases are activated and subsequently cleave gasdermins (primarily GSDMD) and pro-cytokines IL-1β and IL-18. Pore-formation and release of cytokines and tissue factor lead to cell lysis, inflammation, coagulation, and tissue damage. Created with biorender.com.
Several types of inflammasomes have been found to be involved in pyroptosis, including NLRC4 (NOD-like receptor family, CARD domain containing 4), NLRP3 (NOD-like receptor family, pyrin domain containing 3), NLRP1, NLRP6, NLRP9, AIM2, and PYRIN [25]. The NLRP3 inflammasome is unique in that it can be activated by internal stress signals and a large number of metabolites, responding to both DAMPs and PAMPs [26, 27], and is involved in infections as well as several inflammatory disorders [28–30]. Activation of the NLRP3 in macrophages is a two-step process involving priming and receptors including TLRs, IL-1R, and tumor necrosis factor receptor that signals via NF-kB-activating pathways [31, 32]. In certain cell types (such as monocyte), priming alone can activate NLRP3 inflammasome [33, 34]. How NLRP3 detects intracellular stimuli remains elusive. Direct binding to the stimuli seemed unlikely, since a variety of stimuli with drastically different structure can activate the NLRP3 inflammasome [35]. It has been suggested that NLPR3 could sense a common “cellular event” induced by its stimuli. While certain stimuli share a common feature such as the stimulation of K+ efflux or Ca2+ signaling, an event that is shared by all stimuli is yet to be determined [28]. ROS has been suggested as a mediator of NLRP3 activation, as ROS inhibitors were found to potently inhibit NLRP3 activation [36, 37]. Later it was found that ROS inhibitors block NLRP3 inflammasome activation through blocking the upregulation of NLRP3 expression, or the priming step instead [38]. In 2018 Chen and Chen proposed a role of the trans-Golgi network (TGN) as a common feature in mediating NLRP3 activation [39]. They found that stimulation of various NLRP3 stimuli led to the dissemble of the TGN into small vesicles, which they termed dTGN. Formation of dTGN was upstream of NLRP3 binding and activation. NLRP3 was recruited to bind and aggregate on the surface of dTGN through ionic interactions between a region containing polybasic residues in NLRP3 and the negatively charged phospholipid PtdIns4P on dTGN, forming multiple small puncta. These small puncta were then incorporated to form a large speck with the adaptor protein ASC, followed by the subsequent downstream inflammasome activation cascade. How these stimuli trigger the dissembling of the TGN remains elusive.
The NLRC4 inflammasome is mainly triggered by PAMPs from various microbial pathogens [40]. Oligomerization and assembly of NLRC4 inflammasome are triggered by PAMP binding to NAIPs [41]. While a mouse carries seven NAIPs, a human only has one [42]. Mouse NAIPs recognize and bind to specific ligands. For example, NAIP5 and NAIP6 detect flagellin, and NAIP1 and NAIP2 recognize T3SS needle and rod proteins, respectively [41, 43, 44]. Human NAIP binds with both flagellin and T3SS proteins [41, 45]. Binding of the PAMP signal leads to conformational changes in the corresponding NAIP, exposing a previously hidden surface, which promotes the interaction with NLRC4 and activation of the NAIP/NLRC4 inflammasome. NLRC4 contains an N-terminal CARD domain, which can recruit pro-caspase-1 and assemble inflammasome.
Mice carry three NLRP1 paralogs (NLRP1a-c), while human only carry one. Among them NLRP1b is the most well characterized and is activated by bacterial and parasitic toxins and virulent factors [46–48]. Activation of the NLRP1b inflammasome is initiated by the autoproteolysis within the Function to Find Domain (FIIND) domain, which splits NLRP1b into a N-terminal and a C-terminal domain that remain noncovalently bound [48–50]. Several currently identified activators, including the anthrax lethal toxin protease and IpaH7.8, a Shigella flexneri ubiquitin ligase secreted effector, have been shown to initiate the ubiquitination and proteasomal degradation of the N-terminal domain, releasing the C-terminal domain to initiate inflammasome assembly [48]. This is consistent with the observation that NLRP1b activation can be blocked by proteasome inhibition in murine macrophages [51]. Thus, N-terminal degradation is likely a common mechanism of NLRP1b inflammasome activation.
Other well characterized inflammasomes involved in pyroptosis include the AIM2 and Pyrin inflammasomes. AIM2 directly binds to cytosolic double stranded DNA at least 70 bp in length in a sequence-independent manner [52–54]. DNA binding triggers inflammasome assembly. The Pyrin inflammasome indirectly senses inactivating modifications of host Rho GTPases by bacterial toxins. Rho-modifying proteins from pathogens induce pyrin-dependent activation of pyroptosis in macrophages [55–57]. In the resting state pyrin is phosphorylated at two sites and remains bound with its endogenous inhibitor. RhoA plays an important role in maintaining pyrin inhibited by facilitating phosphorylation through the recruitment of specific kinases [58]. Thus, bacterial toxins that inactivate RhoA lead to pyrin activation through disrupting the phosphorylation of pyrin and subsequently binding with its inhibitor [59].
As a direct consequence of inflammasome activation, pro-caspases are recruited and activated. Caspases that have been identified to be involved in pyroptosis include 1, 3, 4, 5, 8, and 11. Pro-inflammatory cytokines and gasdermins, primarily GSDMD, are digested by specific proteases. Gasdermin-mediated pore-formation is a key feature of pyroptosis. Activated caspases cleave GSDMD in the loop connecting the N-terminal and C-terminal domains, which lifts the autoinhibition of the C-terminal domain on the N-terminal domain. The N-terminal domain then migrates and inserts into the plasma membrane, oligomerizes and forms pores. While early studies reported a range of pore sizes, a recent cryo-EM study revealed a large pore of 21.5 nm inner diameter, with 33-fold symmetry [60]. Pore formation and cytokine release initiate a cascade of downstream consequences.
3. Consequence of pyroptosis at the cellular and subcellular level
Since its discovery in early 2000, pyroptosis has been observed to occur in many different cell types. The wide occurrence of this programed cell death process indicates its involvement in various disorders and disease conditions. Pyroptosis first occurs at the cellular level upon detection of pathogen invasion or cell stress/danger signals. Pyroptotic cells then release a large amount of pro-inflammatory cytokines and other mediators, triggering inflammatory responses from neighboring cells, similar as sparks that eventually lead to the burning of the entire forest. Distinct features of pyroptosis include the dependence on protease activation and cleavage of gasdermins, pore-formation in the plasma membrane, rapid cell swelling and lysis, and release of proinflammatory cytokines and microvesicles [3, 61]. Among them, pore formation is the first step of the physical changes of the pyroptotic cells. Pore formation leads to the release of pro-inflammatory cytokines, disruption of the ionic gradients and osmostasis, and eventually cell lysis and release of cellular contents [62]. Release of the inflammatory cellular contents and the cascade of reactions lead to tissue damage, and sometimes organ failure and host death.
Two pro-inflammatory cytokines generated directly in pyroptosis are IL-1β and IL-18. IL-1β and IL-18 lack secretion signals and their mechanism of release has been attributed to the pore-formation in the plasma membrane [62]. In a recent study, Xia et al. reported that the GSDMD pore conduit is predominantly negatively charged, which serves as an electrostatic gate to favor the passage of mature IL-1s over that of their precursors [60]. IL-1 precursors have a negatively charged acidic domain, which is proteolytically removed upon activation [63]. Mutation of acidic residues lining the conduit of the GSDMD pore was found to abolish this preference, suggesting that both charge and size are important factors determining the passage through the GSDMD pore. Similarly, a role in protein secretion has been previously proposed for caspase-1, which is likely the result of pore-formation [64]. Both cytokines play crucial roles in the pathogenesis of a range of inflammatory and autoimmune diseases [65, 66]. IL-1β binds to IL-1 receptors on the surface of immune cells and triggers various processes including fever, vasodilation, hematopoiesis, leukocyte tissue migration, antibody synthesis, and expression of cytokines and chemokines [66, 67]. IL-18 plays an important role in immune responses through inducing IFNγ production, activating T cells, macrophages, and natural killer cells, and plays a role in angiogenesis [65, 68, 69]. Both cytokines play crucial roles in promoting the beneficiary inflammatory process as well as the pathogenesis of a range of inflammatory and autoimmune diseases.
TNFα and IFNγ together had been found to induce pyroptosis and amplify pro-inflammatory responses [70]. Co-administration of TNFα and IFNγ caused a lethal cytokine shock in mice, leading to tissue damage and lethal inflammation, while treatment with neutralizing antibodies against TNFα and IFNγ protected mice from mortality during SARS-CoV-2 infection and sepsis. Additional mechanistic studies in vitro indicated co-treatment with TNFα and IFNγ induced the cleavage of GSDME and subsequent pyroptosis in BMDMs through the RIPK1/FADD/CASP8 Axis.
Activation of caspases is an important step in pyroptosis. While some substrates that are processed by activated caspases have been linked to pyroptosis, including IL-18, IL-1β, and GSDMD, other less documented substrates may also play important roles in inflammation and tissue damage. Through a proteomic study, 41 proteins were discovered that were directly cleaved by caspase-1, including chaperones, cytoskeletal and translation machinery proteins, cytokines, and interestingly, several proteins along the glycolysis pathway [71]. It was then confirmed that digestion by caspase-1 reduced the catalytic activity of these metabolic enzymes in vitro, and caspase-1 activation led to a significant digestion of these proteins in cells including macrophages. A more comprehensive set of caspase-1 substrates were identified through mass spectrometry-based proteomic studies coupled with a N-terminal enrichment method [72]. Analysis of THP-1 monocytic cell lysates treated with caspase-1 revealed 82 putative substrates, while activation of caspase-1 in THP-1 cells with proinflammatory stimuli led to the identification of 46 putative substrates, with 23 of them overlapping with the previous 82 substrates. Other than the well-established caspase-1 substrates GSDMD and IL-1β, a few substrates were identified in both studies, including the cytoskeleton actin and a few proteins involved in protein translation. The overall overlap between substrates identified through the two studies is small. This could be due to differences in sample handling and incomplete sampling.
4. Pyroptosis induced tissue damage
While pyroptosis is a host response that is evolved to protect against infection and cell stress, overactivation of pyroptosis can be detrimental, leading to tissue damage and even host death. Inflammation is initiated by harmful stimuli, as a means of self-protection by the cells. The production, activation, and release of cytokines, transcription factors, and proteases collectively promote the elimination of the harmful stimuli and protection of the host. Thus when under careful control, inflammation is a critical defense mechanism beneficial for the survival of the organism [73]. However, many molecules released as a result of inflammation could be toxic to the host cells as well, acting as a double-edged sword. When overactivated, this beneficial mechanism could cause damage to tissues and evolve into chronic inflammatory disease. IL-1β has been shown to trigger the generation of several cytokines in vivo [74]. Therefore, although IL-1β and IL-18 are the only known proinflammatory cytokines generated directly from inflammasome activation, inflammasome activation in vivo may lead to the generation of multiple proinflammatory cytokines indirectly, including TNF-α and IL-6, resulting in cytokine storm and tissue damage.
Recently, we and another research group reported that pyroptosis can trigger disseminated intravascular coagulation (DIC) [61, 75, 76], an acquired syndrome characterized by widespread intravascular activation of coagulation resulting in the formation of thrombi throughout the vasculature. We found that tissue factor, a protein playing an important role in blood coagulation, released from pyroptotic monocytes and macrophages was the initiator of coagulation cascade during sepsis. DIC can result from both infectious insults (such as COVID-19 and sepsis) and non-infectious insults (such as trauma and deep vein thrombosis) [77, 78]. DIC often leads to bleeding due to the consumption of coagulation factors and platelets, and formation of microvascular thrombi that inevitably results in multiple organ dysfunction.
Pyroptosis can occur in many cell types and affect almost all vital systems in the human body. In this special issue, the impact of pyroptosis and inflammation on liver, kidney, and the central nervous system have been reviewed, thus we will focus on system and organs that are not discussed, including the hematopoietic, cardiovascular, respiratory, digestive, and reproductive systems (Figure 2).
Figure 2.
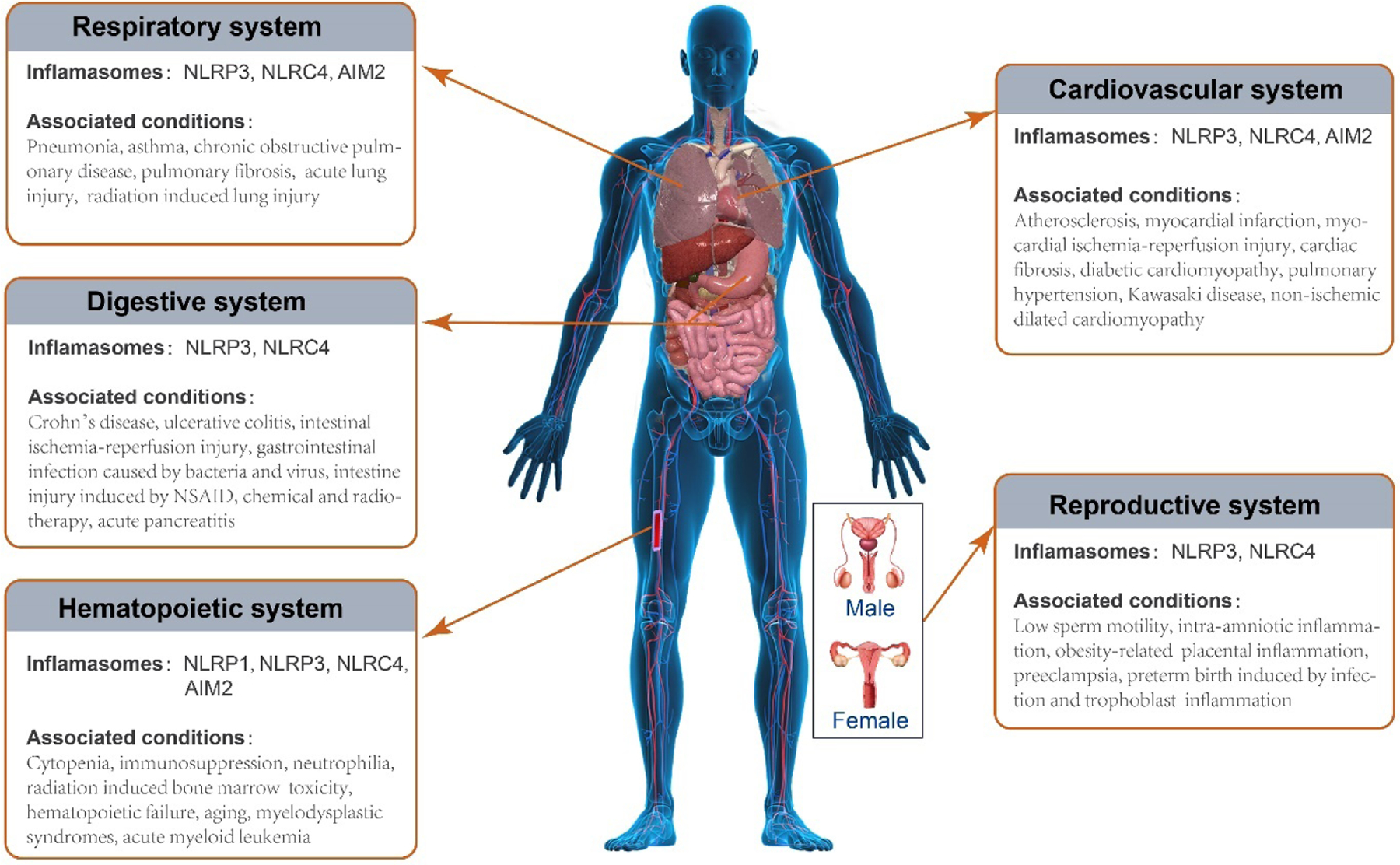
Inflammasomes activation and pyroptosis induced organ damages and disease conditions. NLRP1, NOD-like receptor family, pyrin domain containing 1; NLRP3, NOD-like receptor family, pyrin domain containing 3; NLRC4, NOD-like receptor family, CARD containing 4; AIM2, absent in melanoma 2; NSAID, nonsteroidal anti-inflammatory drug.
4.1. Hematopoiesis
Inflammasome activation during the inflammatory response plays an important role in balancing multiple stages of hematopoietic homeostasis [79, 80]. Proper level of inflammasome activation is critical for the development of stem cell mobilization, homing and engraftment. On the other hand, pathological activation of inflammasome complex results in abnormal hematopoiesis. Early insights into the pathogenesis of abnormal hematopoiesis highlighted elevations of inflammatory cytokines as a mechanism to explain abnormal responses under infection, microenvironmental stress, or sterile inflammation [81–83]. It has been realized only recently that pyroptosis may also contribute to hematopoietic damage following inflammasome activation. Activation of inflammasome in hematopoietic cells (HSCs) may lead to irreversible pyroptotic cell death and contribute to the origin of hematopoietic malignancies. Here, we will focus on recent advances that demonstrate the impact of pyroptosis during infection and selected hematological diseases.
4.1.1. Bacterial and viral Infection
Systemic infection can result in profound alterations in bone marrow, leading to bone marrow failure. A hematological feature of many infections is peripheral blood cytopenia [84]. The development of cytopenia results in immunosuppression and can predict poor prognosis in septic patients. Direct killing of hematopoietic stem and progenitor cells (HSPCs) is effective in clearance of infected cells to limit dissemination of infection during hematopoiesis [85, 86]. Inappropriate inflammasome activation and pyroptotic cell death, however, may lead to cytopenia and immunosuppression during infection. Inflammatory cytokines such as IL-1β and IL-18 released from pyroptotic cells further impact emergency hematopoiesis [81, 82, 87, 88]. Masters et al. demonstrated that NLRP1a activation by lymphocytic choriomeningitis virus (LCMV) infection induced the death of hematopoietic progenitor cells, leading to neutrophilia and lethal inflammatory disease [86]. NLRP1a and caspase-1 deficiency in mice decreased the pyroptotic cell death of progenitor cells upon infection and led to faster recovery [86]. In consistence with this finding, deletion of caspase-1 is associated with elevated number of HSCs [89, 90]. Bone marrow cells from caspase-1 deficient mice had significantly increased longevity compared with control cells [89]. Caspase1/11 deficient neonatal mice had improved survival following bacterial challenge. This reduction in mortality is associated with increased number of HSCs in the bone marrow and spleen following bacterial challenge, suggesting a role of pyroptosis in immunosuppression during sepsis [90].
Several recent papers highlighted the role of pyroptosis during SARS-CoV-2 infection. SARS-CoV-2 infection may lead to a severe complication known as a cytokine storm, which results in uncontrolled hyperactivation of the innate immune response and organ damage [91]. It has been well established that SARS-CoV binds to the angiotensin-converting enzyme 2 (ACE2) receptor through its spike protein to invade human cells [92, 93]. A recent study showed that the SARS-CoV-2 spike protein activated NLRP3 inflammasome in HSCs and small embryonic-like stem cells, which may induce cell pyroptosis [94]. The expression of mRNA of NLPR3, IL-1β,IL-18 and ASC increased after exposure to the recombinant spike protein. Elevated levels of secreted IL-1β were detected using ELISA assay in the conditioned media from cells stimulated by spike protein. These data suggest that inflammasome was activated by the spike protein and pyroptosis likely occurred in these cells, which may contribute to the pathogenesis of SARS-CoV-2 infection.
4.1.2. Radiation induced hematopoietic damage
In addition to infection, pyroptosis also plays an important role in radiation induced hematopoietic damage. Several studies show that inflammasomes including AIM2 and NLRP3 are implicated in radiation induced bone marrow toxicity [95–98]. Stoecklein et al. reported the occurrence of pyroptosis in many types of immune cells in mice underwent whole body radiation [95]. Later, Hu et al. provided evidence of AIM2 inflammasome activation and subsequent pyroptotic cell death as a mechanism of radiation induced bone marrow toxicity [96]. AIM2 mediates pyroptosis of intestinal epithelial cells and bone marrow cells in response to double-strand DNA fragments resulted from ionizing radiation or chemotherapeutic agents. Consistent with the in vitro data, AIM2 and caspase-1 deficient mice showed decreased hematopoietic failure after total body irradiation due to lack of pyroptosis [96]. 5-Androstenediol, a natural steroid hormone, has been found to inhibit pyroptosis through disruption of the interaction between AIM2 and ASC and thus enhanced the recovery of the hematopoietic system after whole body irradiation in mice [98]. A recent study further confirmed that GSDMD, downstream of the NLRP3 and AIM2 inflammasomes, triggered signaling cascades in bone marrow in response to radiation [99].
4.1.3. Aging
The increased basal level of sterile inflammation developed in hematopoietic tissues with advanced aging is associated with increased risk of clonal hematopoiesis of indeterminate potential, myeloid neoplasia and anemia. Enhanced myelopoiesis with impaired lymphopoiesis is a hallmark of bone marrow aging [100]. Defective erythropoiesis that can progress to anemia is also common with aging [101, 102]. This alteration in hematopoiesis has been termed in the literature as “inflammaging”, which is a chronic and low-grade sterile inflammation that develops with advanced age [103]. Mitochondrial stress and DNA damage are two important mechanisms of HSC aging that result in cell death [104, 105]. Pyroptosis has been shown to play an important role in aging. Fali et al. reported an increase of pyroptosis in old HPCs, together with elevated levels of P2X7 transcripts and cleaved caspase-1 [106]. Similarly, Luo et al. showed significantly reduced HSC death in the presence of NAD-dependent deacetylase sirtuin 2 (SIRT2), an NAD+ (nicotinamide adenine dinucleotide)-dependent deacetylase [107]. In an earlier study SIRT2 was identified as among the most significantly repressed genes in old HSCs compared with young HSCs [108]. Deficiency of SIRT2 was found to increase pyroptosis and caspase-1 cleavage upon LPS and ATP induction in aged HSCs in vitro [107]. Specifically, the NLRP3 inflammasome activation was involved in this pyroptotic cell death, as inactivation of NLRP3 and caspase-1 in aged HSCs increased the reconstitution capacity and improved the differentiation into the lymphoid lineage. Furthermore, overexpression of SIRT3 and/or SIRT7, which are supposed to reduce mitochondrial oxidative stress, [104, 109] reduced caspase-1 activation. Thus in aged HSCs, reduction of SIRT2 likely enhances pyroptosis through increased mitochondrial stresses.
4.1.4. Myelodysplastic syndromes (MDS)
MDS is characterized by the clonal proliferation of HSCs, ineffective hematopoiesis, and peripheral blood cytopenia with high-risk of acute myeloid leukemia (AML) development [110]. Pyroptosis is an integrated part of this clonally heterogeneous driven process. Basiorka et al. described a common pathway that DAMP signals and/or somatic mutation were able to induce NLRP3 inflammasome activation and subsequent pyroptosis in HSPCs, resulting in the phenotypic features of MDS [111]. The expression levels of the inflammasome components, including NLRP3, caspase-1, IL-1β and IL-18, were elevated in hematopoietic stem and progenitor cells isolated from the MDS patients. Knockdown of NLRP3 or caspase-1 suppressed pyroptosis and restored effective hematopoiesis. Pyroptosis of the HSCs was triggered by an excess of S100 calcium binding protein A9 (S100A9) and subsequent generation of reactive oxygen species (ROS). During inflammation, S100A8/A9 is actively released from immunocytes, such as neutrophils and macrophages, and exerts a critical role in modulating the inflammatory response by stimulating leukocyte recruitment and inducing cytokine secretion. In a separate study, increased plasma circulation of A100A8/9 was also observed in MDS patients [112]. Overexpression of S100A9 in an S100A9 transgenic mouse model recapitulated progressive cytopenia mediated by NLRP3 inflammasome-induced pyroptosis [111]. Inhibition of inflammasome assembly suppressed pyroptosis, increased HSPCs numbers, and restored effective hematopoiesis in S100A9 transgenic mouse model. The S100A9/NOX/ROS/NLRP3 axis has been identified as the regulator of the Wnt/β-catenin signaling and pyroptotic cell death of HSPCs in MDS. Pyroptosis is clearly responsible for many of the hallmark features of MDS including macrocytosis and ineffective hematopoiesis. ASC specks in the peripheral blood and/or bone marrow plasma from patients with MDS is directly correlated with plasma concentrations of S100A8/A9, confirming the relationship to pyroptosis [113].
4.1.5. Acute myeloid leukemia (AML)
In addition to MDS, the NLPR3 inflammasome and pyroptosis have also been implicated in AML [114]. A recent study shows that the oncogenic KrasG12D mutation, which occurs in various leukemias, has an inflammation-related effect contributing to the disease [115]. KrasG12D mutation caused NLRP3 inflammasome activation, leading to increased caspase-1 cleavage and IL-1β release, and increased cell death, which subsequently promoted myeloproliferation and cytopenia in vivo. Inhibitors of the IL-1 receptor or NLRP3 activation interfered with KRAS-driven myeloproliferation. The oncogenic KRAS triggered pyroptosis through ROS from the RAC1/NADPH axis [115]. In agreement with the observations in mice, ROS production increased in human Krasmut cells derived from AML patients, and blocking RAC1 in the Kras mutant cells derived from AML patients inhibited ROS production and IL-1β expression. Thus, these findings highlight the important role of NLRP3 inflammasome and pyroptosis in hematological disorders.
Transforming growth factor β-activated kinase 1 (TAK1), a member of the mitogen-activated protein kinase kinase kinase (MAP3K) family, is required for NF-κB and MAPK signaling downstream of several cytokine receptors and pattern recognition receptors [116, 117]. TAK1 is also a crucial component in maintaining homeostasis, as TAK1 deletion or inhibition results in spontaneous cell death in a number of different cell types, including hematopoietic cells [118, 119]. Malireddi et al. observed spontaneous receptor interacting serine/threonine Kinase 1 (RIPK1)-dependent NLRP3 inflammasome activation and cell death in mouse bone marrow derived macrophages when TAK1 was absent [120], suggesting that TAK1 played a key role in maintaining NLRP3 inflammasome quiescence and preserving cellular homeostasis and survival. TAK1 appears to be a negative regulator of p38 and IKK activation, since TAK1-deficient neutrophils enhanced the phosphorylation of the kinases IKK, p38, and JNK and the production of IL-1β, IL-6, TNF-α, and ROS after LPS stimulation [121]. Interestingly, specific deletion of TAK1 in the myeloid compartment led to clonal myelomonocytic cell expansion, splenomegaly, and myelomonocytic leukemia [122]. It is not clear whether this phenomenon is associated with increased pyroptosis.
4.2. Cardiovascular system
Cardiovascular disease is a leading cause of death and disability worldwide [123]. Pyroptosis mediated by NLRP3 inflammasome can occur in various vascular cells, such as vascular endothelial cells (VECs), vascular smooth muscle cells (VSMCs), cardiomyocytes, and cardiac fibroblasts [124, 125]. Pyroptosis has been shown to contribute to the pathogenesis of cardiovascular diseases, including atherosclerosis, myocardial infarction, diabetic cardiomyopathy, and Kawasaki disease [126–129]. Here, we focus on the most recent discoveries about the molecular mechanisms and involvement of pyroptosis in cardiovascular diseases.
4.2.1. Atherosclerosis (AS)
AS is a chronic progressive disease characterized by the accumulation of lipids and infiltration of inflammatory cells in the arterial wall [130]. Endothelial cell death is a crucial and initial stage in the process of AS. VSMCs, normally located in the vascular media layer, play an important role in arterial wall remodeling. The prominent role of VSMCs in AS has been established since early 1970s [131, 132]. Recent studies indicate that pyroptosis of endothelial cells, VSMCs, and macrophages plays a critical role in the initiation and development of AS [133, 134]. In the Canakinumab Anti-inflammatory Thrombosis Outcome Study (CANTOS), a human IL-1β neutralized antibody, Canakinumab, was shown to significantly reduce the rate of recurrent adverse cardiovascular events in patients with prior myocardial infarction, suggesting a link between inflammasome activation and atherosclerosis pathogenesis [135].
Many intracellular and extracellular activators of NLRP3 inflammasome in endothelial cells are associated with the pathogenesis of AS, including oxidized low-density lipoprotein (ox-LDL) [136], cholesterol crystal [137], nicotine [138], cadmium [139], trimethylamine N‐oxide (TMAO) [140], Lysophosphatidylcholine (LPC) [141], decabromodiphenyl ethane [142], and polychlorinated biphenyls (PCBs) [143]. Most of those activators mediate cell pyroptosis through the ROS/NLRP3/caspase-1 pathway [137–139, 142]. Briefly, the activators trigger induction of ROS production, which activates NLRP3 inflammasome, leading to the activation of caspase‐1. Activated caspase-1 triggers pore formation in cell membrane, DNA fragmentation, and release of mature IL-1β, IL-18, and high mobility group box 1 (HMGB1) from cells, causing a sterile inflammation response and further contributing to pyroptotic cell death and subsequently promoting AS. Several recent studies reported the signaling pathways that induced the production of ROS associated with AS. Zhaolin et al. reported that a MicroRNAs miR-125a-5p induced ROS generation, NLRP3 activation, and ultimately pyroptosis of VECs upon stimulation by ox-LDL, a crucial pathogenic factor for AS [136]. Wu et al. demonstrated that TMAO, a product from the phosphatidylcholine metabolism of gut flora [144], was able to promote the progression of atherosclerotic lesions in apolipoprotein E-deficient (ApoE-/-) mice fed a high-fat diet by activating vascular endothelial cell pyroptosis [140]. They found that TMAO enhanced expression of succinate dehydrogenase complex subunit B (SDHB) in the vascular endothelial cells of ApoE-/- mice and in cultured human umbilical vein endothelial cells (HUVECs).
Overexpression of SDHB in HUVECs impaired mitochondria and increased ROS levels. A ROS scavenger N-acetyl-L-cysteine (NAC) inhibited pyroptosis of the SDHB overexpressed endothelial cells.
Except for ROS production, several other signals, including potassium efflux, lysosomal rupture and mitochondrial dysfunction were also reported to activate NLRP3 inflammasome in VECs [141, 145, 146]. LPC is a major lipid component of the plasma membrane and plays a crucial role in the formation of atherosclerotic lesions [147]. Corrêa et al. reported that LPC induced pyroptosis of monocytes and endothelial cells by increasing potassium efflux and lysosomal damage [141]. In another study, potassium efflux was shown to play a role in the mixed lineage kinase domain-like (MLKL) protein triggered endothelial cell pyroptosis by ox-LDL stimulation [146]. MLKL is an executor of necrosis [148]. The expression of MLKL significantly increased in the pyroptotic endothelial cells induced by ox-LDL. Overexpression of MLKL induced an increase in NLRP3 inflammasome activation. Blocking of cell potassium efflux significantly attenuated the activation of caspase-1 and IL-1β.
Recently VSMC pyroptosis was reported to occur in mouse and human atherosclerotic plaques [149]. Distribution of active pyroptotic indicators, including cleaved caspase-1 and IL-1β, largely overlapped with α-smooth muscle actin, a marker of smooth muscle cells, especially near the necrosis core, at the plaque surface and in intra-plaque hemorrhage area. Importantly, VX-765, a specific inhibitor of caspase-1, reduced the pyroptosis of mouse VSMCs and IL-1β processing induced by ox-LDL, and inhibited progression of established atheroma and the development of AS [149].
AIM2 promoted pyroptosis of VSMCs has recently been reported to play a role in the progression of atherosclerotic plaques [150]. Oxidized low-density lipoprotein (ox-LDL) is a major risk factor of AS. Incubation of VSMCs with ox-LDL increased AIM2 expression and GSDMD-NT levels, and cell death. In consistent with the in vitro results, high-fat diet increased expression of AIM2 and the level of GSDMD-NT, and death of smooth muscle cells in ApoE-/- mice. Importantly, overexpression of AIM2 in the ApoE-/- mice by injection with lentivirus-AIM2 further increased high-fat diet-induced atherosclerotic lesions, while inhibition of AIM2 expression by injection with hRNA-AIM2 reduced atherosclerotic lesions.
Pyroptosis also occurred in macrophages during AS. ox-LDL induced macrophage pyroptosis through the ROS/NLRP3/caspase-1 pathway [151]. Caspase-11/GSDMD was shown to be involved in ox-LDL-induced macrophage pyroptosis [152]. Depletion of caspase-11 or suppressing GSDMD attenuated the volume and macrophage infiltration of atherosclerotic lesions in the ApoE-/- mice fed with a high-fat/high-cholesterol diet. Nicotine has been reported to play a role in accelerating AS by promoting macrophage pyroptosis [153]. Nicotine exacerbated atherosclerotic lesions in ApoE(-/-) mice, which was mediated by the thioredoxin-interacting protein (TXNIP), a protein binds to thioredoxin and modulates its antioxidant functions. Inhibition of TXNIP expression in mice reversed the effects of nicotine on macrophage invasion and vascular injury, and thus improved atherosclerotic plaque lesions [153]. Nicotine may induce macrophage pyroptosis via the histone deacetylase 6 (HDAC6)/NLRP3 signaling pathway [154]. HDAC6 is required for NLRP3 and pyrin inflammasome activation through microtubule transport and assembly of these inflammasomes [155]. HDAC6 modifies proteins by removing acetyls to regulate diverse biological functions [156, 157]. In vitro studies revealed that nicotine upregulated HDAC6, which deacetylated NF-κB p65. Enhanced nuclear translocation of NF-κB p65 mediated the transcription of NLRP3 and macrophage pyroptosis [154].
4.2.2. Myocardial infarction (MI)
MI is a severe coronary artery related disease, mainly resulted from disruption of coronary atherothrombosis or imbalance of myocardial oxygen supply-demand [158]. Both in vivo and in vitro studies have shown that pyroptosis in cardiomyocytes and cardiac fibroblasts play a significant role in MI [128, 159]. Recently, several studies reported that negative regulators of pyroptosis protected against heart injury after MI. Han et al. reported that CXADR‐like membrane protein (CLMP), a member of the CTX family, was highly expressed in the fibroblasts of the ischemic heart [160]. Since CLMP deficient mice died within 4 weeks after birth, they used Clmp+/− mice to investigate the role of CLMP in MI. Pyroptosis of cardiac fibroblasts was more severe in the Clmp +/− mice. Accordingly, myocardial fibrosis and ventricular dysfunction post‐MI was more severe in the Clmp+/− mice than wild‐type mice. Li and coworkers reported that the growth differentiation factor 11 (GDF11) contributed to cardio protection by preventing cardiomyocyte pyroptosis via the homeobox transcription factor A3 (HOXA3)/NLRP3 signaling pathway in MI mice [161]. The data showed that the expression of GDF11 was markedly lower in heart tissues of MI. Meanwhile, GDF11 overexpression significantly improved heart function in MI mice and also decreased the pyroptosis of hypoxic cardiomyocytes. In support of a role of cardiomyocyte pyroptosis in MI, Li et al. showed that miR-135b protected cardiomyocytes from infarction through the inhibition of the NLRP3/caspase-1/IL-1β pathway [162]. Han et al. showed that long noncoding RNA (lncRNA) H19 and CYP1B1 were associated with the MI progression and pyroptosis of cardiomyocytes [163]. The level of H19 was downregulated while CYP1B1 was upregulated in the peripheral blood in MI patients compared to those in healthy controls. Overexpression of LncRNA H19 suppressed pyroptosis of cardiomyocytes and thus attenuated myocardial infarction, suggesting that cardiomyocyte pyroptosis contributes to MI.
Coronary reperfusion is a main treatment method for acute MI. However, reperfusion itself may lead to accelerated and additional myocardial injury which is the so-called myocardial ischemia/reperfusion injury (MI/RI) [164]. MI/RI induces caspase-1-mediated pyroptosis by activating the NLRP3 inflammasome [165–167]. Importantly, several compounds alleviate MI/RI through targeting the NLRP3/GSDMD pathway, including sweroside [168], metformin [169], piperine [170], emodin [171], Kanglexin (a novel anthraquinone compound) [172], beta-asarone [173], and dexmedetomidine [174]. Nie and coworkers showed that hydrogen gas inhalation could also alleviate MI/RI through the inhibition of oxidative stress and NLRP3-mediated pyroptosis in rats [175].
4.2.3. Pulmonary hypertension
Pulmonary hypertension is a clinically common cardiopulmonary disease, which is characterized by pulmonary vascular remodeling and right ventricular hypertrophy [176]. The NLRP3 inflammasome has been shown to contribute to hypoxic pulmonary hypertension [177]. Consistently, deletion of the inflammasome adaptor ASC reduced hypoxia-induced pulmonary hypertension in mice [178]. ASC deficient mice displayed reduced muscularization and collagen deposition around arteries. He et al. reported that pulmonary artery smooth muscle cell (PASMC) pyroptosis played a role in chronic hypoxia-mediated pulmonary hypertension progression [179]. They also found that Glioma-associated oncogene family zinc finger 1 (GLI1), a transcriptional activator involved in many diseases, was involved in hypoxia-induced pyroptosis and pulmonary hypertension. GLI1 siRNA reversed pyroptotic cell death of pulmonary artery smooth muscle cells in vitro and specific GLI1 inhibition attenuates hypoxia-induced pulmonary hypertension and pulmonary vascular remodeling in vivo. Zhang et al. reported that pyroptosis occurred in the pulmonary arteries in both rat pulmonary hypertension model and hypoxic human pulmonary arterial smooth muscle cells. Importantly, caspase-1 inhibition attenuated pulmonary fibrosis and the pathogenesis of pulmonary hypertension, as assessed by vascular remodeling [180]. Circular RNAs (circRNAs), a unique class of noncoding RNAs, has emerged as important regulators of pulmonary hypertension. Zhang et al. reported the upregulation of a novel circRNA, circ-Calm4, in PASMCs in a mouse model of hypoxia-induced pulmonary hypertension [181]. Later, circ-Calm4 was found to contribute to hypoxia-induced PASMCs pyroptosis [182]. Inhibition of circ-Calm4 expression in mouse lungs abrogated pyroptosis of PASMCs and thus improved pulmonary hypertension [182].
4.2.4. Kawasaki disease (KD)
KD is a rare systemic inflammatory disease that predominately affects children younger than 5 years old, and endothelial cell damage and inflammation are two essential processes resulting in the coronary endothelial dysfunction in KD [183]. Jia et al. recently reported that endothelial cell pyroptosis played an important role in the pathogenesis of KD [184]. The coronary endothelial damage observed in KD was found to be associated with endothelial cell pyroptosis via the NLRP3 inflammasome activation. The high levels of high mobility group box 1 (HMGB1) lead to elevated expression of RAGE (receptor for advanced glycation end-products) and cathepsin B activity, which resulted in NLRP3 inflammasome-dependent caspase-1-mediated pyroptotic cell death in the ECs [184].
4.2.5. Non-ischemic dilated cardiomyopathy
Dilated cardiomyopathy is defined by the presence of left ventricular dilatation and contractile dysfunction [185]. In the myocardial tissues of patients suffering from non-ischemic dilated cardiomyopathy, hyperactivated NLRP3 inflammasome with pyroptotic cell death of cardiomyocytes has been observed [186]. Doxorubicin, an anthracycline antibiotic with anti-tumor activity, was reported to induce dilated cardiomyopathy in patients [187]. Deficiency of either NLRP3 or caspase-1 protected against dilated cardiomyopathy in a Doxorubicin-induced dilated cardiomyopathy mouse model, demonstrating an important role of inflammasome activation and pyroptosis in thiss disease condition[186]. Doxorubicin enhanced expressions of NOX1 and NOX4, which subsequently induced translocation of dynamin-related protein 1 (Drp1) into mitochondria and mitochondrial fission in myocardial tissues, leading to NLRP3 activation and pyroptosis via ROS generation.
4.2.6. Diabetic cardiomyopathy (DCM)
DCM is a common complication of diabetes, which is associated with inflammation in heart [188]. Several studies have demonstrated the participation of cardiomyocyte pyroptosis in the process of DCM. Chemerin and its receptor CMKLR1 (a G-protein-coupled receptor) have been shown to play an important role in DCM. Xie et al. showed that chemerin promoted DCM through NLRP3-dependent pyroptosis [189]. Yang et al. reported a role of a long non-coding RNA (lncRNA) Kcnq1ot1 in DCM [190]. The expression of Kcnq1ot1 increased in the cardiac tissues from patients with diabetes and diabetic mice, and silencing of Kcnq1ot1 alleviated pyroptosis and improved cardiac function and morphology in vivo. In contrast, overexpression of another lncRNA, GAS5, suppressed NLRP3 inflammasome-mediated pyroptosis and alleviated DCM [191]. All these data indicate that pyroptosis downstream from NLRP3 inflammasome plays an important role in diabetic cardiomyopathy.
4.3. Respiratory system
The human respiratory tract is constantly exposed to harmful microorganisms and air pollutants. In patients with community acquired pneumonia, asthma, chronic obstructive pulmonary disease (COPD), pulmonary fibrosis, and acute lung injury (ALI), inflammasome activation regulates respiratory infections and pathological airway inflammation. The PAMPs and DAMPs resulted from these disease situations induce inflammasome activation, leading to secretion of IL-1β and IL-18 as well as pyroptosis [192]. It is known that several inflammasomes, including NLRP3, NLRC4, and AIM2, are involved in lung damage in different diseases. Inhaled environmental stimuli such as smoke, asbestos, silica, particulate matter, allergens, and air pollution can also activate the NLRP3 inflammasome, leading to pyroptosis [36, 193–196].
4.3.1. Pneumonia
Pneumonia is caused by infection and subsequent inflammation of the respiratory tract. S. pneumonia infection is the most common cause of community acquired pneumonia, which is the leading cause of death in many countries [197]. Using a S. pneumonia infection murine model, Witzenrath et al. identified streptococcal exotoxin “pneumolysin” as a key activator of the NLRP3 inflammasome [198]. Pneumolysin destroyed plasma membrane in cells via forming of pores on the cell membrane and caused cell lysis, and the release of cell components led to activation of the NLRP3 inflammasome and caused cell pyroptosis [199]. Influenza A virus and adenovirus infection stimulated ATP and ROS release from respiratory epithelial cells, suggesting a mechanism of NLRP3 inflammasome activation and pyroptosis [200–202]. Lee et al. reported that influenza A infection triggered respiratory epithelial cells pyroptosis and apoptosis in a mutually exclusive manner. While apoptosis played an important role in the early influenza A infection phase, pyroptosis was dominant at the late phase of infection. Type I interferon signaling regulated the epithelial cell apoptosis shift to pyroptosis to promote proinflammatory cytokine production and initiate proinflammatory response [203].
4.3.2. Asthma
Approximately 10% of people in western countries and 300 million people worldwide currently suffer from asthma, a chronic airway inflammatory disease [204]. Increasing clinical and experimental studies have shown the activation of the NLRP3 inflammasome and/or the production of excessive IL-1β are related to the pathogenesis of asthma [205–212]. Baines et al. studied the gene expression profile of inflammatory cells in induced sputum [206]. The expression of IL-1β signaling pathway-related genes, such as IL-1β, IRAK2, IRAK3, and IL-1R2, significantly increased in the sputum of patients with neutrophilic asthma [206]. Simpson et al. reported that the expressions of NLRP3, caspase-1/4/5 and IL-1β in the airways of patients with neutrophilic asthma increased, and macrophages and neutrophils were the main cell sources of NLRP3 and caspase-1 in this cohort [212]. The expression levels of TLR2, TLR4 and IL-8 in these asthmatic patients also elevated, suggesting abnormal inflammasome activation in asthma [212].
A large amount of clinical evidence suggests that bacterial respiratory infections are related to steroid-insensitive asthma. Chlamydia pneumoniae is an obligate intracellular bacterial pathogen and is associated with severe, steroid-insensitive asthma [213–215]. H. Influenza bacillus is a Gram-negative bacteria and is the most commonly isolated bacteria from the airways of steroid-insensitive asthma patients [216, 217]. Essilfie et al. reported that infections by Chlamydia and H. influenza caused an increase of neutrophils, as well as T helper lymphocyte type (Th) 1 and/or Th17 responses and leading to severe, steroid-insensitive neutrophilic allergic airways disease [218]. Both Chlamydia and Haemophilus respiratory tract infections induced the release of active IL-1β in an NLRP3 inflammasome-dependent and caspase-1-mediated manner [219, 220].
4.3.3. COPD
COPD is a progressive and obstructive lung disease, including chronic bronchitis and emphysema. It has become the fourth leading cause of death world-wide [221, 222]. Clinical studies have shown that cigarette smoking, a leading cause of COPD, induced the release of IL-1β in lung tissues [223–225]. The concentration of HMGB1 significantly increased in sputum and bronchoalveolar lavage fluid in patients with COPD [226, 227]. HMGB1 activated NLRP3 inflammasomes in a TLR4-dependent manner in hemorrhagic shock mice model, mediated the activation of NAD(P)H oxidase and ROS generation, which subsequently promoted NLRP3 inflammasome activation and subsequently induced active IL-1β secretion in endothelial cells [228]. HMGB1-mediated inflammasome activation plays a clear role in the pathogenesis of COPD.
4.3.4. Pulmonary fibrosis
Pulmonary fibrosis refers to a series of lung diseases, which is characterized by irreversible destruction and remodeling of the lung structure due to excessive deposition of collagen and extracellular matrix proteins. The role of inflammasomes in the pathogenesis of pulmonary fibrosis remains elusive. However, fibrosis-inducing irritants (such as silica, asbestos, cigarette smoke, and bleomycin) are known to damage the lung epithelium through directly activating NLRP3 inflammasome [229, 230].
4.3.5. ALI
ALI is a main cause of acute respiratory distress syndrome (ARDS) [231]. ALI/ARDS is identified by severe inflammation, leading to diffuse alveolar injury, varying degrees of ventilation/perfusion differentiation, poor lung compliance, and severe hypoxemia [232]. Many factors can lead to ALI, such as severe shock, infection, mechanical damage, and bacterial infection. LPS is a major component of the outer membrane of Gram-negative bacteria and LPS-induced acute lung injury has been extensively studied [233]. Pyroptosis has been revealed as a critical step in LPS-induced inflammation [234–236].
Several studies have shown that alveolar macrophages (AMs) had a profound impact on the occurrence and prognosis of ALI by releasing proinflammatory cytokines and regulating other immune cells [237–239]. Pyroptosis of AMs has been shown to be involved in the development of ALI and the NLRP3/ASC inflammasome was essential in this process [240–242]. Knocking down of ASC or inhibiting the function of NLRP3 reduced AM pyroptosis and release of HMGB1, subsequently reduced the inflammatory response in ALI [241].
IL-1R1 up-regulates the sensitivity of AMs to IL-1β, causing elaboration of pyroptosis of AMs, and subsequent aggravation of ALI [243]. In addition, myeloid differentiation protein 2 (MD-2), an essential factor required for TLR4 binding of LPS, was required for the expression of NLRP3 and IL-1β [244]. Interferon regulatory factor-1 (IRF-1) played an essential role in mediating the pyroptosis of AMs in LPS-induced ALI [245]. MyD88 is also known to recruit IRF-1 transcription factors to activate the TLR4 pathway [246]. Thus, IRF-1 may be the bridge between the LPS-TLR4 signaling pathway and the caspase-1-dependent pyroptosis pathway in AM.
Liu et al. suggested a new mechanism in macrophages to explain how mitochondrial DNA (mtDNA) activated the pyroptosis pathway, thereby aggravating the inflammatory response in ALI [247]. Specifically, the sensory receptor of cytosolic DNA (cyclic GMP-AMP synthase, cGAS) detected mtDNA leaked into the cytoplasm and activated stimulator of interferon gene (STING) [247, 248]. LPS also upregulated the expression level of the transcription factor c-Myc, which directly promoted the expression level of STING. The upregulation and activation of STING activated the NLRP3 inflammasome and caspase-1, which ultimately leads to cell pyroptosis and ALI [247, 248]. STING deficiency blocked LPS‐induced pyroptosis in lung tissues, evidenced by the reduced expression levels of cytokines (IL‐1β and IL‐18) and GSDMD, which was restored by NLRP3 overexpression. However, the exact mechanism by which STING regulate the activation of NLRP3 remains elusive [247].
4.3.6. Radiation lung injury (RILI)
RILI is a common and serious complication in the treatment of thoracic tumors. It not only limits additional fractional radiotherapy, but also seriously affects the prognosis of patients. The development of RILI is divided into an early phase of radiation pneumonitis and a late stage of radiation fibrosis. Acute inflammation occurs within a few weeks after radiation and is characterized by the release of pro-inflammatory cytokines and the accumulation of immune cells in the lung tissue, while chronic fibrosis occurs months to years later, and ultimately leads to permanent damage of lung function [249–252]. More and more evidences indicate that AIM2 inflammasome-mediated pyroptosis of macrophages and epithelial cells play a key role in radiation-induced tissue damage [96, 97]. Under radiation, AIM2 recognizes the double-strand DNA fragments resulted from ionizing radiation and chemotherapeutic agents, forming specks and recruits ASC, thereby activating caspase-1 and leading to pyroptosis [253–257]. Gao et al. demonstrated that inhibition of AIM2 activation attenuated the inflammatory cascade, acute pneumonitis, and subsequent chronic fibrosis, illustrating the role of AIM2 in RILI [258].
4.4. Digestive system
Inflammasome activation and pyroptosis play a significant role in immunopathogenesis of several different conditions related to gastrointestinal tract. The effector molecules of pyroptosis, the Gasdermin proteins, were actually first discovered in cells lining the gastrointestinal tract in early 2000s [259]. However, only recently the role of Gasdermin proteins in pyroptosis was identified [6, 253]. Gastrointestinal tract has been attributed as the biggest immunological organ in the body, hence, the role of innate immune system is pivotal in this system [260]. Si Ming Man, in a review in 2018, listed the expression profile of inflammasome components in cells lining the gastrointestinal tract [261]. The majority of inflammasome components are expressed in these cells with a slight difference between human and mice. For example, human enterocytes express caspase-1, -4,- 5, IL-18, NLRP6, and NLRP9 whereas murine enterocytes express AIM2, ASC, Caspase-1, -11, GSDMD, Il-1β, IL-18, NAIPs, NLRC3, NLRC4, NLRP6 and NLRP9b [261]. Similarly, human gastric cells express AIM2, ASC, Caspase-1, NLRC4, NLRP1,3, and 6 whereas murine gastric cells express ASC, IL-1β and IL-18 [261]. Gastrointestinal system is a site where there is a need of perfect balance between gut microbiome and immune system. Imbalance, either through overactivation or through decreased activation of inflammasome, leads to severe pathogenic conditions. Below we discuss some of the conditions where inflammation and pyroptosis are involved in the immunopathogenesis in gastrointestinal tract and pancreas.
4.4.1. Bacterial and viral infection
Gastrointestinal lining acts as a barrier and maintains a perfect homeostasis between the gut microbiome and the host body [260, 262]. Intestinal epithelial cells (IECs) lie at the interface of gut microbiota and host systemic sites. Unchecked inflammation and pyroptosis of IECs causes disruption of this intestinal homeostasis between the host immune system and gut microbiome [263]. Unregulated disruption of intestinal barrier promote invasion by gut microbiome, leading to systemic infection and sepsis, and excessive inflammation and cell death in the intestinal lining cause tissue damage [263, 264]. However, suboptimal inflammation leads to improper colonization of the intestinal environment by microbes, rendering the host susceptible for systemic infection [265].
NLRP3 and ASC have been found to be important in restricting bacterial colonization in a mice model infected orally with Citrobacter rodentium. During the early stage of infection, NLRP3 activation in intestinal epithelial cells acted as a defense barrier to limit bacterial colonization and systemic infection [266]. Similarly, Crowley et al. demonstrated that caspase-1 and caspase-11 were essential to restrict the intracellular bacteria Salmonella typhimurium within the gut [267]. These caspases were activated after the detection of bacterial virulence factors and triggered pyroptotic cell death, leading to shedding of infected cells and expulsion of bacteria in the gut. Similarly, NAIP and NLRC4 inflammasome has also been shown to be important to restrict the intracellular replication of Salmonella during gastrointestinal infection. Lack of these inflammasome components led to increased bacterial colonization of the intestinal epithelial cells in oral Salmonella infection model mice [268–270]. However, protection from bacterial dissemination into deeper tissues by IEC pyroptosis and expulsion is a very controlled process coordinated by IL-18 and eicosanoids. In addition, extracellular pathogens cause pyroptosis of IECs causing intestinal inflammation and diarrhea like symptoms. For example, enteropathogenic E. coli causes the pyroptosis of IECs in Ca2+ influx dependent LPS internalization [271]. Excessive activation of IEC pyroptosis and expulsion led to severe damage in the epithelial layer and compromise the intestinal barrier to expose the systemic sites [270].
In addition to bacterial infection, pyroptosis is also important during gastrointestinal viral infection. It has been shown that pyroptosis has both positive and negative consequences in terms of viral mediated lethality depending on the mouse genotype and extent of activation [272, 273]. Pyroptosis contributes towards the detrimental effects during gastrointestinal murine norovirus infection in STAT1-/- mice. STAT1 has been found to be protective and block norovirus replication and infection, making the deficient mice more susceptible to infection [272, 274]. Pyroptosis played a detrimental role in STAT1-/- mice but not in WT mice during norovirus infection. Norovirus infection was observed to be causing the NLRP3 inflammasome activation and pyroptosis. In WT mice, STAT1 was shown to control the expression of pro-IL-1β, so that during STAT1 deficiency, murine norovirus induced inflammasome activation and pyroptosis caused excess release of IL-1β [272]. Transmissible gastroenteritis virus (TGEV), which causes fatal severe diarrhea, has recently been found to induce NLRP3 inflammasome and pyroptosis in porcine intestinal epithelial cells [275]. Pyroptosis could also be protective during gastrointestinal viral infection. For instance, Zhu et. al. showed that NLRP9b/ASC/Caspase-1 inflammasome mediated pyroptosis played a protective role during intestinal rotavirus infection [273]. They demonstrated NLRP9b deficient mice were more susceptible to rotavirus infection. Similarly, GSDMD deficiency but not IL-18 deficiency also showed increased susceptibility independent of intestinal microbiota, suggesting a protective role of pyroptosis against rotavirus infection. Thus, similar to bacterial infection, inflammasome activation and pyroptosis may play roles in controlling microbe intracellular replication, however, excessive inflammation could be detrimental.
4.4.2. Inflammatory bowel disease (IBD)
One of the major diseases involving the imbalanced inflammation in intestine is the IBD, which generally comprises of two conditions, Crohn’s disease (CD) and Ulcerative colitis (UC) [276]. UC is more limited in the colon, whereas CD can be present in any part of gastrointestinal tract. IBD can be triggered by various genetic causes, environmental influences, imbalance in gut microbiome and immune system dysregulation [265, 277, 278]. Complete loss of inflammasome can exacerbate the IBD, making the mice more susceptible to experimental colitis [279]. However, overactivation of these inflammasome also creates problem [278, 279]. The role played by inflammasome and pyroptosis remains elusive as both protective and detrimental effects have been observed in various studies [278, 280–283]. Although the reason for the discrepancy between different studies is not fully understood, plenty of evidence suggests that pyroptosis contributes to the pathogenesis of IBD and its proper regulation is one of the keys for the treatment of IBD.
Several studies using dextran sodium sulfate (DSS)-induced colitis models suggested a protection role of pyroptosis. Elinav et al. reported that NLRP6 and ASC deficiency were correlated with the severity of DSS-induced colitis [284]. Allen et. al. reported cancer progression associated with DSS-induced colitis increased in mice lacking NLRP3, ASC or caspase-1 [281]. Deficiency in NLRP3, ASC, caspase-1, and caspase-11 has been shown to promote epithelial cell damage and morbidity, and administration of IL-1β and/or IL-18 attenuated the disease progression [285, 286].
However, a large number of studies reported an opposite effect of pyroptosis in IBD. Some early studies reported a correlation of increased IL-1β, IL-18 among other pro-inflammatory cytokines with the severity of the inflammatory bowel disease [287–289]. Similarly, inhibition of caspase-1 was found to be protective against the DSS-induced acute experimental colitis suggesting a role of pyroptosis during colitis [290, 291]. Besides, neutralization of IL-18 also helped to ameliorate the DSS-induced acute colitis [292]. Data from these studies suggest that inflammatory cytokines from inflammasome activation contribute to the pathogenesis of IBD. Recently, Xu et. al. reported that CD147, a transmembrane protein which induces matrix metalloproteinases and angiogenic factors, exacerbated the IBD in patients by increasing pyroptosis in intestinal epithelial cells [293]. CD147 promotes phosphorylation of NF-kB, leading to upregulation of NLRP3 inflammasome components and increased pyroptosis [293]. Xiong et. al. demonstrated that the protective effect of cholecalciferol cholesterol emulsion for IBD was through downregulation of pyroptosis, specifically by inhibiting the NF-kB mediated expression of pro-forms of inflammasome components and cytokines [278].
Similarly, Chen et al. found that DSS-induced chronic colitis was significantly reduced upon knockdown of NEK7 (NIMA related kinase 7), an important component of macrophage’s NLRP3 inflammasome [277]. Reduction of expression of genes involved in pyroptosis including NLRP3, ASC and GSDMD has been shown to be protective in IBD mice models [294, 295].
4.4.3. Intestinal ischemia-reperfusion (I/R) injury
Another condition where pyroptosis plays a role in tissue damage in gastrointestinal tract is the intestinal I/R injury. I/R injury is a condition where there is obstruction in proper blood flow in certain organs leading to tissue/organ damage and failure [296]. Yang et al. reported the occurrence of pyroptosis during renal I/R injury [297]. Recently, Jia et al. reported the activation of inflammasome and pyroptosis as a downstream process during intestinal I/R injury [298]. Pyroptosis contributed toward the prognosis of injury by cell death and disruption of intestinal barrier, which may further lead to sepsis. Metformin was found to be protective against intestinal I/R injury by inhibiting pyroptosis and inflammatory response by blocking the interaction between TXNIP and NLRP3, which otherwise would activate the NLRP3 inflammasome [298].
4.4.4. Non-steroidal anti-inflammatory drugs (NSAID) induced intestinal injury
Use of NSAID may cause severe injury and bleeding in gastrointestinal tract. This was ameliorated by using the acid suppressants and histamine H2 receptor antagonists in the stomach but not in the intestinal injury, where they can even worsen the case [299, 300]. Recently, Otani et al. demonstrated that NSAID-induced intestinal injury could be averted by colchicine through the inhibition of NLRP3 inflammasome [299]. In NSAID-induced small intestinal injury, NF-kB upregulation through TLR4 signaling and extracellular ATP caused the activation of NLRP3 and pro-IL-1β expression [301, 302]. The release of IL-1β has been shown to aggravate the pathogenesis of this intestinal injury and use of IL-1β neutralizing antibodies ameliorated the injury. Otani et. al. observed that the levels of NLRP3, caspase-1 and IL-1β significantly increase in the small intestine upon indomethacin induced small intestine injury, and colchicine could inhibit the activation of NLRP3 inflammasome [299].
4.4.5. Pancreatic diseases
Prolonged exposure to inorganic arsenic has been shown to cause diabetes and recently pyroptosis of pancreatic beta cells has been found to contribute to the pathogenesis of arsenic induced diabetes [303, 304]. By using insulin secreting beta cell derived cell line (INS-1 cells), Pei et. al. demonstrated that prolonged exposure to As2O3 caused elevated expression of ER stress sensor IRE1α and TNF-α, activating the NLRP3 inflammasome and caspase-1 cleavage [305]. Inhibition of IRE1α, TNF-α and NLRP3 by using their inhibitors ameliorated the As2O3 induced pyroptosis and β-cell dysfunction.
Pyroptosis was also observed to contribute to the pathogenesis of acute pancreatitis. Acute pancreatitis is associated with large-scale damage and death of pancreatic acinar cells. Although several different mechanisms can cause the death of acinar cells, pyroptotic death has been shown to be related with the worst prognosis of the disease [306]. Wang et al. showed that a circular RNA, circHIPK3, stimulated the pyroptotic cell death of acinar cells by regulating the miR-193a-5p/GSDMD axis [306]. In the plasma of acute pancreatitis patients, level of circHIPK3 was elevated. CircHIPK3 regulates the expression of multiple miRNAs, including miR-193a-5p, the level of which reduced in the samples from acute pancreatitis patients. MiR-193a-5p negatively regulates the GSDMD expression, thus enhanced expression of circHIPK3 led to an increased expression of GSDMD, and subsequently pyroptosis.
Pyroptosis is also involved in the intestinal injury during severe AP. GSDMD mediated pyroptosis causes cell deaths in the intestinal mucosal lining, and the downregulation of intestinal epithelial cell tight junction proteins, further weakening the intestinal barrier [307]. Recently, Lin et. al. demonstrated that downregulating the GSDMD expression by using siRNA protected the intestinal injury and helped to alleviate the symptoms during acute pancreatitis in mice model [308].
4.5. Reproductive system
Activation of inflammasome has been associated with many diseases in the reproductive system, including infectious diseases such as vaginitis and sextually transmitted infections (STIs), and non-infectious disease including female and male infertility, recurrent miscarriages, endometriosis, preeclampsia, placental inflammation, and preterm birth [309, 310]. Inhibitors targeting the inflammasome activation have been shown to benefit the treatment of certain reproductive diseases. For example, neutralization of ASC by adding anti-ASC polyclonal antibody to semen significantly increased mean sperm motility by 50% in men with spinal cord injury [311]. Using a mouse model for sterile intra-amniotic inflammation, Gomez-Lopez et al. showed that inhibition of the NLRP3 inflammasome via the injection of a specific inhibitor MCC950 prevented preterm labor/birth by 35.7% and reduced neonatal mortality by 26.7% [312]. Omega-3 fatty acid has been shown to reduce the rate of preterm birth induced by infection and trophoblast inflammation through inhibiting inflammasome-related molecule synthesis and cathepsin S (CTSS) and caspase-1 activation [313].
While a clear role of inflammation has been established early on in many pathological conditions related to the reproductive system, the contribution of pyroptosis was not as clear since mechanistic studies have been limited [309, 314]. Many studies focused on the examination of elevated level or activity of biomarkers related to inflammasome activation, including NLRP3, caspase-1, ASC, IL-1β, and IL-18 in patient, while the underlining mechanism remains elusive. Here we focus our discussion of several recent papers aiming at probing the connection between the molecular trigger and the pathogenesis of disease conditions.
Maternal obesity has been identified as a major risk factor for adverse pregnancy complications and associated with pro-inflammatory cytokine release in the placenta [315]. In the plasma of obese patients, the levels of free fatty acid, including palmitic acids (PAs), are elevated. Using a human trophoblast cell line, Shirasuna et al. discovered that PA could activate the NLRP3 inflammasomes, leading to significant caspase-1 activation and IL-1β secretion, and disruption of caspase-1 activity diminished PA-induced IL-1β release, clearly indicating the involvement of pyroptosis in this process [316]. Furthermore, when production of ROS was disrupted by the addition of antioxidant, the PA-induced cytokine release was abolished. While the role of ROS in the process remains elusive, these studies suggest that free fatty acid such as PA plays an important role in the pathogenesis of obesity-related placental inflammation and subsequent pregnancy complications.
Yang et al. investigated the mechanisms linking inflammatory dysregulation and endoplasmic reticulum (ER) stress to preeclampsia [317]. In preeclampsia, trophoblast cells express inflammatory cytokines including IL-1β, a biomarker of inflammasome activation [318, 319]. Since TXNIP has been reported to activate the NLRP3 inflammasome [320, 321], and NLRP3 inflammasome activation has been shown to activate IL-1β in the placenta of patients with preeclampsia, the authors hypothesize that TXNIP was the linkage between ER stress and NLRP3 inflammasomes to activate IL-1β production in preeclampsia [322]. Consistent with their hypothesis, ER stress was found to enhance TXNIP activation and induce NLRP3 inflammasome formation; suppression of the TXNIP expression using siRNA and lentivirus or treatment with ER stress inhibitors both reduced NLRP3 inflammasome activation. Furthermore, TXNIP has been shown to be involved in compromised trophoblast invasion, a feature involved in the pathogenesis of PE [314, 323, 324]. Through a series of studies, they first established that it is pyroptosis, not necroptosis, that lead to the inflammatory response in the placenta from women with early onset preeclampsia. Then using an ER stress cellular models, they further examined the signaling pathways leading to pyroptosis in human trophoblasts, and observed increased expression of TXNIP and NLRP3 in both human primary trophoblast cells and in placental tissues from early onset preeclampsia deliveries. Based on these results, a mechanism was derived in which excessive ER stress and unfolded protein response lead to activation of the NLRP3-pyroptotic inflammatory pathway through TXNIP, which is pivotal to the inflammation response in early onset PE pathology.
5. Future direction and perspectives
While the importance of pyroptosis has been reported in the pathogenesis of various disease conditions, many studies are focused on the examination of activation of inflammasome and generation of pro-inflammatory cytokines associated with pyroptosis. Studies focusing on mechanistic insights have been limited. Additional insight from mechanistic studies are necessary to lay the foundation for the development of better therapies and to identify new biomarkers for diagnosis and monitoring.
As discussed extensively above, inflammasome activation and pyroptosis could be both protective and detrimental in various inflammatory disorders and disease conditions, depending on many factors. Thus, inhibition of inflammasome as a therapeutic strategy can be both promising and challenging. For example, while the direct outcome of inhibiting the inflammasome activation and pyroptosis had been proven beneficial in many disease models including sepsis [325–327], preterm birth [312, 328], PE [329], AS [148], AML [114], MI/RI [159–165], pulmonary hypertension [170, 173], the overall clinical outcome demands thorough evaluation and careful consideration of the entire body as inhibiting of normal house-keeping inflammasome activation could lead to unintentional immunosuppressive effects.
Highlights.
Pyroptosis occurs in many cell types and affect almost all vital systems in the human body.
Pyroptosis is involved in pathogenesis in the hematopoietic, cardiovascular, respiratory, digestive, and reproductive systems
Inflammasome activation is a double-edged sword with both protective and detrimental effect to host wellbeing
Acknowledgements
Y.W. is supported by NSF CHE-1709381, NIH/NIAID R56 AI137020 and R21 AI142063, NIH/NHLBI R01 HL142640, and NIH/NIGMS R01 GM132443. Z.L. is supported by NIH/NHLBI R01 HL146744, R01 HL142640, and NIH/NIGMS R01 GM132443.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declarations of interest
None.
References
- [1].Miao EA, Leaf IA, Treuting PM, Mao DP, Dors M, Sarkar A, et al. Caspase-1-induced pyroptosis is an innate immune effector mechanism against intracellular bacteria. Nat Immunol 2010;11:1136–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Cookson BT, Brennan MA. Pro-inflammatory programmed cell death. Trends Microbiol 2001;9:113–4. [DOI] [PubMed] [Google Scholar]
- [3].Bergsbaken T, Fink SL, Cookson BT. Pyroptosis: host cell death and inflammation. Nat Rev Microbiol 2009;7:99–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Shi J, Zhao Y, Wang Y, Gao W, Ding J, Li P, et al. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature 2014;514:187–92. [DOI] [PubMed] [Google Scholar]
- [5].Yang J, Zhao Y, Shao F. Non-canonical activation of inflammatory caspases by cytosolic LPS in innate immunity. Curr Opin Immunol 2015;32:78–83. [DOI] [PubMed] [Google Scholar]
- [6].Kayagaki N, Stowe IB, Lee BL, O’Rourke K, Anderson K, Warming S, et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 2015;526:666–71. [DOI] [PubMed] [Google Scholar]
- [7].Kayagaki N, Wong MT, Stowe IB, Ramani SR, Gonzalez LC, Akashi-Takamura S, et al. Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science 2013;341:1246–9. [DOI] [PubMed] [Google Scholar]
- [8].Hagar JA, Powell DA, Aachoui Y, Ernst RK, Miao EA. Cytoplasmic LPS activates caspase-11: implications in TLR4-independent endotoxic shock. Science 2013;341:1250–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Aachoui Y, Leaf IA, Hagar JA, Fontana MF, Campos CG, Zak DE, et al. Caspase-11 protects against bacteria that escape the vacuole. Science 2013;339:975–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Sarhan J, Liu BC, Muendlein HI, Li P, Nilson R, Tang AY, et al. Caspase-8 induces cleavage of gasdermin D to elicit pyroptosis during Yersinia infection. Proc Natl Acad Sci U S A 2018;115:E10888–E97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Mascarenhas DPA, Cerqueira DM, Pereira MSF, Castanheira FVS, Fernandes TD, Manin GZ, et al. Inhibition of caspase-1 or gasdermin-D enable caspase-8 activation in the Naip5/NLRC4/ASC inflammasome. PLoS Pathog 2017;13:e1006502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Wang Y, Gao W, Shi X, Ding J, Liu W, He H, et al. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature 2017;547:99–103. [DOI] [PubMed] [Google Scholar]
- [13].Jiang M, Qi L, Li L, Li Y. The caspase-3/GSDME signal pathway as a switch between apoptosis and pyroptosis in cancer. Cell Death Discov 2020;6:112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Rogers C, Fernandes-Alnemri T, Mayes L, Alnemri D, Cingolani G, Alnemri ES. Cleavage of DFNA5 by caspase-3 during apoptosis mediates progression to secondary necrotic/pyroptotic cell death. Nat Commun 2017;8:14128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Shi J, Gao W, Shao F. Pyroptosis: Gasdermin-Mediated Programmed Necrotic Cell Death. Trends Biochem Sci 2017;42:245–54. [DOI] [PubMed] [Google Scholar]
- [16].Chen X, He WT, Hu L, Li J, Fang Y, Wang X, et al. Pyroptosis is driven by non-selective gasdermin-D pore and its morphology is different from MLKL channel-mediated necroptosis. Cell Res 2016;26:1007–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Kovacs SB, Miao EA. Gasdermins: Effectors of Pyroptosis. Trends Cell Biol 2017;27:673–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Aglietti RA, Dueber EC. Recent Insights into the Molecular Mechanisms Underlying Pyroptosis and Gasdermin Family Functions. Trends Immunol 2017;38:261–71. [DOI] [PubMed] [Google Scholar]
- [19].Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol 2010;11:373–84. [DOI] [PubMed] [Google Scholar]
- [20].Pinaud L, Sansonetti PJ, Phalipon A. Host Cell Targeting by Enteropathogenic Bacteria T3SS Effectors. Trends Microbiol 2018;26:266–83. [DOI] [PubMed] [Google Scholar]
- [21].Shenoy AR, Furniss RCD, Goddard PJ, Clements A. Modulation of Host Cell Processes by T3SS Effectors. Curr Top Microbiol Immunol 2018;416:73–115. [DOI] [PubMed] [Google Scholar]
- [22].Cossart P, Helenius A. Endocytosis of viruses and bacteria. Cold Spring Harb Perspect Biol 2014;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Rathinam VA, Fitzgerald KA. Inflammasome Complexes: Emerging Mechanisms and Effector Functions. Cell 2016;165:792–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Malik A, Kanneganti TD. Inflammasome activation and assembly at a glance. J Cell Sci 2017;130:3955–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Xue Y, Enosi Tuipulotu D, Tan WH, Kay C, Man SM. Emerging Activators and Regulators of Inflammasomes and Pyroptosis. Trends Immunol 2019;40:1035–52. [DOI] [PubMed] [Google Scholar]
- [26].Elliott EI, Sutterwala FS. Initiation and perpetuation of NLRP3 inflammasome activation and assembly. Immunol Rev 2015;265:35–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Zhao Y, Shao F. The NAIP-NLRC4 inflammasome in innate immune detection of bacterial flagellin and type III secretion apparatus. Immunol Rev 2015;265:85–102. [DOI] [PubMed] [Google Scholar]
- [28].Kelley N, Jeltema D, Duan Y, He Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int J Mol Sci 2019;20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Swanson KV, Deng M, Ting JP. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol 2019;19:477–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].He Y, Hara H, Nunez G. Mechanism and Regulation of NLRP3 Inflammasome Activation. Trends Biochem Sci 2016;41:1012–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Bauernfeind FG, Horvath G, Stutz A, Alnemri ES, MacDonald K, Speert D, et al. Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J Immunol 2009;183:787–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Franchi L, Eigenbrod T, Nunez G. Cutting edge: TNF-alpha mediates sensitization to ATP and silica via the NLRP3 inflammasome in the absence of microbial stimulation. J Immunol 2009;183:792–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Mariathasan S, Weiss DS, Newton K, McBride J, O’Rourke K, Roose-Girma M, et al. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 2006;440:228–32. [DOI] [PubMed] [Google Scholar]
- [34].Piccini A, Carta S, Tassi S, Lasiglie D, Fossati G, Rubartelli A. ATP is released by monocytes stimulated with pathogen-sensing receptor ligands and induces IL-1beta and IL-18 secretion in an autocrine way. Proc Natl Acad Sci U S A 2008;105:8067–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Lamkanfi M, Kanneganti TD. Nlrp3: an immune sensor of cellular stress and infection. Int J Biochem Cell Biol 2010;42:792–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Dostert C, Petrilli V, Van Bruggen R, Steele C, Mossman BT, Tschopp J. Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science 2008;320:674–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Cassel SL, Eisenbarth SC, Iyer SS, Sadler JJ, Colegio OR, Tephly LA, et al. The Nalp3 inflammasome is essential for the development of silicosis. Proc Natl Acad Sci U S A 2008;105:9035–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Bauernfeind F, Bartok E, Rieger A, Franchi L, Nunez G, Hornung V. Cutting edge: reactive oxygen species inhibitors block priming, but not activation, of the NLRP3 inflammasome. J Immunol 2011;187:613–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Chen J, Chen ZJ. PtdIns4P on dispersed trans-Golgi network mediates NLRP3 inflammasome activation. Nature 2018;564:71–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Duncan JA, Canna SW. The NLRC4 Inflammasome. Immunol Rev 2018;281:115–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Zhao Y, Yang J, Shi J, Gong YN, Lu Q, Xu H, et al. The NLRC4 inflammasome receptors for bacterial flagellin and type III secretion apparatus. Nature 2011;477:596–600. [DOI] [PubMed] [Google Scholar]
- [42].Endrizzi MG, Hadinoto V, Growney JD, Miller W, Dietrich WF. Genomic sequence analysis of the mouse Naip gene array. Genome Res 2000;10:1095–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Kofoed EM, Vance RE. Innate immune recognition of bacterial ligands by NAIPs determines inflammasome specificity. Nature 2011;477:592–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Lightfield KL, Persson J, Brubaker SW, Witte CE, von Moltke J, Dunipace EA, et al. Critical function for Naip5 in inflammasome activation by a conserved carboxy-terminal domain of flagellin. Nat Immunol 2008;9:1171–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Reyes Ruiz VM, Ramirez J, Naseer N, Palacio NM, Siddarthan IJ, Yan BM, et al. Broad detection of bacterial type III secretion system and flagellin proteins by the human NAIP/NLRC4 inflammasome. Proc Natl Acad Sci U S A 2017;114:13242–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Levinsohn JL, Newman ZL, Hellmich KA, Fattah R, Getz MA, Liu S, et al. Anthrax lethal factor cleavage of Nlrp1 is required for activation of the inflammasome. PLoS Pathog 2012;8:e1002638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Chavarria-Smith J, Vance RE. Direct proteolytic cleavage of NLRP1B is necessary and sufficient for inflammasome activation by anthrax lethal factor. PLoS Pathog 2013;9:e1003452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Sandstrom A, Mitchell PS, Goers L, Mu EW, Lesser CF, Vance RE. Functional degradation: A mechanism of NLRP1 inflammasome activation by diverse pathogen enzymes. Science 2019;364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Chui AJ, Okondo MC, Rao SD, Gai K, Griswold AR, Johnson DC, et al. N-terminal degradation activates the NLRP1B inflammasome. Science 2019;364:82–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Frew BC, Joag VR, Mogridge J. Proteolytic processing of Nlrp1b is required for inflammasome activity. PLoS Pathog 2012;8:e1002659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Okondo MC, Rao SD, Taabazuing CY, Chui AJ, Poplawski SE, Johnson DC, et al. Inhibition of Dpp8/9 Activates the Nlrp1b Inflammasome. Cell Chem Biol 2018;25:262–7 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Roberts TL, Idris A, Dunn JA, Kelly GM, Burnton CM, Hodgson S, et al. HIN-200 proteins regulate caspase activation in response to foreign cytoplasmic DNA. Science 2009;323:1057–60. [DOI] [PubMed] [Google Scholar]
- [53].Jin T, Perry A, Jiang J, Smith P, Curry JA, Unterholzner L, et al. Structures of the HIN domain:DNA complexes reveal ligand binding and activation mechanisms of the AIM2 inflammasome and IFI16 receptor. Immunity 2012;36:561–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Matyszewski M, Morrone SR, Sohn J. Digital signaling network drives the assembly of the AIM2-ASC inflammasome. Proc Natl Acad Sci U S A 2018;115:E1963–E72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Xu H, Yang J, Gao W, Li L, Li P, Zhang L, et al. Innate immune sensing of bacterial modifications of Rho GTPases by the Pyrin inflammasome. Nature 2014;513:237–41. [DOI] [PubMed] [Google Scholar]
- [56].Chung LK, Park YH, Zheng Y, Brodsky IE, Hearing P, Kastner DL, et al. The Yersinia Virulence Factor YopM Hijacks Host Kinases to Inhibit Type III Effector-Triggered Activation of the Pyrin Inflammasome. Cell Host Microbe 2016;20:296–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Ratner D, Orning MP, Proulx MK, Wang D, Gavrilin MA, Wewers MD, et al. The Yersinia pestis Effector YopM Inhibits Pyrin Inflammasome Activation. PLoS Pathog 2016;12:e1006035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Park YH, Wood G, Kastner DL, Chae JJ. Pyrin inflammasome activation and RhoA signaling in the autoinflammatory diseases FMF and HIDS. Nat Immunol 2016;17:914–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Gao W, Yang J, Liu W, Wang Y, Shao F. Site-specific phosphorylation and microtubule dynamics control Pyrin inflammasome activation. Proc Natl Acad Sci U S A 2016;113:E4857–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Xia S, Zhang Z, Magupalli VG, Pablo JL, Dong Y, Vora SM, et al. Gasdermin D pore structure reveals preferential release of mature interleukin-1. Nature 2021;593:607–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Wu C, Lu W, Zhang Y, Zhang G, Shi X, Hisada Y, et al. Inflammasome Activation Triggers Blood Clotting and Host Death through Pyroptosis. Immunity 2019;50:1401–11 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Fink SL, Cookson BT. Caspase-1-dependent pore formation during pyroptosis leads to osmotic lysis of infected host macrophages. Cell Microbiol 2006;8:1812–25. [DOI] [PubMed] [Google Scholar]
- [63].Monteleone M, Stanley AC, Chen KW, Brown DL, Bezbradica JS, von Pein JB, et al. Interleukin-1beta Maturation Triggers Its Relocation to the Plasma Membrane for Gasdermin-D-Dependent and -Independent Secretion. Cell Rep 2018;24:1425–33. [DOI] [PubMed] [Google Scholar]
- [64].Sun Q, Scott MJ. Caspase-1 as a multifunctional inflammatory mediator: noncytokine maturation roles. J Leukoc Biol 2016;100:961–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Nakanishi K, Yoshimoto T, Tsutsui H, Okamura H. Interleukin-18 regulates both Th1 and Th2 responses. Annu Rev Immunol 2001;19:423–74. [DOI] [PubMed] [Google Scholar]
- [66].Delaleu N, Bickel M. Interleukin-1 beta and interleukin-18: regulation and activity in local inflammation. Periodontol 2000 2004;35:42–52. [DOI] [PubMed] [Google Scholar]
- [67].Garlanda C, Dinarello CA, Mantovani A. The interleukin-1 family: back to the future. Immunity 2013;39:1003–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Novick D, Kim S, Kaplanski G, Dinarello CA. Interleukin-18, more than a Th1 cytokine. Semin Immunol 2013;25:439–48. [DOI] [PubMed] [Google Scholar]
- [69].Fabbi M, Carbotti G, Ferrini S. Context-dependent role of IL-18 in cancer biology and counter-regulation by IL-18BP. J Leukoc Biol 2015;97:665–75. [DOI] [PubMed] [Google Scholar]
- [70].Karki R, Sharma BR, Tuladhar S, Williams EP, Zalduondo L, Samir P, et al. Synergism of TNF-alpha and IFN-gamma Triggers Inflammatory Cell Death, Tissue Damage, and Mortality in SARS-CoV-2 Infection and Cytokine Shock Syndromes. Cell 2021;184:149–68 e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Shao W, Yeretssian G, Doiron K, Hussain SN, Saleh M. The caspase-1 digestome identifies the glycolysis pathway as a target during infection and septic shock. J Biol Chem 2007;282:36321–9. [DOI] [PubMed] [Google Scholar]
- [72].Agard NJ, Maltby D, Wells JA. Inflammatory stimuli regulate caspase substrate profiles. Mol Cell Proteomics 2010;9:880–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Nathan C, Ding A. Nonresolving inflammation. Cell 2010;140:871–82. [DOI] [PubMed] [Google Scholar]
- [74].Skelly DT, Hennessy E, Dansereau MA, Cunningham C. A systematic analysis of the peripheral and CNS effects of systemic LPS, IL-1beta, [corrected] TNF-alpha and IL-6 challenges in C57BL/6 mice. PLoS One 2013;8:e69123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Yang X, Cheng X, Tang Y, Qiu X, Wang Y, Kang H, et al. Bacterial Endotoxin Activates the Coagulation Cascade through Gasdermin D-Dependent Phosphatidylserine Exposure. Immunity 2019;51:983–96 e6. [DOI] [PubMed] [Google Scholar]
- [76].Zhang Y, Cui J, Zhang G, Wu C, Abdel-Latif A, Smyth SS, et al. Inflammasome activation promotes venous thrombosis through pyroptosis. Blood Adv 2021;5:2619–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Iba T, Levy JH, Levi M, Thachil J. Coagulopathy in COVID-19. J Thromb Haemost 2020;18:2103–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Gando S, Levi M, Toh CH. Disseminated intravascular coagulation. Nat Rev Dis Primers 2016;2:16037. [DOI] [PubMed] [Google Scholar]
- [79].Ratajczak MZ, Bujko K, Cymer M, Thapa A, Adamiak M, Ratajczak J, et al. The Nlrp3 inflammasome as a “rising star” in studies of normal and malignant hematopoiesis. Leukemia 2020;34:1512–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Yang L, Hu M, Lu Y, Han S, Wang J. Inflammasomes and the Maintenance of Hematopoietic Homeostasis: New Perspectives and Opportunities. Molecules 2021;26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Qian L, Xiang D, Zhang J, Zhu S, Gao J, Wang X, et al. Recombinant human interleukin-1 receptor antagonist reduces acute lethal toxicity and protects hematopoiesis from chemotoxicity in vivo. Biomed Pharmacother 2013;67:108–15. [DOI] [PubMed] [Google Scholar]
- [82].Zhang J, Xiang D, Zhu S, Mao W, Lu H, Wu M, et al. Interleukin 1 receptor antagonist inhibits normal hematopoiesis and reduces lethality and bone marrow toxicity of 5-fluouracil in mouse. Biomed Pharmacother 2009;63:501–8. [DOI] [PubMed] [Google Scholar]
- [83].Baldridge MT, King KY, Boles NC, Weksberg DC, Goodell MA. Quiescent haematopoietic stem cells are activated by IFN-gamma in response to chronic infection. Nature 2010;465:793–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Venet F, Davin F, Guignant C, Larue A, Cazalis MA, Darbon R, et al. Early assessment of leukocyte alterations at diagnosis of septic shock. Shock 2010;34:358–63. [DOI] [PubMed] [Google Scholar]
- [85].Croker BA, O’Donnell JA, Gerlic M. Pyroptotic death storms and cytopenia. Curr Opin Immunol 2014;26:128–37. [DOI] [PubMed] [Google Scholar]
- [86].Masters SL, Gerlic M, Metcalf D, Preston S, Pellegrini M, O’Donnell JA, et al. NLRP1 inflammasome activation induces pyroptosis of hematopoietic progenitor cells. Immunity 2012;37:1009–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Radujkovic A, Kordelas L, Dai H, Schult D, Majer-Lauterbach J, Beelen D, et al. Interleukin-18 and outcome after allogeneic stem cell transplantation: A retrospective cohort study. EBioMedicine 2019;49:202–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Wu Z, Giudice V, Chen J, Sun W, Lin Z, Keyvanfar K, et al. Interleukin-18 plays a dispensable role in murine and likely also human bone marrow failure. Exp Hematol 2019;69:54–64 e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Epperly M, Berhane H, Cao S, Shields D, Franicola D, Goff JP, et al. Increased longevity of hematopoiesis in continuous marrow cultures and radiation resistance of marrow stromal and hematopoietic progenitor cells from caspase-1 homozygous recombinant-negative (knockout) mice. In Vivo 2013;27:419–30. [PMC free article] [PubMed] [Google Scholar]
- [90].Gentile LF, Cuenca AL, Cuenca AG, Nacionales DC, Ungaro R, Efron PA, et al. Improved emergency myelopoiesis and survival in neonatal sepsis by caspase-1/11 ablation. Immunology 2015;145:300–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Ratajczak MZ, Kucia M. SARS-CoV-2 infection and overactivation of Nlrp3 inflammasome as a trigger of cytokine “storm” and risk factor for damage of hematopoietic stem cells. Leukemia 2020;34:1726–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Li W, Moore MJ, Vasilieva N, Sui J, Wong SK, Berne MA, et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 2003;426:450–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Kuba K, Imai Y, Rao S, Gao H, Guo F, Guan B, et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nat Med 2005;11:875–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Ratajczak MZ, Bujko K, Ciechanowicz A, Sielatycka K, Cymer M, Marlicz W, et al. SARS-CoV-2 Entry Receptor ACE2 Is Expressed on Very Small CD45(−) Precursors of Hematopoietic and Endothelial Cells and in Response to Virus Spike Protein Activates the Nlrp3 Inflammasome. Stem Cell Rev Rep 2021;17:266–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Stoecklein VM, Osuka A, Ishikawa S, Lederer MR, Wanke-Jellinek L, Lederer JA. Radiation exposure induces inflammasome pathway activation in immune cells. J Immunol 2015;194:1178–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Hu B, Jin C, Li HB, Tong J, Ouyang X, Cetinbas NM, et al. The DNA-sensing AIM2 inflammasome controls radiation-induced cell death and tissue injury. Science 2016;354:765–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Liu YG, Chen JK, Zhang ZT, Ma XJ, Chen YC, Du XM, et al. NLRP3 inflammasome activation mediates radiation-induced pyroptosis in bone marrow-derived macrophages. Cell Death Dis 2017;8:e2579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Wu T, Liu W, Fan T, Zhong H, Zhou H, Guo W, et al. 5-Androstenediol prevents radiation injury in mice by promoting NF-kappaB signaling and inhibiting AIM2 inflammasome activation. Biomed Pharmacother 2020;121:109597. [DOI] [PubMed] [Google Scholar]
- [99].Xiao J, Wang C, Yao JC, Alippe Y, Yang T, Kress D, et al. Radiation causes tissue damage by dysregulating inflammasome-gasdermin D signaling in both host and transplanted cells. PLoS Biol 2020;18:e3000807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Geiger H, de Haan G, Florian MC. The ageing haematopoietic stem cell compartment. Nat Rev Immunol 2013;13:376–89. [DOI] [PubMed] [Google Scholar]
- [101].Guralnik JM, Eisenstaedt RS, Ferrucci L, Klein HG, Woodman RC. Prevalence of anemia in persons 65 years and older in the United States: evidence for a high rate of unexplained anemia. Blood 2004;104:2263–8. [DOI] [PubMed] [Google Scholar]
- [102].Price EA. Aging and erythropoiesis: current state of knowledge. Blood Cells Mol Dis 2008;41:158–65. [DOI] [PubMed] [Google Scholar]
- [103].Franceschi C, Bonafe M, Valensin S, Olivieri F, De Luca M, Ottaviani E, et al. Inflammaging. An evolutionary perspective on immunosenescence. Ann N Y Acad Sci 2000;908:244–54. [DOI] [PubMed] [Google Scholar]
- [104].Brown K, Xie S, Qiu X, Mohrin M, Shin J, Liu Y, et al. SIRT3 reverses aging-associated degeneration. Cell Rep 2013;3:319–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Rossi DJ, Bryder D, Seita J, Nussenzweig A, Hoeijmakers J, Weissman IL. Deficiencies in DNA damage repair limit the function of haematopoietic stem cells with age. Nature 2007;447:725–9. [DOI] [PubMed] [Google Scholar]
- [106].Fali T, Fabre-Mersseman V, Yamamoto T, Bayard C, Papagno L, Fastenackels S, et al. Elderly human hematopoietic progenitor cells express cellular senescence markers and are more susceptible to pyroptosis. JCI Insight 2018;3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Luo H, Mu WC, Karki R, Chiang HH, Mohrin M, Shin JJ, et al. Mitochondrial Stress-Initiated Aberrant Activation of the NLRP3 Inflammasome Regulates the Functional Deterioration of Hematopoietic Stem Cell Aging. Cell Rep 2019;26:945–54 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Chambers SM, Shaw CA, Gatza C, Fisk CJ, Donehower LA, Goodell MA. Aging hematopoietic stem cells decline in function and exhibit epigenetic dysregulation. PLoS Biol 2007;5:e201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Mohrin M, Shin J, Liu Y, Brown K, Luo H, Xi Y, et al. Stem cell aging. A mitochondrial UPR-mediated metabolic checkpoint regulates hematopoietic stem cell aging. Science 2015;347:1374–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Schanz J, Tuchler H, Sole F, Mallo M, Luno E, Cervera J, et al. New comprehensive cytogenetic scoring system for primary myelodysplastic syndromes (MDS) and oligoblastic acute myeloid leukemia after MDS derived from an international database merge. J Clin Oncol 2012;30:820–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Basiorka AA, McGraw KL, Eksioglu EA, Chen X, Johnson J, Zhang L, et al. The NLRP3 inflammasome functions as a driver of the myelodysplastic syndrome phenotype. Blood 2016;128:2960–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Giudice V, Wu Z, Kajigaya S, Fernandez Ibanez MDP, Rios O, Cheung F, et al. Circulating S100A8 and S100A9 protein levels in plasma of patients with acquired aplastic anemia and myelodysplastic syndromes. Cytokine 2019;113:462–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Basiorka AA, McGraw KL, Abbas-Aghababazadeh F, McLemore AF, Vincelette ND, Ward GA, et al. Assessment of ASC specks as a putative biomarker of pyroptosis in myelodysplastic syndromes: an observational cohort study. Lancet Haematol 2018;5:e393–e402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Urwanisch L, Luciano M, Horejs-Hoeck J. The NLRP3 Inflammasome and Its Role in the Pathogenicity of Leukemia. Int J Mol Sci 2021;22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Hamarsheh S, Osswald L, Saller BS, Unger S, De Feo D, Vinnakota JM, et al. Oncogenic Kras(G12D) causes myeloproliferation via NLRP3 inflammasome activation. Nat Commun 2020;11:1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Ninomiya-Tsuji J, Kishimoto K, Hiyama A, Inoue J, Cao Z, Matsumoto K. The kinase TAK1 can activate the NIK-I kappaB as well as the MAP kinase cascade in the IL-1 signalling pathway. Nature 1999;398:252–6. [DOI] [PubMed] [Google Scholar]
- [117].Wang C, Deng L, Hong M, Akkaraju GR, Inoue J, Chen ZJ. TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature 2001;412:346–51. [DOI] [PubMed] [Google Scholar]
- [118].Omori E, Matsumoto K, Sanjo H, Sato S, Akira S, Smart RC, et al. TAK1 is a master regulator of epidermal homeostasis involving skin inflammation and apoptosis. J Biol Chem 2006;281:19610–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Tang M, Wei X, Guo Y, Breslin P, Zhang S, Zhang S, et al. TAK1 is required for the survival of hematopoietic cells and hepatocytes in mice. J Exp Med 2008;205:1611–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Malireddi RKS, Gurung P, Mavuluri J, Dasari TK, Klco JM, Chi H, et al. TAK1 restricts spontaneous NLRP3 activation and cell death to control myeloid proliferation. J Exp Med 2018;215:1023–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Ajibade AA, Wang Q, Cui J, Zou J, Xia X, Wang M, et al. TAK1 negatively regulates NF-kappaB and p38 MAP kinase activation in Gr-1+CD11b+ neutrophils. Immunity 2012;36:43–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Lamothe B, Lai Y, Hur L, Orozco NM, Wang J, Campos AD, et al. Deletion of TAK1 in the myeloid lineage results in the spontaneous development of myelomonocytic leukemia in mice. PLoS One 2012;7:e51228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Mensah GA, Roth GA, Fuster V. The Global Burden of Cardiovascular Diseases and Risk Factors: 2020 and Beyond. J Am Coll Cardiol 2019;74:2529–32. [DOI] [PubMed] [Google Scholar]
- [124].Zhou W, Chen C, Chen Z, Liu L, Jiang J, Wu Z, et al. NLRP3: A Novel Mediator in Cardiovascular Disease. J Immunol Res 2018;2018:5702103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Hoseini Z, Sepahvand F, Rashidi B, Sahebkar A, Masoudifar A, Mirzaei H. NLRP3 inflammasome: Its regulation and involvement in atherosclerosis. J Cell Physiol 2018;233:2116–32. [DOI] [PubMed] [Google Scholar]
- [126].Wang Q, Wu J, Zeng Y, Chen K, Wang C, Yang S, et al. Pyroptosis: A pro-inflammatory type of cell death in cardiovascular disease. Clin Chim Acta 2020;510:62–72. [DOI] [PubMed] [Google Scholar]
- [127].Zeng C, Wang R, Tan H. Role of Pyroptosis in Cardiovascular Diseases and its Therapeutic Implications. Int J Biol Sci 2019;15:1345–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Zhaolin Z, Guohua L, Shiyuan W, Zuo W. Role of pyroptosis in cardiovascular disease. Cell Prolif 2019;52:e12563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].He B, Nie Q, Wang F, Han Y, Yang B, Sun M, et al. Role of pyroptosis in atherosclerosis and its therapeutic implications. J Cell Physiol 2021. [DOI] [PubMed]
- [130].Libby P, Ridker PM, Maseri A. Inflammation and atherosclerosis. Circulation 2002;105:1135–43. [DOI] [PubMed] [Google Scholar]
- [131].Benditt EP. Evidence for a monoclonal origin of human atherosclerotic plaques and some implications. Circulation 1974;50:650–2. [DOI] [PubMed] [Google Scholar]
- [132].Bennett MR, Sinha S, Owens GK. Vascular Smooth Muscle Cells in Atherosclerosis. Circ Res 2016;118:692–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Grootaert MOJ, Bennett MR. Vascular smooth muscle cells in atherosclerosis:Time for a reassessment. Cardiovasc Res 2021. [DOI] [PMC free article] [PubMed]
- [134].Basatemur GL, Jorgensen HF, Clarke MCH, Bennett MR, Mallat Z. Vascular smooth muscle cells in atherosclerosis. Nat Rev Cardiol 2019;16:727–44. [DOI] [PubMed] [Google Scholar]
- [135].Hassan M CANTOS: A breakthrough that proves the inflammatory hypothesis of atherosclerosis. Glob Cardiol Sci Pract 2018;2018:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Zhaolin Z, Jiaojiao C, Peng W, Yami L, Tingting Z, Jun T, et al. OxLDL induces vascular endothelial cell pyroptosis through miR-125a-5p/TET2 pathway. J Cell Physiol 2019;234:7475–91. [DOI] [PubMed] [Google Scholar]
- [137].Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG, et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 2010;464:1357–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Wu X, Zhang H, Qi W, Zhang Y, Li J, Li Z, et al. Nicotine promotes atherosclerosis via ROS-NLRP3-mediated endothelial cell pyroptosis. Cell Death Dis 2018;9:171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Chen H, Lu Y, Cao Z, Ma Q, Pi H, Fang Y, et al. Cadmium induces NLRP3 inflammasome-dependent pyroptosis in vascular endothelial cells. Toxicol Lett 2016;246:7–16. [DOI] [PubMed] [Google Scholar]
- [140].Wu P, Chen J, Chen J, Tao J, Wu S, Xu G, et al. Trimethylamine N-oxide promotes apoE(−/−) mice atherosclerosis by inducing vascular endothelial cell pyroptosis via the SDHB/ROS pathway. J Cell Physiol 2020;235:6582–91. [DOI] [PubMed] [Google Scholar]
- [141].Correa R, Silva LFF, Ribeiro DJS, Almeida RDN, Santos IO, Correa LH, et al. Lysophosphatidylcholine Induces NLRP3 Inflammasome-Mediated Foam Cell Formation and Pyroptosis in Human Monocytes and Endothelial Cells. Front Immunol 2019;10:2927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Zheng D, Shi Z, Yang M, Liang B, Zhou X, Jing L, et al. NLRP3 inflammasome-mediated endothelial cells pyroptosis is involved in decabromodiphenyl ethane-induced vascular endothelial injury. Chemosphere 2021;267:128867. [DOI] [PubMed] [Google Scholar]
- [143].Long Y, Liu X, Tan XZ, Jiang CX, Chen SW, Liang GN, et al. ROS-induced NLRP3 inflammasome priming and activation mediate PCB 118- induced pyroptosis in endothelial cells. Ecotoxicol Environ Saf 2020;189:109937. [DOI] [PubMed] [Google Scholar]
- [144].Senthong V, Li XS, Hudec T, Coughlin J, Wu Y, Levison B, et al. Plasma Trimethylamine N-Oxide, a Gut Microbe-Generated Phosphatidylcholine Metabolite, Is Associated With Atherosclerotic Burden. J Am Coll Cardiol 2016;67:2620–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Petrilli V, Papin S, Dostert C, Mayor A, Martinon F, Tschopp J. Activation of the NALP3 inflammasome is triggered by low intracellular potassium concentration. Cell Death Differ 2007;14:1583–9. [DOI] [PubMed] [Google Scholar]
- [146].Wu Q, He X, Wu LM, Zhang RY, Li LM, Wu CM, et al. MLKL Aggravates Ox-LDL-Induced Cell Pyroptosis via Activation of NLRP3 Inflammasome in Human Umbilical Vein Endothelial Cells. Inflammation 2020;43:2222–31. [DOI] [PubMed] [Google Scholar]
- [147].Schmitz G, Ruebsaamen K. Metabolism and atherogenic disease association of lysophosphatidylcholine. Atherosclerosis 2010;208:10–8. [DOI] [PubMed] [Google Scholar]
- [148].Quarato G, Guy CS, Grace CR, Llambi F, Nourse A, Rodriguez DA, et al. Sequential Engagement of Distinct MLKL Phosphatidylinositol-Binding Sites Executes Necroptosis. Mol Cell 2016;61:589–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Li Y, Niu X, Xu H, Li Q, Meng L, He M, et al. VX-765 attenuates atherosclerosis in ApoE deficient mice by modulating VSMCs pyroptosis. Exp Cell Res 2020;389:111847. [DOI] [PubMed] [Google Scholar]
- [150].Pan J, Han L, Guo J, Wang X, Liu D, Tian J, et al. AIM2 accelerates the atherosclerotic plaque progressions in ApoE−/− mice. Biochem Biophys Res Commun 2018;498:487–94. [DOI] [PubMed] [Google Scholar]
- [151].Peng X, Chen H, Li Y, Huang D, Huang B, Sun D. Effects of NIX-mediated mitophagy on ox-LDL-induced macrophage pyroptosis in atherosclerosis. Cell Biol Int 2020;44:1481–90. [DOI] [PubMed] [Google Scholar]
- [152].Jiang M, Sun X, Liu S, Tang Y, Shi Y, Bai Y, et al. Caspase-11-Gasdermin D-Mediated Pyroptosis Is Involved in the Pathogenesis of Atherosclerosis. Front Pharmacol 2021;12:657486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Mao C, Li D, Zhou E, Zhang J, Wang C, Xue C. Nicotine exacerbates atherosclerosis through a macrophage-mediated endothelial injury pathway. Aging (Albany NY) 2021;13:7627–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Xu S, Chen H, Ni H, Dai Q. Targeting HDAC6 attenuates nicotine-induced macrophage pyroptosis via NF-kappaB/NLRP3 pathway. Atherosclerosis 2021;317:1–9. [DOI] [PubMed] [Google Scholar]
- [155].Magupalli VG, Negro R, Tian Y, Hauenstein AV, Di Caprio G, Skillern W, et al. HDAC6 mediates an aggresome-like mechanism for NLRP3 and pyrin inflammasome activation. Science 2020;369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Hubbert C, Guardiola A, Shao R, Kawaguchi Y, Ito A, Nixon A, et al. HDAC6 is a microtubule-associated deacetylase. Nature 2002;417:455–8. [DOI] [PubMed] [Google Scholar]
- [157].Valenzuela-Fernandez A, Cabrero JR, Serrador JM, Sanchez-Madrid F. HDAC6: a key regulator of cytoskeleton, cell migration and cell-cell interactions. Trends Cell Biol 2008;18:291–7. [DOI] [PubMed] [Google Scholar]
- [158].Thygesen K, Alpert JS, Jaffe AS, Chaitman BR, Bax JJ, Morrow DA, et al. Fourth Universal Definition of Myocardial Infarction (2018). J Am Coll Cardiol 2018;72:2231–64. [DOI] [PubMed] [Google Scholar]
- [159].Jia C, Chen H, Zhang J, Zhou K, Zhuge Y, Niu C, et al. Role of pyroptosis in cardiovascular diseases. Int Immunopharmacol 2019;67:311–8. [DOI] [PubMed] [Google Scholar]
- [160].Han X, Zhao ZA, Yan S, Lei W, Wu H, Lu XA, et al. CXADR-like membrane protein protects against heart injury by preventing excessive pyroptosis after myocardial infarction. J Cell Mol Med 2020;24:13775–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].Li Z, Xu H, Liu X, Hong Y, Lou H, Liu H, et al. GDF11 inhibits cardiomyocyte pyroptosis and exerts cardioprotection in acute myocardial infarction mice by upregulation of transcription factor HOXA3. Cell Death Dis 2020;11:917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].Li A, Yu Y, Ding X, Qin Y, Jiang Y, Wang X, et al. MiR-135b protects cardiomyocytes from infarction through restraining the NLRP3/caspase-1/IL-1beta pathway. Int J Cardiol 2020;307:137–45. [DOI] [PubMed] [Google Scholar]
- [163].Han Y, Dong B, Chen M, Yao C. LncRNA H19 suppresses pyroptosis of cardiomyocytes to attenuate myocardial infarction in a PBX3/CYP1B1-dependent manner. Mol Cell Biochem 2021;476:1387–400. [DOI] [PubMed] [Google Scholar]
- [164].Braunwald E, Kloner RA. Myocardial reperfusion: a double-edged sword? J Clin Invest 1985;76:1713–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].Ding S, Liu D, Wang L, Wang G, Zhu Y. Inhibiting MicroRNA-29a Protects Myocardial Ischemia-Reperfusion Injury by Targeting SIRT1 and Suppressing Oxidative Stress and NLRP3-Mediated Pyroptosis Pathway. J Pharmacol Exp Ther 2020;372:128–35. [DOI] [PubMed] [Google Scholar]
- [166].Yao L, Song J, Meng XW, Ge JY, Du BX, Yu J, et al. Periostin aggravates NLRP3 inflammasome-mediated pyroptosis in myocardial ischemia-reperfusion injury. Mol Cell Probes 2020;53:101596. [DOI] [PubMed] [Google Scholar]
- [167].Mo G, Liu X, Zhong Y, Mo J, Li Z, Li D, et al. IP3R1 regulates Ca(2+) transport and pyroptosis through the NLRP3/Caspase-1 pathway in myocardial ischemia/reperfusion injury. Cell Death Discov 2021;7:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [168].Li J, Zhao C, Zhu Q, Wang Y, Li G, Li X, et al. Sweroside Protects Against Myocardial Ischemia-Reperfusion Injury by Inhibiting Oxidative Stress and Pyroptosis Partially via Modulation of the Keap1/Nrf2 Axis. Front Cardiovasc Med 2021;8:650368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [169].Zhang J, Huang L, Shi X, Yang L, Hua F, Ma J, et al. Metformin protects against myocardial ischemia-reperfusion injury and cell pyroptosis via AMPK/NLRP3 inflammasome pathway. Aging (Albany NY) 2020;12:24270–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [170].Guo X, Hu S, Liu JJ, Huang L, Zhong P, Fan ZX, et al. Piperine protects against pyroptosis in myocardial ischaemia/reperfusion injury by regulating the miR-383/RP105/AKT signalling pathway. J Cell Mol Med 2021;25:244–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [171].Ye B, Chen X, Dai S, Han J, Liang X, Lin S, et al. Emodin alleviates myocardial ischemia/reperfusion injury by inhibiting gasdermin D-mediated pyroptosis in cardiomyocytes. Drug Des Devel Ther 2019;13:975–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [172].Bian Y, Li X, Pang P, Hu XL, Yu ST, Liu YN, et al. Kanglexin, a novel anthraquinone compound, protects against myocardial ischemic injury in mice by suppressing NLRP3 and pyroptosis. Acta Pharmacol Sin 2020;41:319–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [173].Xiao B, Huang X, Wang Q, Wu Y. Beta-Asarone Alleviates Myocardial Ischemia-Reperfusion Injury by Inhibiting Inflammatory Response and NLRP3 Inflammasome Mediated Pyroptosis. Biol Pharm Bull 2020;43:1046–51. [DOI] [PubMed] [Google Scholar]
- [174].Zhong Y, Li YP, Yin YQ, Hu BL, Gao H. Dexmedetomidine inhibits pyroptosis by down-regulating miR-29b in myocardial ischemia reperfusion injury in rats. Int Immunopharmacol 2020;86:106768. [DOI] [PubMed] [Google Scholar]
- [175].Nie C, Ding X, A R, Zheng M, Li Z, Pan S, et al. Hydrogen gas inhalation alleviates myocardial ischemia-reperfusion injury by the inhibition of oxidative stress and NLRP3-mediated pyroptosis in rats. Life Sci 2021;272:119248. [DOI] [PubMed] [Google Scholar]
- [176].Lau EMT, Giannoulatou E, Celermajer DS, Humbert M. Epidemiology and treatment of pulmonary arterial hypertension. Nat Rev Cardiol 2017;14:603–14. [DOI] [PubMed] [Google Scholar]
- [177].Villegas LR, Kluck D, Field C, Oberley-Deegan RE, Woods C, Yeager ME, et al. Superoxide dismutase mimetic, MnTE-2-PyP, attenuates chronic hypoxia-induced pulmonary hypertension, pulmonary vascular remodeling, and activation of the NALP3 inflammasome. Antioxid Redox Signal 2013;18:1753–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [178].Cero FT, Hillestad V, Sjaastad I, Yndestad A, Aukrust P, Ranheim T, et al. Absence of the inflammasome adaptor ASC reduces hypoxia-induced pulmonary hypertension in mice. Am J Physiol Lung Cell Mol Physiol 2015;309:L378–87. [DOI] [PubMed] [Google Scholar]
- [179].He S, Ma C, Zhang L, Bai J, Wang X, Zheng X, et al. GLI1-mediated pulmonary artery smooth muscle cell pyroptosis contributes to hypoxia-induced pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 2020;318:L472–L82. [DOI] [PubMed] [Google Scholar]
- [180].Zhang M, Xin W, Yu Y, Yang X, Ma C, Zhang H, et al. Programmed death-ligand 1 triggers PASMCs pyroptosis and pulmonary vascular fibrosis in pulmonary hypertension. J Mol Cell Cardiol 2020;138:23–33. [DOI] [PubMed] [Google Scholar]
- [181].Zhang J, Li Y, Qi J, Yu X, Ren H, Zhao X, et al. Circ-calm4 Serves as an miR-337–3p Sponge to Regulate Myo10 (Myosin 10) and Promote Pulmonary Artery Smooth Muscle Proliferation. Hypertension 2020;75:668–79. [DOI] [PubMed] [Google Scholar]
- [182].Jiang Y, Liu H, Yu H, Zhou Y, Zhang J, Xin W, et al. Circular RNA Calm4 Regulates Hypoxia-Induced Pulmonary Arterial Smooth Muscle Cells Pyroptosis via the Circ-Calm4/miR-124–3p/PDCD6 Axis. Arterioscler Thromb Vasc Biol 2021;41:1675–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [183].Rife E, Gedalia A. Kawasaki Disease: an Update. Curr Rheumatol Rep 2020;22:75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [184].Jia C, Zhang J, Chen H, Zhuge Y, Chen H, Qian F, et al. Endothelial cell pyroptosis plays an important role in Kawasaki disease via HMGB1/RAGE/cathespin B signaling pathway and NLRP3 inflammasome activation. Cell Death Dis 2019;10:778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [185].Weintraub RG, Semsarian C, Macdonald P. Dilated cardiomyopathy. Lancet 2017;390:400–14. [DOI] [PubMed] [Google Scholar]
- [186].Zeng C, Duan F, Hu J, Luo B, Huang B, Lou X, et al. NLRP3 inflammasome-mediated pyroptosis contributes to the pathogenesis of non-ischemic dilated cardiomyopathy. Redox Biol 2020;34:101523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [187].Chatterjee K, Zhang J, Honbo N, Karliner JS. Doxorubicin cardiomyopathy. Cardiology 2010;115:155–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [188].Boudina S, Abel ED. Diabetic cardiomyopathy, causes and effects. Rev Endocr Metab Disord 2010;11:31–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [189].Xie Y, Huang Y, Ling X, Qin H, Wang M, Luo B. Chemerin/CMKLR1 Axis Promotes Inflammation and Pyroptosis by Activating NLRP3 Inflammasome in Diabetic Cardiomyopathy Rat. Front Physiol 2020;11:381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [190].Yang F, Qin Y, Wang Y, Li A, Lv J, Sun X, et al. LncRNA KCNQ1OT1 Mediates Pyroptosis in Diabetic Cardiomyopathy. Cell Physiol Biochem 2018;50:1230–44. [DOI] [PubMed] [Google Scholar]
- [191].Xu Y, Fang H, Xu Q, Xu C, Yang L, Huang C. LncRNA GAS5 inhibits NLRP3 inflammasome activation-mediated pyroptosis in diabetic cardiomyopathy by targeting miR-34b-3p/AHR. Cell Cycle 2020;19:3054–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [192].Brusselle GG, Provoost S, Bracke KR, Kuchmiy A, Lamkanfi M. Inflammasomes in respiratory disease: from bench to bedside. Chest 2014;145:1121–33. [DOI] [PubMed] [Google Scholar]
- [193].Hirota JA, Gold MJ, Hiebert PR, Parkinson LG, Wee T, Smith D, et al. The nucleotide-binding domain, leucine-rich repeat protein 3 inflammasome/IL-1 receptor I axis mediates innate, but not adaptive, immune responses after exposure to particulate matter under 10 mum. Am J Respir Cell Mol Biol 2015;52:96–105. [DOI] [PubMed] [Google Scholar]
- [194].Hirota JA, Knight DA. Human airway epithelial cell innate immunity: relevance to asthma. Curr Opin Immunol 2012;24:740–6. [DOI] [PubMed] [Google Scholar]
- [195].Kim RY, Pinkerton JW, Gibson PG, Cooper MA, Horvat JC, Hansbro PM. Inflammasomes in COPD and neutrophilic asthma. Thorax 2015;70:1199–201. [DOI] [PubMed] [Google Scholar]
- [196].Hirota JA, Hirota SA, Warner SM, Stefanowicz D, Shaheen F, Beck PL, et al. The airway epithelium nucleotide-binding domain and leucine-rich repeat protein 3 inflammasome is activated by urban particulate matter. J Allergy Clin Immunol 2012;129:1116–25 e6. [DOI] [PubMed] [Google Scholar]
- [197].Lanks CW, Musani AI, Hsia DW. Community-acquired Pneumonia and Hospital-acquired Pneumonia. Med Clin North Am 2019;103:487–501. [DOI] [PubMed] [Google Scholar]
- [198].Witzenrath M, Pache F, Lorenz D, Koppe U, Gutbier B, Tabeling C, et al. The NLRP3 inflammasome is differentially activated by pneumolysin variants and contributes to host defense in pneumococcal pneumonia. J Immunol 2011;187:434–40. [DOI] [PubMed] [Google Scholar]
- [199].Kadioglu A, Weiser JN, Paton JC, Andrew PW. The role of Streptococcus pneumoniae virulence factors in host respiratory colonization and disease. Nat Rev Microbiol 2008;6:288–301. [DOI] [PubMed] [Google Scholar]
- [200].Aeffner F, Traylor ZP, Yu EN, Davis IC. Double-stranded RNA induces similar pulmonary dysfunction to respiratory syncytial virus in BALB/c mice. Am J Physiol Lung Cell Mol Physiol 2011;301:L99–L109. [DOI] [PubMed] [Google Scholar]
- [201].Allen IC, Scull MA, Moore CB, Holl EK, McElvania-TeKippe E, Taxman DJ, et al. The NLRP3 inflammasome mediates in vivo innate immunity to influenza A virus through recognition of viral RNA. Immunity 2009;30:556–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [202].Muruve DA, Petrilli V, Zaiss AK, White LR, Clark SA, Ross PJ, et al. The inflammasome recognizes cytosolic microbial and host DNA and triggers an innate immune response. Nature 2008;452:103–7. [DOI] [PubMed] [Google Scholar]
- [203].Lee S, Hirohama M, Noguchi M, Nagata K, Kawaguchi A. Influenza A Virus Infection Triggers Pyroptosis and Apoptosis of Respiratory Epithelial Cells through the Type I Interferon Signaling Pathway in a Mutually Exclusive Manner. J Virol 2018;92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [204].Hansbro NG, Horvat JC, Wark PA, Hansbro PM. Understanding the mechanisms of viral induced asthma: new therapeutic directions. Pharmacol Ther 2008;117:313–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [205].Pinkerton JW, Kim RY, Robertson AAB, Hirota JA, Wood LG, Knight DA, et al. Inflammasomes in the lung. Mol Immunol 2017;86:44–55. [DOI] [PubMed] [Google Scholar]
- [206].Baines KJ, Simpson JL, Wood LG, Scott RJ, Gibson PG. Transcriptional phenotypes of asthma defined by gene expression profiling of induced sputum samples. J Allergy Clin Immunol 2011;127:153–60, 60 e1–9. [DOI] [PubMed] [Google Scholar]
- [207].Besnard AG, Togbe D, Couillin I, Tan Z, Zheng SG, Erard F, et al. Inflammasome-IL-1-Th17 response in allergic lung inflammation. J Mol Cell Biol 2012;4:3–10. [DOI] [PubMed] [Google Scholar]
- [208].Kim HY, Lee HJ, Chang YJ, Pichavant M, Shore SA, Fitzgerald KA, et al. Interleukin-17-producing innate lymphoid cells and the NLRP3 inflammasome facilitate obesity-associated airway hyperreactivity. Nat Med 2014;20:54–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [209].Kim RY, Rae B, Neal R, Donovan C, Pinkerton J, Balachandran L, et al. Elucidating novel disease mechanisms in severe asthma. Clin Transl Immunology 2016;5:e91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [210].Kim SR, Kim DI, Kim SH, Lee H, Lee KS, Cho SH, et al. NLRP3 inflammasome activation by mitochondrial ROS in bronchial epithelial cells is required for allergic inflammation. Cell Death Dis 2014;5:e1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [211].Konno S, Gonokami Y, Kurokawa M, Kawazu K, Asano K, Okamoto K, et al. Cytokine concentrations in sputum of asthmatic patients. Int Arch Allergy Immunol 1996;109:73–8. [DOI] [PubMed] [Google Scholar]
- [212].Simpson JL, Phipps S, Baines KJ, Oreo KM, Gunawardhana L, Gibson PG. Elevated expression of the NLRP3 inflammasome in neutrophilic asthma. Eur Respir J 2014;43:1067–76. [DOI] [PubMed] [Google Scholar]
- [213].Cho YS, Kim TB, Lee TH, Moon KA, Lee J, Kim YK, et al. Chlamydia pneumoniae infection enhances cellular proliferation and reduces steroid responsiveness of human peripheral blood mononuclear cells via a tumor necrosis factor-alpha-dependent pathway. Clin Exp Allergy 2005;35:1625–31. [DOI] [PubMed] [Google Scholar]
- [214].Patel KK, Vicencio AG, Du Z, Tsirilakis K, Salva PS, Webley WC. Infectious Chlamydia pneumoniae is associated with elevated interleukin-8 and airway neutrophilia in children with refractory asthma. Pediatr Infect Dis J 2010;29:1093–8. [DOI] [PubMed] [Google Scholar]
- [215].Wark PA, Johnston SL, Simpson JL, Hensley MJ, Gibson PG. Chlamydia pneumoniae immunoglobulin A reactivation and airway inflammation in acute asthma. Eur Respir J 2002;20:834–40. [DOI] [PubMed] [Google Scholar]
- [216].Simpson JL, Grissell TV, Douwes J, Scott RJ, Boyle MJ, Gibson PG. Innate immune activation in neutrophilic asthma and bronchiectasis. Thorax 2007;62:211–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [217].Wood LG, Simpson JL, Hansbro PM, Gibson PG. Potentially pathogenic bacteria cultured from the sputum of stable asthmatics are associated with increased 8-isoprostane and airway neutrophilia. Free Radic Res 2010;44:146–54. [DOI] [PubMed] [Google Scholar]
- [218].Essilfie AT, Horvat JC, Kim RY, Mayall JR, Pinkerton JW, Beckett EL, et al. Macrolide therapy suppresses key features of experimental steroid-sensitive and steroid-insensitive asthma. Thorax 2015;70:458–67. [DOI] [PubMed] [Google Scholar]
- [219].He X, Mekasha S, Mavrogiorgos N, Fitzgerald KA, Lien E, Ingalls RR. Inflammation and fibrosis during Chlamydia pneumoniae infection is regulated by IL-1 and the NLRP3/ASC inflammasome. J Immunol 2010;184:5743–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [220].Rotta Detto Loria J, Rohmann K, Droemann D, Kujath P, Rupp J, Goldmann T, et al. Nontypeable Haemophilus Influenzae Infection Upregulates the NLRP3 Inflammasome and Leads to Caspase-1-Dependent Secretion of Interleukin-1beta - A Possible Pathway of Exacerbations in COPD. PLoS One 2013;8:e66818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [221].Chapman KR, Mannino DM, Soriano JB, Vermeire PA, Buist AS, Thun MJ, et al. Epidemiology and costs of chronic obstructive pulmonary disease. Eur Respir J 2006;27:188–207. [DOI] [PubMed] [Google Scholar]
- [222].Keely S, Talley NJ, Hansbro PM. Pulmonary-intestinal cross-talk in mucosal inflammatory disease. Mucosal Immunol 2012;5:7–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [223].Kuschner WG, D’Alessandro A, Wong H, Blanc PD. Dose-dependent cigarette smoking-related inflammatory responses in healthy adults. Eur Respir J 1996;9:1989–94. [DOI] [PubMed] [Google Scholar]
- [224].Pauwels NS, Bracke KR, Dupont LL, Van Pottelberge GR, Provoost S, Vanden Berghe T, et al. Role of IL-1alpha and the Nlrp3/caspase-1/IL-1beta axis in cigarette smoke-induced pulmonary inflammation and COPD. Eur Respir J 2011;38:1019–28. [DOI] [PubMed] [Google Scholar]
- [225].Beckett EL, Stevens RL, Jarnicki AG, Kim RY, Hanish I, Hansbro NG, et al. A new short-term mouse model of chronic obstructive pulmonary disease identifies a role for mast cell tryptase in pathogenesis. J Allergy Clin Immunol 2013;131:752–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [226].Ferhani N, Letuve S, Kozhich A, Thibaudeau O, Grandsaigne M, Maret M, et al. Expression of high-mobility group box 1 and of receptor for advanced glycation end products in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2010;181:917–27. [DOI] [PubMed] [Google Scholar]
- [227].Hou C, Zhao H, Liu L, Li W, Zhou X, Lv Y, et al. High mobility group protein B1 (HMGB1) in Asthma: comparison of patients with chronic obstructive pulmonary disease and healthy controls. Mol Med 2011;17:807–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [228].Xiang M, Shi X, Li Y, Xu J, Yin L, Xiao G, et al. Hemorrhagic shock activation of NLRP3 inflammasome in lung endothelial cells. J Immunol 2011;187:4809–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [229].Gasse P, Mary C, Guenon I, Noulin N, Charron S, Schnyder-Candrian S, et al. IL-1R1/MyD88 signaling and the inflammasome are essential in pulmonary inflammation and fibrosis in mice. J Clin Invest 2007;117:3786–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [230].Hornung V, Bauernfeind F, Halle A, Samstad EO, Kono H, Rock KL, et al. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat Immunol 2008;9:847–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [231].Butt Y, Kurdowska A, Allen TC. Acute Lung Injury: A Clinical and Molecular Review. Arch Pathol Lab Med 2016;140:345–50. [DOI] [PubMed] [Google Scholar]
- [232].Liu B, He R, Zhang L, Hao B, Jiang W, Wang W, et al. Inflammatory Caspases Drive Pyroptosis in Acute Lung Injury. Front Pharmacol 2021;12:631256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [233].Mowery NT, Terzian WTH, Nelson AC. Acute lung injury. Curr Probl Surg 2020;57:100777. [DOI] [PubMed] [Google Scholar]
- [234].Zhao LR, Xing RL, Wang PM, Zhang NS, Yin SJ, Li XC, et al. NLRP1 and NLRP3 inflammasomes mediate LPS/ATPinduced pyroptosis in knee osteoarthritis. Mol Med Rep 2018;17:5463–9. [DOI] [PubMed] [Google Scholar]
- [235].Qiu Z, He Y, Ming H, Lei S, Leng Y, Xia ZY. Lipopolysaccharide (LPS) Aggravates High Glucose- and Hypoxia/Reoxygenation-Induced Injury through Activating ROS-Dependent NLRP3 Inflammasome-Mediated Pyroptosis in H9C2 Cardiomyocytes. J Diabetes Res 2019;2019:8151836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [236].Muendlein HI, Jetton D, Connolly WM, Eidell KP, Magri Z, Smirnova I, et al. cFLIPL protects macrophages from LPS-induced pyroptosis via inhibition of complex II formation. Science 2020;367:1379–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [237].Tang L, Zhang H, Wang C, Li H, Zhang Q, Bai J. M2A and M2C Macrophage Subsets Ameliorate Inflammation and Fibroproliferation in Acute Lung Injury Through Interleukin 10 Pathway. Shock 2017;48:119–29. [DOI] [PubMed] [Google Scholar]
- [238].Hung CM, Peng CK, Wu CP, Huang KL. Bumetanide attenuates acute lung injury by suppressing macrophage activation. Biochem Pharmacol 2018;156:60–7. [DOI] [PubMed] [Google Scholar]
- [239].Kojima M, Gimenes-Junior JA, Chan TW, Eliceiri BP, Baird A, Costantini TW, et al. Exosomes in postshock mesenteric lymph are key mediators of acute lung injury triggering the macrophage activation via Toll-like receptor 4. FASEB J 2018;32:97–110. [DOI] [PubMed] [Google Scholar]
- [240].Wu DD, Pan PH, Liu B, Su XL, Zhang LM, Tan HY, et al. Inhibition of Alveolar Macrophage Pyroptosis Reduces Lipopolysaccharide-induced Acute Lung Injury in Mice. Chin Med J (Engl) 2015;128:2638–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [241].Hou L, Yang Z, Wang Z, Zhang X, Zhao Y, Yang H, et al. NLRP3/ASC-mediated alveolar macrophage pyroptosis enhances HMGB1 secretion in acute lung injury induced by cardiopulmonary bypass. Lab Invest 2018;98:1052–64. [DOI] [PubMed] [Google Scholar]
- [242].Qu L, Chen C, Chen Y, Li Y, Tang F, Huang H, et al. High-Mobility Group Box 1 (HMGB1) and Autophagy in Acute Lung Injury (ALI): A Review. Med Sci Monit 2019;25:1828–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [243].He X, Qian Y, Li Z, Fan EK, Li Y, Wu L, et al. TLR4-Upregulated IL-1beta and IL-1RI Promote Alveolar Macrophage Pyroptosis and Lung Inflammation through an Autocrine Mechanism. Sci Rep 2016;6:31663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [244].Luo M, Hu L, Li D, Wang Y, He Y, Zhu L, et al. MD-2 regulates LPS-induced NLRP3 inflammasome activation and IL-1beta secretion by a MyD88/NF-kappaB-dependent pathway in alveolar macrophages cell line. Mol Immunol 2017;90:1–10. [DOI] [PubMed] [Google Scholar]
- [245].Wu D, Pan P, Su X, Zhang L, Qin Q, Tan H, et al. Interferon Regulatory Factor-1 Mediates Alveolar Macrophage Pyroptosis During LPS-Induced Acute Lung Injury in Mice. Shock 2016;46:329–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [246].Harroch S, Gothelf Y, Revel M, Chebath J. 5’ upstream sequences of MyD88, an IL-6 primary response gene in M1 cells: detection of functional IRF-1 and Stat factors binding sites. Nucleic Acids Res 1995;23:3539–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [247].Ning L, Wei W, Wenyang J, Rui X, Qing G. Cytosolic DNA-STING-NLRP3 axis is involved in murine acute lung injury induced by lipopolysaccharide. Clin Transl Med 2020;10:e228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [248].Jena KK, Mehto S, Nath P, Chauhan NR, Sahu R, Dhar K, et al. Autoimmunity gene IRGM suppresses cGAS-STING and RIG-I-MAVS signaling to control interferon response. EMBO Rep 2020;21:e50051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [249].Kainthola A, Haritwal T, Tiwari M, Gupta N, Parvez S, Tiwari M, et al. Immunological Aspect of Radiation-Induced Pneumonitis, Current Treatment Strategies, and Future Prospects. Front Immunol 2017;8:506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [250].Carver JR, Shapiro CL, Ng A, Jacobs L, Schwartz C, Virgo KS, et al. American Society of Clinical Oncology clinical evidence review on the ongoing care of adult cancer survivors: cardiac and pulmonary late effects. J Clin Oncol 2007;25:3991–4008. [DOI] [PubMed] [Google Scholar]
- [251].Zhang XJ, Sun JG, Sun J, Ming H, Wang XX, Wu L, et al. Prediction of radiation pneumonitis in lung cancer patients: a systematic review. J Cancer Res Clin Oncol 2012;138:2103–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [252].Im J, Lawrence J, Seelig D, Nho RS. FoxM1-dependent RAD51 and BRCA2 signaling protects idiopathic pulmonary fibrosis fibroblasts from radiation-induced cell death. Cell Death Dis 2018;9:584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [253].Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015;526:660–5. [DOI] [PubMed] [Google Scholar]
- [254].Man SM, Kanneganti TD. Regulation of inflammasome activation. Immunol Rev 2015;265:6–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [255].Vanaja SK, Rathinam VA, Fitzgerald KA. Mechanisms of inflammasome activation: recent advances and novel insights. Trends Cell Biol 2015;25:308–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [256].Rathinam VA, Vanaja SK, Fitzgerald KA. Regulation of inflammasome signaling. Nat Immunol 2012;13:333–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [257].Strowig T, Henao-Mejia J, Elinav E, Flavell R. Inflammasomes in health and disease. Nature 2012;481:278–86. [DOI] [PubMed] [Google Scholar]
- [258].Gao J, Peng S, Shan X, Deng G, Shen L, Sun J, et al. Inhibition of AIM2 inflammasome-mediated pyroptosis by Andrographolide contributes to amelioration of radiation-induced lung inflammation and fibrosis. Cell Death Dis 2019;10:957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [259].Saeki N, Kuwahara Y, Sasaki H, Satoh H, Shiroishi T. Gasdermin (Gsdm) localizing to mouse Chromosome 11 is predominantly expressed in upper gastrointestinal tract but significantly suppressed in human gastric cancer cells. Mamm Genome 2000;11:718–24. [DOI] [PubMed] [Google Scholar]
- [260].Takiishi T, Fenero CIM, Camara NOS. Intestinal barrier and gut microbiota: Shaping our immune responses throughout life. Tissue Barriers 2017;5:e1373208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [261].Man SM. Inflammasomes in the gastrointestinal tract: infection, cancer and gut microbiota homeostasis. Nat Rev Gastroenterol Hepatol 2018;15:721–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [262].Zheng D, Liwinski T, Elinav E. Interaction between microbiota and immunity in health and disease. Cell Res 2020;30:492–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [263].Subramanian S, Geng H, Tan XD. Cell death of intestinal epithelial cells in intestinal diseases. Sheng Li Xue Bao 2020;72:308–24. [PMC free article] [PubMed] [Google Scholar]
- [264].Opipari A, Franchi L. Role of inflammasomes in intestinal inflammation and Crohn’s disease. Inflamm Bowel Dis 2015;21:173–81. [DOI] [PubMed] [Google Scholar]
- [265].Ringel-Scaia VM, McDaniel DK, Allen IC. The Goldilocks Conundrum: NLR Inflammasome Modulation of Gastrointestinal Inflammation during Inflammatory Bowel Disease. Crit Rev Immunol 2016;36:283–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [266].Song-Zhao GX, Srinivasan N, Pott J, Baban D, Frankel G, Maloy KJ. Nlrp3 activation in the intestinal epithelium protects against a mucosal pathogen. Mucosal Immunol 2014;7:763–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [267].Crowley SM, Han X, Allaire JM, Stahl M, Rauch I, Knodler LA, et al. Intestinal restriction of Salmonella Typhimurium requires caspase-1 and caspase-11 epithelial intrinsic inflammasomes. PLoS Pathog 2020;16:e1008498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [268].Sellin ME, Muller AA, Felmy B, Dolowschiak T, Diard M, Tardivel A, et al. Epithelium-intrinsic NAIP/NLRC4 inflammasome drives infected enterocyte expulsion to restrict Salmonella replication in the intestinal mucosa. Cell Host Microbe 2014;16:237–48. [DOI] [PubMed] [Google Scholar]
- [269].Hausmann A, Bock D, Geiser P, Berthold DL, Fattinger SA, Furter M, et al. Intestinal epithelial NAIP/NLRC4 restricts systemic dissemination of the adapted pathogen Salmonella Typhimurium due to site-specific bacterial PAMP expression. Mucosal Immunol 2020;13:530–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [270].Rauch I, Deets KA, Ji DX, von Moltke J, Tenthorey JL, Lee AY, et al. NAIP-NLRC4 Inflammasomes Coordinate Intestinal Epithelial Cell Expulsion with Eicosanoid and IL-18 Release via Activation of Caspase-1 and −8. Immunity 2017;46:649–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [271].Zhong Q, Roumeliotis TI, Kozik Z, Cepeda-Molero M, Fernandez LA, Shenoy AR, et al. Clustering of Tir during enteropathogenic E. coli infection triggers calcium influx-dependent pyroptosis in intestinal epithelial cells. PLoS Biol 2020;18:e3000986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [272].Dubois H, Sorgeloos F, Sarvestani ST, Martens L, Saeys Y, Mackenzie JM, et al. Nlrp3 inflammasome activation and Gasdermin D-driven pyroptosis are immunopathogenic upon gastrointestinal norovirus infection. PLoS Pathog 2019;15:e1007709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [273].Zhu S, Ding S, Wang P, Wei Z, Pan W, Palm NW, et al. Nlrp9b inflammasome restricts rotavirus infection in intestinal epithelial cells. Nature 2017;546:667–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [274].Karst SM, Wobus CE, Lay M, Davidson J, Virgin HWt. STAT1-dependent innate immunity to a Norwalk-like virus. Science 2003;299:1575–8. [DOI] [PubMed] [Google Scholar]
- [275].Wei G, Luo S, Wu W, Hu J, Zhou R. Activation of Interleukin-1beta Release and Pyroptosis by Transmissible Gastroenteritis Virus Is Dependent on the NOD-Like Receptor Protein 3 Inflammasome in Porcine Intestinal Epithelial Cell Line. Viral Immunol 2021. [DOI] [PubMed]
- [276].Goyette P, Labbe C, Trinh TT, Xavier RJ, Rioux JD. Molecular pathogenesis of inflammatory bowel disease: genotypes, phenotypes and personalized medicine. Ann Med 2007;39:177–99. [DOI] [PubMed] [Google Scholar]
- [277].Chen X, Liu G, Yuan Y, Wu G, Wang S, Yuan L. NEK7 interacts with NLRP3 to modulate the pyroptosis in inflammatory bowel disease via NF-kappaB signaling. Cell Death Dis 2019;10:906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [278].Xiong Y, Lou Y, Su H, Fu Y, Kong J. Cholecalciterol cholesterol emulsion ameliorates experimental colitis via down-regulating the pyroptosis signaling pathway. Exp Mol Pathol 2016;100:386–92. [DOI] [PubMed] [Google Scholar]
- [279].Yuan YY, Xie KX, Wang SL, Yuan LW. Inflammatory caspase-related pyroptosis: mechanism, regulation and therapeutic potential for inflammatory bowel disease. Gastroenterol Rep (Oxf) 2018;6:167–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [280].Bauer C, Duewell P, Lehr HA, Endres S, Schnurr M. Protective and aggravating effects of Nlrp3 inflammasome activation in IBD models: influence of genetic and environmental factors. Dig Dis 2012;30 Suppl 1:82–90. [DOI] [PubMed] [Google Scholar]
- [281].Allen IC, TeKippe EM, Woodford RM, Uronis JM, Holl EK, Rogers AB, et al. The NLRP3 inflammasome functions as a negative regulator of tumorigenesis during colitis-associated cancer. J Exp Med 2010;207:1045–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [282].Zhen Y, Zhang H. NLRP3 Inflammasome and Inflammatory Bowel Disease. Front Immunol 2019;10:276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [283].Bauer C, Duewell P, Mayer C, Lehr HA, Fitzgerald KA, Dauer M, et al. Colitis induced in mice with dextran sulfate sodium (DSS) is mediated by the NLRP3 inflammasome. Gut 2010;59:1192–9. [DOI] [PubMed] [Google Scholar]
- [284].Elinav E, Strowig T, Kau AL, Henao-Mejia J, Thaiss CA, Booth CJ, et al. NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell 2011;145:745–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [285].Williams TM, Leeth RA, Rothschild DE, McDaniel DK, Coutermarsh-Ott SL, Simmons AE, et al. Caspase-11 attenuates gastrointestinal inflammation and experimental colitis pathogenesis. Am J Physiol Gastrointest Liver Physiol 2015;308:G139–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [286].Zaki MH, Boyd KL, Vogel P, Kastan MB, Lamkanfi M, Kanneganti TD. The NLRP3 inflammasome protects against loss of epithelial integrity and mortality during experimental colitis. Immunity 2010;32:379–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [287].Ishiguro Y Mucosal proinflammatory cytokine production correlates with endoscopic activity of ulcerative colitis. J Gastroenterol 1999;34:66–74. [DOI] [PubMed] [Google Scholar]
- [288].Monteleone G, Trapasso F, Parrello T, Biancone L, Stella A, Iuliano R, et al. Bioactive IL-18 expression is up-regulated in Crohn’s disease. J Immunol 1999;163:143–7. [PubMed] [Google Scholar]
- [289].Sivakumar PV, Westrich GM, Kanaly S, Garka K, Born TL, Derry JM, et al. Interleukin 18 is a primary mediator of the inflammation associated with dextran sulphate sodium induced colitis: blocking interleukin 18 attenuates intestinal damage. Gut 2002;50:812–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [290].Siegmund B, Lehr HA, Fantuzzi G, Dinarello CA. IL-1 beta -converting enzyme (caspase-1) in intestinal inflammation. Proc Natl Acad Sci U S A 2001;98:13249–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [291].Bauer C, Loher F, Dauer M, Mayer C, Lehr HA, Schonharting M, et al. The ICE inhibitor pralnacasan prevents DSS-induced colitis in C57BL/6 mice and suppresses IP-10 mRNA but not TNF-alpha mRNA expression. Dig Dis Sci 2007;52:1642–52. [DOI] [PubMed] [Google Scholar]
- [292].Siegmund B, Fantuzzi G, Rieder F, Gamboni-Robertson F, Lehr HA, Hartmann G, et al. Neutralization of interleukin-18 reduces severity in murine colitis and intestinal IFN-gamma and TNF-alpha production. Am J Physiol Regul Integr Comp Physiol 2001;281:R1264–73. [DOI] [PubMed] [Google Scholar]
- [293].Xu Z, Liu R, Huang L, Xu Y, Su M, Chen J, et al. CD147 Aggravated Inflammatory Bowel Disease by Triggering NF-kappaB-Mediated Pyroptosis. Biomed Res Int 2020;2020:5341247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [294].Chao L, Li Z, Zhou J, Chen W, Li Y, Lv W, et al. Shen-Ling-Bai-Zhu-San Improves Dextran Sodium Sulfate-Induced Colitis by Inhibiting Caspase-1/Caspase-11-Mediated Pyroptosis. Front Pharmacol 2020;11:814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [295].Neudecker V, Haneklaus M, Jensen O, Khailova L, Masterson JC, Tye H, et al. Myeloid-derived miR-223 regulates intestinal inflammation via repression of the NLRP3 inflammasome. J Exp Med 2017;214:1737–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [296].Kalogeris T, Baines CP, Krenz M, Korthuis RJ. Ischemia/Reperfusion. Compr Physiol 2016;7:113–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [297].Yang JR, Yao FH, Zhang JG, Ji ZY, Li KL, Zhan J, et al. Ischemia-reperfusion induces renal tubule pyroptosis via the CHOP-caspase-11 pathway. Am J Physiol Renal Physiol 2014;306:F75–84. [DOI] [PubMed] [Google Scholar]
- [298].Jia Y, Cui R, Wang C, Feng Y, Li Z, Tong Y, et al. Metformin protects against intestinal ischemia-reperfusion injury and cell pyroptosis via TXNIP-NLRP3-GSDMD pathway. Redox Biol 2020;32:101534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [299].Otani K, Watanabe T, Shimada S, Takeda S, Itani S, Higashimori A, et al. Colchicine prevents NSAID-induced small intestinal injury by inhibiting activation of the NLRP3 inflammasome. Sci Rep 2016;6:32587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [300].Wallace JL, Syer S, Denou E, de Palma G, Vong L, McKnight W, et al. Proton pump inhibitors exacerbate NSAID-induced small intestinal injury by inducing dysbiosis. Gastroenterology 2011;141:1314–22, 22 e1–5. [DOI] [PubMed] [Google Scholar]
- [301].Higashimori A, Watanabe T, Nadatani Y, Takeda S, Otani K, Tanigawa T, et al. Mechanisms of NLRP3 inflammasome activation and its role in NSAID-induced enteropathy. Mucosal Immunol 2016;9:659–68. [DOI] [PubMed] [Google Scholar]
- [302].Watanabe T, Higuchi K, Kobata A, Nishio H, Tanigawa T, Shiba M, et al. Non-steroidal anti-inflammatory drug-induced small intestinal damage is Toll-like receptor 4 dependent. Gut 2008;57:181–7. [DOI] [PubMed] [Google Scholar]
- [303].Paul DS, Devesa V, Hernandez-Zavala A, Adair BM, Walton FS, Drobna Z, et al. Environmental arsenic as a disruptor of insulin signaling. Met Ions Biol Med 2008;10:1–7. [PMC free article] [PubMed] [Google Scholar]
- [304].Wu W, Yao X, Jiang L, Zhang Q, Bai J, Qiu T, et al. Pancreatic islet-autonomous effect of arsenic on insulin secretion through endoplasmic reticulum stress-autophagy pathway. Food Chem Toxicol 2018;111:19–26. [DOI] [PubMed] [Google Scholar]
- [305].Pei P, Yao X, Jiang L, Qiu T, Wang N, Yang L, et al. Inorganic arsenic induces pyroptosis and pancreatic beta cells dysfunction through stimulating the IRE1alpha/TNF-alpha pathway and protective effect of taurine. Food Chem Toxicol 2019;125:392–402. [DOI] [PubMed] [Google Scholar]
- [306].Wang J, Li X, Liu Y, Peng C, Zhu H, Tu G, et al. CircHIPK3 Promotes Pyroptosis in Acinar Cells Through Regulation of the miR-193a-5p/GSDMD Axis. Front Med (Lausanne) 2020;7:88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [307].Lin T, Pan X, Wan Y, Wu Z, Lyu S, Wang Y, et al. [Mechanism of gasdermin D on intestinal injury in severe acute pancreatitis by mediating pyroptosis]. Zhonghua Wei Zhong Bing Ji Jiu Yi Xue 2021;33:89–94. [DOI] [PubMed] [Google Scholar]
- [308].Lin T, Song J, Pan X, Wan Y, Wu Z, Lv S, et al. Downregulating Gasdermin D Reduces Severe Acute Pancreatitis Associated with Pyroptosis. Med Sci Monit 2021;27:e927968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [309].de Rivero Vaccari JP. The Inflammasome in Reproductive Biology: A Promising Target for Novel Therapies. Front Endocrinol (Lausanne) 2020;11:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [310].Stodle GS, Silva GB, Tangeras LH, Gierman LM, Nervik I, Dahlberg UE, et al. Placental inflammation in pre-eclampsia by Nod-like receptor protein (NLRP)3 inflammasome activation in trophoblasts. Clin Exp Immunol 2018;193:84–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [311].Ibrahim E, Castle SM, Aballa TC, Keane RW, de Rivero Vaccari JP, Lynne CM, et al. Neutralization of ASC improves sperm motility in men with spinal cord injury. Hum Reprod 2014;29:2368–73. [DOI] [PubMed] [Google Scholar]
- [312].Gomez-Lopez N, Romero R, Garcia-Flores V, Leng Y, Miller D, Hassan SS, et al. Inhibition of the NLRP3 inflammasome can prevent sterile intra-amniotic inflammation, preterm labor/birth, and adverse neonatal outcomesdagger. Biol Reprod 2019;100:1306–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [313].Chen CY, Chen CY, Liu CC, Chen CP. Omega-3 polyunsaturated fatty acids reduce preterm labor by inhibiting trophoblast cathepsin S and inflammasome activation. Clin Sci (Lond) 2018;132:2221–39. [DOI] [PubMed] [Google Scholar]
- [314].Cheng SB, Nakashima A, Huber WJ, Davis S, Banerjee S, Huang Z, et al. Pyroptosis is a critical inflammatory pathway in the placenta from early onset preeclampsia and in human trophoblasts exposed to hypoxia and endoplasmic reticulum stressors. Cell Death Dis 2019;10:927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [315].Marchi J, Berg M, Dencker A, Olander EK, Begley C. Risks associated with obesity in pregnancy, for the mother and baby: a systematic review of reviews. Obes Rev 2015;16:621–38. [DOI] [PubMed] [Google Scholar]
- [316].Shirasuna K, Takano H, Seno K, Ohtsu A, Karasawa T, Takahashi M, et al. Palmitic acid induces interleukin-1beta secretion via NLRP3 inflammasomes and inflammatory responses through ROS production in human placental cells. J Reprod Immunol 2016;116:104–12. [DOI] [PubMed] [Google Scholar]
- [317].Yang Y, Li J, Han TL, Zhou X, Qi H, Baker PN, et al. Endoplasmic reticulum stress may activate NLRP3 inflammasomes via TXNIP in preeclampsia. Cell Tissue Res 2020;379:589–99. [DOI] [PubMed] [Google Scholar]
- [318].Amash A, Holcberg G, Sapir O, Huleihel M. Placental secretion of interleukin-1 and interleukin-1 receptor antagonist in preeclampsia: effect of magnesium sulfate. J Interferon Cytokine Res 2012;32:432–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [319].Siljee JE, Wortelboer EJ, Koster MP, Imholz S, Rodenburg W, Visser GH, et al. Identification of interleukin-1 beta, but no other inflammatory proteins, as an early onset pre-eclampsia biomarker in first trimester serum by bead-based multiplexed immunoassays. Prenat Diagn 2013;33:1183–8. [DOI] [PubMed] [Google Scholar]
- [320].Lerner AG, Upton JP, Praveen PV, Ghosh R, Nakagawa Y, Igbaria A, et al. IRE1alpha induces thioredoxin-interacting protein to activate the NLRP3 inflammasome and promote programmed cell death under irremediable ER stress. Cell Metab 2012;16:250–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [321].Guo M, Wang X, Zhao Y, Yang Q, Ding H, Dong Q, et al. Ketogenic Diet Improves Brain Ischemic Tolerance and Inhibits NLRP3 Inflammasome Activation by Preventing Drp1-Mediated Mitochondrial Fission and Endoplasmic Reticulum Stress. Front Mol Neurosci 2018;11:86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [322].Fu J, Zhao L, Wang L, Zhu X. Expression of markers of endoplasmic reticulum stress-induced apoptosis in the placenta of women with early and late onset severe pre-eclampsia. Taiwan J Obstet Gynecol 2015;54:19–23. [DOI] [PubMed] [Google Scholar]
- [323].Kumar A, Mittal R. Mapping Txnip: Key connexions in progression of diabetic nephropathy. Pharmacol Rep 2018;70:614–22. [DOI] [PubMed] [Google Scholar]
- [324].I CW, Romao-Veiga M, Matias ML, Fioratti EG, Peracoli JC, Borges VT, et al. Increased expression of NLRP3 inflammasome in placentas from pregnant women with severe preeclampsia. J Reprod Immunol 2017;123:40–7. [DOI] [PubMed] [Google Scholar]
- [325].Rathkey JK, Zhao J, Liu Z, Chen Y, Yang J, Kondolf HC, et al. Chemical disruption of the pyroptotic pore-forming protein gasdermin D inhibits inflammatory cell death and sepsis. Sci Immunol 2018;3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [326].Humphries F, Shmuel-Galia L, Ketelut-Carneiro N, Li S, Wang B, Nemmara VV, et al. Succination inactivates gasdermin D and blocks pyroptosis. Science 2020;369:1633–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [327].Hu JJ, Liu X, Xia S, Zhang Z, Zhang Y, Zhao J, et al. FDA-approved disulfiram inhibits pyroptosis by blocking gasdermin D pore formation. Nat Immunol 2020;21:736–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [328].Tamura K, Ishikawa G, Yoshie M, Ohneda W, Nakai A, Takeshita T, et al. Glibenclamide inhibits NLRP3 inflammasome-mediated IL-1beta secretion in human trophoblasts. J Pharmacol Sci 2017;135:89–95. [DOI] [PubMed] [Google Scholar]
- [329].Liu Z, Zhao X, Shan H, Gao H, Wang P. microRNA-520c-3p suppresses NLRP3 inflammasome activation and inflammatory cascade in preeclampsia by downregulating NLRP3. Inflamm Res 2019;68:643–54. [DOI] [PubMed] [Google Scholar]