

Summary
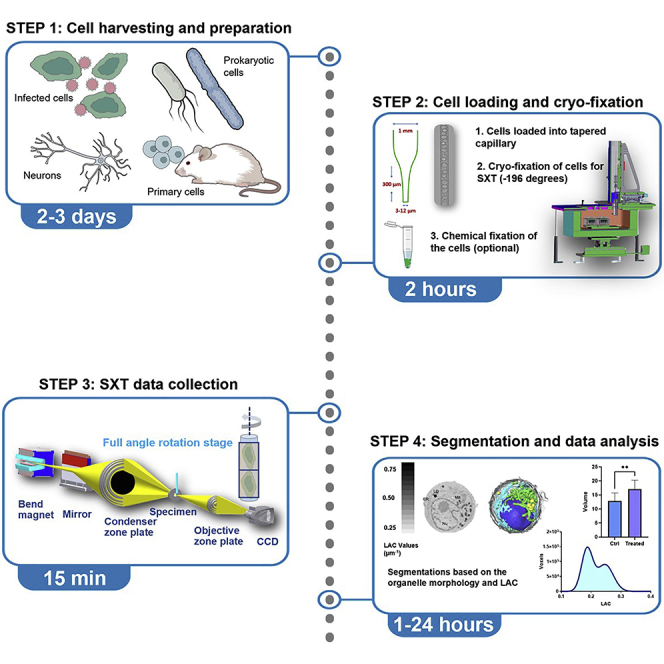
The protocol describes step-by-step sample preparation, data acquisition, and segmentation of cellular organelles with soft X-ray tomography. It is designed for microscopes built to perform full-rotation data acquisition on specimens in cylindrical sample holders, such as the XM-2 microscope at the Advanced Light Source, LBNL; however, it might be generalized for similar sample holder designs for both synchrotron and table-top microscopes.
For complete details on the use and execution of this profile, please refer to Loconte et al. (2021).
Subject areas: Cell Biology, Single Cell, Microscopy
Graphical abstract
Highlights
-
•
Rapid full-rotation soft X-ray tomography of single cells
-
•
Cryopreservation of biological cells in glass capillaries
-
•
Quantitative calibration of X-ray linear absorption coefficient
-
•
Step-by-step segmentation of main cellular organelles
The protocol describes step-by-step sample preparation, data acquisition, and segmentation of cellular organelles with soft X-ray tomography. It is designed for microscopes built to perform full-rotation data acquisition on specimens in cylindrical sample holders, such as the XM-2 microscope at the Advanced Light Source, LBNL; however, it might be generalized for similar sample holder designs for both synchrotron and table-top microscopes.
Before you begin
Technological advancements in light, electron and X-ray microscopy techniques have brought researchers to the discovery of cellular ultrastructural rearrangements thanks to the ever-increasing spatial resolution of the microscope in use. Among these techniques, soft X-ray Tomography (SXT) allows for the imaging of hydrated and intact cells at the so-called X-ray “water window” (within 284 and 583 eV of photon energy) (Larabell and Nugent 2010) with enhanced and quantitative absorption signal for the cell carbon-rich components. SXT is suitable to image many different cell types, such as immortalized cell lines, yeast, bacteria, algae, primary cells and viruses (Guo and Larabell 2019; Harkiolaki et al., 2018). Pertinent to the current global pandemic, we describe specific steps to image human lung epithelial cells infected with SARS-CoV-2 virus. The handling of SARS-CoV-2 and many other viruses is one of the challenging scenarios for SXT imaging, as neutralization or inactivation of virus is required prior to imaging. In the past, we have used this protocol (simplified in terms of sample preparation) on other cell cultures as well as primary cells (Le Gros et al., 2012).
Glass capillary preparation
Timing: 10 min to 1 h
The imaging of cells grown on flat sample holders is limited to a certain region of interest (ROI) of the whole cell, due to the spreading of the cell on the flat surface and to the limited field of view (FOV), due to the gradual increase in X-ray absorption. In this protocol, we acquire data from cells placed in thin-walled glass capillaries to optimize the acquisition time (see Data Acquisition paragraph) and perform full-rotation tomography without “missing wedge” artifacts.
-
1.
The borosilicate glass capillaries consisting of two capillary types glued together are available from Hilgenberg GmbH (Cat.No . 4023088) (Figure 1A).
Note: Other glass capillaries can be used as well. Choose material and thickness of glass capillaries with highest X-ray transmission.
-
2.
Set the glass puller parameters. In our specific case, the parameters are set on the commercial instrument Sutter P-97. In the display (Figure 1B), set the program to 2, performing 3 cycles of pulling; the pressure (P) of the air used to cool down the glass is set to 500; the HEAT controls the current in the filament and is set to 340; PULL which controls the force of the hard puller is set on 5; the VELOCITY of the glass carriage is determined as the glass softens and pulls apart and is set on 25; finally, TIME determines the exposure of the glass to air cooling stream and is set on 95, which represents about 48 ms.
-
3.
One by one pull capillaries on a glass puller. The glass puller will heat the glass to a temperature of about 600°C and then will rapidly pull horizontally from two directions (see Figure 1A), thus thinning glass wall and generating two capillaries for sample loading.
Pause point: Pulled capillaries can be stored in a closed box for a few months.
-
4.
Using a custom-made trimmer machine (see Figure 1C), cut the capillary tip to the size matching the cell diameter. For example, 15 μm cells used in this study require capillaries with 8–10 μm diameter.
Note: Alternatively, to the trimmer machine, capillaries can be cut under a standard light microscope with tweezers. Though not as precise, these capillaries can be used as well.
CRITICAL: Glass capillaries are sharp and can easily break. Use appropriate personal protective equipment, for example goggles and gloves, when preparing capillaries.
Figure 1.

Instruments and steps for sample holder preparation
(A) The borosilicate glass capillaries consist of two capillary types glued together.
(B) The puller with two parts of the capillary (outlined in white dashed circles) after heating by filament (white arrow) and pulling.
(C) Trimming of the capillary (white arrow) realized by viewing (magnifying lens) and cutting with diamond blade the capillary tip to the specific tip size (magnified). The scale bar is 10 μm.
Cell preparation
Here, we present the protocol to describe the harvest and the preparation of SARS-CoV-2 infected samples prior to freezing down.
-
5.
SARS-CoV-2 permissive cells are grown in 6 cm2 dishes at 37°C and in 5% CO2 atmosphere. Either HEK-293 and A549 overexpressing SARS-CoV-2 receptor protein ACE-2 (293-ACE2 and A549-ACE2, respectively) or Calu-3 lung epithelial cells have been used.
-
6.
After 24 h infect cells with SARS-CoV-2 at high multiplicity of infection (MOI), that is 10 or 20, to ensure high level of infection.
-
7.
At different time points after infection detach the infected cells with trypsin, transfer them in a 1.5-mL tube and centrifuge the cells at 300×g for 5 min.
-
8.
Wash the pellet once in PBS 1×.
-
9.
Centrifuge the cells at 300×g and resuspend the pellet in fixative solution composed of 2.5% glutaraldehyde in PBS for 30 min.
-
10.
Pellet the cells and resuspend the pellet in 6% paraformaldehyde in PBS for 30 min.
-
11.
Pellet the cells and wash them once in PBS 1×.
-
12.
Pellet the cells and resuspend them 20–30 μL of PBS(1×). The cells are now ready for loading.
Note: Use of high MOI will synchronize the infection and increase the chances to analyze infected over uninfected cells.
Note: If not required for biosafety purposes, the fixation step can be omitted. For shipping purposes, particularly of sensitive samples, chemical fixation will help to preserve cell integrity (Loconte et al., 2021).
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and virus strains | ||
| SARS-CoV-2 (isolate Bavpat1/2020) | European Virology Archive | Ref-SKU: 026V-03883 |
| Chemicals, peptides, and recombinant proteins | ||
| PBS | Thermo Scientific | Cat.#10010023 |
| 4000 Series monosized microbeads in polystyrene | Thermo Scientific | Cat.#4206A (6 μm) Cat.#4208A (8 μm) Cat.#4210A (10 μm) |
| Cell Strainer | pluriSelect | SKU 43-10040-40 (40 μm) SKU 43-10020-40 (20 μm) |
| Paraformaldehyde (optional, for chemical fixation) | Thermo Scientific | Cat.#J61899-AP |
| Formaldehyde (optional, for chemical fixation) | Thermo Scientific | Cat.#28906 |
| Glutaraldehyde EM grade | Electron Microscopy Sciences | Cat. #E16210 |
| Experimental models: Cell lines | ||
| Calu-3 | ATCC | Cat.#HTB-55 |
| A549-ACE2 | (Klein et al., 2020) | N/A |
| HEK293T-ACE2 | Gift by Prof. Becker | N/A |
| Human B lymphocytes | Coriell Institute of Medical Research | GM12878 |
| Software and algorithms | ||
| ImageJ – Fiji 1.53 c | (Schneider et al., 2012) | https://imagej.nih.gov/ij/ |
| GraphPad Prism 9 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| Amira 2019.1 | Thermo Scientific | https://thermofisher.com/amira-avizo |
| IMOD – 4.10.42 | (Kremer et al., 1996) | https://bio3d.colorado.edu/imod/ |
| AREC reconstruction | (Parkinson et al., 2012) | N/A |
Step-by-step method details
Preparation of calibration samples
To guarantee a high quality of the data collected on the microscope, a calibration routine is usually performed on the X-ray microscope, typically after shutdown of the synchrotron, that is twice per year. Calibration protocols allow for reproducible measurements of the effective pixel size and X-ray linear absorption coefficient, also called LAC (which is at the basis for segmentation and analysis of the reconstructed data). The calibration process uses polystyrene microbeads of different sizes. Depending on spatial resolution in the X-ray microscope and the size of a specimen, we use beads of 6 μm, 8 μm, and 10 μm with a size variation of less than 1% and with a chemical composition that meets ultra-high purity standards (see key resources table).
-
1.
To ensure full penetration of X-rays, choose beads of <8 μm in size.
-
2.
Spin microbeads at 10000×g for 2 min using a centrifuge. Remove supernatant and resuspend beads in PBS 1×.
-
3.
Repeat washing in PBS 1× twice.
-
4.
Calibration standards were generated by filling a specimen capillary with a suspension of polystyrene beads (as delivered by the manufacturer).
-
5.
Load 1.1–3.0 μL of beads mixture into a glass capillary using a micro-loading tip.
-
6.
Repeat steps 5 until a few capillaries containing beads are prepared.
-
7.
Proceed with cryofixation.
Optional: If specimens are prepared with addition of calibration beads, the calibration step can be performed on capillaries containing both – beads and cells.
Specimen loading
This step shows how to load cells in glass capillaries and prepare the samples for rapid plunge-freezing. The addition of beads to cells serves two purposes: 1 – beads have greater mass and thus “shield” cells from propagation of pressure during plunge-freezing and reduce deformation of cells; 2 – they serve as calibration samples to calculate the effective pixel size.
-
8.
Prepare trimmed capillaries with a tip size suiting the cell diameter as described in Glass capillary preparation.
-
9.
Count cell density using hemocytometer or an automated cell counter.
-
10.
Choose beads of the same size as capillary tip and calculate volume to ensure 1:1 ratio of cells and beads. For example, 15mL of 8μm beads with concentration of solids 0.3%, contains about 4.5 million beads per mL.
-
11.
Spin microbeads at 10000×g for 2 min using a centrifuge. Remove supernatant and resuspend beads in PBS 1×.
-
12.
Repeat washing in PBS 1× twice.
Note: The bead washing is to guarantee that the beads will not affect physiological state of cells.
-
13.
Spin cells in a 15-mL tube at 300×g for 10 min. Remove supernatant and resuspend cells in 1.5mL tube together with microbeads.
-
14.
Spin mix of microbeads and cells at 300×g for 10 min. Remove supernatant, resuspend beads/cells mixture in 20–30 μL PBS 1× and mix it by tapping with your index finger.
-
15.
Load 1.1–3.0 μL of beads/cells mixture into a glass capillary using a micro-loading tip.
Note: Cells and beads go into the glass tip due to capillary forces, thus it is recommended to wait a few seconds before removing micro-loading tip from the glass tube. Check the quality of the prepared capillary under a light microscope (see (Smith et al., 2014) for example of equally distributed cells in a tip). Capillaries with low number of cells or lower ratio of cells and beads are usually discarded.
-
16.
Repeat step 15 until the desired number of capillaries containing specimens is prepared.
Optional: It is helpful to document approximate number of cells and beads in each capillary visible under a light microscope.
Cryofixation
All specimens, whether live or chemically fixed, must be frozen prior to X-ray imaging to prevent radiation damage. Cryo-fixation can be performed with high-pressure freezer or plunge-freezer. Both techniques are optimized to prevent ice crystal formation and preserve cellular ultrastructure. This step describes rapid plunge-freezing of cells in liquid propane with the in-house developed automatic plunge freezer.
-
17.
Put a puck and storage box in the freezing apparatus (Figure 2A) and cool down the freezing machine by liquid nitrogen.
Optional: Due to symmetry of a puck, it is helpful to insert colored plastic pins in a few capillary slots (see Figures 2A and 2B) to fix the orientation of the puck and retrieve capillary of interest.
-
18.
Home the robot, fill and put the propane well in position for plunging and attach tubes for dry nitrogen.
-
19.
Set up the plunger according to the type of samples to freeze down to allow proper vitrification of the sample. Make sure the height of the propane well is set correctly.
-
20.
To create a temperature gradient for the plunging, set up the heater for dry nitrogen at 30°C. Set up heater for the viewing glass window at 30°C to protect from ice buildup and ensure visibility of capillaries (Figure 2B).
-
21.
Mount the capillary on the grabber (Figure 2C). Plunge the capillary in the propane well and automatically transfer the capillary from the propane well to an empty position in the storage puck.
Figure 2.

Components for the cryofixation of capillaries
(A) Empty puck and a storage box inside the freezing apparatus before addition of liquid nitrogen.
(B) Top: Puck filled with capillaries (blue) and pins (orange) for fixed orientation of a puck in a storage box filled with liquid nitrogen. Bottom: Propane well (at 20°C–24°C temperature) in which the capillary is plunged positioned on the propane heater to avoid overcooling of propane.
(C) The grabber firmly holding a capillary before plunging down.
X-ray beam alignment and the microscope setup
The transmission soft X-ray microscope (XM-2) is located at the beamline 2.1 of the Advanced Light Source (ALS), Lawrence Berkeley National Laboratory. The description of the beamline, its optical components, microscope design and cooling system was described elsewhere (Le Gros et al., 2014). This step describes alignment of illumination optics and imaging objective to achieve maximum illumination flux.
-
22.
Check the beam status of the ALS (https://als.lbl.gov/beam-status/). If the beam is available for user operation, open the Branchline Equipment Protective System (EPS) (Figure 3A) and reset the pressure on the system.
-
23.
Ensure that the pressure of the pressure gauge IG207 is around 10−5 Torr, IG206 is around 10−7 Torr, IG205 is around 10−8 Torr, IG204 and IG203 are around 10−10 Torr (Figure 3A).
Note: The electronic logbook activated by the user at the beginning of the measurement asks to document these values. If out of range the logbook will give warning by depicting values in red, thus ensuring safe handling of the microscope.
-
24.
Open the vacuum valves from right to the left, that is first VVR204 , followed byVVR203 and VVR202 to let the X-ray beam come through into the microscope. The black line turns purple showing the path of the X-ray beam in the microscope.
Note: Once the X-ray beam is open, the pressure on the gauges IG203 and IG204 will drop to 10−9 Torr.
-
25.
Open the GUI and turn on the microscope. The GUI of the microscope turns on thermocouples, mechanical stages, their controllers for the condenser zone plate (KZP), micro-zone plate (MZP), and the X-ray camera (Figure 3B).
-
26.
Fill the liquid nitrogen cold traps of the KZP and MZP box.
-
27.Align the microscope:
-
a.Home the sample stage, the filters, and the camera of the microscope.
-
b.Move the X-ray camera towards the sample stage to get a big field-of-view and to see the intactness of the silicon nitrate window (Figure 4A).Note: To enable a quick alignment of the beam, a 2×2 binning (512×512 pixels) combined with 100% filter transmission in non-scanning mode is used.
-
c.Align the microscope at ambient (20°C–24°C) temperature by translating the MZP and the KZP to the center of the FOV. After several displacements, the X-ray beam will look like on Figure 4A.Note: With regular use of the microscope, adjustment of the pinhole is not required.
-
a.
-
28.Cool down the sample stage and re-align the beam
-
a.Initialize the nitrogen and helium flow.
-
b.To cool down helium, fill the zeolite tank with liquid nitrogen.Note: Helium flow purges away humid air in the sample chamber.
-
c.Start cooling down the cryobox with liquid nitrogen and turn on the heaters on top of the cryobox to avoid ice accumulation in the sample chamber.
-
d.Re-align the microscope after cooling down as described in Step 27 The result of this alignment should look like Figure 4B.
-
a.
-
29.
After alignment, drive the camera back to its initial position and check the homogeneity of the illumination by making a snapshot with the scanning mode (pattern generator, Figure 4C). If not, re-adjust the MZP and, possibly, the KZP. The scanning image should look similar to the one in Figure 4D.
Note: The pattern generator allows to scan the X-ray beam in a zig zag like pattern (see Figure 4D). It is defined by the amplitude in X and Y directions, the exposure time (time to make the scan), the number of steps and samples. These last two parameters are rarely modified. Here the result is displayed with a 100 ms exposure time and pattern of 18 x18 μm with helium flow. The exposure time is adjusted to not saturate the camera.
Optional: To ensure “water window” regime, the operating energy of the microscope can be calibrated using the K-shell absorption edges of oxygen (543 eV) and nitrogen (410 eV), by translating KZP and MZP along the x-ray beam path effectively scanning over whole spectral energy range. Position with highest X-ray transmission corresponds to ideal microscope settings just below oxygen absorption edge is identified.
Figure 3.

Main components of the XM-2 microscope
(A) Schematic overview of the Branchline EPS (Equipment Protective System) showing all valves (orange), gauges (blue), optical components and the path taken by the X-rays along the beamline and into the experimental end-station (outlined in purple).
(B) Photograph of the experimental end-station. The optical table is 5m long and accommodates KZP, MZP boxes, sample chamber, X-ray camera, cryo-transfer device and controllers for stages, temperature and gas flow (N and He).
Figure 4.

Alignment of the X-ray beam to ensure maximum intensity and homogeneous illumination of the FOV
(A) Low magnification (ca. 2150×) X-ray image after alignment of MZP, KZP and a pinhole at ambient (20°C–24°C) temperature.
(B) X-ray image after MZP and KZP alignment after cooling down in high (ca. 3100×) magnification.
(C) X-ray image of a homogeneous illumination with the scanning of KZP. (D) Pattern generator shows a line scanned pattern defined by the amplitude in X and in Y, the exposure time of the image (time to make the scan), the number of steps and samples.
Transferring the specimen in the microscope
The loaded capillaries are stored in liquid nitrogen until data collection. This step describes transfer of the capillary in the sample chamber of the SXT microscope using a custom-made cryo-transfer device.
-
30.
Initiate the specimen transfer procedure in the GUI of the microscope. The sample stage will automatically move away from the chamber where the capillary is positioned, providing room for the cryo-transfer device. A little column of cold helium can be observed coming out of the sample chamber.
Note: As for most tomography systems, the sample stage is equipped with several motors, specifically translational axes X, Y, Z, two inclination stages for rotation in planes XY and XZ, and the rotation around the Y-axis. These set of motors allows positioning of the specimen in the center of rotation and positioning of the rotation axis in the center of the X-ray camera.
-
31.
Add the sample to the database (name of the principal investigator, cell type and cell condition).
Note: The capillary ID will be generated and assigned to all datasets automatically.
-
32.
Maximize the cold helium flow of the cryo-transfer device in order to keep the sample vitrified and transfer the capillary to the sample chamber.
Note: The specimen is suspended in the sample chamber by clamps.
-
33.
Grab the capillary by translating the sample stage in Y and Z axis (X is typically not required) and attaching capillary to a goniometer by a pin (Figure 5A).
-
34.
Press sample transfer complete, which will automatically release the clamps and send the sample in the sample chamber for alignment with a Borescope.
-
35.
Align the capillary tip in the center of X-ray beam under Borescope (low magnification) which is fiducially aligned with the optical path of the X-ray microscope (Figure 5B).
-
36.
Select alignment done.
Note: The capillary will be transferred down into the X-ray beam path.
Optional: If ice buildup on the capillary surface is observed under the microscope, de-ice the capillary (Figure 6). The de-ice function of the microscope will increase the cryobox temperature to -90°C for 60 seconds and then cool down the microscope to −150°C.
-
37.
Find the tip of the capillary under the X-rays and align the capillary with higher resolution until the capillary is in the center of the FOV (Figure 5C).
-
38.
By translating capillary vertically, find the cell within the FOV. Check rotation center again. If not centered, repeat step 37.
Note: To avoid multiple exposure of a cell and thus high X-ray dose, we recommend to perform fine alignment of the capillary rotation below the cell of interest.
Figure 5.

Transfer and alignment of the capillary in the X-ray microscope
(A) Microscopy image of the goniometer and attached (by pin) to its capillary.
(B) Sparse alignment of a capillary tip to the center of the FOV (green circle) using a Borescope.
(C) X-ray image of the capillary with cell when performing fine alignment under X-rays.
Figure 6.

Graphical User Interface for the tomographic data acquisition
On the left X-ray projection imaging of a capillary with a cell. On the right modules for control of capillary positioning in X, Y and Z (in blue), rotation of capillary tip to align capillary in the FOV (orange), settings for tomography acquisition (purple), automatic correction of capillary to the center of the FOV (dashed circle), selection of MZP (dashed rectangular) and others (image histogram, beam alignment, de-ice, etc.).
Tomographic data acquisition
The tomographic data acquisition module ensures automatic control of the capillary position within FOV.
-
39.
Set data acquisition parameters to 2 × 2 binning, 2° per step and 92 projections, corresponding to 180° data acquisition (Figure 6).
Optional: For sensitive cells we recommend to distribute X-ray radiation dose more evenly (Loconte et al., 2021) and acquire tomogram over 360° by setting step size to 4°.
-
40.
Set the exposure time between 200 ms and 500 ms, depending on the sample thickness and the beam flux (see troubleshooting).
-
41.
Move the sample out of the X-ray beam and select X-ray absorption filter (67%, 23%, and 4%) for reference (also called flat field) images to not saturate the X-ray camera.
-
42.
Move the sample back to the center of the FOV.
-
43.
Switch on auto-correction procedure that, if capillary shifts out of the FOV, will automatically bring it back to the center of the FOV (Figure 6).
-
44.
Start the data acquisition.
Note: Two sets of 10 flat-field images are collected automatically before and after the acquisition of projection images of the specimen.
Optional: If a cell is larger than the field of view horizontally, shift the X-ray camera closer to the capillary. Changing the specimen-camera distance will reduce pixel size and increase field of view. If a cell is larger than the field of view vertically, shift specimen vertically by 80% of the field of view (20% overlapping area for stitching of 3D datasets) and repeat tomography acquisition.
-
45.
Find the next cell in the capillary and repeat tomographic data acquisition, i.e., step 39–44.
Removing specimen from the microscope
-
46.
When all cells in the capillary are imaged, move capillary up (Y axis) and remove the capillary from sample stage.
-
47.
Transfer the next specimen in the microscope, repeat alignment and tomography acquisition step 30–44.
Note: In a typical working time of 8 h about 40 samples can be imaged.
Shutting down the microscope
When all specimens are imaged, the microscope can be shut down and slowly brought to ambient (20°C–24°C) temperature.
-
48.
Reduce the flow of dry nitrogen.
-
49.
Set de-ice of the microscope to 40°C for 360 s. Repeat de-ice procedure until liquid nitrogen in sample chamber has evaporated.
-
50.
Close the nitrogen and helium flow.
-
51.
Set the heaters on the sample box to continue heating to 24°C for 6 h after shutting down.
Note: The heaters will continue to warm up the microscope after shutting down to ensure slow transition to ambient (20°C–24°C) temperature and avoiding buildup of ice on the microscope.
Tomographic data reconstruction
After the data acquisition, the tomographic data reconstruction will be performed automatically (Parkinson et al., 2012). Given the data collection was successful there are no manual steps involved. The reconstruction is available within approximately 5 min of the acquisition in its corresponding data folder.
Calibration of the microscope
To enable quantitative analysis of the SXT data, the microscope magnification and LAC values are calibrated with beads.
-
52.
To calibrate the effective pixel size (pixel size of the camera changed by the microscope magnification) open SXT dataset with for example Fiji (Schneider et al. 2012) of polystyrene beads with known size.
-
53.
Take measurements of bead size in pixels. Calculate the effective pixel size.
-
54.
If necessary, repeat step 53 for different X-ray camera positions, along the beam.
Note: The XM-2 specimen stage contains a photolithographic, interpolated graticule that provides 1% positional accuracy over a 10 μm range. Consequently, the microscope magnification is measured with very high confidence. Therefore, the effective pixel size for different magnifications is measured and calculated once per year and can be used for any SXT dataset acquired with the same zone plate.
-
55.
To calibrate LAC values, open tomographic data of beads collected under the same conditions as cells.
-
56.
Using thresholding (see segmentation and data analysis) segment 80–100 polystyrene beads with a 6 μm of diameter. Multiply X-ray absorption (intensity) by the pixel area, i.e., 1/(2·pixel size) in μm.
-
57.
Compare the X-ray LAC of the beads with expected 0.81 ± 0.03 μm−1.
Optional: In the case of mismatch, it is possible to estimate the X-ray energy window used with SXT imaging by comparing experimental LAC with expected absorption for polystyrene (C8H8) from the atomic scattering database maintained by the Center for X-Ray Optics (CXRO) at the Lawrence Berkeley National Laboratory (http://cxro.lbl.gov/x-ray-data-booklet).
Note: After calibration steps, each voxel in the 3D reconstruction of the cell has an associated X-ray LAC value. The voxel LAC value directly correlates with the concentration of molecules in the corresponding space in the specimen.
Segmentation and data analysis
To analyze and quantify volume and mutual distances of cellular organelles, the procedure of isolating individual components, known as segmentation, is performed on reconstructed data. Currently segmentation of soft x-ray tomography datasets, particularly for samples of unknown variability like virally infected cells, is performed manually. Reconstructed tomograms are saved in .mrc format and are segmented and analyzed, using the commercial software Amira-Avizo (Thermo Fisher Scientific) as described here.
Optional: Segmentation of SXT datasets can be performed with other segmentation editors, for example in Fiji (Schneider et al. 2012), MITK (Nolden et al., 2013), Slicer3D (Fedorov et al., 2012), Chimera (Pettersen et al., 2004) or others.
-
58.
Open tomographic data (.mrc file).
-
59.
Normalize the LAC value of each pixel as described in step 56. The normalization allows interpreting x-ray absorption per μm (not per pixel as reconstructed) assuring accurate measurements of both organelle chemical composition and volume.
Optional: Perform edge-preserving smoothing to simplify outlining of organelles and to smooth visualization of the SXT data.
-
60.
Go to segmentation editor and define a set of materials (Figure 7), that is nucleus (if desired heterochromatin and euchromatin), mitochondria, endoplasmic reticulum (ER), cytosol and lipid droplets. For infected cells, we recommend adding unknown materials for organelles associated with virus, like double membrane vesicles. These unknown organelles can be identified later.
-
61.
Adjust the contrast to enhance the edge of the structure of interest (i.e., nucleus, nucleolus, mitochondria, endoplasmic reticulum - ER, intracellular vesicles, plasma membrane).
-
62.Segment cytosol using magic wand tool (see Tools Figure 7)
-
a.select “same material only” and “all slices”.
-
b.adjust the range of LAC values until all parts of the cells are selected and add to material (cytosol).
-
a.
-
63.Segment a nucleus
-
a.Using the tool Brush outline the edge of the nucleus, fill outlined region (by pressing F) and add to the nucleus material.
-
b.Repeat outlining of the nucleus for every tenth orthoslices.
-
c.Select all slices and interpolate the labels (Selection - interpolate)
-
d.If desired heterochromatin and euchromatin can be segmented semi-automatically based on their bimodal distribution by thresholding (nucleus volume only).
-
a.
-
64.Segment mitochondria, ER, lipid droplets, and intracellular vesicles semi-automatically
-
a.Select Magic Wand tool
-
b.Define threshold for X-ray absorption threshold for the analyzed organelle. From previous studies (McDermott et al., 2009; Loconte et al., 2021), for lipid droplets 0.75 ± 0.09 μm−1, mitochondria 0.45 ± 0.03 μm−1, ER 0.24 ± 0.04 μm−1.
-
c.Select voxel corresponding to the organelle.
-
d.Add it to the material.
-
e.Repeat until all organelles are segmented.
-
a.
Note: LAC values have relatively small variability, depending on the size and biological features of the analyzed cell type.
Optional: Tomographic data collection is conducted in thin-walled glass capillaries, which show an average X-ray absorption of 0.70 μm−1. When the selection of a certain organelle does not omit the selection of the capillary, the capillary can be removed by extracting its internal content. The procedure is performed in Amira using the “Volume Edit” option.
-
65.
Retrieve physical features and values of each set of organelles by selecting Menu bar, Segmentation and Material Statistics (Mode: materials).
Note: The immediate output of the segmentation provides information about either the cumulative or the individual volume (in pixels), surface area, and the average X-ray absorption of individual label fields. In general each label field can be exported and used as an input in custom-made analysis pipelines.
Figure 7.

Segmentation of cellular organelles using Amira
On the left: segmentation editor panel including materials and tools. On the right: virtual slice through the reconstructed SXT volume with manually segmented nucleus (blue) and based on the absorption threshold (0.28 - 0.45 μm-1) semi-automatically segmented mitochondria (red). The insert shows expected outcome of segmentation procedure, i.e., 3D rendering of the cell, showing heterochromatin (blue) and euchromatin (magenta) defining the nucleus, mitochondria (magenta), lipid droplets (yellow), ER (cyan), and plasma membrane (grey).
Expected outcomes
The anticipated outcome of the full-rotation SXT of single cells includes the visualization of whole cell morphology (such as nucleus and chromatin distribution, mitochondria, lipid droplets, endoplasmic reticulum, endosomes, lysosomes, and intracellular vesicles), accounting for the X-ray absorption signal of individual subcellular components. Among different groups of cells, the analyzed results provide information about organelle spatial distribution, changes in the cell ultrastructure organization upon viral infection (or other conditions), and alteration in the biochemical composition upon viral infection (or other conditions). The method provides qualitative information regarding the phenotype of a single cell. The acquisition of multiple tomograms from the same cell type and condition allows to map and quantify the mutual arrangement of the subcellular components, providing an average phenotype - detailed description of the biophysical features of the cell type. Such features provide information on the 3D subcellular reorganization induced by viral infections (or other pathological conditions), telling about infection pathway and ultimately helping in the development of specific drugs.
Limitations
Living organisms are composed of many cells. Only in humans, rough estimate put more than 400 distinct cell types (Vickaryous and Hall 2006). Since cell types are diverse in shape and size, it remains difficult to image their internal structure with one imaging protocol. It has to be noted that the protocol above is designed and ideally suited for cells in suspension and adherent cells, such as human lung epithelial cells have to be trypsinized prior to capillary loading. Similarly, imaging of tissue sections is not yet possible with XM-2.
To prevent X-ray radiation damages and immobilize the specimens in their native state we preserve specimens by cryo-fixation. To assure safe handling of viruses at the reduced biosafety level of the synchrotron, infected cells must be neutralized or inactivated with fixatives prior to imaging. While chemical fixation prior to cryo-fixation helps to preserve pressure-sensitive cells, it introduces alteration of cell morphology, such as cell shrinking, membrane blebbing, and deformation of lipid droplets (Loconte et al., 2021).
In our current protocol, segmentation is a primarily manual task, which is tedious and time consuming. Despite specific X-ray LAC values and known organelle morphology, the manual segmentation might result in user-dependent biases.
Automatic segmentation pipelines based on convolutional neuronal networks (Pelt and Sethian 2018; Plautz et al., 2017) can segment some organelles, like nucleus, lipid droplets and mitochondria, in human B cells. However, segmentation of cells with more varying morphology like during viral infection is yet a difficult task due to limited availability of already-labeled data.
Troubleshooting
Problem 1
Step 8–15
No cells are observed in the capillary after cell loading.
Potential solutions
Increase the cell density (spin at 300×g for 10 min and resuspend in smaller volume) to make sure there is a cell pellet visible prior to loading.
Problem 2
Step 8–15
The cell preparation causes too much cell debris, which makes the loading difficult and that could affect the quality of the data acquisition.
Potential solutions
Use cell strainer of 40 or 20 μm, depending on the cell size, to filter cells from debris and homogenize cell size distributions.
For adherent cells, use a milder way to detach cells from the substrate, for example: use highly diluted detaching agents.
Problem 3
Step 8–15
Difficulties loading cells into the capillary.
Potential solutions
Gently aspirate the cell pellet to break the aggregations/clumping or flick the tube to have individual cells before loading into the capillaries.
Filter the cells through cell strainer before loading them into the capillaries.
Problem 4
Step 8–15
Low quality of cellular vitrification.
Potential solutions
Check the settings of the plunger, particularly speed. The speed of the plunger can be adjusted to the type of cells as a function of their size; for instance, for larger cells, we recommend higher speed.
Check the level and temperature of the liquid propane and nitrogen. When the environmental temperature goes up during cryofixation, the quality of the vitrification is reduced.
Ensure that the heater of the propane well is on. Liquid propane will become slushy if liquid nitrogen overcools propane chamber.
Use a mix of propane and ethane. It is reported that this mix can result in better vitreous ice while plunge freezing the samples, on top it can also produce less damage to the specimens mounted on the thin, fragile carbon substrate (Tivol et al., 2008).
Problem 5
Step 8–15
Distortions of fragile cells.
Potential solutions
Fix cells chemically prior to cryo fixation as described in Cell preparation.
Problem 6
Step 30–38
Vitrification damage.
Potential solutions
Check if the purged helium is cold enough during cryo-transfer in SXT from the storage box to the microscope to keep the cells frozen.
Keep the temperature of the sample chamber low to avoid re-vitrification of the capillary.
Problem 7
Step 30–38
Ice around the capillary.
Potential solutions
Purge cooled dry helium gas during sample transfer to keep the temperature of the capillary low enough and avoid ice formation.
Run the de-icing protocol to warm up the specimen and sublimate the ice flakes.
Problem 8
Step 39–45
Projection image is black, no object visible.
Potential solutions
Check if the shutter is open that allows the X-ray beam to come through.
Make sure there are no glass debris as obstacles on the beam path.
Problem 9
Step 39–45
Inhomogeneous flat field which reduces the sub-cellular contrast.
Potential solutions
Optimize the beam profile by aligning the position of KZP, MZP, and pinhole.
Problem 10
Step 39–45
Low number of counts on X-ray camera or longer than usual exposure time.
Potential solutions
Optimize the beam profile making sure that the beam energy is within the “water window”.
Check if the vacuum conditions are correct for KZP, MZP, and CCD chambers.
Check if the helium flow is enough to purge away the air in the sample chamber.
Make sure there is no glass and ice debris along the beam path that may absorb the X-ray beam.
If big chunks of ice block the beam path, heat up the microscope and repeat startup procedure properly.
Problem 11
Step 39–45
Capillary with sample shifts out of the field of view.
Potential solutions
Repeat alignment of the capillary to the rotation center.
Check settings for helium and nitrogen gases. Wrong settings might cause fluctuations of the capillary within the chamber.
Switch on auto-correction procedure to keep capillary in the field of view during tomographic acquisition.
Problem 12
Step 52–57
Reconstructed dataset has structures which are not recognizable as the specimen.
Potential solution
Check projection data. If capillary drifted outside of the field-of-view during data acquisition, remove (up to 7) unwanted images as well as the corresponding angles from the angle file and reset reconstruction job by deleting the 'queued' file in the database.
Problem 13
Step 52–57
X-ray LAC values for well-described organelles, like lipid droplets, mitochondria and nucleus, or glass wall of the capillary.
This could happen after long period of shutdown, when the mutual position of CCD camera and MZP need recalibration.
Potential solutions
Calibrate with standard BSA solution with different concentrations (Parkinson et al., 2012; Hanssen et al., 2012).
Calibrate the practical pixel size using known size beads (2–5 μm), as reported in the paragraph” Microscope alignment.”
Check on the saturation of the pixel values in the dynamic range to control the over exposure of the region of interest (ROI) within the capillary area.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Venera Weinhardt (venera.weinhardt@cos.uni-heidelberg.de).
Materials availability
This study did not generate new reagents.
Acknowledgments
We thank Dr. Manfred Frey, Mannheim; Prof. Stephan Becker, University of Marburg; and Prof. Christian Drosten, Charité Berlin, Germany for their kind gift of cell cultures used in this study. V.W. is supported by German Research Foundation research fellowship WE 6221/2-1. The National Center for X-ray Tomography is supported by NIH NIGMS (P41GM103445, P30GM138441) and the DOE’s Office of Biological and Environmental Research (DE-AC02-5CH11231). V.W. was supported by the ERC Synergy Grant IndiGene (no. 810172). V.W. and R.B. are supported by the CoCID project (no. 101017116) funded within EU Research and Innovation act.
Author contributions
J-H.C., B.V., and V.L. performed experiments and formal analysis. M.C. provided samples. A.E., M A.L.G., and G.M. provided methodology, software, and data curation. R.B. and M.C. provided expertise and feedback. V.W., R.B., and C.L. conceived experiments and secured funding. All authors wrote the manuscript.
Declaration of interests
The authors declare no competing interests
Data and code availability
This paper does not report original data or code
References
- Fedorov A., Beichel R., Kalpathy-Cramer J., Finet J., Fillion-Robin J.-C., Pujol S., Bauer C., et al. 3D slicer as an image computing platform for the quantitative imaging network. Magn. Reson. Imaging. 2012;30:1323–1341. doi: 10.1016/j.mri.2012.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo J., Larabell C.A. Soft x-ray tomography: virtual sculptures from cell cultures. Curr. Opin. Struct. Biol. 2019;58:324–332. doi: 10.1016/j.sbi.2019.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanssen E., Knoechel C., Dearnley M., Dixon M.W.A., Le Gros M., Larabell C., Tilley L. Soft x-ray microscopy analysis of cell volume and hemoglobin content in erythrocytes infected with asexual and sexual stages of plasmodium falciparum. J. Struct. Biol. 2012;177:224–232. doi: 10.1016/j.jsb.2011.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harkiolaki M., Darrow M.C., Spink M.C., Kosior E., Dent K., Duke E. “Cryo-soft x-ray tomography: using soft x-rays to explore the ultrastructure of whole cells.” Edited by Marcellus Ubbink and Anastassis Perrakis. Emerging Top. Life Sci. 2018;2:81–92. doi: 10.1042/ETLS20170086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein S., Cortese M., Winter S.L., Wachsmuth-Melm M., Neufeldt C.J., Cerikan B., Stanifer M.L., Boulant S., Bartenschlager R., Chlanda P. SARS-CoV-2 structure and replication characterized by in situ cryo-electron tomography. Nat. Commun. 2020;11:5885. doi: 10.1038/s41467-020-19619-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kremer J.R., Mastronarde D.N., McIntosh J.R. Computer Visualization of Three-Dimensional Image Data Using IMOD. J. Struct. Biol. 1996;116:71–76. doi: 10.1006/jsbi.1996.0013. [DOI] [PubMed] [Google Scholar]
- Larabell C.A., Nugent K.A. Imaging cellular architecture with x-rays. Curr. Opin. Struct. Biol. 2010;20:623–631. doi: 10.1016/j.sbi.2010.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Gros M.A., Knoechel C.G., Uchida M., Parkinson D.Y., McDermott G., Larabell C.A. In: Comprehensive Biophysics. Egelman E.H., editor. Elsevier; 2012. 2.6 visualizing sub-cellular organization using soft x-ray tomography; pp. 90–110. [DOI] [Google Scholar]
- Le Gros M.A., McDermott G., Cinquin B.P., Smith E.A., Do M., Chao W.L., Naulleau P.P., Larabell C.A. Biological soft x-ray tomography on beamline 2.1 at the advanced light source. J. Synchrotron Radiat. 2014;21:1370–1377. doi: 10.1107/S1600577514015033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loconte V., Chen J.-H., Cortese M., Ekman A., Le Gros M.A., Larabell C., Bartenschlager R., Weinhardt V. Using soft x-ray tomography for rapid whole-cell quantitative imaging of SARS-CoV-2-infected cells. Cell Rep. Methods. 2021:100117. doi: 10.1016/j.crmeth.2021.100117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDermott G., Le Gros M.A., Knoechel C.G., Uchida M., Larabell C.A. Soft x-ray tomography and cryogenic light microscopy: the cool combination in cellular imaging. Trends Cell Biol. 2009;19:587–595. doi: 10.1016/j.tcb.2009.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolden M., Zelzer S., Seitel A., Wald D., Müller M., Franz A.M., Maleike D., et al. The medical imaging interaction toolkit: challenges and advances: 10 years of open-source development. Int. J. Computer Assisted Radiol. Surg. 2013;8:607–620. doi: 10.1007/s11548-013-0840-8. [DOI] [PubMed] [Google Scholar]
- Parkinson D.Y., Knoechel C., Yang C., Larabell C.A., Le Gros M.A. Automatic alignment and reconstruction of images for soft x-ray tomography. J. Struct. Biol. 2012;177:259–266. doi: 10.1016/j.jsb.2011.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson E.I., Prince T., Anderson E.R., Casas-Sanchez A., Smith S.L., Cansado-Utrilla C., Solomon T., et al. Methods of inactivation of SARS-CoV-2 for downstream biological assays. J. Infect. Dis. 2020;222:1462–1467. doi: 10.1093/infdis/jiaa507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelt D.M., Sethian J.A. A mixed-scale dense convolutional neural network for image analysis. Proc. Natl. Acad. Sci. U S A. 2018;115:254–259. doi: 10.1073/pnas.1715832114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen E.F., Goddard T.D., Huang C.C., Couch G.S., Greenblatt D.M., Meng E.C., Ferrin T.E. UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Plautz T., Boudreau R., Chen J.-H., Ekman A., LeGros M., McDermott G., Larabell C. Progress toward automatic segmentation of soft x-ray tomograms using convolutional neural networks. Microsc. Microanalysis. 2017;23:984–985. doi: 10.1017/S143192761700558X. [DOI] [Google Scholar]
- Schneider C.A., Rasband W.S., Eliceiri K.W. NIH image to ImageJ: 25 years of image analysis. Nat. Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith E.A., McDermott G., Do M., Leung K., Panning B., Le Gros M.A., Larabell C.A. Quantitatively imaging chromosomes by correlated cryo-fluorescence and soft x-ray tomographies. Biophysical J. 2014;107:1988–1996. doi: 10.1016/j.bpj.2014.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tivol W.F., Briegel A., Jensen G.J. An improved cryogen for plunge freezing. Microsc. Microanal. 2008;14:375–379. doi: 10.1017/S1431927608080781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vickaryous M.K., Hall. B.K. Human cell type diversity, evolution, development, and classification with special reference to cells derived from the neural crest. Biol. Rev. Cambridge Philosophical Soc. 2006;81:425–455. doi: 10.1017/S1464793106007068. [DOI] [PubMed] [Google Scholar]
- Weinhardt V., Chen J.-H., Ekman A.A., Guo J., Remesh S.G., Hammel M., McDermott G., Chao W., Oh S., Le Gros M.A., Larabell C.A. Switchable resolution in soft X-ray tomography of single cells. PLoS One. 2020;15:e0227601. doi: 10.1371/journal.pone.0227601. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This paper does not report original data or code