

Abstract
The tremendous success of stereogenic carbon compounds has never ceased to inspire researchers to explore the potentials of stereogenic silicon compounds. Intermolecular C–H silylation thus represents the most versatile and straightforward strategy to construct C–Si bonds, however, its enantioselective variant has been scarcely reported to date. Herein we report a protocol that allows for the enantioselective intermolecular C–H bond silylation, leading to the construction of a wide array of acyclic stereogenic Si–H compounds under simple and mild reaction conditions. Key to the success is (1) a substrate design that prevents the self-reaction of prochiral silane and (2) the employment of a more reactive rhodium hydride ([Rh]-H) catalyst as opposed to the commonly used rhodium chloride ([Rh]-Cl) catalyst. This work unveils opportunities in converting simple arenes into value-added stereogenic silicon compounds.
Subject terms: Synthetic chemistry methodology, Asymmetric catalysis
Construction of chiral organosilicon compounds could have implications in photophysical, biological, and chemical fields, as silicon is isoelectronic with carbon, and can mimic carbon atoms while providing slightly different properties. Here the authors present an intermolecular, enantioselective C–H silylation of heterocycles via rhodium catalysis.
Introduction
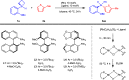
Catalytic functionalizations of C–H bonds including their asymmetric variants are at the center stage of modern chemistry1–4. Catalytic C–H silylations5–10 thus represent the most straightforward strategy to access value-added silicon compounds from simple arenes or heteroarenes (Fig. 1a). Along this line, a plethora of catalyst systems including Lewis acids11–16, bases17–19, or transition metals20–43 had been shown to affect intermolecular C–H silylations under various conditions. In particular, Hartwig and coworkers28 have successfully developed a rhodium/bisphosphine catalyzed reaction between MeSiH(OTMS)2 and various arenes, which demonstrated a broad C–H bond scope and an excellent regioselectivity. Despite the enormous knowledge and important progresses, enantioselective intermolecular C–H silylation has been scarcely reported to date.
Fig. 1. Direct C–H silylation for the synthesis of silicon-stereogenic silanes.

a Racemic catalytic intermolecular C–H silylation. b Enantioselective intramolecular C–H silylation (well established). c Enantioselective intermolecular C–H silylation (this work).
In sharp contrast, enantioselective intramolecular C–H silylation44–50 has met with great successes (Fig. 1b). Takai, Kuminobu and coworkers51 were the pioneers to demonstrate the asymmetric induction by a Rh/BINAP system in their intramolecular double C–H silylations. Building on this concept, a number of research groups including us have developed various Rh catalyzed enantioselective intramolecular C–H silylation reactions52–60 to construct stereogenic silicons constrained in a ring (cyclic stereogenic silicons). In these intramolecular reactions, the C–H bonds were placed in well-designed positions such that they were predisposed to intercept the Si-[Rh] intermediate, thus overcoming the low reactivity and poor regioselectivity of the C–H bonds.
Since such a pre-organized reaction pattern enjoyed by the intramolecular CH silylation is not available in the intermolecular C–H silylation, the low reactivity of the C–H bonds brings up significant challenges. Because the C–H bonds are not readily available to react with the Si-[Rh] intermediate, the Si-[Rh] tends to react with itself, leading exclusively to Si–Si (dehydrogenative coupling)61–63 or Si–C metathesis (silane redistribution)64–66 side products. Such low stabilities of the Si–H precursors had been repeatedly documented in literature examples. For instance, in the reports from the Grubbs and Stoltz groups17 as well as the Hartwig group28, excess amount of precursor silanes were used. Moreover, only a few of the existing C–H silylation reactions12,14,15,38,43 had produced an intrinsically chiral silicon (i.e., a silicon bearing three different groups), suggesting that the development of enantioseletive C–H silylation was not merely to find a proper chiral catalyst system. Instead, it is clear that three prerequisites have to be met: (1) a prochiral silane that can be converted into a stereogenic Si in an enantioselective fashion; (2) a substrate design that circumvents the self-reaction of the prochiral silane, and (3) a catalyst system that has high reactivity towards the intermolecular C–H bonds, thus further alleviating the self-reaction of the prochiral silane.
Herein, we report an example of enantioselective intermolecular C-H silylation (Fig. 1c), which stems from our long-term research in silicon chemistry. First, we choose silacyclobutanes (SCBs) as the prochiral silanes based on our earlier findings54,67. Second, we employ [Rh]-H as the catalyst, since our combined computational and experimental study68 have identified it as the true and more reactive catalyst for C–H silylation. Third, we uncover that a blocking group (but not a directing group) on the aryl substituent effectively prevents the oligomerization of SCBs. These considerations collectively lead to the current protocol, which allows the access to a wide array (77 examples) of acyclic stereogenic silanes in high yields and excellent enantioselectivities (up to 96% ee).
Results and discussion
Optimization of reaction conditions
We commenced our study by using silacyclobutane 1a and 2-methylthiophene 2a as the model substrates. Since our earlier work67 showed that the catalyst and solvent of choices were Rh(cod)Cl ([Rh]-Cl) and toluene, respectively, we investigated a panel of diphosphine ligands using this system first. Importantly, no reaction took place in the absence of a ligand (Table 1, entry 1), dispelling the concern about background reactions. When (S)-DTBM-BINAP (L1, entry 2) was employed, the desired tertiary hydrosilane 3aa was obtained in a low yield (22%) with a moderate enantioselectivity (74% ee). The MeO-Biphep ligands L2 and L3 that have smaller dihedral angles exhibited a decreased reactivity yet an enhanced enantioselectivity, giving poorer yields (13%) and good enantioselectivities (80% and 78% ee, respectively). Segphos ligands L4 and L5 (entries 5 and 6) that worked well in our intramolecular C-H silylations54,57,69 gave even poorer yields (12% and 10%, respectively). In order to improve the reaction, we tested the more reactive Rh precatalyst Rh(C2H4)2Cl (entry 7). This catalyst showed a slightly improved yield (19%) and comparable enantioselectivity (78% ee). We performed in situ 1H NMR monitoring on the reaction and observed that the substrate 1a suffered rapid oligomerization while the thiophene remained intact. This observation was consistent with the prior findings70 that the C–H bond partner was not capable of intercepting the [Rh]-Si intermediate before it was engaged in self-reaction.
Table 1.
Optimization of reaction conditionsa,b.
 | ||||
|---|---|---|---|---|
| Entry | [Rh] | Ligand | Yield/%c | ee/%d |
| 1 | Rh(cod)Cl | — | — | — |
| 2 | Rh(cod)Cl | L1 | 22 | 74 |
| 3 | Rh(cod)Cl | L2 | 13 | 80 |
| 4 | Rh(cod)Cl | L3 | 13 | 78 |
| 5 | Rh(cod)Cl | L4 | 12 | 80 |
| 6 | Rh(cod)Cl | L5 | 10 | 78 |
| 7 | Rh(C2H4)2Cl | L4 | 19 | 78 |
| 8 | Rh(L2)-H | 54 | 84 | |
| 9 | Rh(L3)-H | 48 | 88 | |
| 10 | Rh(L4)-H | 61 | 84 | |
| 11 | Rh(L5)-H | 68 | 76 | |
| 12e | Rh(L3)-H | 57(46)f | 92 | |
aReaction conditions: 1a (0.1 mmol), 2a (0.2 mmol), [Rh] catalyst (10 mol%), Ligand (10 mol%), toluene (1.0 mL) in a sealed Schlenk tube. bFor the [Rh(L)-H] species, [Rh(C2H4)2Cl]2 (5 mol%), Ligand (5 mol%), Et3SiH (0.5 equiv.) were employed. cYield of isolated product. dEnantioexcesses determined by HPLC analysis on a chiral stationary phase. eThe reaction was conducted at room temperature (r.t.) for 48 h. f[Rh(C2H4)2Cl]2 (2.5 mol%), Ligand (2.5 mol%), Et3SiH (0.25 equiv.) were employed.
We thus turned our attention to [Rh]-H catalyst based on our earlier findings that it was the true catalyst for the intramolecular C–H silylation of SCB with aryl C–H bonds68. To this end, we prepared a number of pre-formed [Rh]-H catalysts (Supplementary Figs. 1–8). Even at a lower catalyst loading (5 mol%) than the [Rh]-Cl catalyst (10 mol%), the [Rh]-H catalyzed reactions showed systemically improved yields (cf. entries 2–6 vs. entries 8–11). Among them, Rh(L3)-H gave the best enantioselectivity of 88% ee (entry 9). There was probably a tradeoff between the reaction yield and enantioselectivity. For example, the bulkiest ligand L5 saw the highest yield (68%) but the lowest enantioselectivity (76% ee) (entry 11), while ligand L3 gave a balanced performance in terms of yield and ee (entry 9). Importantly, the employment of [Rh]-H allowed the reaction to proceed at room temperature, albeit a longer reaction time (48 h) was required. Under this new set of conditions with Rh(L3)-H as the catalyst, the reaction showed a better yield (57%) and excellent enantioselectivity (92% ee) (entry 12).
Substrate scope
We then demonstrated the scope of C–H bond partners in the forms of thiophenes and benzothiopene under the optimized reaction conditions (Fig. 2). First, a number of 2-substituted thiophenes with different electronic and steric demands were tested, which all successfully produced the corresponding chiral silanes (3ab-3ag). Second, the unsubstituted thiophene reacted smoothly to give the desired product 3ah in excellent enantioselectivty (90% ee). Third, alike the 2-substituted thiophenes, the 3-subsituted thiophenes showed good compatibility with the reaction, affording the products 3ai-3al in good yields and high enantioselectivities (80–90% ee). It was seen that collectively the substitution on the 3-position had a less impact on the reaction outcomes than that on the 2-position which was probably attributed to the initial coordination of thiophenes to the pre-formed [Rh]-H catalyst (Supplementary Fig. 14, 15). Finally, di-substituted thiophene and benzothiophene reacted smoothly to furnish the desired products 3am and 3an in excellent enantioselectivies (90% and 86% ee, respectively). However, 3,4-dimethylthiophene was not compatible with current transformation probably due to the steric effect of methyl group, and other heterocycles such as furan, pyrrole, pyridine, and indole appeared to be unsuccessful C-H partners in this reaction. We also attempted SCBs bearing unsubstituted phenyl (i.e., 1a devoid of the –OBn group), but the reaction substrates suffered complete oligomerization and no silylation products were detected. This observation suggests that the substitution ortho- to the phenyl-Si bond is essential for the success of the reaction, which likely prevents the [Rh]-Si self-reaction by virtue of steric hindrance.
Fig. 2. Reactions with different C–H partners.

aReaction conditions: 1a (0.1 mmol), 2 (0.2 mmol), [Rh(C2H4)2Cl]2 (5 mol%), L3 (5 mol%), Et3SiH (0.5 equiv.), toluene (1.0 mL) in a sealed Schlenk tube. bYield of isolated product. cEnantiomer ratio determined by HPLC analysis on a chiral stationary phase. dThe reaction was conducted on 1.0 mmol scale. r.t. = room temperature.
Next, we showed that a number of blocking groups instead of benzyl ethers were well tolerated (Fig. 3). For illustration, we employed three representative C-H partners, namely, 2-methylthiopene (2a), 3-methylthoiphene (2i) and benzothiophene (2n). Four alkyl ethers, methyl- (3b), ethyl- (3c), n-butyl- (3d) and isopropyl- (3e) were examined first. Across the panel these substrates showed consistent yields (50–69%) and high enantioselectivities (86–94% ee). It was observed that the 3e accumulatively performed better than other ethers, probably due to its biggest steric hindrance derived from the isopropyl group. The insensitivity of the reaction outcomes to the ether substitution suggests that many other types of ether substitution should be compatible with this reaction if desired. We also replaced the ethers with simple alkyls as seen in the cases of 1 f and 1 g. The desired products 3fa-3gn were obtained in even higher yields (76–84%) and excellent enantioselectivities (90–95% ee). These two examples indicate that many alkyl substitutions ortho- to the Si should be tolerated by our method. Interestingly, ortho-phenyl substitution was also compatible if the available intramolecular C–H silylation sites were sterically shielded. As shown in the cases of 3 ha, 3hi and 3hn, the desired products were obtained in ~60% yields and ~80% ee. Such a pattern was successfully extended to small heterocycle framework such as in the case of 1i. Thus, acyclic stereogenic silicons 3ia, 3ii and 3in with two heterocycle substituents were obtained in 72–74% yields with 90–93% ee. The different choices of the groups on the ortho-position indicate that they are blocking groups but not directing groups. Many other functionalities that are stable towards Si-H, such as F-, Cl-, CF3-, carbocycles and so on, should be compatible with this reaction.
Fig. 3. Scope of silacyclobutanes (SCBs) in terms of blocking groups.

aReaction conditions: 1 (0.1 mmol), 2 (0.2 mmol), [Rh(C2H4)2Cl]2 (5 mol%), L3 (5 mol%), Et3SiH (0.5 equiv.), toluene (1.0 mL) in a sealed Schlenk tube. bYield of isolated product. cEnantiomer ratio determined by HPLC analysis on a chiral stationary phase. dee of 3 ha was determined by its silanol derivative. r.t. = room temperature.
We further demonstrated that poly-substitutions on the aryl ring were tolerated (Fig. 4). For illustration purpose, we used ethyl-ether when appropriate. Again, 2-methylthiophene (2a), 3-methylthiophene (2i) and benzothiophene (2n) were used as the representative C-H partners. This matrix of 11×3 reactions (3ja-3tn) gave moderate to good yields and high enantioselectivities. (1) A number of tri-substituted phenyls (1j-1r) on the SCBs were explored. In this class of substrates, we first walked a methyl group on the phenyl ring. It was seen that the position of the methyl group did not undermine the reaction. We then examined a number of different groups para to the Si (1n-1r). Again, the electronic demands of these groups did not significantly affect the efficiency and enantioselectivity of the reaction. Thus, electron-withdrawing F- (1n), Cl- (1o), electron-donating MeO- (1p), CF3O- (1q) and electron neutral Ph- (1r) substitutions all saw yields in 49–68% range and enantioselectivities around 90% ee. The absolutely configuration of product 3pn was established by X-ray crystallography; (2) Frameworks other than the phenyl ethers were also successful. The 1-naphthalene (1t) and 1-ethylether-2-naphthalene (1 s) frameworks provided the corresponding products in good yields and high enantioselectivities (89–96% ee). Importantly, the acyclic stereogenic Si-H products are shown in Figs. 2–4 had never been reported. The structures of 3 h and 3i marked a fresh level of complexity of stereogenic silicons available to date. The poly-substitution pattern shown in Fig. 4 (e.g., 3n and 3o) suggests that our products should be amenable to peripheral elaboration via standard reactions such as Suzuki couplings and SNAr reactions.
Fig. 4. Substrate scope on the aryl ring.

aReaction conditions: 1 (0.1 mmol), 2 (0.2 mmol), [Rh(C2H4)2Cl]2 (5 mol%), L3 (5 mol%), Et3SiH (0.5 equiv.), toluene (1.0 mL) in a sealed Schlenk tube. bYield of isolated product. cEnantiomer ratio determined by HPLC analysis on a chiral stationary phase. d[Rh(C2H4)2Cl]2 (10 mol%), L3 (10 mol%), toluene (1.0 mL) were employed. r.t. = room temperature.
Synthetic elaborations and control experiments
To showcase the synthetic utility of our method, we first carried out a gram-scale synthesis of compound 3aa, which smoothly afforded the desired product in good yield (60%) and excellent enantioselectivity (90% ee). We then explored the elaboration of the stereogenic Si–H in compound 3aa under two set of conditions (Fig. 5a): (1) Pd/C catalyzed silane oxidation afforded silanol 3aa-1 in a 92% yield with 90% ee; (2) Hydrosilylation with 3-hexyne under Karstedt catalyst produced the vinylsilane 3aa-2 in a 93% yield with 89% ee. In both cases the enantio-purity of product was not compromised, suggesting that the Si–H was amenable to stereospecific transformations to access other stereogenic silicons. Besides, we also conducted control experiments for the direct construction of chiral tetraorganosilicons (Fig. 5b, c). No reaction took place in the case of methyl-substituted SCB 1 u with benzothiophene 2n under standard conditions. When the reaction mixture was heated to 80 °C, only trace intermolecular silylation product 3un could be detected by GC-MS analysis. Similar results were also obtained in the reaction of diphenylsilacyclobutane (Supplementary Fig. 13), which underscored the different reactivities between the current work with our previous report67,68. Significantly, when a possible intramolecular silylation site of SCB substrate was available, as opposed to the cases of 1 h and 1i wherein the sites were blocked, intramolecular C–H silylation prevailed as seen in the cases of 1 v and 1w. The tandem intramolecular C-H silylation/intermolecular dehydrogenative coupling products 3va~3wn were consistent with our earlier work54, albeit lower enantioselectivities were observed.
Fig. 5. Preliminary elaborations and control experiments.

a Synthetic transformations of monohydrosilane 3aa. b Desymmetrization of methyl-substituted silacyclobutane (SCB). Reaction conditions: 1 u (0.1 mmol), 2n (0.1 mmol), [Rh(C2H4)2Cl]2 (5 mol%), L3 (5 mol%), Et3SiH (0.5 equiv.), toluene (1.0 mL) in a sealed Schlenk tube. 3un was detected by GC-MS analysis. T = temperature. c Competitive intramolecular silylation versus intermolecular silylation. Reaction conditions: 1 (0.1 mmol), 2 (0.2 mmol), [Rh(C2H4)2Cl]2 (5 mol%), L3 (5 mol%), Et3SiH (0.5 equiv.), toluene (1.0 mL) in a sealed Schlenk tube. Isolated yields were given. Enantiomer ratio determined by HPLC analysis on a chiral stationary phase. r.t. = room temperature.
Mechanism Investigation
We then carried out deuterium labeling experiments to shed light on the reaction mechanism (Fig. 6). First, the reaction of SCB 1c with deuterated benzothiophene D-2n proceeded smoothly under [Rh]-H catalysis to afford the desired product D-3cn in 58% yield. H/D scrambling from 1H and 2H NMR spectra showed that Si-H of D-3cn was partially deuterated (0.35 D), and control experiments suggested H/D exchange was facilitated by [Rh]-H catalyst outside of the catalytic cycle between D-2n and [Si]-H source (Fig. 7a, Supplementary Fig. 18–20). Deuteration merely on the terminal carbon atom of n-propyl group of D-3cn was also detected. These results indicated that the β-hydride elimination was unlikely to be operative in the process. Second, the relative ratio of the initial rates of two parallel reactions using 1c with 2n, 1c with D-2n, respectively, was determined to be 2.0, indicating that C–H bond cleavage might be involved in the rate-determining step. In our earlier intramolecular C–H silylation67,68, C–H bond activation was found to be energetically irreversible but not rate-limiting because the KIE of parallel reactions was determined to be 1.0. The fact that the C–H activation is involved in the rate-limiting step in this intermolecular C–H silylation underscores the vast difference between these two seemingly related reactions. Thirdly, a competition experiment of 2a and 2e was carried out and the corresponding products 3ca and 3ce were obtained in a 1:6 ratio. The faster reaction of thiophene bearing electron-withdrawing group suggested a faster oxidative addition process for the C–H bond cleavage, which further indicated C–H bond activation was involved in the rate-limiting step.
Fig. 6. Mechanistic studies.

a Deuterium labeling experiment. Reaction conditions: 1c (0.1 mmol), D-2n (0.1 mmol), [Rh(C2H4)2Cl]2 (5 mol%), L4 (5 mol%), Et3SiH (0.5 equiv.), toluene (1.0 mL) in a sealed Schlenk tube. Isolated yield was given. b Determination of kinetic isotope effect from two parallel reactions. c Intermolecular competition experiment of thiophenes. Reaction conditions: 1c (0.1 mmol), 2a (0.2 mmol), 2e (0.2 mmol), [Rh(C2H4)2Cl]2 (5 mol%), L4 (5 mol%), Et3SiH (0.5 equiv.), toluene (1.0 mL) in a sealed Schlenk tube. The ratio of 3ca to 3ce was determined by 1H NMR analysis. r.t. = room temperature.
Fig. 7. Proposed reaction mechanism.

a Putative pathway for the generation of D-3cn. b Plausible mechanism for intermolecular C–H silylation. [Rh*] = diphosphine coordinated rhodium.
Based on the above results a plausible mechanism is depicted in Fig. 7b. The reaction initiates with the coordination of thiophene to the pre-formed [Rh]-H catalyst, which was identified as the catalyst resting state, followed by oxidative addition of silacyclobutane (SCB) onto Rh(I), generating a five-membered rhodacycle A. This is the enantio-determining step based on our previous DFT calculations68. Reductive elimination occurs on Rh(III) cycle A, forming the Rh(I) intermediate B that is capable of C–H silylation. Subsequent C-H activation by Rh(I) B is perhaps the rate-limiting step. In a substrate that lacks steric hindrance around the Si atom, the Rh(I) intermediate B would react with the silane precursor, leading to oligomerization side reaction of the SCBs. However, in virtue of the steric shielding of the R group in Rh(I) intermediate B, C–H bond activation becomes competitive to give oxidative addition intermediate C. A final reductive elimination would produce the desired acyclic stereogenic Si-H product 3.
In conclusion, we have developed a protocol that enables enantioselective intermolecular C–H silylation reactions, producing a wide scope of acyclic stereogenic silanes in high efficiency and excellent enantioselectivity. This work clearly indicates that how to match the reactivity of the C–H bonds with the reactivity of the prochiral silanes is the most important question. In this study, it is achieved by placing a steric blocking group next to the Si center, and employing a highly reactive [Rh]-H catalyst. Future efforts should be paid to the expansion of the scope of both the silylation reagents and the C–H partners, such that more general prochiral silicons and C–H bonds could be applied to the access of almost all kinds of acyclic stereogenic Si–H compounds, which in turn can be elaborated into many kinds of stereogenic silicons.
Methods
General procedure for the preparation of [Rh]-H catalyst
[Rh(C2H4)2Cl]2 (7.8 mg, 0.02 mmol, 1.0 equiv.), DTBM-MeO-biphep (23.0 mg, 0.02 mmol, 1.0 equiv.) and toluene (2.0 mL) were added into a sealed tube in N2-flushed glove box. The reaction mixture was stirred at room temperature for 30 minutes. Then Et3SiH (32 µL, 0.2 mmol, 10.0 equiv.) was added in one portion and the stirring continued for 4 h at 50 °C to afford the stock solution of Rh(L3)-H which was used directly without further purification.
General procedure for the synthesis of stereogenic monohydrosilane 3
SCB substrate 1 (0.1 mmol), thiophene 2 (0.2 mmol) and Rh(L3)-H stock solution (0.5 mL) were added into a sealed tube equipped with magnetic stirring bar in N2-flushed glove box, and the total volume of toluene solution was adjusted to be 1.0 mL. The tube was removed from glovebox and stirred at room temperature for 48 h. After that the reaction mixture was diluted with dichloromethane (2.0 mL), and the organic layer was concentrated under reduced pressure. The residue was purified by preparative TLC to afford the corresponding monohydrosilane 3.
Supplementary information
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 21625104, W.H.), (No. 21901235, Q.Z.), (No. 21971133, W.H.) and the National Key Research and Development Program of China (No. 2017YFA0505203, W.H.).
Source data
Author contributions
K.A. and W.M. contributed equally to this work. W.H. conceived of the project. Q.Z. designed the experiments. K.A., W.M., L.L., T.H., and G.G. performed the research. Q.Z. and W.H. wrote the manuscript.
Peer review
Peer review information
Nature Communications thanks Chuan He and the anonymous reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Data availability
All data generated in this study are provided in the Supplementary Information/Source Data file. The X-ray crystallographic data used in this study are available in the Cambridge Crystallographic Data Center (CCDC) under accession code 2053377 (3pn) [www.ccdc.cam.ac.uk/data_request/cif].
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Kun An, Wenpeng Ma.
Contributor Information
Qing-Wei Zhang, Email: qingweiz@ustc.edu.cn.
Wei He, Email: whe@mail.tsinghua.edu.cn.
Supplementary information
The online version contains supplementary material available at 10.1038/s41467-022-28439-w.
References
- 1.Hartwig JF, Larsen MA. Undirected, homogeneous C−H bond functionalization: challenges and opportunities. ACS Cent. Sci. 2016;2:281–292. doi: 10.1021/acscentsci.6b00032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Newton CG, Wang S-G, Oliveira CC, Cramer N. Catalytic enantioselective transformations involving C-H bond cleavage by transition-metal complexes. Chem. Rev. 2017;117:8908–8976. doi: 10.1021/acs.chemrev.6b00692. [DOI] [PubMed] [Google Scholar]
- 3.Diesel J, Cramer N. Generation of heteroatom stereocenters by enantioselective C–H functionalization. ACS Catal. 2019;9:9164–9177. [Google Scholar]
- 4.Achar TK, Maiti S, Jana S, Maiti D. Transition metal catalyzed enantioselective C(sp2)–H bond functionalization. ACS Catal. 2020;10:13748–13793. [Google Scholar]
- 5.Hartwig JF. Borylation and silylation of C-H bonds: a platform for diverse C-H bond functionalizations. Acc. Chem. Res. 2012;45:864–873. doi: 10.1021/ar200206a. [DOI] [PubMed] [Google Scholar]
- 6.Cheng C, Hartwig JF. Catalytic silylation of unactivated C-H bonds. Chem. Rev. 2015;115:8946–8975. doi: 10.1021/cr5006414. [DOI] [PubMed] [Google Scholar]
- 7.Xu Z, Xu L-W. Silylations of arenes with hydrosilanes: from transition-metal-catalyzed C-X bond cleavage to environmentally benign transition-metal-free C-H bond activation. ChemSusChem. 2015;8:2176–2179. doi: 10.1002/cssc.201500467. [DOI] [PubMed] [Google Scholar]
- 8.Yang Y, Wang C. Direct silylation reactions of inert C-H bonds via transition metal catalysis. Sci. China Chem. 2015;58:1266–1279. [Google Scholar]
- 9.Richter SC, Oestreich M. Emerging strategies for C-H silylation. Trends Chem. 2020;2:13–27. [Google Scholar]
- 10.Li B, Dixneuf PH. Metal-catalyzed silylation of sp3 C-H bonds. Chem. Soc. Rev. 2021;50:5062–5085. doi: 10.1039/d0cs01392g. [DOI] [PubMed] [Google Scholar]
- 11.Curless LD, Clark ER, Dunsford JJ, Ingleson MJ. E-H (E = R3Si or H) bond activation by B(C6F5)3 and heteroarenes; competitive dehydrosilylation, hydrosilylation and hydrogenation. Chem. Commun. 2014;50:5270–5272. doi: 10.1039/c3cc47372d. [DOI] [PubMed] [Google Scholar]
- 12.Ma Y, Wang B, Zhang L, Hou Z. Boron-catalyzed aromatic C−H bond silylation with hydrosilanes. J. Am. Chem. Soc. 2016;138:3663–3666. doi: 10.1021/jacs.6b01349. [DOI] [PubMed] [Google Scholar]
- 13.Han Y, Zhang S, He J, Zhang Y. B(C6F5)3-catalyzed (convergent) disproportionation reaction of indoles. J. Am. Chem. Soc. 2017;139:7399–7407. doi: 10.1021/jacs.7b03534. [DOI] [PubMed] [Google Scholar]
- 14.Ma Y, et al. B(C6F5)3/amine-catalyzed C(sp)-H silylation of terminal alkynes with hydrosilanes: experimental and theoretical studies. Angew. Chem. Int. Ed. 2018;57:15222–15226. doi: 10.1002/anie.201809533. [DOI] [PubMed] [Google Scholar]
- 15.Zhang J, Park S, Chang S. Catalytic access to bridged sila-N-heterocycles from piperidines via cascade sp3 and sp2 C-Si bond formation. J. Am. Chem. Soc. 2018;140:13209–13213. doi: 10.1021/jacs.8b08733. [DOI] [PubMed] [Google Scholar]
- 16.Fang H, Xie K, Kemper S, Oestreich M. Consecutive β,β‘-selective C(sp3)-H silylation of tertiary amines with dihydrosilanes catalyzed by B(C6F5)3. Angew. Chem. Int. Ed. 2021;60:8542–8546. doi: 10.1002/anie.202016664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Toutov AA, et al. Silylation of C−H bonds in aromatic heterocycles by an earth-abundant metal catalyst. Nature. 2015;518:80–84. doi: 10.1038/nature14126. [DOI] [PubMed] [Google Scholar]
- 18.Toutov AA, et al. Alkali metal-hydroxide-catalyzed C(sp)-H bond silylation. J. Am. Chem. Soc. 2017;139:1668–1674. doi: 10.1021/jacs.6b12114. [DOI] [PubMed] [Google Scholar]
- 19.Gu Y, Shen Y, Zarate C, Martin R. A mild and direct site-selective sp2 C-H silylation of (poly)azines. J. Am. Chem. Soc. 2019;141:127–132. doi: 10.1021/jacs.8b12063. [DOI] [PubMed] [Google Scholar]
- 20.Uchimaru Y, El Sayed AMM, Tanaka M. Selective arylation of a Si−H bond in o-bis(dimethylsilyl)benzene via C−H bond activation of arenes. Organometallics. 1993;12:2065–2069. [Google Scholar]
- 21.Kakiuchi F, Igi K, Matsumoto M, Chatani N, Murai S. Ruthenium-catalyzed dehydrogenative silylation of aryloxazolines with hydrosilanes via C−H bond cleavage. Chem. Lett. 2001;30:422–423. [Google Scholar]
- 22.Tsukada N, Hartwig JF. Intermolecular and intramolecular, platinum-catalyzed, acceptorless dehydrogenative coupling of hydrosilanes with aryl and aliphatic methyl C−H Bonds. J. Am. Chem. Soc. 2005;127:5022–5023. doi: 10.1021/ja050612p. [DOI] [PubMed] [Google Scholar]
- 23.Lu B, Falck JR. Efficient iridium-catalyzed C–H functionalization/silylation of heteroarenes. Angew. Chem. Int. Ed. 2008;47:7508–7510. doi: 10.1002/anie.200802456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ihara H, Suginome M. Easily attachable and detachable ortho-directing agent for arylboronic acids in ruthenium-catalyzed aromatic C–H silylation. J. Am. Chem. Soc. 2009;131:7502–7503. doi: 10.1021/ja902314v. [DOI] [PubMed] [Google Scholar]
- 25.Klare HFT, et al. Cooperative catalytic activation of Si–H bonds by a polar Ru–S bond: regioselective low-temperature C–H silylation of indoles under neutral conditions by a Friedel−Crafts mechanism. J. Am. Chem. Soc. 2011;133:3312–3315. doi: 10.1021/ja111483r. [DOI] [PubMed] [Google Scholar]
- 26.Oyamada J, Nishiura M, Hou Z. Scandium-catalyzed silylation of aromatic C-H bonds. Angew. Chem. Int. Ed. 2011;50:10720–10723. doi: 10.1002/anie.201105636. [DOI] [PubMed] [Google Scholar]
- 27.Choi G, Tsurugi H, Mashima K. Hemilabile N-xylyl-N’-methylperimidine carbene iridium complexes as catalysts for C-H activation and dehydrogenative silylation: dual role of N-xylyl moiety for ortho-C-H bond activation and reductive bond cleavage. J. Am. Chem. Soc. 2013;135:13149–13161. doi: 10.1021/ja406519u. [DOI] [PubMed] [Google Scholar]
- 28.Cheng C, Hartwig JF. Rhodium-catalyzed intermolecular C−H silylation of arenes with high steric regiocontrol. Science. 2014;343:853–857. doi: 10.1126/science.1248042. [DOI] [PubMed] [Google Scholar]
- 29.Cheng C, Hartwig JF. Iridium-catalyzed silylation of aryl C–H bonds. J. Am. Chem. Soc. 2015;137:592–595. doi: 10.1021/ja511352u. [DOI] [PubMed] [Google Scholar]
- 30.Lee K-S, Katsoulis D, Choi J. Intermolecular C−H silylation of arenes and heteroarenes with HSiEt3 under operationally diverse conditions: neat/stoichiometric and acceptor/acceptorless. ACS Catal. 2016;6:1493–1496. [Google Scholar]
- 31.Liu Y-J, et al. Divergent and stereoselective synthesis of β-silyl-α-amino acids through palladium-catalyzed intermolecular silylation of unactivated primary and secondary C−H bonds. Angew. Chem. Int. Ed. 2016;55:13859–13862. doi: 10.1002/anie.201607766. [DOI] [PubMed] [Google Scholar]
- 32.Pan J-L, et al. Palladium-catalyzed direct intermolecular silylation of remote unactivated C(sp3)−H bonds. Chem. Commun. 2016;52:13151–13154. doi: 10.1039/c6cc07885k. [DOI] [PubMed] [Google Scholar]
- 33.Rubio-Pérez L, et al. A well-defined NHC−Ir(III) catalyst for the silylation of aromatic C−H bonds: substrate survey and mechanistic insights. Chem. Sci. 2017;8:4811–4822. doi: 10.1039/c6sc04899d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fukumoto Y, Hirano M, Chatani N. Iridium-catalyzed regioselective C(sp3)−H silylation of 4-alkylpyridines at the benzylic position with hydrosilanes leading to 4-(1-silylalkyl)pyridines. ACS Catal. 2017;7:3152–3156. doi: 10.1021/acs.joc.7b02375. [DOI] [PubMed] [Google Scholar]
- 35.Maji A, et al. Experimental and computational exploration of para-selective silylation with a hydrogen-bonded template. Angew. Chem. Int. Ed. 2017;56:14903–14907. doi: 10.1002/anie.201708449. [DOI] [PubMed] [Google Scholar]
- 36.Elsby MR, Johnson SA. Nickel-catalyzed C−H silylation of arenes with vinylsilanes: rapid and reversible β-Si elimination. J. Am. Chem. Soc. 2017;139:9401–9407. doi: 10.1021/jacs.7b05574. [DOI] [PubMed] [Google Scholar]
- 37.Fang HQ, Hou WJ, Liu GX, Huang Z. Ruthenium-catalyzed site-selective intramolecular silylation of primary C-H bonds for synthesis of sila-heterocycles. J. Am. Chem. Soc. 2017;139:11601–11609. doi: 10.1021/jacs.7b06798. [DOI] [PubMed] [Google Scholar]
- 38.Luo Y, Teng H-L, Xue C, Nishiura M, Hou Z. Yttrium-catalyzed regioselective α-C–H silylation of methyl sulfides with hydrosilanes. ACS Catal. 2018;8:8027–8032. [Google Scholar]
- 39.Karmel C, Chen Z, Hartwig JF. Iridium-catalyzed silylation of C-H bonds in unactivated arenes: a sterically encumbered phenanthroline ligand accelerates catalysis. J. Am. Chem. Soc. 2019;141:7063–7072. doi: 10.1021/jacs.9b01972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Esteruelas MA, Martínez A, Oliván M, Oñate E. Kinetic analysis and sequencing of Si–H and C–H bond activation reactions: direct silylation of arenes catalyzed by an iridium-polyhydride. J. Am. Chem. Soc. 2020;142:19119–19131. doi: 10.1021/jacs.0c07578. [DOI] [PubMed] [Google Scholar]
- 41.Wen J, Dong B, Zhu J, Zhao Y, Shi Z. Revealing silylation of C(sp2)/C(sp3)-H bonds in arylphosphines by ruthenium catalysis. Angew. Chem. Int. Ed. 2020;59:10909–10912. doi: 10.1002/anie.202003865. [DOI] [PubMed] [Google Scholar]
- 42.Wang D, et al. Phosphorus(III)-assisted regioselective C–H silylation of heteroarenes. Nat. Commun. 2021;12:524. doi: 10.1038/s41467-020-20531-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.He T, Li B, Liu L, Ma W, He W. Rhodium-catalyzed intermolecular silylation of Csp-H by silacyclobutanes. Chem. Eur. J. 2021;27:5648–5652. doi: 10.1002/chem.202100084. [DOI] [PubMed] [Google Scholar]
- 44.Xu L-W, Li L, Lai G-Q, Jiang J-X. The recent synthesis and application of silicon-stereogenic silanes: a renewed and significant challenge in asymmetric synthesis. Chem. Soc. Rev. 2011;40:1777–1790. doi: 10.1039/c0cs00037j. [DOI] [PubMed] [Google Scholar]
- 45.Xu L-W. Desymmetrization catalyzed by transition-metal complexes: enantioselective formation of silicon-stereogenic silanes. Angew. Chem. Int. Ed. 2012;51:12932–12934. doi: 10.1002/anie.201207932. [DOI] [PubMed] [Google Scholar]
- 46.Bauer, J. O. & Strohmann, C. Recent progress in asymmetric synthesis and application of difunctionalized silicon‐stereogenic silanes. Eur. J. Inorg. Chem. 2016, 2868–2881 (2016).
- 47.Shintani R. Recent progress in catalytic enantioselective desymmetrization of prochiral organosilanes for the synthesis of silicon-stereogenic compounds. Synlett. 2017;29:388–396. [Google Scholar]
- 48.Kumar R, Hoshimoto Y, Yabuki H, Ohashi M, Ogoshi S. Nickel(0)-catalyzed enantio- and diastereoselective synthesis of benzoxasiloles: ligand-controlled switching from inter- to intramolecular aryl-transfer process. J. Am. Chem. Soc. 2015;137:11838–11845. doi: 10.1021/jacs.5b07827. [DOI] [PubMed] [Google Scholar]
- 49.Zhang G, et al. Asymmetric synthesis of silicon‐stereogenic silanes by copper‐catalyzed desymmetrizing protoboration of vinylsilanes. Angew. Chem. Int. Ed. 2020;59:11927–11931. doi: 10.1002/anie.202005341. [DOI] [PubMed] [Google Scholar]
- 50.Wang XB, et al. Controllable Si−C bond activation enables stereocontrol in the palladium‐catalyzed [4+2] annulation of cyclopropenes with benzosilacyclobutanes. Angew. Chem. Int. Ed. 2020;59:790–797. doi: 10.1002/anie.201913060. [DOI] [PubMed] [Google Scholar]
- 51.Kuninobu Y, Yamauchi K, Tamura N, Seiki T, Takai K. Rhodium‐catalyzed asymmetric synthesis of spirosilabifluorene derivatives. Angew. Chem. Int. Ed. 2013;52:1520–1522. doi: 10.1002/anie.201207723. [DOI] [PubMed] [Google Scholar]
- 52.Murai M, Takeshima H, Morita H, Kuninobu Y, Takai K. Acceleration effects of phosphine ligands on the rhodium-catalyzed dehydrogenative silylation and germylation of unactivated C(sp3)-H bonds. J. Org. Chem. 2015;80:5407–5414. doi: 10.1021/acs.joc.5b00920. [DOI] [PubMed] [Google Scholar]
- 53.Murai M, Takeuchi Y, Yamauchi K, Kuninobu Y, Takai K. Rhodium‐catalyzed synthesis of chiral spiro‐9‐silabifluorenes by dehydrogenative silylation: mechanistic insights into the construction of tetraorganosilicon stereocenters. Chem. Eur. J. 2016;22:6048–6058. doi: 10.1002/chem.201504718. [DOI] [PubMed] [Google Scholar]
- 54.Zhang QW, et al. Construction of chiral tetraorganosilicons by tandem desymmetrization of silacyclobutanes/intermolecular dehydrogenative silylation. Angew. Chem. Int. Ed. 2017;56:1125–1129. doi: 10.1002/anie.201609022. [DOI] [PubMed] [Google Scholar]
- 55.Mu D, et al. Streamlined construction of silicon-stereogenic silanes by tandem enantioselective C–H silylation/alkene hydrosilylation. J. Am. Chem. Soc. 2020;142:13459–13468. doi: 10.1021/jacs.0c04863. [DOI] [PubMed] [Google Scholar]
- 56.Yang B, Yang W, Guo Y, You L, He C. Enantioselective silylation of aliphatic C−H bonds for the synthesis of silicon-stereogenic dihydrobenzosiloles. Angew. Chem. Int. Ed. 2020;59:22217–22222. doi: 10.1002/anie.202009912. [DOI] [PubMed] [Google Scholar]
- 57.Ma W, Liu LC, An K, He T, He W. Rh-catalyzed syntheses of chiral monohydrosilanes via intramolecular C–H functionalization of dihydrosilanes. Angew. Chem. Int. Ed. 2021;60:4245–4251. doi: 10.1002/anie.202013041. [DOI] [PubMed] [Google Scholar]
- 58.Yuan W, et al. Asymmetric synthesis of silicon-stereogenic monohydrosilanes by dehydrogenative C–H silylation. Org. Lett. 2021;23:1367–1372. doi: 10.1021/acs.orglett.1c00029. [DOI] [PubMed] [Google Scholar]
- 59.Chen S, et al. Enantioselective construction of six- and seven-membered triorgano-substituted silicon-stereogenic heterocycles. Nat. Commun. 2021;12:1249. doi: 10.1038/s41467-021-21489-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Guo Y, Liu MM, Zhu X, Zhu L, He C. Catalytic asymmetric synthesis of silicon-stereogenic dihydrodibenzosilines: silicon-central to axial chirality relay. Angew. Chem. Int. Ed. 2021;60:13887–13891. doi: 10.1002/anie.202103748. [DOI] [PubMed] [Google Scholar]
- 61.Rosenberg L, Davis CW, Yao J. Catalytic dehydrogenative coupling of secondary silanes using Wilkinson’s catalyst. J. Am. Chem. Soc. 2001;123:5120–5121. doi: 10.1021/ja015697i. [DOI] [PubMed] [Google Scholar]
- 62.Corey JY. Dehydrocoupling of hydrosilanes to polysilanes and silicon oligomers: a 30 year overview. Adv. Organomet. Chem. 2004;51:1–52. [Google Scholar]
- 63.Mucha NT, Waterman R. Iridium pincer catalysts for silane dehydrocoupling: ligand effects on selectivity and activity. Organometallics. 2015;34:3865–3872. [Google Scholar]
- 64.Curtis, M. D. & Epstein, P. S. Redistribution reactions on silicon catalyzed by transition metal complexes. In Adv. Organomet. Chem. Elsevier: 19, 213–255 (1981).
- 65.Park MJ, Lee SJ, Park MG, Han BH. Redistribution of (aryl, benzyl, octyl) silane and dehydrogenative coupling of methylphenylsilane using an activated metal catalysts prepared by the reduction of transition metal chlorides with lithium metal powder. Bull. Korean Chem. Soc. 2000;21:336–338. [Google Scholar]
- 66.Park S, Kim BG, Göttker-Schnetmann I, Brookhart M. Redistribution of trialkyl silanes catalyzed by iridium silyl complexes. ACS Catal. 2012;2:307–316. [Google Scholar]
- 67.Zhang QW, et al. Rhodium-catalyzed intramolecular C-H silylation by silacyclobutanes. Angew. Chem. Int. Ed. 2016;55:6319–6323. doi: 10.1002/anie.201602376. [DOI] [PubMed] [Google Scholar]
- 68.Zhang L, et al. A combined computational and experimental study of Rh-catalyzed C–H silylation with silacyclobutanes: insights leading to a more efficient catalyst system. J. Am. Chem. Soc. 2021;143:3571–3582. doi: 10.1021/jacs.0c13335. [DOI] [PubMed] [Google Scholar]
- 69.Zhang QW, An K, Liu LC, Yue Y, He W. Rhodium-catalyzed enantioselective intramolecular C-H silylation for the syntheses of planar-chiral metallocene siloles. Angew. Chem. Int. Ed. 2015;54:6918–6921. doi: 10.1002/anie.201502548. [DOI] [PubMed] [Google Scholar]
- 70.Cundy C, Eaborn C, Lappert M. The role of the transition metal in the homogeneous catalytic polymerisation of strained organosilicon heterocycles. J. Organomet. Chem. 1972;44:291–297. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated in this study are provided in the Supplementary Information/Source Data file. The X-ray crystallographic data used in this study are available in the Cambridge Crystallographic Data Center (CCDC) under accession code 2053377 (3pn) [www.ccdc.cam.ac.uk/data_request/cif].