

Abstract
Genes involved in spermatogenesis tend to evolve rapidly, but we lack a clear understanding of how protein sequences and patterns of gene expression evolve across this complex developmental process. We used fluorescence-activated cell sorting (FACS) to generate expression data for early (meiotic) and late (postmeiotic) cell types across 13 inbred strains of mice (Mus) spanning ∼7 My of evolution. We used these comparative developmental data to investigate the evolution of lineage-specific expression, protein-coding sequences, and expression levels. We found increased lineage specificity and more rapid protein-coding and expression divergence during late spermatogenesis, suggesting that signatures of rapid testis molecular evolution are punctuated across sperm development. Despite strong overall developmental parallels in these components of molecular evolution, protein and expression divergences were only weakly correlated across genes. We detected more rapid protein evolution on the X chromosome relative to the autosomes, whereas X-linked gene expression tended to be relatively more conserved likely reflecting chromosome-specific regulatory constraints. Using allele-specific FACS expression data from crosses between four strains, we found that the relative contributions of different regulatory mechanisms also differed between cell types. Genes showing cis-regulatory changes were more common late in spermatogenesis, and tended to be associated with larger differences in expression levels and greater expression divergence between species. In contrast, genes with trans-acting changes were more common early and tended to be more conserved across species. Our findings advance understanding of gene evolution across spermatogenesis and underscore the fundamental importance of developmental context in molecular evolutionary studies.
Keywords: gene expression, allele-specific expression, faster-X evolution, fluorescence activated cell sorting (FACS), phylogenetic contrasts
Introduction
Mature sperm are the most morphologically diverse animal cell type, likely as a consequence of intense selection on sperm form and function (Pitnick et al. 2009). Genes involved in spermatogenesis also tend to evolve rapidly (Swanson et al. 2003; Good and Nachman 2005; Turner et al. 2008; Larson et al. 2016; Finseth and Harrison 2018), suggesting that pervasive sexual selection also shapes molecular evolution (Swanson and Vacquier 2002; Harrison et al. 2015). However, direct genotype-to-phenotype connections remain elusive for primary sexually selected traits, and there are additional evolutionary forces acting during spermatogenesis that shape overall patterns of molecular evolution (Good and Nachman 2005; Burgoyne et al. 2009; Dean et al. 2009; Larson et al. 2016; Schumacher and Herlyn 2018). For example, many spermatogenesis genes are highly specialized (Eddy 2002; Chalmel et al. 2007; Green et al. 2018), which can relax pleiotropic constraint and contribute to rapid evolution even in the absence of positive directional selection (Winter et al. 2004; Larracuente et al. 2008; Meisel 2011). Other components of spermatogenesis are highly conserved because small disruptions can lead to infertility (Burgoyne et al. 2009). Thus, spermatogenesis genes are likely to experience strong and sometimes contradictory evolutionary pressures. Understanding how these processes interact to shape molecular evolution across spermatogenesis is essential to understanding how natural selection shapes the genetic determinants of male fertility.
There are many components or levels of molecular evolution, spanning from protein sequence changes to differences in gene expression level, timing, and developmental specificity (King and Wilson 1975; Wray et al. 2003; Larracuente et al. 2008; Kaessmann 2010; Piasecka et al. 2013; Cridland et al. 2020). Many of these components have been shown to evolve relatively rapidly during spermatogenesis (Meiklejohn et al. 2003; Khaitovich et al. 2005; Voolstra et al. 2007; Brawand et al. 2011; Harrison et al. 2015; Vicens et al. 2017; Cridland et al. 2020; Sánchez-Ramírez et al. 2021), and generally trend toward increased divergence during the later stages of development (Good and Nachman 2005; Piasecka et al. 2013; Larson et al. 2016). Novel genes disproportionately arise with testis-specific expression (Levine et al. 2006; Zhao et al. 2014; Cridland et al. 2020; Schroeder et al. 2020; Lange et al. 2021), likely as a consequence of the more permissive regulatory environment of the later stages of sperm development (Kaessmann 2010; Soumillon et al. 2013). Likewise, the later stages of spermatogenesis tend to be enriched for novel testis-specific genes (Eddy 2002; Chalmel et al. 2007; Green et al. 2018). These developmental signatures of novelty and specialization are further reflected in patterns of increased divergence of protein sequences (Good and Nachman 2005; Kousathanas et al. 2014) and expression levels (Larson et al. 2016) between species during the later stages of sperm development. Parallel signatures of rapid molecular evolution likely reflect both relaxed constraints during the late stages of spermatogenesis, and enhanced positive selection on late-developing sperm phenotypes (Eddy 2002; Good and Nachman 2005; Larracuente et al. 2008; Larson et al. 2016; Cutter and Bundus 2020). However, it remains unclear how strongly different forms of molecular evolution are correlated. For example, changes in gene expression may often be cell or stage-specific and therefore may be less pleiotropic than protein-coding changes. This pleiotropic constraint hypothesis primarily applies to cis-regulatory changes, which likely affect one gene, whereas trans-regulatory changes can affect many genes across multiple cell types (Wray et al. 2003; Carroll 2008; Cutter and Bundus 2020).
The X chromosome provides a compelling example of how the conflicting selective pressures acting on spermatogenesis may shape different components of molecular evolution. Theory predicts that the X chromosome should evolve more rapidly than the autosomes, particularly if most beneficial mutations are recessive, because X-linked recessive beneficial mutations will always be exposed to selection in males (Charlesworth et al. 1987; Vicoso and Charlesworth 2009). Differences in effective population size (Ne) on the X chromosome may also affect relative rates of fixation on the X chromosome and autosomes due to genetic drift, but the relative differences in Ne depend on the relative reproductive success of different sexes in a population (Vicoso and Charlesworth 2009). Consistent with more efficient X-linked selection, protein-coding evolution tends to be faster on the X chromosome compared with the autosomes in several taxa, and this effect is often strongest for genes with male-biased expression (Khaitovich et al. 2005; Baines and Harr 2007; Baines et al. 2008; Meisel and Connallon 2013; Parsch and Ellegren 2013; Larson et al. 2016). Novel genes tend to arise more often on the X chromosome, and these are often expressed during spermatogenesis (Levine et al. 2006; Kaessmann 2010). There is also some evidence for rapid expression evolution on the X chromosome in flies and mammals (Khaitovich et al. 2005; Brawand et al. 2011; Meisel et al. 2012; Coolon et al. 2015), but X-linked expression in mice appears conserved relative to autosomal genes expressed during the later stages of spermatogenesis (Larson et al. 2016). Stage-specific differences in relative rates of expression evolution on the X chromosome may result from the unique regulatory pattern that the sex chromosomes undergo during mammalian spermatogenesis. In males, the X chromosome is inactivated early in meiosis (i.e., meiotic sex chromosome inactivation, MSCI; McKee and Handel 1993) and remains partially repressed during the postmeiotic haploid stages of sperm development (i.e., postmeiotic sex chromosome repression, PSCR; Namekawa et al. 2006). The theory underlying faster-X protein-coding evolution may also apply to cis-regulatory gene expression evolution, but X chromosome expression divergence is likely also affected by trans-regulatory changes on other chromosomes and regulatory constraints unique to the X chromosome (e.g., MSCI and PSCR, Meisel et al. 2012). Thus, comparing relative expression divergence on the X chromosome compared with the autosomes can give insight into the types of mutations and selective forces affecting X chromosome expression.
These stage-specific patterns highlight the importance of studying specific components of molecular evolution in a developmental framework (fig. 1A; Larson, Kopania, et al. 2018; Cutter and Bundus 2020). However, studies of molecular evolution have primarily focused on pairwise contrasts across nuanced aspects of tissue development (Good and Nachman 2005; Larson et al. 2016), or examined protein-coding versus regulatory evolution in whole tissues (Khaitovich et al. 2005; Voolstra et al. 2007; Mack et al. 2016; Vicens et al. 2017; Cridland et al. 2020), without combining both in a phylogenetic framework (but see Murat F, Mbengue N, Winge SB, Trefzer T, Leushkin E, Sepp M, Cardoso-Moreira M, Schmidt J, Schneider C, Mößinger K, Brüning T, Lamanna F, Belles MR, Conrad C, Kondova I, Bontrop R, Behr R, Khaitovich P, Pääbo S, Marques-Bonet T, Grützner F, Almstrup K, Schierup MH, Kaessmann H, 2021, unpublished data, https://www.biorxiv.org/content/10.1101/2021.11.08.467712v1, last accessed November 30, 2021). Relying on whole tissue expression comparisons may be particularly problematic for spermatogenesis, because differences in testis composition are expected to evolve rapidly between species (Ramm and Schärer 2014; Yapar E, Saglican E, Dönertaş HM, Özkurt E, Yan Z, Hu H, Guo S, Erdem B, Rohlfs RV, Khaitovich P, Somel M, 2021, unpublished data, https://www.biorxiv.org/content/10.1101/010553v2, last accessed July 12, 2021) and may confound patterns of expression level divergence (Good et al. 2010; Larson et al. 2016; Hunnicutt et al. 2021). Nonetheless, collection of stage or cell-specific expression data remains technically demanding (da Cruz et al. 2016; Green et al. 2018), likely limiting widespread use in comparative studies. As a consequence, most evolutionary studies of gene expression have relied on whole tissue comparisons between closely related species pairs, instead of using more powerful phylogenetic approaches (Rohlfs and Nielsen 2015; Dunn et al. 2018).
In this study, we use a comparative developmental approach to gain a more comprehensive understanding of molecular evolution across spermatogenesis in house mice (Mus). Mice are the predominant laboratory model for mammalian reproduction (Phifer-Rixey and Nachman 2015; Firman 2020), with abundant genomic resources (Keane et al. 2011; Thybert et al. 2018), and established wild-derived inbred strains that can be crossed to resolve mechanisms underlying expression divergence (i.e., cis- vs. trans-regulatory changes; Mack et al. 2016). Mice also show divergence in sperm head morphologies across closely related species (Skinner et al. 2019) and experience sperm competition in the wild (Dean et al. 2006), providing a compelling system for understanding the evolution of spermatogenesis.
We used fluorescence-activated cell sorting (FACS) to resolve patterns of gene expression in two enriched spermatogenic cell populations across several mouse strains, species, and cross types (fig. 1A). Our study used two main comparisons. First, we evaluated divergence in spermatogenic protein sequences and gene expression levels across thirteen inbred strains of mice, including two subspecies of the house mouse (Mus musculus) and two other Mus species spanning 7 My of evolution (fig. 1B; Chevret et al. 2005). Second, we used published data from reciprocal crosses between a subset of these inbred strains to resolve the relative contribution of cis- versus trans-regulatory changes to expression divergence. We used these data to address five main questions: 1) Is gene expression more lineage-specific during late spermatogenesis? 2) Do protein-coding sequences and gene expression levels evolve faster during the later stages of spermatogenesis? 3) Is the rate of molecular evolution elevated on the X chromosome compared with the autosomes, and does this relationship change across spermatogenesis? 4) To what extent are protein-coding and gene expression divergence correlated, and does this relationship change across developmental stages? 5) Are there differences in the relative contributions of regulatory mechanisms (cis- vs. trans-regulatory changes) across spermatogenesis?
Fig. 1.

(A) Predictive framework depicting the major stages of spermatogenesis and expected relative expression levels of the X chromosome and autosomes at each stage (Namekawa et al. 2006). The two cell populations used in this study are leptotene-zygotene (“early,” second from left, orange) and round spermatids (“late,” second from right, blue). The relative thickness of the gray bar represents the predicted cell type specificity at each stage (Eddy 2002; Chalmel et al. 2007; Larson et al. 2016; Green et al. 2018). (B) Maximum likelihood tree of concatenated exome data from the four Mus species or subspecies used in this study: Mus musculus musculus (mus), Mus musculus domesticus (dom), Mus spretus (spr), Mus pahari (pah). Tips are labeled with the inbred strains from each lineage, with select crosses used to generate F1 hybrids indicated with arrows. Number of individuals sampled for each strain indicated in parentheses. Approximate divergence times are placed at each major node (Chevret et al. 2005). All nodes had 100% bootstrap support.
Results
Spermatogenesis Gene Expression by Cell Type and Lineage
We collected spermatogenesis expression data from 34 mice representing four different species or subspecies: Mus musculus musculus, Mus musculus domesticus, Mus spretus, and Mus pahari. We will use the abbreviations mus, dom, spr, and pah to reference the four major groups, and refer to all taxa as “lineages” for concision (fig. 1B). For each sample, we generated expression data for two spermatogenic cell types, an early meiotic cell type (leptotene-zygotene cells from early prophase of meiosis I, hereafter “early”) and a postmeiotic cell type (round spermatids, hereafter “late”). We identified 23,164 one-to-one orthologs, including both protein-coding and nonprotein-coding genes, that were annotated in all four mouse lineages and the mouse reference (GRCm38). From this set, we defined expressed genes as those with an FPKM > 1 in all samples of a given cell type. Expression variance cleanly separated samples by cell type and lineage (supplementary fig. S1, Supplementary Material online), indicating successful enrichment of different cell types. Most expressed genes were detected in both cell types (table 1). However, approximately one third of the detected genes were preferentially expressed or “induced” in a given cell type (transcripts with > 2× median expression level in one cell type across all lineages; table 1). We also identified expressed genes that show testis-specific expression based on published multi-tissue expression data (Chalmel et al. 2007). We found that 493 testis-specific genes were induced late, whereas only 65 testis-specific genes were induced early (table 1), consistent with increased specificity late in spermatogenesis (Eddy 2002; Larson et al. 2016; Green et al. 2018). To distinguish experimental noise from biologically meaningful expression, we also used a Bayesian approach to determine if a gene was “active” in a tissue or cell type (Thompson et al. 2020) and found broad overlap with genes in the expressed data set (table 1). Using the same framework, we identified genes showing evidence for lineage-specific expression (“active” in a single lineage or subset of lineages). We tested for lineage-specificity in each cell type separately, so a gene that we considered lineage-specific in one cell type may be expressed in other lineages during other spermatogenesis stages.
Table 1.
Counts of Genes in Each Data Set and Cell Type across Spermatogenesis.
| Early | Late | Both Early and Late | |
|---|---|---|---|
| Expressed | 9,570 | 8,986 | 7,670 |
| Induced | 3,375 | 2,769 | 0 |
| Testis-specific (TS)a | 544 | 655 | 524 |
| Induced and TS | 65 | 493 | 0 |
| Active (dom) | 8,206 (98.2%) | 8,581 (90.4%) | 6,355 |
| Active (mus) | 8,782 (97.5%) | 10,098 (83.4%) | 7,289 |
| Active (spr) | 8,728 (97.1%) | 9,509 (86.0%) | 7,227 |
| Active (pah) | 8,124 (97.6%) | 9,563 (83.9%) | 6,682 |
Note.—Numbers in parentheses represent the percent of genes in the “active” data sets that were also in the “expressed” data set. Early, spermatocytes (leptotene/zygotene); Late, round spermatids.
Testis-specific inferred from Chalmel et al. (2007).
We found that lineage-specificity was rare overall, but more common for autosomal genes active during late spermatogenesis (Pearson’s χ2 test; dom: P ≪ 0.0001, mus: P ≪ 0.0001, spr: P ≪ 0.0001, dom-mus common ancestor: P ≪ 0.0001; fig. 2A). X-linked genes showed no significant differences in lineage-specificity between early and late cell types (fig. 2B), which could reflect a lack of specialization on the sex chromosomes, or reduced power to detect differences between cell types given small sample sizes. Few genes were lineage-specific in both cell types, and all were autosomal (dom: 9 genes, mus: 24 genes, spr: 24 genes, dom-mus: 21 genes). We found similar results using a log fold-change (logFC) approach with different logFC cutoff values to identify lineage-specific genes (supplementary fig. S2 and table S1, Supplementary Material online). Lineage-specific genes were not enriched for any processes specifically related to male reproduction. We also tested if lineage-specific genes tended to have higher or lower associations with coexpression networks using weighted gene coexpression network analysis (WGCNA, Langfelder and Horvath 2008). We did not see a general pattern across all lineage-specific genes, but genes specific to a given lineage tended to have higher association with coexpression modules associated with that lineage (supplementary fig. S3A, Supplementary Material online). Our results suggest that lineage-specific expression of spermatogenic genes is relatively uncommon at these shallow phylogenetic scales, but more likely to arise later in spermatogenesis.
Fig. 2.
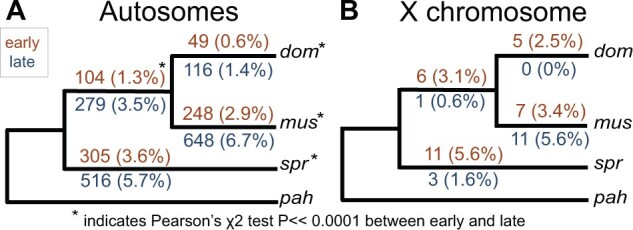
Number of genes that were lineage-specific on each internal branch of the mouse phylogeny used in this study. Numbers in parentheses are the percent of active genes that were lineage-specific. Results are presented separately for the autosomes (A) and X chromosome (B). Orange values above each branch represent the early cell type and blue values below represent the late cell type. Asterisks indicate a significant difference between early and late on that branch based on a Pearson’s χ2 test.
Greater Protein-Coding and Gene Expression Divergence during Late Spermatogenesis
Having detected subtle increases in lineage specificity late in spermatogenesis, we next tested if rates of protein sequence evolution (dN/dS) and expression level divergence were also elevated during the postmeiotic stage, as has been reported previously (Larson et al. 2016). Genes induced late in spermatogenesis showed significantly higher rates of protein-coding divergence on both the autosomes (n = 2,046 genes induced early, median dN/dS = 0.11; n = 1,711 genes induced late, median dN/dS = 0.20; Wilcoxon rank sum test P ≪ 0.0001) and the X chromosome (n = 54 genes induced early, median dN/dS = 0.25; n = 61 genes induced late, median dN/dS = 0.41; Wilcoxon rank sum test P = 0.049; fig. 3A, supplementary tables S2 and S3, Supplementary Material online). The 489 testis-specific genes showed elevated dN/dS overall, but most testis-specific genes were expressed in both cell types and there was no significant difference between genes expressed early and late for the autosomes (n = 350 genes expressed early, median dN/dS = 0.28; n = 424 genes expressed late, median dN/dS = 0.30; Wilcoxon rank sum test P = 1) or the X chromosome (n = 16 genes expressed early; median dN/dS = 0.59; n = 24 genes expressed late, median dN/dS = 0.58; Wilcoxon rank sum test P = 1). However, 348 testis-specific genes were preferentially expressed in the late cell type, representing ∼20% of all genes induced late for which we were able to calculate dN/dS. Taken together, these results confirm that tissue specificity plays an important role in the rapid protein-coding divergence of spermatogenic genes, and that most of this signature involves genes induced during postmeiotic spermatogenesis.
Fig. 3.
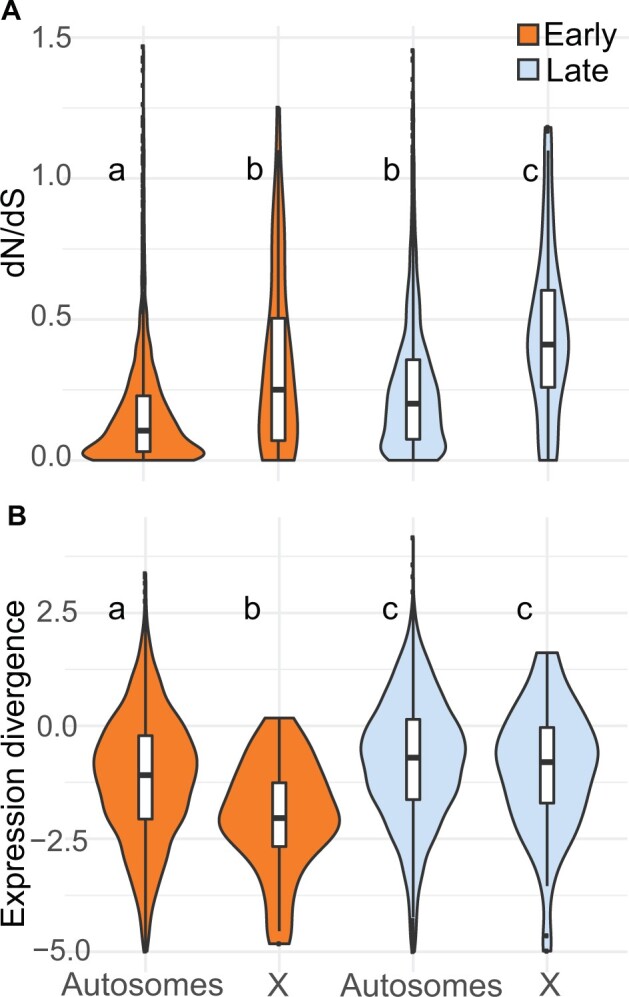
(A) Protein-coding and (B) expression divergence on the autosomes and X chromosome for genes induced in each cell type. Expression divergence values on the y-axis are , where betai is the measure of expression divergence from EVE. Higher values on the y-axis represent higher divergence. The center of each violin plot is a standard boxplot, with the center horizontal line representing the median divergence value. The violins show the probability density of divergence values for each group. A wider part of the violin at a given value means genes expressed in that group are more likely to have that divergence value. The letters above each violin indicate significant differences between the cell types and chromosome types based on a Wilcoxon rank sum test.
We used a phylogenetic ANOVA to estimate expression divergence while controlling for phylogenetic relatedness and variance within lineages (i.e., the expression variance and evolution [EVE] model; Rohlfs and Nielsen 2015). We report expression divergence from EVE as , where betai is a metric from EVE that represents the ratio of within-lineage variance to between-lineage evolutionary divergence, and higher positive values correspond to greater divergence between lineages. Expression divergence was higher for genes induced late in spermatogenesis on both the autosomes (n = 2,461 genes induced early, median EVE divergence = −1.09; n = 2,305 genes induced late, median EVE divergence = −0.70; Wilcoxon rank sum test P ≪ 0.0001) and the X chromosome (n = 44 genes induced early, median EVE divergence = −2.04; n = 68 genes induced late, median EVE divergence = −0.80; Wilcoxon rank sum test P = 0.00019; fig. 3B). This pattern held for all expressed genes, testis-specific genes, and different threshold cutoffs for considering genes induced (supplementary tables S4 and S5, Supplementary Material online). We also found higher divergence late for expressed and induced autosomal genes (supplementary table S5, Supplementary Material online) based on pairwise expression divergences using logFC and the metric from (Meisel et al. 2012); however, the pairwise framework did not give a consistent pattern on the X chromosome. When looking at all genes, most pairwise comparisons showed higher divergence late, but induced genes showed no difference between early and late spermatogenesis for most comparisons. However, the dom versus spr comparison had lower divergence late for all expressed genes and induced genes (supplementary table S5, Supplementary Material online).
Next, we tested if pleiotropic constraint imposed by protein–protein interactions contributed to less divergence during early spermatogenesis. We compared EVE expression divergence and dN/dS protein sequence divergence to the number of protein–protein interactions for genes in the mouse interactome database (MIPPIE, Alanis-Lobato et al. 2020). We found that genes induced early had fewer high-scoring protein–protein interactions (FDR-corrected Wilcoxon rank sum P ≪ 0.0001, supplementary fig. S4, Supplementary Material online), suggesting that these genes may actually be less constrained by protein–protein interactions. However, this difference was subtle, and protein-protein interactions are only one measure of potential pleiotropy, so genes induced early may still be constrained by their roles in other tissues or cell types. For both cell types, the number of protein–protein interactions was significantly negatively correlated with dN/dS (early: ρ = −0.122, Spearman’s rank correlation P ≪ 0.001; late: ρ = −0.143, Spearman’s rank correlation P ≪ 0.001), but not EVE divergence (early: ρ = −0.032, Spearman’s rank correlation P = 0.5; late: ρ = −0.060, Spearman’s rank correlation P = 0.5), consistent with hypotheses that protein sequence evolution is more constrained by pleiotropy and protein–protein interactions compared with gene expression evolution (Carroll 2008).
Collectively, we found strong evidence for more rapid protein-coding and gene expression level divergence during postmeiotic spermatogenesis, suggesting that these general patterns hold after controlling for phylogeny and at deeper divergence levels than had previously been shown in mice (Larson et al. 2016). Despite our expanded phylogenetic sample, we still lacked the power to determine if more rapid expression and protein-coding divergence is due to positive directional selection (supplementary fig. S5, Supplementary Material online).
Weak Positive Correlation between Gene Expression and Protein-Coding Divergence
We next tested for more general relationships between protein-coding and expression divergence across sets of genes expressed or induced during spermatogenesis (supplementary fig. S6 and table S6, Supplementary Material online). Across all autosomal genes expressed early, there was a weak positive correlation between dN/dS and pairwise expression divergence (ρ = 0.13–0.17, Spearman’s rank correlation P ≪ 0.0001). For induced genes, this correlation was weaker but still significant (ρ = 0.07–0.11, Spearman’s rank correlation P < 0.05). For the late cell type, there was also a weak positive correlation between pairwise expression divergence and dN/dS on the autosomes, but the correlation was weaker than that seen in the early cell type (ρ = 0.03–0.05, Spearman’s rank correlation P < 0.05). There was no correlation for the set of genes induced late. When looking only at genes with evidence for positive directional selection at the protein-coding level after correction for multiple tests (366 genes), the correlation was stronger on the autosomes late for the dom versus spr (n = 250 genes, ρ = 0.17, Spearman’s rank correlation P = 0.02) and mus versus spr comparisons (n = 249 genes, ρ = 0.18, Spearman’s rank correlation P ≪ 0.0001). When comparing dN/dS to EVE expression divergence, we only saw a significant positive correlation for genes expressed late that were also under positive selection at the protein-coding level (n = 160 genes, ρ = 0.18, Spearman’s rank correlation P = 0.04). We also tested if dN/dS was correlated with module eigengene values in our WGCNA. There was a weak positive correlation for eigengene values in the late cell type module (ρ = 0.033, FDR-corrected P = 0.03, supplementary fig. S3C, Supplementary Material online), but not the early cell type module (ρ = 0.026, FDR-corrected P = 0.07). In summary, we tended to observe a positive relationship between protein-coding and expression level divergence, but the strength of this relationship was weak and varied by gene set and divergence metric.
Faster-X Protein-Coding but Not Gene Expression Evolution
In addition to comparisons between spermatogenesis cell types, we compared relative rates of molecular evolution between X-linked and autosomal genes within a cell type. We found that protein-coding divergence was higher on the X chromosome, both early and late, across all gene sets (fig. 3A, supplementary tables S3 and S4, Supplementary Material online) consistent with several previous studies (Khaitovich et al. 2005; Baines et al. 2008; Meisel and Connallon 2013; Kousathanas et al. 2014; Larson et al. 2016). For expression evolution, we found lower divergence on the X chromosome early using EVE (n = 2,461 autosomal genes, median EVE divergence = −1.09; n = 44 X-linked genes, median EVE divergence = −2.04; Wilcoxon rank sum test P = 0.00015; fig. 3B), but higher X-linked divergence when using pairwise comparisons (supplementary table S5, Supplementary Material online). A major difference between these approaches was that EVE calculates divergence across a phylogeny, so genes that show divergent expression levels in one lineage may still be conserved across the entire phylogeny. We detected significant correlations between pairwise divergence values for different pairwise comparisons on the autosomes, and during late spermatogenesis, but lower or nonsignificant correlations on the X early (table 2). Thus, many genes on the X chromosome expressed early showed relatively high divergence between two particular lineages, but lower divergence across other pairwise comparisons and across the phylogeny as a whole. This lineage-specific variance underscores the importance of evaluating gene expression divergence in a phylogenetic framework (Rohlfs and Nielsen 2015; Dunn et al. 2018).
Table 2.
Correlation between Pairwise Expression Divergence Values for All Possible Pairwise Comparisons.
| dom versus mus | dom versus spr | mus versus spr | dom versus pah | mus versus pah | ||
|---|---|---|---|---|---|---|
| Early, X-linked | dom versus spr | 0.34 | ||||
| mus versus spr | 0.07 | 0.28 | ||||
| dom versus pah | 0.07 | 0.14 | 0.19 | |||
| mus versus pah | 0.16 | 0.10 | 0.03 | 0.62 | ||
| spr versus pah | 0.14 | 0.27 | 0.16 | 0.58 | 0.67 | |
| Early, autosomal | dom versus spr | 0.32 | ||||
| mus versus spr | 0.32 | 0.61 | ||||
| dom versus pah | 0.28 | 0.28 | 0.27 | |||
| mus versus pah | 0.29 | 0.26 | 0.30 | 0.74 | ||
| spr versus pah | 0.24 | 0.32 | 0.34 | 0.55 | 0.57 | |
| Late, X-linked | dom versus spr | 0.36 | ||||
| mus versus spr | 0.50 | 0.45 | ||||
| dom versus pah | 0.20 | 0.23 | 0.22 | |||
| mus versus pah | 0.28 | 0.28 | 0.36 | 0.74 | ||
| spr versus pah | 0.15 | 0.20 | 0.20 | 0.73 | 0.72 | |
| Late, autosomal | dom versus spr | 0.35 | ||||
| mus versus spr | 0.37 | 0.59 | ||||
| dom versus pah | 0.30 | 0.33 | 0.30 | |||
| mus versus pah | 0.30 | 0.30 | 0.33 | 0.76 | ||
| spr versus pah | 0.25 | 0.32 | 0.33 | 0.64 | 0.63 |
Note.—Numbers presented are ρ values from a Spearman’s rank correlation test. We tested for correlations in pairwise expression divergence value among induced genes in each stage and chromosome group (early X, early autosomal, late X, and late autosomal). Gray boxes indicate no significant correlation between pairwise divergence values after FDR correction (Spearman’s rank correlation P > 0.05). Italic values indicate the lowest Spearman’s ρ value for each pairwise comparison across the four stages and chromosome groups.
In late spermatogenic cells (i.e., round spermatids), X-linked expression divergence was similar to or lower than on the autosomes depending on the contrast and approach. Using EVE, we found similar divergence on the X chromosome and autosomes late (n = 2,305 autosomal genes, median EVE divergence = −0.70; n = 68 X-linked genes, median EVE divergence = −0.80; Wilcoxon rank sum test P = 0.34; fig. 3B), whereas pairwise comparisons gave mixed results, depending on which two lineages were compared (supplementary table S5, Supplementary Material online). There were proportionally fewer differentially expressed genes on the X chromosome (fig. 4, supplementary fig. S7, Supplementary Material online), and this pattern was strongest for the more closely related comparisons (hypergeometric test; mus vs. dom P ≪ 0.0001, spr vs. dom P ≪ 0.0001, spr vs. mus P ≪ 0.0001). Across all metrics of expression divergence and both developmental stages, there was no evidence for pervasive faster-X gene expression level evolution. We also asked if there were differences in the degree of module association for X chromosome and autosomal genes based on WGCNA. X-linked genes tended to have higher eigengene values for the early cell type module (Wilcoxon rank sum test P ≪ 0.001), but lower values for the late cell type module (Wilcoxon rank sum test P ≪ 0.001, supplementary fig. S3B, Supplementary Material online). Because the X chromosome is repressed during late spermatogenesis, these differences in module association are likely a consequence of overall differences in expression level.
Fig. 4.

Observed versus expected number of genes differentially expressed (DE) in late spermatogenesis for three pairwise comparisons at different levels of evolutionary divergence: (A) dom versus mus, (B) spr versus mus, and (C) pah versus mus. Each point represents a different chromosome. The diagonal line is the one-to-one line at which the observed number of DE genes equals the expected number. P values are shown for the X chromosome only. They are based on a hypergeometric test for enrichment and corrected for multiple tests using a false discovery rate correction. A significant P value indicates that the observed number of DE genes is different from the expected number.
Relative Contributions of cis- and trans-Regulatory Evolution Vary across Spermatogenesis
Having shown differences in expression divergence between cell types, we next asked if there were differences in the types of regulatory mutations (e.g., cis- vs. trans-regulatory changes) underlying expression divergence of autosomal genes in each cell type. Note that allele-specific expression cannot be examined for X-linked genes in hemizygous males. We used whole testis (Mack et al. 2016) and FACS-sorted (Larson et al. 2017) data from reciprocal crosses between house mouse subspecies (dom × mus) to estimate allele-specific expression (ASE) and assign genes to eight different regulatory categories: cis, trans, cis × trans, compensatory, cis + trans opposite, cis + trans same, other, and conserved (Coolon et al. 2014; Mack et al. 2016).
Across all cell types and genotypes, 50–90% of genes were conserved. Comparing the two spermatogenic stages, we saw striking differences in the proportions of nonconserved genes within each regulatory category (fig. 5, supplementary table S7, Supplementary Material online). Trans was more common than cis early, whereas trans and cis made up a similar proportion of regulatory changes late (fig. 5, supplementary table S7, Supplementary Material online). Compensatory changes (compensatory and cis+trans opposite) were more common than reinforcing (cis+trans same) in both cell types, but there was a higher relative proportion of reinforcing late (fig. 5, supplementary table S7, Supplementary Material online). Correlated error can lead to an overestimation of compensatory effects in some instances; therefore we verified our result showing a bias towards compensatory changes using a subtraction approach with cross-replicate analysis (Fraser 2019; see supplementary methods for details, Supplementary Material online). We found significant negative correlations between cis and trans effects, with a trend towards more negative correlations early (early: r = −0.13 to −0.16, P ≪ 0.0001; late: r = −0.12 to −0.15, P ≪ 0.0001). We also asked if genes tended to be assigned to the same regulatory category or switch categories between the two cell types. Overall, most genes assigned to a given regulatory category in one cell type were either not expressed or conserved in the other cell type (supplementary table S8, Supplementary Material online). Of the 1,052 genes that were assigned to a regulatory category in both cell types, 501 remained in the same category and 551 switched categories, indicating that different types of mutations may shift the regulation of the same genes in different cell types.
Fig. 5.

Regulatory category results for the fertile F1 hybrid (LEWES♀ X PWK♂). (A) Percent of nonconserved genes in each regulatory category both early and late. (B and C) Expression logFC between alleles within the fertile F1 (y-axis) plotted against the expression logFC between the parental subspecies (x-axis). Each point represents a single gene. Colors correspond to (A) and indicate the regulatory category to which that gene was assigned. cXt = cis X trans; comp = compensatory; c + t opp = cis + trans opposite; c + t same = cis + trans same.
We focused on results for the dom (LEWES)♀ × mus (PWK)♂ cross (fig. 5) because these F1 hybrids are more fertile and therefore less likely to have misexpressed genes due to hybrid incompatibilities (Good et al. 2010). However, the subfertile reciprocal hybrids also showed similar overall proportions of genes in each regulatory category. The proportions of different regulatory mechanisms in whole testes were more similar to the late cell type (supplementary table S7, Supplementary Material online), consistent with previous studies showing high overlap in expression profiles between whole testes and spermatid stage cells (Soumillon et al. 2013). We further verified our results using pure strain (LEWES and PWK) expression data from our phylogenetic expression data set to determine differences in parental strain expression levels (supplementary table S7, Supplementary Material online). Finally, we evaluated the relative contributions of regulatory mechanisms contributing to expression differences between strains within each M. musculus subspecies using expression data from within-subspecies F1s (WSB X LEWES and CZECHII X PWK) and from the respective parental inbred strains. Consistent with results from the more divergent F1 hybrids, there was more trans than cis early but some variation depending on subspecies and cross-type (cis early: 8–14%, trans early: 46–59%, cis late: 12–22%, trans late: 28–29%; supplementary table S7, Supplementary Material online). In summary, early and late spermatogenesis differed in the types of regulatory mutations contributing to expression divergence, with a proportionally higher contribution of trans-regulatory changes early. This pattern was consistent across different degrees of evolutionary divergence and between reciprocal crosses.
cis-Regulatory Changes Tended to Have Larger Effects on Expression Level Divergence
Given that trans-regulatory changes were proportionally more common during early spermatogenesis (fig. 5), and that expression levels tended to be more conserved early (fig. 3), we hypothesized that trans-regulatory changes would have smaller effect sizes (Coolon et al. 2014; Hill et al. 2021). Consistent with this, genes with trans changes showed lower median divergence than those with cis changes (fig. 6). We saw higher divergence for reinforcing mutations based on logFC, but not EVE (fig. 6), suggesting that genes with reinforcing changes specific to the dom and mus comparison may not accumulate more divergence at deeper phylogenetic levels. For the early cell type, 26% of genes in the reinforcing category overlapped with genes that had high pairwise divergence between dom and mus, whereas only 10–16% of genes in this category overlapped with high divergence genes in other pairwise comparisons (supplementary table S9, Supplementary Material online). Similar patterns were observed for late cell type genes, with 22% of genes in the reinforcing category overlapping those with high divergence between dom and mus but only 10–14% overlapping with genes showing high divergence in other pairwise comparisons (supplementary table S9, Supplementary Material online). Collectively, cis-regulatory changes tended to have larger effects on expression divergence than trans-regulatory changes, and reinforcing mutations tended to have large effects on expression divergence between mus and dom, but not at deeper levels of evolutionary divergence.
Fig. 6.

Expression divergence violin plots by regulatory category for the fertile hybrid. Expression divergence is calculated using the value from EVE (A and B) and as the absolute value of the logFC in expression between parental subspecies (C and D). Plots (A) and (C) correspond to the early cell type and plots (B) and (D) correspond to the late cell type. Letters indicate significant differences between categories based on a pairwise Wilcoxon rank sum test.
Discussion
Developmental stage and context play an important role in shaping the molecular evolution of reproductive genes (Dean et al. 2009; Larson et al. 2016; Finseth and Harrison 2018; Schumacher and Herlyn 2018), with genes expressed in later developmental stages evolving more rapidly (Good and Nachman 2005; Larson et al. 2016). However, comparing gene expression and protein divergence across developmental stages has rarely been done in a phylogenetic framework. In this study, we combined comparative genomics with cell sorting in four species to understand mouse spermatogenesis evolution across a common developmental framework. Our results give insight into how evolution proceeds at different stages of sperm development, at different molecular levels, and on different chromosome types.
Molecular Divergence across Development
There is a long-standing prediction that early developmental stages should be more constrained, with evolutionary divergence gradually increasing across development (Abzhanov 2013), which likely contributes to more rapid molecular evolution during the later stages of sperm development. In addition, the postmeiotic stages are enriched for genes with narrower expression profiles or highly specific biological functions and are therefore expected to experience relaxed pleiotropic constraint (Eddy 2002; Good and Nachman 2005; Green et al. 2018), also motivating our general hypothesis that the postmeiotic round spermatid stage would diverge more rapidly. Sexual selection is also likely to be a primary determinant of spermatogenic evolution, but variation in the intensity of sexual selection across spermatogenesis is not well understood (White-Cooper et al. 2009). Sperm competition and cryptic female choice can select for changes in sperm production rate, form, or function, and many aspects of sperm morphology correlate with the intensity of postmating sexual selection (Lüpold et al. 2016; McLennan et al. 2017; Pahl et al. 2018). Rates of mitotic and initial meiotic divisions during early spermatogenesis can control the overall rate of sperm production (Ramm and Schärer 2014). Therefore, selection for increased sperm production likely acts during the development of spermatogonia (diploid mitotic cells; White-Cooper et al. 2009). In contrast, sexual selection shaping the form and function of mature sperm (e.g., sperm swimming speed and fertilization ability) likely acts on later developmental stages such as haploid spermatids (Alavioon et al. 2017). However, many genes involved in mature spermatozoa functions are also highly expressed during early meiosis (da Cruz et al. 2016), suggesting that spermatozoa may be shaped by regulatory networks operating throughout spermatogenesis.
All aspects of molecular evolution that we considered showed more divergence when considering genes induced in late spermatogenesis: lineage-specific expression (fig. 2), protein-coding divergence, and expression level divergence (fig. 3). On first principles, these likely result from a combination of positive selection and relaxed developmental and pleiotropic constraint (Eddy 2002; Swanson and Vacquier 2002; Winter et al. 2004; Good and Nachman 2005; Abzhanov 2013; Green et al. 2018). However, our study was underpowered to formally test for positive selection using likelihood ratio test approaches (Anisimova et al. 2001; Rohlfs and Nielsen 2015). Thus, the relative contributions of positive selection and relaxed constraint to rapid spermatogenesis evolution remain unclear, especially for gene expression phenotypes.
Induced genes provided strong evidence for rapid evolution late, but results were less clear when looking at other genes. Spermatogenesis is a transcriptionally complex process, with most genes in the genome expressed in the testes (Soumillon et al. 2013) and high overlap between genes expressed early and late in our data set (table 1). For protein-coding divergence, we saw more rapid evolution late only when looking at the induced data set, but not when looking at all expressed genes, likely because most genes in our data set were expressed in both cell types. For expression divergence, there was more rapid evolution late even when looking at all expressed genes. This suggests that even genes with broader (i.e., noninduced) expression patterns tended to show more conserved expression early in spermatogenesis.
Testis-specific genes tended to be both induced late and rapidly evolving at the protein-coding level. Testis-specific and male-biased gene sequences often evolve rapidly, which could be the result of positive selection on genes with specific spermatogenesis functions as well as relaxed constraint because these genes tend to have highly specific functions (Meiklejohn et al. 2003; Baines et al. 2008; Meisel 2011; Parsch and Ellegren 2013). However, we did not see a significant faster late pattern for protein-coding or pairwise expression divergence when looking only at testis-specific genes. Although there were relatively few testis-specific genes, it appears that they tended to be rapidly evolving regardless of which spermatogenesis stage they were expressed in. If generally true, more rapid divergence late in spermatogenesis may partially reflect a higher proportion of testis-specific genes induced in the late cell type (table 1).
In addition to these broad patterns of molecular evolution, we explored the potential functional relevance of rapid divergence for specific genes (supplementary table S10, Supplementary Material online). We detected 20 genes with high (>2.5) EVE divergence in either cell type, and of these 15 were broadly expressed, but five may have specific roles in spermatogenesis (The UniProt Consortium 2020). For example, Rnf19a had an EVE value of 4.2 in the late cell type and has a known role in the formation of the sex body, which isolates the sex chromosomes in the nucleus during meiosis, a process that is required for proper spermatogenesis (Párraga and del Mazo 2000) and appears to be disrupted in sterile hybrid mice (Bhattacharyya et al. 2013).
Gene Expression versus Protein-Coding Divergence
Protein-coding changes alter a gene in every tissue and developmental stage in which it is expressed, whereas expression changes have the potential to be more specific (Wray et al. 2003; Carroll 2008). Expression changes, specifically cis-regulatory changes, should be less constrained by pleiotropy and may underlie evolutionary changes when purifying selection acts more strongly against protein-coding divergence (Wray et al. 2003; Carroll 2008). Under this model, we might expect to see less pronounced differences in relative expression levels when comparing early versus late stages. However, more recent work has shown that cis-regulatory elements such as enhancers can be highly pleiotropic, so cis-regulatory changes may be more constrained than once thought (Sabarís et al. 2019; Hill et al. 2021). If gene expression and protein-coding are subject to similar constraints, we would expect them to show similar evolutionary patterns across spermatogenesis, as we observed for autosomal genes (fig. 3).
Interestingly, despite parallel trends in relative divergence across spermatogenesis, expression level divergence and protein-coding divergence were not strongly correlated across genes, suggesting that these two types of molecular changes mostly evolve independently (Khaitovich et al. 2005). Perhaps surprisingly, there was no overlap between genes with very rapid protein-coding divergence (dN/dS > 1.5) and high expression divergence (EVE divergence > 2.5). Likewise, only 26 genes with high pairwise expression divergence in at least one comparison (pairwise divergence metric > 1) also had high protein-coding divergence (dN/dS > 1.5; supplementary table S10, Supplementary Material online). Whether expression or protein-coding is more rapid for a particular gene may depend on factors such as expression breadth and protein function, but rarely did spermatogenic genes appear to be rapidly evolving for both gene expression and protein sequences.
We also investigated the evolution of lineage-specificity. Testes and sperm tend to be enriched for lineage-specific genes (Brawand et al. 2011) and novel genes (Cridland et al. 2020; Schroeder et al. 2020; Lange et al. 2021). Lineage-specific and novel genes may be common in spermatogenesis because testes are highly transcriptionally active and have a high tissue-specific expression profile, which may allow new genes to arise without disrupting other processes (Levine et al. 2006; Kaessmann 2010; Soumillon et al. 2013; Zhao et al. 2014). We found that late spermatogenesis also had proportionally more lineage-specific genes (fig. 2). Increased lineage-specificity late is consistent with and likely contributed to higher protein and expression level divergence late, as all results suggest that spermatogenesis can tolerate more genetic changes during the late stages without impacting fertility.
X Chromosome Evolution
The X chromosome is predicted to evolve faster than the autosomes because it is hemizygous in males so beneficial recessive mutations will fix more quickly (Charlesworth et al. 1987; Vicoso and Charlesworth 2009). Empirical studies show evidence for a faster-X effect at the protein-coding level in many taxa, particularly for male reproductive genes (Khaitovich et al. 2005; Baines et al. 2008; Meisel and Connallon 2013; Parsch and Ellegren 2013; Larson et al. 2016; but see Whittle et al. 2020). Our data provide strong evidence for faster-X protein-coding evolution for both early and late spermatogenesis, demonstrating that the faster-X effect applies across genes involved in different spermatogenesis stages in mice.
Our results were more complex for expression evolution, with phylogenetic (Rohlfs and Nielsen 2015) and pairwise approaches (Meisel et al. 2012) sometimes yielding contrasting results. In the early cell type, pairwise comparisons supported a faster-X effect, whereas the phylogenetic model did not (fig. 3B, supplementary table S5, Supplementary Material online). Correlations between different pairwise divergence values were relatively low on the X chromosome early, suggesting that X-linked genes with high expression level divergence in one pairwise comparison did not tend to have high divergence in other comparisons (table 2). In the late cell type, both phylogenetic and pairwise divergence metrics supported a similar rate of X-linked and autosomal expression evolution (fig. 3B, supplementary table S5, Supplementary Material online). It is well-established that lineage-specific changes can create false signatures of rapid divergence in pairwise comparisons (Felsenstein 1985), including in studies of gene expression evolution (Dunn et al. 2018). Thus, our results highlight the importance of accounting for shared evolutionary history when inferring general evolutionary trends (Rohlfs and Nielsen 2015; Dunn et al. 2018).
Overall, our results did not support a faster-X effect for testis gene expression evolution, in contrast to several previous studies (Khaitovich et al. 2005; Brawand et al. 2011; Meisel et al. 2012). These studies were in other systems and used whole testes samples, which are made up of different cell types, so signals of expression divergence may partially reflect differences in cell type composition rather than true per cell changes in expression levels (Good et al. 2010; Hunnicutt et al. 2021; Yapar E, Saglican E, Dönertaş HM, Özkurt E, Yan Z, Hu H, Guo S, Erdem B, Rohlfs RV, Khaitovich P, Somel M, 2021, unpublished data, https://www.biorxiv.org/content/10.1101/010553v2, last accessed July 12, 2021). One previous study used cell type-specific data and found that the X chromosome showed fewer differentially expressed genes during late spermatogenesis between mus and dom (Larson et al. 2016), and our phylogenetic sampling demonstrates that this result likely applies across mouse species.
Theoretical predictions for the faster-X effect on protein-coding evolution may also apply to gene expression changes, but only for cis-regulatory changes or trans-regulatory changes where both the causative mutations and affected loci are on the X chromosome (Meisel and Connallon 2013; Larson et al. 2016). The lack of faster-X effect for gene expression could indicate that trans-regulatory changes on other chromosomes play an important role in X chromosome spermatogenesis expression evolution. Unfortunately, we are unable to differentiate allele-specific testis expression for X-linked genes in hemizygous males and thus the contribution of cis- versus trans-regulatory changes remain speculative. Nonetheless, it is plausible that contrasting patterns of expression level and protein sequence divergence on the X chromosome could also reflect the fact that X-linked regulatory phenotypes experience additional constraints during spermatogenesis (Larson et al. 2016). For example, the sex chromosomes undergo MSCI and PSCR, which likely imposes an overall repressive regulatory environment that constrains gene expression levels but not protein-coding changes. Disruption of MSCI and PSCR strongly impairs male fertility, so evolutionary constraints on X chromosome expression during spermatogenesis are expected to be strong (Burgoyne et al. 2009; Good et al. 2010; Larson et al. 2017). These stage-specific mechanisms would not explain lower regulatory divergence early, which we also observed (fig. 3B). Overall, our results support the hypothesis that regulatory constraints reduce X-linked expression level divergence during at least some stages of spermatogenesis, while still allowing rapid protein-coding divergence (Larson et al. 2016; Larson, Kopania, et al. 2018). This finding underscores how different components of molecular evolution may experience unique evolutionary pressures that result in distinct patterns of divergence (Brawand et al. 2011; Halligan et al. 2013; Larson et al. 2016).
Regulatory Mechanisms Underlying Expression Divergence
Resolving the relative contributions of cis- versus trans-acting mutations underlying expression divergence is an important step toward understanding the genetic architecture of expression phenotypes and how different evolutionary forces may act on gene expression (Benowitz et al. 2020; Hill et al. 2021). Although considerable progress has been made in a few key model systems on this important question (Goncalves et al. 2012; Coolon et al. 2014; Mack et al. 2016; Benowitz et al. 2020; Cridland et al. 2020; Sánchez-Ramírez et al. 2021), available data mostly come from whole tissues or organisms. Our results showed that the relative contribution of underlying regulatory mechanisms can differ dramatically between two cell types within a single complex tissue. Genes assigned to a regulatory category in one cell type were often conserved, not expressed, or assigned to a different category in the other cell type, suggesting that most regulatory mutations were cell type-specific in our experiments. This finding supports the hypothesis that regulatory changes may experience less pleiotropic constraint than protein-coding changes, even for genes that are expressed in multiple cell types (Carroll 2008). Although these striking differences are perhaps an expected consequence of different selective pressures acting on cellular function and developmental stage, they also underscore how difficult it is to resolve regulatory phenotypes from complex tissues.
Trans-regulatory changes acting during early development are more likely to cause wide-ranging disruptions to regulatory networks, which are more likely to have detrimental effects on downstream developmental stages. Thus, trans-regulatory changes altering expression during early development are predicted to be removed by purifying selection, whereas cis-regulatory changes are generally thought to be less pleiotropic and therefore more common in early stages (Carroll 2008; Hill et al. 2021). Based on this simple logic, we predicted that cis-regulatory mutations may be proportionally more common in early spermatogenesis, but we found the opposite pattern (fig. 5, supplementary table S7, Supplementary Material online). The relative contributions of cis- and trans-regulatory changes to expression divergence likely depend on other factors, including a tendency of cis mutations to have larger individual effect sizes (Coolon et al. 2014; Hill et al. 2021). We did observe proportionally more cis-regulatory changes of large effect during late spermatogenesis (fig. 6D) underlying higher overall expression divergence at this stage (fig. 3). Thus, differences in individual effect sizes of cis- versus trans-acting changes likely play a central role in shaping regulatory evolution across mouse spermatogenesis.
Cis- and trans-regulatory mutations can combine to affect the expression of a single gene, either in the same direction (reinforcing) or in opposite directions (compensatory; Goncalves et al. 2012; Coolon et al. 2014; Mack et al. 2016). We observed a higher proportion of compensatory mutations than reinforcing mutations across both spermatogenesis cell types and in whole testes. Even after controlling for correlated error (Fraser 2019), we observed a negative correlation between cis- and trans-regulatory effects, supporting our result that compensatory mutations were more common than reinforcing mutations. This was expected given that gene expression tends to evolve under stabilizing selection (Rohlfs and Nielsen 2015), and it is consistent with previous studies across many tissue types in mice (Goncalves et al. 2012; Mack et al. 2016), flies (Coolon et al. 2014; Benowitz et al. 2020), and roundworms (Sánchez-Ramírez et al. 2021). We also saw relatively more reinforcing mutations during postmeiotic spermatogenesis. Reinforcing mutations tended to have a larger effect size based on expression differences (logFC) between mus and dom (fig. 6D), thus large-effect reinforcing changes also likely contribute to higher expression level divergence in late spermatogenesis.
Given the striking differences that we saw between just two cell types, it is likely that complex tissues composed of many cell types may often give different results than isolated cell populations. Consistent with this prediction, our observed proportions of genes in each regulatory category differ from some other published results in house mouse whole tissues (i.e., liver, Goncalves et al. 2012; whole testes, Mack et al. 2016), primarily in that we saw a higher proportion of genes in the trans category. We also found some different patterns when reanalyzing whole testes expression data from (Mack et al. 2016) that likely reflect technical differences in the analytical pipelines used between studies (supplementary table S7, see supplementary methods for details, Supplementary Material online). In general, our analysis used more conservative approaches to test for significant DE or ASE. Thus, only genes showing relatively pronounced differences in expression levels between genotypes or alleles were assigned to regulatory mechanisms in our study.
We also found that the relative proportion of cis- and trans-regulatory changes were similar between whole testes and the late cell type in the fertile F1 hybrid (supplementary table S7, Supplementary Material online), consistent with the observation that postmeiotic spermatids have a disproportionately large contribution to mouse whole testes expression patterns (Hunnicutt et al. 2021). These results suggest that changes in the relative intensities of different selective pressures acting across spermatogenesis not only change the extent of expression level divergence, but also select for different mechanisms of regulatory evolution underlying these expression changes. Given this, analyzing such patterns at the level of whole organisms or tissues seems unlikely to provide a clear understanding of how mechanisms of regulatory evolution proceed in underlying cells. Indeed, even enriched cell populations as we have generated may be limited by relative purities.
By considering both expression divergence across the Mus phylogeny and underlying mechanisms of regulatory divergence between two lineages (mus and dom), our study also provided a novel opportunity to connect different types of regulatory changes to patterns of expression divergence at a deeper phylogenetic scale. Although trans-acting changes were relatively common (fig. 5), genes with cis-regulatory changes between mus and dom tended to have higher phylogeny-wide expression divergence than those with trans-regulatory changes for both cell types (fig. 6A, 6B). This suggests that genes showing cis-regulatory changes were also more likely to accumulate regulatory differences over time, resulting in phylogeny-wide expression divergence, whereas genes showing trans-regulatory changes at relatively shallow evolutionary scales tended to be relatively conserved across the Mus phylogeny. Genes with reinforcing changes also had relatively low phylogeny-wide expression level divergence (fig. 6A and B), in contrast to their high pairwise divergence between mus and dom (fig. 6C and D). Genes in this category likely have large-effect, lineage-specific changes in expression that may be under purifying selection over deeper phylogenetic levels. Finally, our phylogenetic contrast revealed rapid expression level divergence late in spermatogenesis. By combining these data with allele-specific expression data, we further showed that cis-regulatory changes are likely to underlie this rapid phylogeny-wide expression divergence in late spermatogenesis.
Materials and Methods
Mouse Resources
We investigated gene expression and protein-coding evolution in 12 Mus musculus domesticus (dom) individuals from four inbred strains (2 BIK/g, 3 DGA, 3 LEWES/EiJ, 4 WSB/EiJ), 8 M. m. musculus (mus) individuals from three inbred strains (2 CZECHII/EiJ, 3 MBS, 3 PWK/PhJ), 11 M. spretus (spr) individuals from three inbred strains (5 SEG, 2 SFM, 4 STF), and 3 M. pahari (pah) individuals from one inbred strain (3 PAHARI/EiJ; fig. 1B). By using multiple wild-derived inbred strains of dom, mus, and spr, we sampled natural within-species variation while also having biological replicates of genetically similar individuals. These mice were maintained in breeding colonies at the University of Montana (UM) Department of Laboratory Animal Resources (IACUC protocol 002-13). These colonies were initially established from mice purchased from The Jackson Laboratory, Bar Harbor, ME (CZECHII/EiJ, PWK/PhJ, WSB/EiJ, LEWES/EiJ, PAHARI/EiJ) or acquired from Matthew Dean’s colonies at the University of Southern California which were derived from François Bonhomme’s stocks at the University of Montpellier, Montpellier, France (MBS, BIK, DGA, STF, SFM, SEG). We weaned males at ∼21 days postpartum (dpp) into same sex sibling groups and caged males individually at least 15 days prior to euthanization to avoid dominance effects on testes expression. We euthanized mice at 60–160 dpp by CO2 followed by cervical dislocation.
For expression data from reciprocal F1 males, we used FACS enriched expression data from (Larson et al. 2017). These data include males from reciprocal F1 crosses between different inbred strains within each M. musculus subspecies (mus: CZECHII females X PWK males, dom: WSB females X LEWES males), as well as reciprocal mus and dom F1 hybrids (LEWES females X PWK males and PWK females X LEWES males), allowing us to compare results at two different levels of divergence (i.e., within and between lineages). We also analyzed whole testes expression data from (Mack et al. 2016) to compare FACS-enriched cell types to whole testes, including crosses between different strains within each M. musculus subspecies (LEWES females X WSB males and PWK females X CZECHII males) and the same reciprocal F1 hybrid crosses to those in (Larson et al. 2017).
Testis Cell Sorting and RNAseq
We collected testes from mice immediately following euthanization and isolated cells at different stages of spermatogenesis using FACS (Getun et al. 2011). The full FACS protocol is available on GitHub (https://github.com/goodest-goodlab/good-protocols/tree/main/protocols/FACS, last accessed June 16, 2021). Briefly, we decapsulated testes and washed them twice with 1 mg/ml collagenase (Worthington Biochemical), 0.004 mg/ml DNase I (Qiagen), and GBSS (Sigma), followed by disassociation with 1 mg/ml trypsin (Worthington Biochemical) and 0.004 mg/ml DNase I. We then inactivated trypsin with 0.16 mg/ml fetal calf serum (Sigma). For each wash and disassociation step, we incubated and agitated samples at 33 °C for 15 min on a VWR minishaker at 120 rpm. We stained cells with 0.36 mg/ml Hoechst 33324 (Invitrogen) and 0.002 mg/ml propidium iodide, filtered with a 40 μm cell filter, and sorted using a FACSAria IIu cell sorter (BD Biosciences) at the UM Center for Environmental Health Sciences Fluorescence Cytometry Core. We periodically added 0.004 mg/ml DNase I as needed during sorting to prevent DNA clumps from clogging the sorter. We sorted cells into 15 μl beta-mercaptoethanol (Sigma) per 1 ml of RLT lysis buffer (Qiagen) and kept samples on ice whenever they were not in the incubator or the cell sorter. For this study, we focused on two cell populations: early meiotic spermatocytes (leptotene/zygotene) and postmeiotic round spermatids. We extracted RNA using the Qiagen RNeasy Blood and Tissue Kit and checked RNA integrity with a Bioanalyzer 2000 (Agilent) or TapeStation 2200 (Agilent). All samples except one had RIN ≥ 7 (supplementary table S11, Supplementary Material online). We prepared RNAseq libraries using the Agilent SureSelect protocol and sequenced samples at the Hudson Alpha Institute for Biotechnology using Illumina NextSeq (75 bp single end). All sample libraries were prepared and sequenced together to minimize batch effects.
Mus Strain Phylogeny
We generated the phylogeny in figure 1B using available exome (Chang et al. 2017; Sarver et al. 2017) and whole genome (Keane et al. 2011; Thybert et al. 2018) sequence data (PRJNA326865, PRJNA323493, PRJEB2003, PRJEB14896). Genotypes were based on iterative mapping assemblies relative to the house mouse reference genome (mm10) conducted using pseudo-it v3.0 (Sarver et al. 2017) that restricts genotyping to targeted exons. We ran pseudo-it with one iteration to generate consensus fasta files for each sample. We then extracted exons, aligned these regions using MAFFT v7.271 (Katoh and Standley 2013), converted to PHYLIP format using AMAS (Borowiec 2016), and inferred a maximum likelihood concatenated tree using IQ-TREE v2.1.4-beta (Nguyen et al. 2015).
Processing of Gene Expression Data
We used R version 3.6.3 and Bioconductor version 3.10 for all analyses. We trimmed raw reads for adaptors and low-quality bases using expHTS (Streett et al. 2015) and mapped trimmed reads with TopHat version 2.1.0 (Kim et al. 2013). Genome assemblies were previously published for all four lineages (Keane et al. 2011; Thybert et al. 2018), allowing us to map reads to the correct assembly and reduce reference bias (Sarver et al. 2017). Mapping rates were consistent across lineages (supplementary table S11, Supplementary Material online). To select orthologous genes among the four lineages, we used BiomaRt (Durinck et al. 2005, 2009) to identify one-to-one Ensembl orthologs and retained only those that were present in all genome assemblies and the mouse reference build GRCm38.
We counted reads using featureCounts and included multiply-mapping reads (Liao et al. 2014). We used edgeR 3.28.1 (Robinson et al. 2010) to normalize expression data, calculate fragments per kilobase per million reads (FPKM), and perform differential expression (DE) analyses. A gene was defined as “expressed” in our data set if it had an FPKM > 1 in at least eight samples. We tested different FPKM cutoffs for considering a gene “expressed” as well as different ways of handling multiply-mapped reads, and our results were consistent across these approaches (supplementary table S4, Supplementary Material online). A gene was expressed in a particular lineage and cell type if it had an FPKM > 1 in all samples of that lineage and cell type. A gene was considered induced in a particular cell type if its median FPKM in that cell type across all lineages was greater than two times its median FPKM in the other cell type across all lineages. We also tested different threshold cutoffs for considering a gene induced. Testis-specific genes were those only expressed in testis based on the mouse tissue expression data from (Chalmel et al. 2007).
We defined lineage-specific genes in two ways. First, we used a log fold-change (logFC) method in which a gene was considered lineage-specific if its median expression level in a lineage was greater than two times its median expression level in any of the other three lineages. We tested different logFC threshold cutoffs ranging from 1.5 to 10 and saw similar results as the logFC > 2 cutoff (supplementary table S1, Supplementary Material online). Second, we used a Bayesian approach to determine if a gene was active or inactive in an expression data set based on transcript levels as implemented with the program Zigzag (Thompson et al. 2020). Genes identified as being active (posterior P > 0.5) in one lineage and inactive (posterior P < 0.5) in the other lineages were considered lineage-specific. We ran Zigzag twice and only included genes with consistent active or inactive assignments between the two runs. Both the logFC and Zigzag analyses were performed for each cell type, so a gene could be lineage-specific in one cell type but not the other. For each lineage, we determined the proportion of expressed (logFC) or active (Zigzag) genes that were lineage-specific and used a Pearson’s χ2 test to determine if one cell type had greater lineage-specificity than the other. We used the R package topGO with the default algorithm and Fisher’s Exact Test to do a gene ontology (GO) enrichment test on lineage-specific genes.
Protein-Coding Divergence
We used the “iqtree-omp” command in IQTree version 1.5.5 (Nguyen et al. 2015) to infer a mouse species tree based on gene trees estimated from the reference sequences for all four mouse lineages (Keane et al. 2011; Thybert et al. 2018). We took the longest transcript for all one-to-one orthologs and aligned these using MAFFT v7.271 (Katoh and Standley 2013) and converted to PHYLIP format using AMAS (Borowiec 2016). We used a custom script to exclude genes that did not begin with a start codon, had early stop codons, or had sequence lengths that were not multiples of three. We then used the Codeml program in the PAML package to calculate protein-coding divergence and test for positive selection on protein-coding genes (Yang 2007). We used the M0 model to calculate phylogeny-wide dN/dS for each gene, which we report as the overall protein-coding divergence values. We also performed a likelihood ratio test between the M8 and M8a site-based models to test for positive directional selection on each gene (Swanson et al. 2003).
Differential Expression
We performed all analyses of expression level divergence for three different gene sets: expressed genes, induced genes, and testis-specific genes. To calculate expression divergence in a phylogenetic framework, we used the EVE model (Rohlfs and Nielsen 2015), which performs a phylogenetic ANOVA using an Ornstein-Uhlenbeck model to evaluate divergence while controlling for evolutionary relatedness. We report expression divergence from EVE as , where betai is a metric from EVE that represents the ratio of within-lineage variance to between-lineage evolutionary divergence. By taking the negative log, higher positive numbers correspond to greater evolutionary divergence. We excluded genes with extremely low divergence values [] because this subset did not show a linear relationship between evolutionary divergence and population variance and therefore violated underlying assumptions of the EVE model (supplementary fig. S8, Supplementary Material online).
We also calculated expression divergence in a pairwise framework (Meisel et al. 2012). This method takes the difference in expression level between two lineages and normalizes based on the average expression of the gene in both lineages:
| (1) |
Da, ij is the divergence of gene a between lineages i and j. Sa, i is the median FPKM of gene a in lineage i, and Sa, j is the median FPKM of gene a in lineage j. We also calculated the logFC in expression between every pairwise comparison of lineages as an additional pairwise divergence metric (Robinson et al. 2010). For the EVE, pairwise divergence, and logFC methods, we compared relative expression divergence between cell types and between the X chromosome and autosomes using a Wilcoxon rank sum test. We tested if certain cell types or chromosome types showed greater correlation among pairwise divergence values using Spearman’s rank correlation.
To compare rates of divergence with number of protein–protein interactions, we downloaded publicly available data from the mouse integrated protein–protein interaction reference (MIPPIE, Alanis-Lobato et al. 2020). We used scripts provided by MIPPIE to calculate the number of protein–protein interactions among genes induced early and among genes induced late based on MIPPIE data, only counting interactions with high (> 0.6) MIPPIE scores. We then compared the median number of interactions between early and late genes using a Wilcoxon rank sum test and tested if the number of interactions was correlated with EVE expression divergence or dN/dS protein sequence divergence using Spearman’s rank correlation tests. We also tested if groups of genes had higher coexpression network association using a coexpression network analysis implemented in the R package WGCNA (Langfelder and Horvath 2008). We tested if WGCNA modules were associated with cell types or lineages using linear models with posthoc Tukey tests implemented in the R package multcomp. We then used Wilcoxon rank sum tests with FDR-correction for multiple tests to compare gene eigenvalues between the X chromosome and autosomes, and between lineage-specific and nonlineage-specific genes to test if certain groups of genes had higher module associations.
We also compared relative expression divergence on the X chromosome versus the autosomes using the proportion of DE genes on each chromosome (Good et al. 2010; Larson et al. 2016). First, we calculated the proportion of expressed genes that are DE across all autosomes. We then multiplied this proportion by the number of genes expressed on each chromosome to calculate the expected number of DE genes for each chromosome. We plotted the observed number of DE genes against the expected number and used a hypergeometric test to evaluate if each chromosome was over- or under-enriched for DE genes.
Allele-Specific Expression and Regulatory Divergence
We used the modtools and lapels-suspenders pipelines (Huang et al. 2014) to reduce mapping bias and to assign the parental origin of reads in F1 individuals (see supplementary methods for details, Supplementary Material online). This approach requires mapping to pseudogenomes generated using modtools to resolve differences in genome coordinates between different references. We used published pseudogenomes for WSB and PWK, which incorporate single nucleotide variants (SNVs) and indels from these strains into the GRCm38 mouse reference build (Huang et al. 2014). For LEWES and CZECHII, we generated our own pseudogenomes with modtools version 1.0.2 using published VCF files (Morgan et al. 2016; Larson, Vanderpool, et al. 2018). We developed a custom pipeline (see supplementary methods for details, Supplementary Material online) to assign autosomal genes to regulatory categories following previous recommendations (Coolon et al. 2014; Mack et al. 2016; Combs and Fraser 2018; Benowitz et al. 2020). To determine significant differences between cell types, we performed a Pearson’s χ2 test followed by false discovery rate correction for multiple tests.
Supplementary Material
Supplementary data are available at Molecular Biology and Evolution online.
Supplementary Material
Acknowledgments
We thank Michael Nachman and three anonymous reviewers for their helpful comments on an earlier version of this manuscript. We would like to thank Pamela K. Shaw and the UM Fluorescence Cytometry Core supported by an Institutional Development Award from the NIGMS (P30GM103338), the UM Genomics Core supported by the M.J. Murdock Charitable Trust, the UM Lab Animal Resources staff, Gregg Thomas for assistance generating the phylogeny in figure 1B, Nathanael Herrera for mouse photos, and Frank Albert and members of the Good Lab for helpful advice. This work was supported by grants from the Eunice Kennedy Shriver National Institute of Child Health and Human Development of the National Institutes of Health (R01-HD073439, R01-HD094787 to JMG). E.E.K.K. was supported by the National Science Foundation Graduate Research Fellowship Program (DGE-1313190). E.L.L. was supported by the National Science Foundation (DEB-2012041). Any opinions, findings, and conclusions or recommendations expressed in this material are those of the author(s) and do not necessarily reflect the views of the National Science Foundation or the National Institutes of Health.
Author Contributions
J.M.G conceived and funded the project. E.E.K.K., E.L.L., and J.M.G. designed the experiments. E.L.L. and S.K. performed the mouse husbandry and breeding experiments. C.C., S.K., and E.L.L. performed mouse dissections and cell sorts. C.C. and E.L.L. prepared the sequencing libraries. E.E.K.K. analyzed the data. E.E.K.K., E.L.L., and J.M.G. wrote the manuscript with input from all authors.
Data Availability
RNAseq data generated for this project are available through the National Center for Biotechnology Information under accession PRJNA735780. Individual sample accessions are in supplementary table S11, Supplementary Material online. A table of genes in our analyses and whether they were considered expressed, induced, or active in each cell type is available in supplementary table S12, Supplementary Material online. Scripts used for expression divergence and allele-specific expression analyses are available on GitHub: https://github.com/ekopania/mus_spermatogenesis_analyses (last accessed November 18, 2021) and https://github.com/ekopania/cis-trans-pipeline (last accessed September 15, 2021).
References
- Abzhanov A. 2013. von Baer's law for the ages: lost and found principles of developmental evolution. Trends Genet. 29(12):712–722. [DOI] [PubMed] [Google Scholar]
- Alanis-Lobato G, Möllmann JS, Schaefer MH, Andrade-Navarro MA.. 2020. MIPPIE: the mouse integrated protein–protein interaction reference. Database. 2020:baaa035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alavioon G, Hotzy C, Nakhro K, Rudolf S, Scofield DG, Zajitschek S, Maklakov AA, Immler S.. 2017. Haploid selection within a single ejaculate increases offspring fitness. Proc Natl Acad Sci U S A. 114(30):8053–8058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anisimova M, Bielawski JP, Yang Z.. 2001. Accuracy and power of the likelihood ratio test in detecting adaptive molecular evolution. Mol Biol Evol. 18(8):1585–1592. [DOI] [PubMed] [Google Scholar]
- Baines JF, Harr B.. 2007. Reduced X-linked diversity in derived populations of house mice. Genetics 175(4):1911–1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baines JF, Sawyer SA, Hartl DL, Parsch J.. 2008. Effects of X-linkage and sex-biased gene expression on the rate of adaptive protein evolution in Drosophila. Mol Biol Evol. 25(8):1639–1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benowitz KM, Coleman JM, Allan CW, Matzkin LM.. 2020. Contributions of cis- and trans-regulatory evolution to transcriptomic divergence across populations in the Drosophila mojavensis larval brain. Genome Biol Evol. 12(8):1407–1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharyya T, Gregorova S, Mihola O, Anger M, Sebestova J, Denny P, Simecek P, Forejt J.. 2013. Mechanistic basis of infertility of mouse intersubspecific hybrids. Proc Natl Acad Sci U S A. 110(6):E468–E477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borowiec ML. 2016. AMAS: a fast tool for alignment manipulation and computing of summary statistics. PeerJ 4:e1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brawand D, Soumillon M, Necsulea A, Julien P, Csardi G, Harrigan P, Weier M, Liechti A, Aximu-Petri A, Kircher M. et al. 2011. The evolution of gene expression levels in mammalian organs. Nature 478(7369):343–348. [DOI] [PubMed] [Google Scholar]
- Burgoyne PS, Mahadevaiah SK, Turner JMA.. 2009. The consequences of asynapsis for mammalian meiosis. Nat Rev Genet. 10(3):207–216. [DOI] [PubMed] [Google Scholar]
- Carroll SB. 2008. Evo-devo and an expanding evolutionary synthesis: a genetic theory of morphological evolution. Cell 134(1):25–36. [DOI] [PubMed] [Google Scholar]
- Chalmel F, Rolland AD, Niederhauser-Wiederkehr C, Chung SSW, Demougin P, Gattiker A, Moore J, Patard J-J, Wolgemuth DJ, Jégou B. et al. 2007. The conserved transcriptome in human and rodent male gametogenesis. Proc Natl Acad Sci U S A. 104(20):8346–8351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang PL, Kopania E, Keeble S, Sarver BAJ, Larson E, Orth A, Belkhir K, Boursot P, Bonhomme F, Good JM. et al. 2017. Whole exome sequencing of wild-derived inbred strains of mice improves power to link phenotype and genotype. Mamm Genome. 28(9–10):416–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth B, Coyne JA, Barton NH.. 1987. The relative rates of evolution of sex chromosomes and autosomes. Am Nat. 130(1):113–146. [Google Scholar]
- Chevret P, Veyrunes F, Britton-Davidian J.. 2005. Molecular phylogeny of the genus Mus (Rodentia: Murinae) based on mitochondrial and nuclear data. Biol J Linn Soc. 84(3):417–427. [Google Scholar]
- Combs PA, Fraser HB.. 2018. Spatially varying cis-regulatory divergence in Drosophila embryos elucidates cis-regulatory logic. PLoS Genet. 14(11):e1007631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coolon JD, McManus CJ, Stevenson KR, Graveley BR, Wittkopp PJ.. 2014. Tempo and mode of regulatory evolution in Drosophila. Genome Res. 24(5):797–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coolon JD, Stevenson KR, McManus CJ, Yang B, Graveley BR, Wittkopp PJ.. 2015. Molecular mechanisms and evolutionary processes contributing to accelerated divergence of gene expression on the Drosophila X chromosome. Mol Biol Evol. 32(10):2605–2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cridland JM, Majane AC, Sheehy HK, Begun DJ.. 2020. Polymorphism and divergence of novel gene expression patterns in Drosophila melanogaster. Genetics 216(1):79–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cutter AD, Bundus JD.. 2020. Speciation and the developmental alarm clock. eLife 9:e56276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Cruz I, Rodríguez-Casuriaga R, Santiñaque FF, Farías J, Curti G, Capoano CA, Folle GA, Benavente R, Sotelo-Silveira JR, Geisinger A.. 2016. Transcriptome analysis of highly purified mouse spermatogenic cell populations: gene expression signatures switch from meiotic-to postmeiotic-related processes at pachytene stage. BMC Genomics. 17:294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean MD, Ardlie KG, Nachman MW.. 2006. The frequency of multiple paternity suggests that sperm competition is common in house mice (Mus domesticus). Mol Ecol. 15(13):4141–4151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean MD, Clark NL, Findlay GD, Karn RC, Yi X, Swanson WJ, MacCoss MJ, Nachman MW.. 2009. Proteomics and comparative genomic investigations reveal heterogeneity in evolutionary rate of male reproductive proteins in mice (Mus domesticus). Mol Biol Evol. 26(8):1733–1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn CW, Zapata F, Munro C, Siebert S, Hejnol A.. 2018. Pairwise comparisons across species are problematic when analyzing functional genomic data. Proc Natl Acad Sci U S A. 115(3):E409–E417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durinck S, Moreau Y, Kasprzyk A, Davis S, De Moor B, Brazma A, Huber W.. 2005. BioMart and Bioconductor: a powerful link between biological databases and microarray data analysis. Bioinformatics 21(16):3439–3440. [DOI] [PubMed] [Google Scholar]
- Durinck S, Spellman PT, Birney E, Huber W.. 2009. Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat Protoc. 4(8):1184–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eddy EM. 2002. Male germ cell gene expression. Recent Prog Horm Res. 57:103–128. [DOI] [PubMed] [Google Scholar]
- Felsenstein J. 1985. Phylogenies and the comparative method. Am Nat. 125(1):1–15. [Google Scholar]
- Finseth FR, Harrison RG.. 2018. Genes integral to the reproductive function of male reproductive tissues drive heterogeneity in evolutionary rates in Japanese Quail. Genes Genom Genet. 8:39–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firman RC. 2020. Of mice and women: advances in mammalian sperm competition with a focus on the female perspective. Philos Trans R Soc Lond B Biol Sci. 375(1813):20200082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser HB. 2019. Improving estimates of compensatory cis–trans regulatory divergence. Trends Genet. 35(1):3–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Getun IV, Torres B, Bois PRJ.. 2011. Flow cytometry purification of mouse meiotic cells. J Vis Exp. (50):2602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goncalves A, Leigh-Brown S, Thybert D, Stefflova K, Turro E, Flicek P, Brazma A, Odom DT, Marioni JC.. 2012. Extensive compensatory cis-trans regulation in the evolution of mouse gene expression. Genome Res. 22(12):2376–2384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Good JM, Giger T, Dean MD, Nachman MW.. 2010. Widespread over-expression of the X chromosome in sterile F1 hybrid mice. PLoS Genet. 6(9):e1001148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Good JM, Nachman MW.. 2005. Rates of protein evolution are positively correlated with developmental timing of expression during mouse spermatogenesis. Mol Biol Evol. 22(4):1044–1052. [DOI] [PubMed] [Google Scholar]
- Green CD, Ma Q, Manske GL, Shami AN, Zheng X, Marini S, Moritz L, Sultan C, Gurczynski SJ, Moore BB. et al. 2018. A comprehensive roadmap of murine spermatogenesis defined by single-cell RNA-seq. Dev Cell. 46(5):651–667.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halligan DL, Kousathanas A, Ness RW, Harr B, Eöry L, Keane TM, Adams DJ, Keightley PD.. 2013. Contributions of protein-coding and regulatory change to adaptive molecular evolution in murid rodents. PLoS Genet. 9(12):e1003995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison PW, Wright AE, Zimmer F, Dean R, Montgomery SH, Pointer MA, Mank JE.. 2015. Sexual selection drives evolution and rapid turnover of male gene expression. Proc Natl Acad Sci U S A. 112(14):4393–4398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill MS, Vande Zande P, Wittkopp PJ.. 2021. Molecular and evolutionary processes generating variation in gene expression. Nat Rev Genet. 22(4):203–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang S, Holt J, Kao C-Y, McMillan L, Wang W.. 2014. A novel multi-alignment pipeline for high-throughput sequencing data. Database 2014(0):bau057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunnicutt KE, Good JM, Larson EL.. 2021. Unraveling patterns of disrupted gene expression across a complex tissue. Evolution. Available from: https://doi.org/10.1111/evo.14420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaessmann H. 2010. Origins, evolution, and phenotypic impact of new genes. Genome Res. 20(10):1313–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K, Standley DM.. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30(4):772–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keane TM, Goodstadt L, Danecek P, White MA, Wong K, Yalcin B, Heger A, Agam A, Slater G, Goodson M. et al. 2011. Mouse genomic variation and its effect on phenotypes and gene regulation. Nature 477(7364):289–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khaitovich P, Hellmann I, Enard W, Nowick K, Leinweber M, Franz H, Weiss G, Lachmann M, Pääbo S.. 2005. Parallel patterns of evolution in the genomes and transcriptomes of humans and chimpanzees. Science 309(5742):1850–1854. [DOI] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL.. 2013. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 14(4):R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King M, Wilson A.. 1975. Evolution at two levels in humans and chimpanzees. Science 188(4184):107–116. [DOI] [PubMed] [Google Scholar]
- Kousathanas A, Halligan DL, Keightley PD.. 2014. Faster-X adaptive protein evolution in house mice. Genetics 196(4):1131–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange A, Patel PH, Heames B, Damry AM, Saenger T, Jackson CJ, Findlay GD, Bornberg-Bauer E.. 2021. Structural and functional characterization of a putative de novo gene in Drosophila. Nat Commun. 12(1):1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langfelder P, Horvath S.. 2008. WGCNA: an R package for weighted correlation network analysis. BMC Bioinform. 9(1):559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larracuente AM, Sackton TB, Greenberg AJ, Wong A, Singh ND, Sturgill D, Zhang Y, Oliver B, Clark AG.. 2008. Evolution of protein-coding genes in Drosophila. Trends Genet. 24(3):114–123. [DOI] [PubMed] [Google Scholar]
- Larson EL, Keeble S, Vanderpool D, Dean MD, Good JM.. 2017. The composite regulatory basis of the large X-effect in mouse speciation. Mol Biol Evol. 34(2):282–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson EL, Kopania EEK, Good JM.. 2018. Spermatogenesis and the evolution of mammalian sex chromosomes. Trends Genet. 34(9):722–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson EL, Vanderpool D, Keeble S, Zhou M, Sarver BAJ, Smith AD, Dean MD, Good JM.. 2016. Contrasting levels of molecular evolution on the mouse X chromosome. Genetics 203(4):1841–1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson EL, Vanderpool D, Sarver BAJ, Callahan C, Keeble S, Provencio LP, Kessler MD, Stewart V, Nordquist E, Dean MD. et al. 2018. The evolution of polymorphic hybrid incompatibilities in house mice. Genetics 209(3):845–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine MT, Jones CD, Kern AD, Lindfors HA, Begun DJ.. 2006. Novel genes derived from noncoding DNA in Drosophila melanogaster are frequently X-linked and exhibit testis-biased expression. Proc Natl Acad Sci U S A. 103(26):9935–9939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, Shi W.. 2014. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30(7):923–930. [DOI] [PubMed] [Google Scholar]
- Lüpold S, Manier MK, Puniamoorthy N, Schoff C, Starmer WT, Luepold SHB, Belote JM, Pitnick S.. 2016. How sexual selection can drive the evolution of costly sperm ornamentation. Nature 533(7604):535–538. [DOI] [PubMed] [Google Scholar]
- Mack KL, Campbell P, Nachman MW.. 2016. Gene regulation and speciation in house mice. Genome Res. 26(4):451–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKee BD, Handel MA.. 1993. Sex chromosomes, recombination, and chromatin conformation. Chromosoma 102(2):71–80. [DOI] [PubMed] [Google Scholar]
- McLennan HJ, Lüpold S, Smissen P, Rowe KC, Breed WG.. 2017. Greater sperm complexity in the Australasian old endemic rodents (Tribe: hydromyini) is associated with increased levels of inter-male sperm competition. Reprod Fertil Dev. 29(5):921–930. [DOI] [PubMed] [Google Scholar]
- Meiklejohn CD, Parsch J, Ranz JM, Hartl DL.. 2003. Rapid evolution of male-biased gene expression in Drosophila. Proc Natl Acad Sci U S A. 100(17):9894–9899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meisel RP. 2011. Towards a more nuanced understanding of the relationship between sex-biased gene expression and rates of protein-coding sequence evolution. Mol Biol Evol. 28(6):1893–1900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meisel RP, Connallon T.. 2013. The faster-X effect: integrating theory and data. Trends Genet. 29(9):537–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meisel RP, Malone JH, Clark AG.. 2012. Faster-X evolution of gene expression in Drosophila. PLoS Genet. 8(10):e1003013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan AP, Didion JP, Doran AG, Holt JM, McMillan L, Keane TM, Pardo-Manuel de Villena F.. 2016. Genome report: whole genome sequence of two wild-derived Mus musculus domesticus inbred strains, LEWES/EiJ and ZALENDE/EiJ, with different diploid numbers. G3. 6(12):4211–4216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namekawa SH, Park PJ, Zhang L-F, Shima JE, McCarrey JR, Griswold MD, Lee JT.. 2006. Postmeiotic sex chromatin in the male germline of mice. Curr Biol. 16(7):660–667. [DOI] [PubMed] [Google Scholar]
- Nguyen L-T, Schmidt HA, von Haeseler A, Minh BQ.. 2015. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 32(1):268–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pahl T, McLennan HJ, Wang Y, Achmadi AS, Rowe KC, Aplin K, Breed WG.. 2018. Sperm morphology of the Rattini - are the interspecific differences due to variation in intensity of intermale sperm competition? Reprod Fertil Dev. 30(11):1434–1442. [DOI] [PubMed] [Google Scholar]
- Párraga M, del Mazo J.. 2000. XYbp, a novel RING-finger protein, is a component of the XY body of spermatocytes and centrosomes. Mech Dev. 90(1):95–101. [DOI] [PubMed] [Google Scholar]
- Parsch J, Ellegren H.. 2013. The evolutionary causes and consequences of sex-biased gene expression. Nat Rev Genet. 14(2):83–87. [DOI] [PubMed] [Google Scholar]
- Phifer-Rixey M, Nachman MW.. 2015. Insights into mammalian biology from the wild house mouse Mus musculus. Elife. 4:e05959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piasecka B, Lichocki P, Moretti S, Bergmann S, Robinson-Rechavi M.. 2013. The hourglass and the early conservation models—co-existing patterns of developmental constraints in vertebrates. PLoS Genet. 9(4):e1003476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitnick S, Hosken DJ, Birkhead TR.. 2009. Sperm morphological diversity. In: Birkhead TR, Hosken DJ, Pitnick S, editors. Sperm biology. London: Academic Press. p. 69–149. [Google Scholar]
- Ramm SA, Schärer L.. 2014. The evolutionary ecology of testicular function: size isn't everything. Biol Rev. 89(4):874–888. [DOI] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, Smyth GK.. 2010. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26(1):139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohlfs RV, Nielsen R.. 2015. Phylogenetic ANOVA: the expression variance and evolution model for quantitative trait evolution. Syst Biol. 64(5):695–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabarís G, Laiker I, Preger-Ben Noon E, Frankel N.. 2019. Actors with multiple roles: pleiotropic enhancers and the paradigm of enhancer modularity. Trends Genet. 35(6):423–433. [DOI] [PubMed] [Google Scholar]
- Sánchez-Ramírez S, Weiss JG, Thomas CG, Cutter AD.. 2021. Widespread misregulation of inter-species hybrid transcriptomes due to sex-specific and sex-chromosome regulatory evolution. PLoS Genet. 17(3):e1009409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarver BAJ, Keeble S, Cosart T, Tucker PK, Dean MD, Good JM.. 2017. Phylogenomic insights into mouse evolution using a pseudoreference approach. Genome Biol Evol. 9(3):726–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder CM, Valenzuela JR, Mejia Natividad I, Hocky GM, Malik HS.. 2020. A burst of genetic innovation in Drosophila actin-related proteins for testis-specific function. Mol Biol Evol. 37(3):757–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumacher J, Herlyn H.. 2018. Correlates of evolutionary rates in the murine sperm proteome. BMC Evol Biol. 18(1):35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skinner BM, Johnson EEP, Bacon J, Affara NA, Rathje CC, Yousafzai G, Ellis PJI, Larson EL, Kopania EEK, Good JM.. 2019. A high-throughput method for unbiased quantitation and categorisation of nuclear morphology. Biol Reprod. 100(5):1250–1260. [DOI] [PMC free article] [PubMed]
- Soumillon M, Necsulea A, Weier M, Brawand D, Zhang X, Gu H, Barthès P, Kokkinaki M, Nef S, Gnirke A. et al. 2013. Cellular source and mechanisms of high transcriptome complexity in the mammalian testis. Cell Rep. 3(6):2179–2190. [DOI] [PubMed] [Google Scholar]
- Streett DA, Petersen KR, Gerritsen AT, Hunter SS, Settles ML.. 2015. expHTS: analysis of high throughput sequence data in an experimental framework. In: Proceedings of the 6th ACM Conference on Bioinformatics, Computational Biology and Health Informatics. Atlanta (GA: ): Association for Computing Machinery. p. 523–524. Available from: https://doi.org/10.1145/2808719.2811442. [Google Scholar]
- Swanson WJ, Nielsen R, Yang Q.. 2003. Pervasive adaptive evolution in mammalian fertilization proteins. Mol Biol Evol. 20(1):18–20. [DOI] [PubMed] [Google Scholar]
- Swanson WJ, Vacquier VD.. 2002. The rapid evolution of reproductive proteins. Nat Rev Genet. 3(2):137–144. [DOI] [PubMed] [Google Scholar]
- The UniProt Consortium. 2020. UniProt: the universal protein knowledgebase in 2021. Nucleic Acids Res. 49:D480–D489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson A, May MR, Moore BR, Kopp A.. 2020. A hierarchical Bayesian mixture model for inferring the expression state of genes in transcriptomes. Proc Natl Acad Sci U S A. 117(32):19339–19346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thybert D, Roller M, Navarro FCP, Fiddes I, Streeter I, Feig C, Martin-Galvez D, Kolmogorov M, Janoušek V, Akanni W. et al. 2018. Repeat associated mechanisms of genome evolution and function revealed by the Mus caroli and Mus pahari genomes. Genome Res. 28(4):448–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner LM, Chuong EB, Hoekstra HE.. 2008. Comparative analysis of testis protein evolution in rodents. Genetics 179(4):2075–2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicens A, Borziak K, Karr TL, Roldan ERS, Dorus S.. 2017. Comparative sperm proteomics in mouse species with divergent mating systems. Mol Biol Evol. 34(6):1403–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicoso B, Charlesworth B.. 2009. Effective population size and the faster-x effect: an extended model. Evolution 63(9):2413–2426. [DOI] [PubMed] [Google Scholar]
- Voolstra C, Tautz D, Farbrother P, Eichinger L, Harr B.. 2007. Contrasting evolution of expression differences in the testis between species and subspecies of the house mouse. Genome Res. 17(1):42–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White-Cooper H, Doggett K, Ellis RE.. 2009. The evolution of spermatogenesis. In: Birkhead TR, Hosken DJ, Pitnick S, editors. Sperm biology. London: Academic Press. p. 151–183. [Google Scholar]
- Whittle CA, Kulkarni A, Extavour CG.. 2020. Absence of a faster-X effect in beetles (Tribolium, Coleoptera). G3 (Bethesda) 10:1125–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter EE, Goodstadt L, Ponting CP.. 2004. Elevated rates of protein secretion, evolution, and disease among tissue-specific genes. Genome Res. 14(1):54–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wray GA, Hahn MW, Abouheif E, Balhoff JP, Pizer M, Rockman MV, Romano LA.. 2003. The evolution of transcriptional regulation in eukaryotes. Mol Biol Evol. 20(9):1377–1419. [DOI] [PubMed] [Google Scholar]
- Yang Z. 2007. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol. 24(8):1586–1591. [DOI] [PubMed] [Google Scholar]
- Zhao L, Saelao P, Jones CD, Begun DJ.. 2014. Origin and spread of de novo genes in Drosophila melanogaster populations. Science 343(6172):769–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNAseq data generated for this project are available through the National Center for Biotechnology Information under accession PRJNA735780. Individual sample accessions are in supplementary table S11, Supplementary Material online. A table of genes in our analyses and whether they were considered expressed, induced, or active in each cell type is available in supplementary table S12, Supplementary Material online. Scripts used for expression divergence and allele-specific expression analyses are available on GitHub: https://github.com/ekopania/mus_spermatogenesis_analyses (last accessed November 18, 2021) and https://github.com/ekopania/cis-trans-pipeline (last accessed September 15, 2021).