

Summary
Semiconductor nanocrystals have become ubiquitous both in scientific research and in applied technologies related to light. When a nanocrystal absorbs a photon an electron-hole pair is created whose fate dictates whether the nanocrystal will be suitable for a particular application. Ultrafast spectroscopy provides a real-time window to monitor the evolution of the electron-hole pair. In this review, we focus on CdSe nanocrystals, the most-studied nanocrystal system to date, and also highlight ultrasmall nanocrystals, “standard nanocrystals” of different binary composition, alloyed nanocrystals, and core/shell nanocrystals and nanorods. We focus on four time-resolved spectroscopies used to interrogate nanocrystals: pump-probe, fluorescence upconversion, time-correlated single photon counting, and non-linear spectroscopies. The basics of the nanocrystals and the spectroscopies are presented, followed by a detailed synopsis of ultrafast spectroscopy studies performed on the various semiconductor nanocrystal systems.
Subject areas: Inorganic chemistry, Inorganic materials, Materials science, Materials chemistry, Optical materials
Graphical abstract
Inorganic chemistry; Inorganic materials; Materials science; Materials chemistry; Optical Materials
Introduction
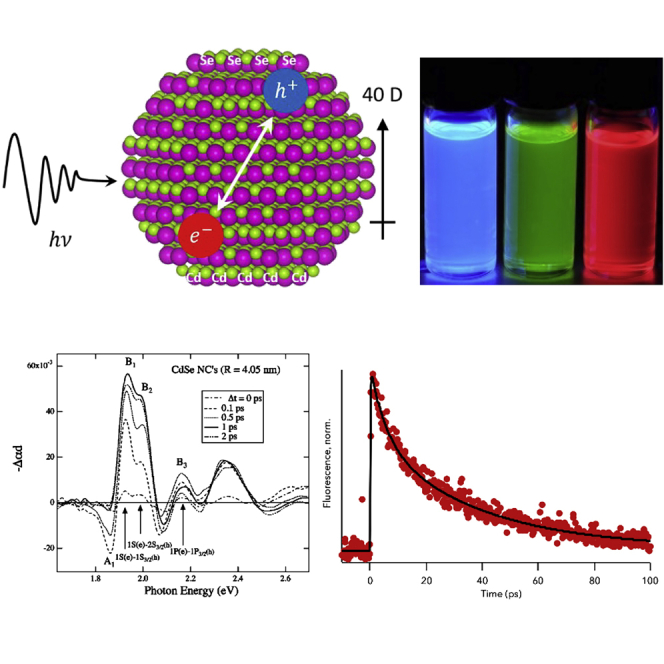
The size-tunable optical properties of semiconductor nanocrystals (NC), or quantum dots, were first explained by Brus (1984). As nanocrystal size falls below the bulk Bohr exciton diameter, the electron and hole become quantum confined and resemble the “particle in a box” studied in undergraduate chemistry and physics (Kippeny et al., 2002). Because their optical properties are dictated by quantum mechanics, semiconductor nanocrystals are referred to as quantum dots. Figure 1 depicts an electron-hole pair creation after the absorption of a photon. The shape and structure of nanocrystals are determined by high resolution transmission electron microscopy (McBride et al., 2004; Shiang et al., 1995). The CdSe nanocrystal depicted in Figure 1 has been determined to be Cd rich (Taylor et al., 2001) and to have an intrinsic dipole moment (Blanton et al., 1997) which can be an internal driving force to separate charge carriers. The chemical synthesis that creates the nanocrystal also leaves organic ligands on the surface, primarily passivating surface cations and leaving the anions bare. Dangling bonds on the surface of nanocrystals form trap states for the photogenerated electron and/or hole, and much of the ultrafast spectroscopy performed on nanocrystals explores these carrier-trapping timescales.
Figure 1.

Model of a CdSe nanocrystal obtained from HRTEM images depicting the inherent dipole of the nanocrystal and the creation and evolution of an electron-hole pair following absorption of a photon.
Adapted from Rosenthal et al. (Rosenthal et al., 2011).
There are numerous demonstrated and proposed uses for semiconductor nanocrystals. As a semiconductor with a quantum-mechanically enhanced ability to absorb light and thus generate a charge pair, nanocrystals are an obvious candidate for solar cells (Kamat, 2008,2013; McDonald et al., 2005; Nozik, 2002; Sargent, 2012; Swafford and Rosenthal, 2003; Underwood et al., 2001a). Similarly, photogenerated charges imply nanocrystals can be used as photocatalysts (Kamat and Dimitrijević, 1990; La Croix et al., 2017; Moroz et al., 2018), photosensitizers (Bakalova et al., 2004; Robel et al., 2006), and photodetectors (Konstantatos et al., 2006; McDonald et al., 2005; Sukhovatkin et al., 2009). Imperative to the successful incorporation of semiconductor nanocrystals in the devices listed above is that the photogenerated charges must separate and leave the nanocrystal. This process can be monitored by ultrafast spectroscopy, which provides necessary feedback to improve nanocrystal design for enhanced device operations.
A second set of applications of semiconductor nanocrystals requires the opposite behavior: The electron-hole pair must not leave the nanocrystal or fall into surface traps but must recombine to emit light from the nanocrystal. Applications with this set of requirements includes the use of nanocrystals in light emitting diodes (Pal et al., 2012; Schreuder et al., 2008, 2010; Shirasaki et al., 2013), lasers (Fan et al., 2017; Klimov, 2000a; Klimov and Bawendi, 2001), and as fluorescent probes in biology (Alivisatos et al., 2005; Kovtun et al., 2011; Rosenthal et al., 2002, 2011). Indeed, improved capabilities in nanocrystal synthesis and control of carrier dynamics have led to two now-widespread commercial applications. Thermo-FischerTM (and other vendors) sell core/shell quantum dots for biological applications and SamsungTM incorporates robust core/shell nanocrystals in QLED (quantum dot light emitting diode) displays. Another company, Nanoco Technologies Ltd., is also developing cadmium-free quantum dots for infrared sensing, display, solar, lighting and bio-imaging applications. Wrapping a semiconductor nanocrystal in a shell of a wider band gap material is the chemist's trick to keep carriers out of surface traps and force carrier recombination. Here, again, ultrafast spectroscopy can interrogate the successfulness of the synthetic objective.
The example applications noted above highlight the practical importance of excitonic properties, specifically, how nanocrystal properties are influenced by controlling the degree of surface trapping and charge transfer to surface moieties. “Wave function engineering” is a term that has been used to emphasize the importance of the structure-property relationship of semiconductor nanomaterials and underscore how synthetic outcomes affect the excitonic properties which ultimately determine the utility of nanocrystals for specific applications (Zhu et al., 2012). This fundamental understanding of the excited state and how it is dependent on nanocrystal size, shape, composition, surface chemistry, and so forth, now allows chemists to design nanocrystals with specific properties tailored to their intended application.
This review is structured in three parts. First, we describe the different types of nanocrystals that have been studied with ultrafast spectroscopy with a focus on CdSe nanocrystals, which are by far the most studied system. Here, we provide information about their band structure and surface states, brief descriptions of their synthesis, and electron microscopy structural data. In the second section of this article, we describe the time resolved spectroscopies used to interrogate carrier dynamics in nanocrystals, discussing strengths of each. Then, at the heart of this review we compile and discuss an extensive body of literature in which the carrier dynamics of nanocrystals have been interrogated by ultrafast spectroscopy. Depth is provided on CdSe-based systems and supplemented with comparison to select other systems that have been studied.
The scope of this review excludes studies on multi exciton generation in nanocrystals (Labrador and Dukovic, 2020; Pandey and Guyot-Sionnest, 2007; Qin et al., 2021; Seiler et al., 2018; Sewall et al., 2008), and electron transfer studies in which the nanocrystal donates a charge (electron or hole) to a surface moiety (Frederick and Weiss, 2010; Peterson et al., 2014; Taheri et al., 2021; Zhu et al., 2010).
Semiconductor nanocrystals
Band structure
The band structure of semiconductor nanocrystals is unique from atoms, molecules, and bulk semiconductors in that the extremely small size of the nanocrystal changes the energies of the valence and conduction bands and also lifts the degeneracy of the band edge states leading to quantization of states near the band edge. This phenomenon is observed as a size-dependence in the absorption and emission of the nanocrystals, as well as discrete peaks or features in the optical absorption spectra, as seen in Figure 2.
Figure 2.

Overview of size dependence and band structure in nanocrystals and core/shells
(A and B) Size-dependence of semiconductor nanocrystals is illustrated in the optical absorption (A, top) and emission (A, bottom) of CdSe nanocrystals as they increase in size from left (2 nm) to right (6 nm). Reproduced with permission from Rosenthal et el (Rosenthal et al., 2007). Electronic transitions (B) that give rise to the features in the optical absorption spectra of CdSe nanocrystals, reproduced with permission from Klimov et al. (Klimov et al., 1999a).
(C) Band structure of CdSe nanocrystals (C) illustrating the quantization of states at the band edge and mid-gap states that can arise from surface states and/or alloying (Freymeyer et al., 2020; Underwood et al., 2001b). Illustration developed from theoretical results of Ekimov, Efros, et al. (Ekimov et al., 1993).
(D) Band alignment (D) in type I, II, and quasi-type II core/shell or other nanoheterostructures (Keene et al., 2014).
A defining feature of semiconductor nanomaterials is their large surface-to-volume ratio that leads to significant overlap of the excited charge carriers with the surface of the nanocrystal. Therefore, not only do the excited charge carriers populate and transition between the electronic states of the semiconductor nanocrystal material, the charge carriers also interact with and populate the electronic states present at the surface of the material. The surface of a nanocrystal includes a wide range of physical and chemical phenomena such as unpassivated surface atoms, dangling bonds, organic ligands or other semiconductor materials, all of which may have electronic energy levels near the band edge or within the band gap of the nanocrystal (Hartley et al., 2021). These surface states are often called ‘trap states’ because they can extract the charge carrier(s) from the nanocrystal and trap them there, preventing radiative recombination and quenching photoluminescence (PL).
In complex heterostructures, the alignment between the conduction and valence bands of one semiconductor material with another provides an additional avenue by which the charge carriers can be manipulated. Illustrated in Figure 2, these are classified as different ‘types’ of heterostructures based on how the band alignment distributes charge carriers between the materials: Type I structures localize both carriers in a single material, type II structures separate each charge to a different material, and quasi-type II structures localize one charge carrier in one material while the other is delocalized throughout both materials.
Synthesis
CdSe remains the quintessential colloidal quantum dot system. By injecting highly reactive organometallic chemical vapor deposition precursors in a high boiling point solvent, production of relatively monodisperse colloidal ‘quantum crystallites’ composed of cadmium and selenium is achieved (Bawendi et al., 1992; Murray et al., 1993). While the selenium precursor has remained relatively fixed as either the powder form of selenium dissolved in tri-n-butyl phosphine or tri-n-octyl phosphine, the source of cadmium has moved on from the highly reactive and toxic dimethylcadmium. Now the most common literature preparation utilizes cadmium oxide complexed with carboxylic or phosphonic acids (Peng and Peng, 2001). The addition of long chain amines imbued improved shape control and high fluorescent quantum yields (Talapin et al., 2001). However, this fluorescence improvement by organic passivants is easily degraded by the displacement of ligands during purification and processing. From the highly spherical CdSe, synthesis strategies for shape control have evolved to produce rods, tetrapods, ultrasmall, bulklike, magic size clusters, and most recently 2D nanosheets (Bowers et al., 2005; Ithurria and Dubertret, 2008; Peng and Peng, 2001).
The chemistry for the synthesis of other semiconductor systems has been developed following CdSe as a model. PbS and PbSe quickly became relatively simple to synthesize with large batches being assembled into structures for potential photovoltaic applications (Okuno et al., 2000). Conversely, other semiconductor systems with band gaps of interest proved far more difficult. Although early works demonstrated that the synthesis of indium phosphide and gallium phosphide was possible, only recently has the development of these materials reached a point where their properties are comparable to their cadmium selenide predecessors (Li et al., 2019; Micic et al., 1994, 1995; Won et al., 2019). The demand for heavy metal free technologies pushed research on indium phosphide chemistry so that most commercial displays are indium-based and cadmium-free.
Beyond “standard” binary nanocrystal systems, alloys of materials afford another mechanism to tune their optical properties. For example, the stoichiometry of homogenously alloyed CdSSe nanocrystals determines their absorption and emission wavelengths independent of their diameter (Swafford et al., 2006). This is a critical step toward improving the brightness of blue and green emitting quantum dots since the molar absorptivity, or the ability of the quantum dot to absorb light, is size dependent. Graded alloys where the composition varies radially from the core, enables a gradual change in the chemical composition to reduce lattice strain and control surface termination (Keene et al., 2014).
However, core/shell structures are required to achieve the highest quantum yields and long term photostability. The shell material typically consists of a wider band gap semiconductor (type I) capable of epitaxial growth necessary to eliminate surface trap sites. Early in the nanocrystal literature, CdSe was typically passivated using ZnS where the zinc source was once again a highly reactive organometallic precursor while the sulfur source was often an equally reactive material (Hines and Guyot-Sionnest, 1996; Peng et al., 1997). It became clear that the ∼12% lattice mismatch between CdSe and ZnS limits the ultimate shell thickness attainable. Better matched CdS shells were able to be grown far thicker and were the first to suppress optical intermittency and to achieve stable, near-unity quantum yields (Chen et al., 2013; Hanifi et al., 2019). Unfortunately, the use of CdS as the shell material leads to a loss of confinement as a result of the band offsets (or a lack thereof) between core and shell. Various alloys in core and shell have regained optical tunability at the expense of greatly increased material complexity (Shen et al., 2011).
Structural characterization
Transmission electron microscopy, or TEM, has played a critical role in most ultrafast spectroscopy studies as any measured behavior requires a physical model to support the conclusions. At its simplest, high-resolution phase contrast TEM (HRTEM) can resolve the size, shape, and crystal structure of individual nanocrystals. The atoms can appear as white or dark spots with their arrangement related to the real atomic arrangement. Figure 3 shows an early example of HRTEM of CdSe nanocrystals exhibiting different orientations (Shiang et al., 1995). Phase contrast imaging is adequate for characterizing simple systems of one type of semiconductor; however, diffraction and thickness effects complicate the interpretation of image contrast of core/shell or other multicomponent particles. Most early core/shell papers relied on powder X-ray diffraction and a change in particle size for confirmation of shell coverage. Atomic number contrast scanning transmission electron microscopy (Z-STEM) forms images by collecting only the highly scattered electrons using a high-angle annular dark-field detector (HAADF) (Nellist and Pennycook, 1999). This gives directly interpretable images and, when paired with the sub-angstrom resolution afforded by aberration-correction, single atom detection (McBride et al., 2004). Core/shell systems such as CdSe/ZnS that exhibit strong Z-contrast can be imaged atomic column by atomic column. Figure 3 shows examples of CdSe cores and CdSe/CdS core/shell nanocrystals (McBride et al., 2006; Pennycook et al., 2012). In the latter case, the Z-contrast that originates from the anion atomic columns and the core/shell interface is revealed by the disappearance of the selenium atomic columns as the material transitions to the purely CdS shell (McBride et al., 2006). Quests to image the surface termination of the nanocrystal surface, information crucial toward understanding physical structure of surface defects, were limited by electron beam induced motion of the atoms. This surface fluctuation creates a fuzzy haze at the nanocrystal surface and persists even down to the ‘gentle STEM’ regime of accelerating voltages of 60 kV and was consistent across different II–IV systems with different organic passivation (Krivanek et al., 2010; McBride et al., 2013; Pennycook et al., 2012).
Figure 3.

Comparison of transmission electron microscopy techniques used to characterize nanocrystal with descriptions of benefits and challenges (in red).
Top: High resolution TEM showing the lattice structure of CdSe from two different alignments (Shiang et al., 1995). Middle: Aberration-corrected Z-STEM directly showing the atomic arrangement of CdSe (Pennycook et al., 2012). Bottom: STEM-EDS of a giant-shelled CdSe/CdS core/shell nanocrystal (McBride and Rosenthal, 2019).
Ultimately, there remains the finite effect of nanocrystal thickness that limits direct contrast interpretation for highly irregular shapes or very thick structures. Analytical STEM affords the ability to correlate chemical composition with atomic structure. Electron Energy Loss Spectroscopy (EELS) and/or Energy Dispersive Spectroscopy (EDS) coupled with the scanning electron beam creates images with chemical information from a few nanometers down to single atomic columns. While EELS can provide information beyond simple chemical identification such as bonding, coordination, and density of states, most compositions of semiconductor quantum dots consist of elements that give rise to weak EELS edges and line shapes (Williams and Carter, 1996). Recently, the advancement of solid-state X-ray detector design has greatly improved the efficacy of STEM-EDS imaging of colloidal quantum dots (Schlossmacher et al., 2010). Prior to ∼2010, low X-ray count rates required high beam currents and long acquisition times that would obliterate the nanocrystal before any reliable chemical information could be obtained. Modern STEM-EDS systems with multiple large area detectors can readily confirm chemical compositions of nanoparticles in one 15-sec scan where subsequent collections can be used to acquire enough counts for single nanocrystal chemical quantification. This technique is proving critical toward confirmation of the complex nanostructures currently being studied. Figure 3 (bottom) shows an example of a STEM-EDS map of a core/shell nanocrystal system that highlights the technique's ability to identify the location of the core of the particle (McBride and Rosenthal, 2019; Reid et al., 2018).
Ultrafast techniques
Various ultrafast techniques have been developed that analyze features such as absorption, emission, and anisotropy. These techniques rely on femtosecond (fs) laser pulses typically generated using a regeneratively amplified Ti:sapphire laser, and time resolution is dependent on the laser pulse width (∼10 fs in the best case). Depending on the experimental conditions, either an optical parametric amplifier (OPA), second harmonic generator (SHG), dye, or sapphire plate (for white light (WL) continuum) are used to generate the desired wavelengths. Most techniques involve multiple pulses serving as excitation and probe pulses. Figure 4 summarizes these ultrafast techniques giving both what is measured and what electronic process those measurements correlate with.
Figure 4.

Overview of ultrafast spectroscopy techniques with processes they monitor and their time scales
Anisotropy measurements
Femtosecond absorption anisotropy probes changes in the symmetry of the electronic structure of NCs after charge carrier evolution. Anisotropy measurements allow for determination of the symmetry of the lowest electronic state. It can also provide information about long time anisotropy in the NC. Wavelength-tuned excitation and probe beams monitor the changes in anisotropy over the course of several picoseconds (Rosenthal et al., 1996). Spectrally resolved pump-probe (SRPP) polarization anisotropy measurements build on these measurements by recording anisotropy changes for a range of detection energies near the band gap using frequency resolved optical gating (FROG) (Park et al., 2016).
Transient absorption
Transient absorption (TA) measurements monitor time-correlated absorption changes resulting from state filling by electrons and the bleaching of interband optical transitions that are proportional to the sum of the electron and hole occupation numbers (Klimov, 2000b). Samples are pumped to photoexcite a fraction of the sample into electronic excited states and absorption changes are probed using various sources depending on the experiment (IR, NIR, visible, or broadband). For example, to separate hole and electron dynamics, an IR probe is used which is tuned in resonance with either electron or hole intraband transitions (Klimov et al., 1999b). Changes in absorption can be measured for either changes in photon energy (at a constant time delay) or with a variable time delay (and constant energy). TA measurements have been performed for femto- to nano-second time scales and are thus able to cover a wide range of charge carrier properties. Example TA data are shown in Figures 5A and 5B.
Figure 5.

Example spectra for several ultrafast techniques
(A) TA spectra for 4.05 nm CdSe NCs at several delay times with the electronic transitions labeled.
(B) TA dynamics at the position of the 1S (thick solid line) and 1P (thick dashed line) transitions for the same 4.05 nm CdSe NCs.
(C) Ultrafast fluorescence upconversion decay curves for InP/ZnSe NCs.
(D) Rise time from the ultrafast fluorescence upconversion experiment.
(E) Total 2D ES for various times.
(F) Measured 2D ES spectra at 300 fs with labels indicating major contributions for the two diagonals and cross peaks in which is the 1Se-1S3/2 exciton state and is the 1Se-2S3/2 exciton state. (A) and (B) Reprinted figure with permission from Klimov et al. (Klimov et al., 1999a), and (E) and (F) are adapted with permission from Turner et al. (Turner et al., 2012).
More involved techniques allow for greater specificity to be obtained including a three-pulse pump-pump-probe (vis + IR + WL probe) where the first pump places an electron in 1Se and the second pumps the electron from 1Se to 1Pe, allowing both the 1Se recovery and 1Pe depopulation to be probed (Pandey and Guyot-Sionnest, 2008). Additionally, circularly polarized pump-probe uses circularly polarized pump and probe beams in a transient bleaching experiment. The RRRR-RRLL signal will give the fine structure of the relaxation rate (Johnson et al., 2008). Femtosecond degenerate pump-probe is a modification of the TA experiment where both the pump and probe come from the same source (or are otherwise equal) (Cho et al., 2010).
Furthermore, an exciton selective pump-probe approach can be used to look at state-to-state exciton transition rates. Here, the pump wavelengths are tuned to specific initial excitonic wavelengths and a white light continuum is used to probe the absorption. ΔΔOD transients, the difference between standard pump-probe ΔOD transients, monitor the survival probability of hot holes or electrons. This technique maintains femtosecond time resolution while also probing the initial and final excitonic states providing direct information about the pathways by which an exciton relaxes (Cooney et al., 2007).
Fluorescence upconversion
While transient absorption monitors changes in absorption over time, fluorescence upconversion spectroscopy allows for changes in PL to be tracked on ultrafast timescales. This fluorescence signal correlates with radiative recombination of the exciton and NC charge carrier trapping appears as a decrease in fluorescence intensity (Keene et al., 2014). This optical gating technique uses a wavelength-tuned pump beam to excite the NCs. Subsequently, their fluorescence is probed by overlapping a gate beam spatially and temporally in a non-linear crystal to create the sum frequency signal (ωsum = ωfluorescence + ωgate). The excitation beam's arrival is delayed with a stepper stage, providing time resolution. In contrast to pump-probe laser spectroscopies, fluorescence upconversion offers a preferential look at the excited state without interference from other processes such as excited-state absorption or ground-state recovery (Underwood et al., 2001b). Example fluorescence upconversion data are shown in Figures 5C and 5D.
Time correlated single photon counting (TCSPC)
Many of the techniques that have been utilized to study the ultrafast processes in nanocrystals are of the pump-probe spectroscopies which, while they provide excellent temporal resolution, can be somewhat limited in the window of time they can extend out to probe. Time correlated single photon counting (TCSPC) can be used to directly measure long-lived fluorescence decays and provides information about the nature of the optical transitions (Gul et al., 2011). Here, samples are laser excited, and their fluorescence is directly recorded using a TCSPC unit which allows for extended length measurements (up to 1000s of nanoseconds). Correlating time resolved PL measurements with an additional variable such as varied temperature allow for more information about specific trap populations to be determined (Jones et al., 2009).
Two-Dimensional electronic spectroscopy
Two-dimensional electronic spectroscopy (2D ES) has been used to measure the decoherence time of a superposition of the two lowest energy excitons and provides information about exciton and biexciton fine structures. 2D ES measurements remove the effect of inhomogeneous broadening due to nanocrystal size variation that can completely obscure or dominate in other types of spectroscopic measurements. 2D ES is carried out similarly to one-dimensional four-wave-mixing measurements, but records both the spectrum of states photoexcited and the spectrum of states after a delay time showing cross peaks and revealing couplings between states (Turner et al., 2012). Example 2D ES data are shown in Figures 5E and 5F.
Femtosecond (3-pulse) photon echo
Femtosecond (3-pulse) photon echo can be used to measure the electronic dephasing, population dynamics, and the dynamics of the vibrational mode (Mittleman et al., 1994). Three-pulse photon echo spectroscopy is preferred over hole burning and two-pulse echo experiments for observing electronic dephasing because it is able to suppress the quantum beats or the effects of the coupling to the phonon mode which is also coupled to the exciton in this time regime, obfuscating the observed signal. It also can distinguish between electronic dephasing due to dynamic processes and linewidth broadening due to phonon line structure (Schoenlein et al., 1993). Three beams of equal intensity are used with independent delay controls to allow the various properties to be measured.
Third-order transient grating spectroscopy and cross-polarized transient grating (CPTG) spectroscopy
Third-order transient grating (3-TG) spectroscopy is again similar to the traditional pump-probe method but allows for further polarization experiments to be performed because each of the three laser beams and an analyzer in the signal path can be independently arranged. This technique monitors the evolution of ground and excited-state density as a function of time delay. In this case, the pump pulse is split into a pair of beams that have different wave vectors that arrive simultaneously to photoexcite. Then a probe beam detects signals in the phase-matched directions as a function of the pump−probe time delay (Huxter et al., 2005). Because of this phase matching, CPTG offers a higher signal-to-noise ratio than transient bleaching (Johnson et al., 2008).
Steady-state photoluminescence
While not an ultrafast technique specifically, steady-state photoluminescence at various temperatures has also been employed as a way of understanding the relevant energy levels in NCs. Changes in spectra at various temperatures both in intensity and wavelength can be used to help determine the trapping mechanisms inside NCs (Mooney et al., 2013).
Carrier dynamics in semiconductor nanocrystals
The vast majority of ultrafast studies on semiconductor nanocrystals are aimed at gaining a fundamental understanding of the behavior of the electron-hole pair as a function of nanocrystal size, shape, composition, and surface chemistry. Fundamentally, there is the question of how the excited state behaves within the nanocrystal, how the charges populate and transition between the electronic states and how long it takes for these transitions to occur as the excited state relaxes back to the ground state. Within the first ∼100 fs after excitation the electron-hole pair experiences rapid loss of initial anisotropy and electronic dephasing or decoupling. These are followed by exciton spin relaxation, carrier cooling or relaxation to the band edge, charge transfer to or from adsorbed molecules at the nanocrystal surface, and charge separation between materials in type II structures on the sub-picosecond timescale. Once the exciton has formed within the first picosecond, interaction of the charge carriers with the surface of the nanocrystal plays an important role as hole trapping and electron trapping at surface states occur on the few-ps to tens-of-ps time scales, respectively. Also in this regime Auger relaxation and charge trapping at internal interfacial lattice defects in heterostructures compete with carrier trapping at the surface. Finally, the charge carriers relax back to the ground state with an exciton lifetime on the tens of nanoseconds timescale or relax from trap states on the order of nanoseconds to microseconds. These processes and their time scales are summarized in Figure 6.
Figure 6.

Overview of processes and timescale for the evolution of the excited state
Anisotropy
The pioneering work of Rosenthal, Alivisatos, Shank, et al. investigated the anisotropy of the exciton in wurtzite CdSe nanocrystals (Rosenthal et al., 1996). Like many of the processes and properties of nanostructures, the symmetry of the lowest electronic state in CdSe nanocrystals is dependent on size. The exciton of larger nanocrystals has E symmetry, arising from a symmetric degenerate planar transition in which the electron is promoted from pxpy linear combination of atomic orbitals. At smaller diameters the exciton has A symmetry, arising from an asymmetric nondegenerate linear dipole transition in which the electron is promoted from a pz orbital. The difference in the exciton state with size is attributed to the structure of the nanocrystals: the nanocrystals are actually non-spherical and are elongated along the C3V axis. The tetrahedral crystal field created by the Cd atoms in the wurtzite crystal lattice lifts the degeneracy of the px, py, and pz orbitals. Femtosecond absorption anisotropy measurements revealed a rapid decay of the initial anisotropy of the CdSe nanocrystals within 20 fs. The extremely fast evolution of the transition dipole was attributed to the rapid evolution of the excited state electron wave function to the surface of the nanocrystal.
The anisotropy of PbS nanocrystals has more recently been investigated by Jonas et al. (Park et al., 2016). The anisotropy at the bandgap for chlorine-passivated PbS nanocrystals was found to be anomalously low, less than 0.1. The low initial anisotropy is potentially due to strong spin-orbit coupling or formation of hot one-electron states by excited state absorption. Additionally, rapid loss of any anisotropy was observed to occur in less than 10 fs, suggesting that coupling to singly excited states beyond the pulse bandwidth occurs faster than the duration of the pulse.
Exciton-phonon dephasing (i.e., quantum beats in TA)
Quantum beats become evident in transient absorption spectra over the first several picoseconds due to the coupling of the exciton to the lattice vibrations (phonons) of the nanocrystal directly after excitation, as illustrated in Figure 7. More specifically, longitudinal-optical (LO) phonons are coherently excited alongside the exciton by the excitation pulse. In other words, in addition to electronic excitation of the nanocrystal, coherent nuclear motion at the phonon frequency is simultaneously initiated by the first laser pulse. Taking a Fourier transform of the time-dependent kinetic traces exhibiting quantum beats allows for elucidation of the frequency of the phonons and yields consistent results as direct measurements of the phonon energies. In particular for CdSe, the LO phonon mode has a wavenumber of ∼205 cm−1 which corresponds to coherent nuclear motion of the crystal lattice with a vibrational period of ∼163 fs (Figure 7). These signals can potentially obfuscate fine details in the decay kinetic spectra obtained via time-resolved spectroscopies with features on this timescale, including investigations of electronic dephasing, exciton spin relaxation, and carrier cooling.
Figure 7.

Three-pulse photon echo on 2.2 nm CdSe nanocrystals as a function of t13 with t12 = 33 fs demonstrating the quantum beats observed in transient absorption spectra
The dashed line is a theoretical fit with one phonon mode (ω = 205 cm−1). Adapted with permission from Schoenlein et al. (Schoenlein et al., 1993).
Shoenlein, Mittleman, Alivisatos, Shank, et al. demonstrated that three-pulse photon-echo experiments can suppress the quantum beats in electronic dephasing studies by tuning the delay of the third (probe) pulse to coincide with exactly one vibrational period (Mittleman et al., 1994; Schoenlein et al., 1993). Under this experimental design, the third (probe) pulse arrives exactly when the nuclei are in their initial equilibrium positions, thereby eliminating any effects from lattice vibrations in the time-resolved spectra.
Like many other processes in semiconductor nanocrystals, the damping time of the phonon oscillations is size-dependent (Mittleman et al., 1994). In three-pulse photon echo experiments, an LO-phonon coupling parameter (Δ) was determined by comparing the echo decay rate at ωt13 = 2π and ωt13 = 3π, illustrated in Figure 7. The coupling of the LO-phonon to the exciton was found to peak at an intermediate size: It is smallest at small (∼2 nm) nanocrystals, increases and peaks for diameter of ∼3 nm, then as diameter continues to increase the coupling decreases to a value resembling that of the bulk.
Electronic dephasing
The electronic dephasing, or electron-electron scattering, in semiconductor nanocrystals is a process in which momentum is changed while energy is conserved. Throughout the literature this process is also referred to to as exciton dephasing, polarization modulation, or polarization dephasing.
Electronic dephasing in CdSe nanocrystals is dependent on both temperature and size. First investigation of electronic dephasing in CdSe nanocrystals was performed by Schoenlein, Shank, et al. and probed 2.2 nm CdSe nanocrystals at varying temperature (Schoenlein et al., 1993). The polarization dephasing was found to be roughly linear with temperature. At 15 K, the timescale for electronic dephasing was found to be 85 fs and it decreased to 25 fs as temperature increased to 240 K. The temperature dependence indicates that electronic dephasing is strongly coupled to low-frequency (∼5–50 cm−1) acoustic phonon modes. The acoustic modes are less populated at lower temperature, leading to a slower rate of polarization dephasing because they are simply not available to assist in the process at low temperatures. At higher temperatures, however, the acoustic phonon modes are thermally populated and thus readily contribute to the electronic dephasing process, which occurs more rapidly. Increased electronic dephasing also contributes to the increased line broadening and linewidth of nanocrystal samples at increased temperature.
Mittleman, Shank, et al. later followed up by additionally probing the size dependence the electronic dephasing in CdSe nanocrystals ranging from 2 to 4 nm in diameter and found the dephasing time to increase with nanocrystal size (Mittleman et al., 1994). Electronic dephasing occurs in 85 fs for smaller 2 nm nanocrystals and increases to 270 fs for larger 4 nm nanocrystals. This is notably much faster than the bulk timescale for electronic dephasing of 50 ps for CdSe. The electronic dephasing and homogeneous linewidth are ultimately attributed to three dynamical processes, with each one exhibiting some degree of size dependence. First is the coupling to low-frequency acoustic phonon modes and is the only process dependent on temperature. The other two processes are dependent on size. The authors describe a lifetime effect, or the decay of the excited state into an electronic configuration that is less strongly coupled to the ground state. This process varies from 400 fs for small 2 nm CdSe up to 8 ps in larger 4 nm CdSe and is suggestive of trapping to a surface state. The last contribution to electronic dephasing is attributed to elastic scattering from defect sites or impurities in the crystal lattice.
Exciton spin relaxation/spin flip
The kinetics of the exciton spin relaxation (or flipping) from one bright exciton state (F = ±1) to the other in CdSe nanocrystals was observed by Scholes et al. using third-order transient grating (3-TG) spectroscopy (Huxter et al., 2005). In addition to first demonstrating that such an experiment is possible, the exciton spin flip was found to be size-dependent, 182 fs for smaller nanocrystals (r = 1.7 nm) and longer (1 ps) for larger nanocrystals (r = 2.5 nm). Its origin is attributed to a long-range contribution to the electron-hole exchange interaction.
Scholes et al. later investigated the transitions between electronic states of F = ±1 in CdSe nanocrystals using two-dimensional electronic spectroscopy after femtosecond excitation of the two lowest exciton states (Turner et al., 2012). They observed cross peaks in the 2D spectra correspond to ‘coupled’ exciton states, which are electronic transitions that involve common electronic orbitals. The two lowest-energy excited states in CdSe nanocrystals both involve an electron in the 1Se orbital: 1Se-1S3/2 for the lowest-energy excited state which has both the electron and hole in 1S orbitals, and 1Se-2S3/2 for the second-lowest-energy excited state which has the electron in a 1S orbital and the hole in a 2S orbital. Thus, both states have an electron in a common 1Se orbital. The dominant coherence of these states is a superposition of F = +1 and F = −1 excitons which have a total angular momentum of F = 0. There are ten possible F = 0 configurations that involve twelve electrons in this superposition state. It was found that this twelve-particle correlation of the electronically coupled superposition of the two lowest energy excitons has a decoherence time of about 50 fs.
Importantly, Collini et al. recently investigated effects of different capping ligands and solvent systems and demonstrated that their 2D ES measurements to investigate the coherent properties in CdSe quantum dots are fairly insensitive to the capping ligand or solvent and therefore the signals are primarily dependent on the CdSe core (Collini et al., 2019). They identified interband coherences among the 2S3/2– 1S3/2 and the 1S3/2– 2S3/2 bands and demonstrated that, not only can 2D ES be utilized to identify the fine structure energy levels in the nanocrystals, but it can also monitor the evolution of their superpositions, even in spite of the inherent inhomogeneity of the sample.
The spin flip relaxation times for lead chalcogenide (PbX, X = S, Se, Te) nanocrystals have also been investigated by Nozik, Scholes, et al. with CPTG spectroscopy and transient bleaching in circularly polarized pump-probe spectroscopy (Johnson et al., 2008). PbS and PbSe exhibit similar spin flip dynamics in that they have both a ‘fast’ (210–290 fs) component and a ‘slow’ (790–860 fs) component with similar time constants between the two materials. The fast decay is assigned to transitions between states split by intervalley scattering (IVS) and the slower decay pathway is attributed to transitions between states split by electron-hole exchange interactions (EI). PbS has a fast decay of 290 fs followed by a slower 790 fs decay whereas PbSe has a fast decay of 210 fs followed by a slower decay of 860 fs. For both PbS and PbSe, the faster decay is dominant in the kinetic traces. The PbTe nanocrystals exhibit much faster rates than PbS and PbSe and only display a single decay of 145 fs. The faster decay in PbTe is attributed to the larger effective mass anisotropy (EMA) leading to increased energy splitting between fine structure states and/or larger spin-orbit coupling (SOC) because of the heavier Te anion, leading to faster interconversion of states with opposite angular momentum. Each of the fine structure relaxation pathways occur in parallel and have their own unique size dependences, attributed to the electronic matrix element for each transition as opposed to electron-phonon coupling.
Temperature dependent experiments on PbX nanocrystals also revealed that the fine structure relaxation processes are sensitive to the presence of lattice phonons (Johnson et al., 2008). At lower temperature, both the amplitude and the rate of the spin flip decreases. Similar to conclusions from electronic dephasing studies in CdSe nanocrystals because one or more optical phonons may be needed to activate the transitions, these phonons are not available at lower temperatures and the transition rates are limited.
In comparison to CdSe, the PbX nanocrystals exhibit slower hole flip and faster exciton spin-flip (or electron spin-flip) because of the larger electron effective mass in PbX than in CdSe.
Electron cooling (1Pe→ 1Se)
Carrier cooling, also known as intraband relaxation or carrier thermalization, in semiconductor nanocrystals is when charge carriers that are excited into a higher-energy state (also known as hot carriers) relax back to the band edge state, or the lowest-energy excited state. The vast majority of intraband relaxation studies for hot electrons in the conduction band of CdSe nanocrystals have investigated the transition from the 1Pe state down to the 1SE band edge state (Burda et al., 2001, 2002; Cooney et al., 2007; Guyot-Sionnest et al., 2005; Guyot-Sionnest and Hines, 1998; Klimov, 2000b; Klimov et al., 1996, 1999a, 1999b, 2000; Kambhampati, 2011a, Kambhampati, 2011b, Klimov and McBranch, 1998; Knowles et al., 2011; Peterson et al., 2014; Saari et al., 2013; Wheeler and Zhang, 2013; Zhu et al., 2010). Electron cooling has been experimentally monitored in multiple ways. The 1Pe-1Se transition has been directly probed with NIR transient absorption spectroscopy in addition to indirectly investigated as a rise time for the population of the 1Se state after excitation of the electron into the 1Pe state, upon which electrons cool from the 1Pe level and relax into the 1Se state (Zhu et al., 2010). Additionally, fluorescence upconversion spectroscopy has indirectly observed the transition as a rise time after excitation into the 1Pe state because the electron must relax to the band edge before it can radiatively recombine.
The most notable breakthrough in the earliest ultrafast spectroscopy studies on CdSe nanocrystals was the lack of observation of a phonon bottleneck in thermalization of the charge carriers (Guyot-Sionnest et al., 2005; Klimov and McBranch, 1998; Saari et al., 2013; Wehrenberg et al., 2002). High-energy electrons in energy states above the band edge must transfer their excess energy in order to relax back to the band edge. It was initially thought that hot electrons transfer their energy to lattice vibrations and this process should be limited by the low availability of phonons in nanocrystalline systems, and would lead to what was termed the phonon bottleneck (Guyot-Sionnest et al., 2005). Specifically in CdSe, the energy separation between the 1Pe-1Se states corresponds to about eight LO phonon energies; therefore, the probability of direct multi-phonon emission is negligibly small (Klimov, 2000b). The low probability of the proposed cooling process was expected to decrease the rate and increase the timescale for this transition. However, instead of a phonon bottleneck, in all cases fast thermalization of electrons in the range of 120–540 fs (dependent on size) was typically observed (Burda et al., 2001, 2002; Cooney et al., 2007; Guyot-Sionnest et al., 2005; Guyot-Sionnest and Hines, 1998, Kambhampati, 2011a, Kambhampati, 2011b; Klimov, 2000b; Klimov et al., 1996, 1999a, 2000; Klimov and McBranch, 1998; Knowles et al., 2011; Peterson et al., 2014; Saari et al., 2013; Wehrenberg et al., 2002; Wheeler and Zhang, 2013; Zhu et al., 2010).
Investigations into the mechanisms of intraband relaxation for hot electrons first probed the effects of surface chemistry on the thermalization of the charges. Early studies suggested sensitivity to surface chemistry, even finding a trend that softer ligands lead to a slower relaxation which provided evidence of electronic-to-vibrational energy transfer influencing electron cooling (Guyot-Sionnest et al., 2005). Additional studies showed that the surface sensitivity for electron cooling was significantly influenced by how the ligands interacted with the hole, such as hole transfer to the ligand from the nanocrystal core. Surface ligands that decrease the electron-hole coupling, such as hole transfer from CdSe to pyridine, were found to slow the electron cooling rate somewhat to the few-ps timescale (Klimov, 2000b). Studies that capped CdSe nanocrystals in ZnS shell found no change in the intraband relaxation of the electron (Cooney et al., 2007; Klimov, 2000b; Klimov et al., 1999a, 1999b, 2000).
As the body of literature has evolved, it is now widely established that the primary or dominant mechanism by which hot electrons cool in semiconductor nanocrystals is an Auger relaxation process in which the excess electron energy is transferred to the hole (Cooney et al., 2007; Kambhampati, 2011a). This ‘up-pumps’ the hole to ‘higher’ energy levels (such as the 2P3/2 state) in the valence band and has implications in carrier cooling studies for the hole.
One notable investigation by Guyot-Sionnest and Pandey decoupled the electron from the hole by separating the charge carriers between materials in a CdSe/ZnSe type-II core/shell structure (Pandey and Guyot-Sionnest, 2008). This system (actually CdSe/ZnS1ML/ZnSexML/CdSe1ML with alkane thiol ligands) localized the electron in the core, separating it from the hole, from trap states at the nanocrystal surface, and from molecular vibrations of the ligands. By decoupling the electron from the ways by which it dissipates energy, the electron cooling time was slowed to longer than 1 ns. Additional work by Kambhampati et al. showed that the 1Pe hot electron lifetime for the 1Pe-1Se process varied linearly with particle radius with lifetimes of 155 fs for r = 2.01 nm up to 250 fs for r = 3.18 nm (Cooney et al., 2007). The unified picture of hot exciton cooling when the initial excitonic state is 1P presented by Kambhampati involves three sequential steps, the first and third related to hole cooling while the second step is as an Auger-based electron-to-hole energy transfer.
Investigations of electron cooling in other Cd-based nanocrystalline materials found similar results to CdSe. One of the earliest studies of ultrafast carrier dynamics performed by Klimov et al. investigated CdS nanocrystals (Klimov et al., 1996). They found that relaxation of photoluminescence intensity at 3 eV occurred in 400 fs and was complementary to photoluminescence buildup dynamics at 2.6 eV, which took 800 fs; these processes were attributed carrier cooling during intraband energy relaxation. Kambhampati et al. observed electron cooling in CdTe nanocrystals, both freshly prepared and aged, and found the 1Pe-1Se transition to be a fast sub-ps process that also breaks the phonon bottleneck (Saari et al., 2013).
Electron cooling in non-Cd-based nanomaterials has also been explored. Electron thermalization in InP nanocrystals has been studied by Lian et al. (Wu et al., 2013). Electron cooling is observed as the first rise component in the bleach signal for InP nanocrystals and occurs in 330 fs. Additionally, Guyot-Sionnest et al. investigated electron cooling in lead chalcogenide nanocrystals and found that the lifetime of the 1Pe,h states in PbSe is less than 4 ps (instrument resolution at the time) and again no phonon bottleneck was observed (Wehrenberg et al., 2002). Jonas et al. investigated PbS nanocrystals (r = 4 nm) and found the relaxation of hot carriers over the first 2 ps is insensitive to nanocrystal surface or even the presence of biexcitons (Cho et al., 2010). Interestingly, the decay of the quantum confined E1 state is bulk-like in nature and quantum confinement is not expected because hot carrier dynamics at three times the band gap is localized on the 1–2 nm length scale. With diameter of 8 nm, the PbS nanocrystals are much larger than the carrier localization and, aside from frequent collisions with the surface, their dynamics closely resemble the relaxation processes found in the bulk. They observed intervalley scattering populated bulk-like states in 100 fs which relax in 240 fs.
Hole cooling
Intraband transitions of the hole are inextricably coupled to the thermalization of the electron. Early studies used NIR TA spectroscopy to directly probe the hole relaxation in CdSe nanocrystals and found the initial hole relaxation occurs in the range of 500 fs – 2 ps, dependent on size but independent of surface properties (Burda et al., 2001, 2002; Klimov, 2000b; Klimov et al., 1999a,1999b; Klimov and McBranch, 1998; Knowles et al., 2011; Peterson et al., 2014). Many of the earliest studies that investigated hole cooling (in addition to electron cooling) were performed by Klimov, McBranch, Leatherdale, Bawendi, Burda, and El-Sayed (Burda et al., 2001; Klimov, 2000b; Klimov et al., 1999a,1999b; Klimov and McBranch, 1998). The various nanocrystal surfaces investigated that do not significantly affect intraband relaxation of the hole include ligands such as trioctylphosphine (TOP), trioctylphosphine oxide (TOPO), and pyridine as well as overcoating with inorganic shells of ZnS. Weiss et al. more recently established that hole cooling to the band edge is the starting point for all subsequent relaxations of the quantum dot, such as carrier trapping and interband relaxation (Knowles et al., 2011).
Kambhampati et al. probed hot carrier dynamics with state-to-state specificity with ΔΔOD transients of pump-probe transient absorption spectroscopy and developed a three-step overall picture of hot exciton cooling when the initial exciton state is 1P (Cooney et al., 2007). First, hot holes relax from the 1P3/2 state to the 1S3/2 state in 10 fs through surface ligand induced nonadiabatic transitions. Next, the electron cools to the band edge through an Auger process in 150–250 fs which ‘up-pumps’ the hole to the 2P3/2 state. Finally, hot hole cooling to the band edge follows sequential kinetics on the 1 ps timescale. That is, the rate of hole cooling slows as it moves through the states and approaches the band edge because the states nearer the band edge have increased energy spacings. They also decomposed the 2S3/2-1S3/2 transition into two sequential steps through an intermediate 1P3/2 state. They measured the sequential kinetics of the 2S3/2-1P3/2 transition followed by the 1P3/2-1S3/2 transition to have time constants of 240 fs and 10 fs, respectively. With the primary mechanism of hole cooling in their new model being nonadiabatic coupling to the surface ligands, they investigated CdSe capped with two monolayers of ZnS shell. They found that while the ZnS shell does not affect the relaxation of the hot electron, it did increase the time for hole cooling from 248 fs to 355 fs, providing evidence of surface ligand influence on hole cooling.
Beyond Cd-based nanocrystals, in fact beyond just CdSe nanocrystals, studies of hole thermalization are very limited. Lian et al. observed hole cooling in InP nanocrystals, in which decay of the hole in the valence band and/or trap states occurs in 550 fs (Wu et al., 2013).
Surface trapping
Nanocrystal surface chemistry has been demonstrated to influence how the charge carriers behave upon their interaction with the surface. Some of the earliest studies on the ultrafast dynamics of the exciton in semiconductor nanocrystals found evidence of hole trapping in the first few ps after excitation and electron trapping in 20–50 ps in both CdSe and CdS nanocrystals (Burda et al., 1999, 2002; Klimov, 2000b; Klimov et al., 1996, 1999b; Logunov et al., 1998). The surface chemistries first explored included ligands such as TOP, TOPO, and pyridine, and ZnS as an inorganic overcoating. Electron trapping at the surface was found to be enhanced in aged samples with native TOPO ligands (increasing by more than three times) and suppressed by about half for ZnS overcoated samples (Klimov et al., 1999b).
The work of Rosenthal et al. studied the ultrafast dynamics of TOPO-passivated CdSe nanocrystals with fluorescence upconversion spectroscopy. Surface trapping was found to occur in 2–6 ps after excitation due to electrons in the Se dangling bonds annihilating the hole in the valence band (deep trap states as illustrated in Figure 2B) (Garrett et al., 2008a; Underwood et al., 2001b). The surface trapping was found to be size-dependent, occurring faster in smaller nanocrystals because of larger surface-to-volume ratio and enhanced carrier overlap with the surface. Radiative recombination was also observed at lower energies from charge carriers after being trapped in the deep trap states. Emission from these states was found to have a rise time of ∼2 ps, consistent with the timescale of carrier trapping at the surface which populates the deep trap states.
Rosenthal et al. later explored other surface chemistries such as hexadecylamine (HDA) which was found to eliminate the size dependence of the exciton dynamics as well as increase the time for trapping to occur in comparison to TOPO-only CdSe nanocrystals (Garrett et al., 2008a; Kippeny et al., 2008). On the mixed Cd and Se (100) facets of the nanocrystal surface, the amine binds to Cd atoms and the methylene protons on the carbon adjacent to the amine then interact with the Se dangling bonds on the surface of the nanocrystal and partially passivate the trap sites. This passivation may occur because of a hydrogen bond-like interaction that passivates the dangling Se orbitals or the interaction may decrease the energy of the mid-gap trap state so it is closer to the band edge state and is indistinguishable from band edge emission. The role of photo-oxidation was also investigated by Rosenthal et al. and nanocrystals completely free of surface Se oxidation do not fluoresce, indicating that nonradiative relaxation dominates the decay pathways (Kippeny et al., 2008). Upon photo-oxidation, however, the nonradiative relaxation mechanisms are diminished and band edge and deep trap emissions are enhanced.
Other notable work on the carrier trapping in semiconductor nanocrystals includes the work by Weiss et al. who studied the exciton dynamics of CdSe nanocrystals with both transient absorption and time-resolved photoluminescence (Knowles et al., 2011). They found a one-to-one correspondence between dynamics observed by each technique which indicates that each lifetime originates from a state that is potentially emissive but is competing with a nonradiative quenching process. Three different major populations were observed in the ensemble CdSe sample, each with distinct radiative and nonradiative pathways for carrier relaxation. In one population fast hole trapping into shallow defect states intrinsic to the nanocrystal lattice was observed to occur in 2.5–4.5 ps, after which the electron in the conduction band is either trapped (the dominant pathway) or it radiatively recombines with the trapped hole on the order of slower than ∼1 ns. The other two populations both have slower hole trapping on the order of 26–48 ps followed by relaxation of the electron into either a trap state or recombination with the trapped hole on the order of 13 or 45 ns for populations two and three, respectively. Of note, holes residing in trap states (as opposed to the valence band) were observed to recombine radiatively with electrons in the conduction band in multiple populations of nanocrystals. Additional work by Weiss et al. demonstrated that hole trapping is affected by hole trapping ligands such as octanethiol and is not affected by electron trapping ligands like benzoquinone (McArthur et al., 2010). While this may appear self-evident, it was found that both hole trapping and electron trapping ligands influence the electron trapping by decreasing its relative amplitude (or contribution to the decay) and increasing its rate. This finding suggests that electron trapping is a surface-mediated decay process that is nonspecific to chemical functionalization of the nanocrystal surface.
Another notable contribution to surface trapping in nanocrystals was by Jones and Scholes who modeled carrier trapping as an electron transfer reaction described with classical Marcus theory, treating each trap state as a distribution of trap states (Jones et al., 2008, 2009). Applying this model to CdSe/CdS/ZnS core/shell/shell and CdSe/CdZnS core/shell structures, they determined two trap state distributions exist. For each trap state they were able to determine the reorganization energy, peak distribution energy level, and even estimate the number of individual trap states in each distribution of trap states. Using Marcus ET theory, they ultimately determined that one trap state population was due to surface traps and the other was due to interfacial trap states near the CdSe core. The surface trap state distribution has a large reorganization energy due mainly to an outer-sphere/solvent component that is linearly related to the molecular polarizability of the solvent. The surface trap distribution is also centered ∼120 meV above the exciton state, consistent with the wider-band gap outermost ZnS shell leading to trap state energies on the surface having average higher energy than the CdSe exciton state. The other trap state distribution is attributed to interfacial trap states near the CdSe core. It has a small reorganization energy since the trap states are within the nanocrystal lattice and involve minimal nuclear reorganization. The distribution is only ∼5 meV above the exciton state, close in energy since the states are near the CdSe core. They later followed up with a temperature-dependent study to relate trap populations to PL decay lifetimes: at low temperature only the low-energy trap states are populated while at higher energies trapping and detrapping processes occur more readily, increasing average PL lifetimes (Jones et al., 2009).
Another model to describe carrier trapping (detrapping) in CdSe nanocrystals was proposed by Kambhampati et al., in which they invoke a semiclassical Marcus-Jortner based model to describe surface photoluminescence over a range of temperatures (Mooney et al., 2013). This approach couples the excitonic states in the core of the nanocrystals to a single surface state. In this sense, and in contrast to many of the viewpoints and considerations in previous studies, the surface state is considered a fundamental electronic state of the system as opposed to a source of defects that must be eliminated or diminished. Kambhampati et al. later utilized time-resolved emission spectroscopy to provide evidence that there are in fact two surface states, one that is in a slow equilibrium with the core state and one that is considered a ‘deep trap’ that is kinetically isolated from the other two states (Palato et al., 2017). This work suggests that electron transfer between the core and surface can be on the order of tens of nanoseconds, much slower than previous studies have demonstrated.
Recently, Lindenberg et al. utilized femtosecond electron diffraction measurements to monitor lattice distortions upon photoexcitation of CdSe nanocrystals and CdSe/CdS core/shells (Guzelturk et al., 2021). Changes in relative diffraction intensity over time were attributed to localized structural deformations caused by hot charge carriers distorting the nanocrystal lattice (i.e. forming a polaron). For CdSe nanocrystals, near-band-edge excitation (which did not generate hot carriers) led to “slow” progression of localized lattice disordering on the order of 167 ps. The holes generated by this excitation did not have enough energy to directly cause localized surface trapping and, instead, Auger relaxation of the photoexcited carriers formed hot holes at later times (see Hole cooling) which were then able to trap to the surface and gave rise to the (slower) formation of the localized lattice distortions. Excitation at higher energies, however, generated hot carriers capable of directly trapping to the surface of the nanocrystals and the localized distortions were much more pronounced (indicating a higher degree of trapping for the hot carriers) and were observed to form in 6.9 ps. In the same study, CdSe/CdS core/shells were also investigated which have a quasi type II band alignment in which electrons are delocalized throughout core and shell material while holes are localized in the core of the material away from the surface (Keene et al., 2014). For near-band-edge excitation, significant localized lattice distortions were not observed which indicated that the deformations were not primarily caused by electron trapping at the surface. However, when excited at higher energy, hot holes were formed which were able to access the nanocrystal surface and become trapped. Localized lattice deformations attributed to hole trapping at the surface were found to form in ∼3.5 ps after excitation.
While CdSe, and to a lesser extent CdS, have been investigated the most in terms of fundamental studies related to surface trapping of charge carriers, other materials such as CdTe and ZnSe have also been explored. CdTe nanocrystals have much faster surface trapping than CdSe nanocrystals, occurring in about ∼2 ps, as opposed to ∼20–50 ps for CdSe (Saari et al., 2013). The trapping in CdTe is also a more significant relative contribution to the overall relaxation pathways experienced by the excited charge carriers in CdTe than in CdSe. Additionally, aged CdTe samples exhibit greater efficiency of surface trapping than do freshly-prepared CdTe. ZnSe has also been investigated with TCSPC and the lifetimes of both shallow and deep trap state emission were observed (Gul et al., 2011). The shallow trap states near the band edge have a longer lifetime than the near-band edge emission, 12 versus 1.1 ns, respectively. The deep trap states, attributed to zinc vacancies or selenium dangling bonds which have energy levels deep within the bandgap (similar to CdSe) have even longer lifetimes of 60 ns.
Charge transfer
While charge transfer to/from adsorbed ligands on nanocrystal surfaces are not the focus of this review article, it must be acknowledged that charge transfer is a process that competes with surface trapping and plays a pivotal role in the fate of the excited state when adsorbed to the nanocrystal (Dworak et al., 2021; Logunov et al., 1998; Zhu et al., 2012). Early work found that electron transfer from the nanocrystal to an electron acceptor such as methyl viologen occurs in 200–300 fs and therefore competes with surface trapping and electron-hole recombination processes, quenching both band gap and deep trap emission and creating a long-lived charge-separated state (Logunov et al., 1998). Electrons have also been found to transfer to other electron acceptors such as adsorbed quinones in 200–400 fs after thermalization of the excited charge carriers and then back transfer to the valence band from the quinones (Burda et al., 1999). Then, in direct contrast to methyl viologen, the quinone electron acceptor bypasses the nanocrystal core and surface trap states and shuttles the excited electron from the conduction band to the hole in the valence band, increasing the rate of charge carrier recombination by about five times. Many other studies investigating the carrier dynamics of nanocrystals have utilized molecules to extract charges from the nanocrystals, not as an end or application in itself but rather to purposefully decouple the charge carriers in order to isolate certain processes such as electron versus hole cooling in nanocrystals, or to validate and attribute spectroscopic signatures to a certain process (i.e. if the hole is removed and a signal still remains, it must be due to the electron) (Burda et al., 1999; Klimov, 2000b; Klimov et al., 2000; La Croix et al., 2017; Peterson et al., 2014; Taheri et al., 2021; Wu et al., 2013; Zhu et al., 2010). That is, instead of being studied as a new phenomenon with its own applications, charge acceptors have been utilized as a means or a tool to provide fundamental information on the behavior of the charge carriers in semiconductor nanocrystals.
One study of note by Weiss et al. exemplifies charge carriers interfacing with the organic ligands at the surface of nanocrystals through adsorption of phenyldithiocarbamate ligands to CdSe (Frederick and Weiss, 2010). The frontier HOMO orbitals of the ligand are resonant with the valence band of the nanocrystal and thus the hole is delocalized into the ligand shell, yielding an effective relaxation of the quantum confinement beyond the limits of the nanocrystalline material. TCSPC results reported for the phenomenon indicate shorter lifetimes getting even faster and dominating the decay processes for the PTC-ligated CdSe. The longest decay observed, however, increased substantially from ∼80 ns to ∼179 ns for the nanocrystals with the delocalized hole.
Exciton lifetime and deep trap emission
Many of the techniques that have been utilized to study the ultrafast processes in nanocrystals are of the pump-probe type with mechanically generated delays which, while they provide excellent temporal resolution, can be somewhat limited in the window of time they can extend out to probe. Therefore, many studies utilize an extra decay that is long-lived and extends beyond the temporal capabilities of the instrument that is necessary to yield an acceptable fit to the data(Bowers et al., 2009; Burda et al., 2002; Fitzmorris et al., 2012b; Garrett et al., 2008b, 2008a; Keene et al., 2014; Kippeny et al., 2008; Klimov et al., 1999a, 1999b; Klimov, 2000b; Klimov and McBranch, 1998; Labrador and Dukovic, 2020; Logunov et al., 1998; McArthur et al., 2010; Pandey and Guyot-Sionnest, 2007, 2008; Underwood et al., 2001b; Wehrenberg et al., 2002). While these particular studies are unable to accurately assign a value to the lifetime, the work of Rosenthal et al., for instance, was able to find a size-dependent trend in the long-lived component attributed to the band edge lifetime that extends beyond instrument capability (Garrett et al., 2008b). Band edge lifetime increases with size and the interband relaxation begins to dominate the decay processes as size increases because of a decrease in the surface-to-volume ratio which decreases the number of trap sites per nanocrystal volume. The long-lived band edge lifetime is attributed to the dark exciton effect (relaxation from the triplet state to the ground state) which is more pronounced for smaller nanocrystals whereas larger nanocrystals recombine from the singlet to the ground state (Garrett et al., 2008b; Underwood et al., 2001b).
Using techniques such as TCSPC and/or electronically generated white light that extend the temporal window to longer time scales than mechanically generated delays are capable of, the full lifetime of the excited state has been observed to be on the order of a few ns to tens of ns for CdSe nanocrystals (Burda et al., 2001; Fitzmorris et al., 2012a; Frederick and Weiss, 2010; Knowles et al., 2011; Zhu et al., 2010). Similar results have been found for other binary nanocrystalline materials such as ZnSe (Gul et al., 2011).
Long-lived emission from nanocrystalline materials has also been observed to extend beyond even that of the band edge lifetime. This observation is attributed to emission from relaxation of charges that have been localized in trap states. The trapped charges, sometimes referred to as charge-separated states, have a much longer lifetime than the charge carriers at the band edge. Termed deep trap emission, this phenomenon occurs at longer wavelengths, corresponding to lower-energy transitions from trap states located deep within the band gap. The deep trap emission has ∼2 ps rise time in CdSe nanocrystals, complementary to the fast initial decay of hole trapping by electrons in Se dangling bonds at the surface of the nanocrystals (Underwood et al., 2001b). In CdSe nanocrystals, multiple levels of trap states have been probed. The lifetime of deep-trap emission at 1000 nm wavelength in the NIR is 250 ns while deeper trap emission at 4.8 μm can be as long at 1 μs (Burda et al., 2001). The observation of longer lifetimes and longer wavelengths implies a trap hopping mechanism in which carriers relax from the band edge to shallow traps, upon which they continue stepwise relaxation to lower energy (or ‘deeper’) trap states. This phenomenon has also been observed in ZnSe nanocrystals, albeit on a somewhat faster (tens of ns) timescale, with shallow trap state emission lifetime of 12 ns and deep trap state emission lifetime of 60 ns (Gul et al., 2011). Kambhampati et al. have also shown that there are multiple surface states that are vibronically coupled to the LO phonon of the CdSe nanocrystal (Mack et al., 2017). Furthermore, they concluded that the surface state exhibits bright-dark splitting similar to the core excitonic state and there must be at least two bright surface electronic states in order to account for the temperature dependence of the emission line widths for the surface emission bands in CdSe nanocrystals.
Carrier dynamics in nontraditional/nonbinary nanocrystalline structures
Ultrasmall nanocrystals
Emission from ultrasmall CdSe nanocrystals is completely dominated by deep trap emission from forbidden surface states. Banin et al. found a size-dependent trend of deep trap emission from ultrasmall CdSe nanoclusters ranging in size from 0.7 nm to 2 nm in diameter (Soloviev et al., 2001). The largest cluster has the shortest deep trap emission lifetime of 0.78 μs whereas the smallest cluster has the longest lifetime of 10 μs. They also noted that the PL decay is only able to be fit with a stretched exponential which is indicative of trapped emission. In addition, the PL increases with a decrease in temperature, which also supports emission related to forbidden surface states.
Upon discovery of white-light emission from ultrasmall CdSe nanocrystals, Rosenthal et al. also investigated the broad emission of the ultrasmall nanocrystals at various wavelengths across the visible spectrum with femtosecond fluorescence upconversion spectroscopy (Bowers et al., 2009). They found that, much like molecules, the radiative lifetimes increase as the spectrum evolves from blue to red, and trapping slows. Specifically, emission at the highest energy peak of the spectrum (459 nm) has a fast 250 fs decay that is mapped to a 270 fs rise time for a lower-energy trap state (552 nm), indicating rapid depopulation of the excited state by carrier trapping that is observed to directly correspond to population of the lower-energy trap state. Additional emission observed at 488 nm is attributed to recombination at surface Se and is unaffected by Cd ligand exchanges. The remaining peaks across the visible spectrum exhibit two decays on the order of 2–7 ps and 40–80 ps, each increasing in time from blue to red.
Core/shells
Many studies have investigated the effects of encapsulating the nanocrystal in an epitaxial shell of a different inorganic semiconductor material. The band gap alignment of the semiconductor shell with respect to the valence and conduction bands of the core electronically influence the behavior of the charge carriers. Type I and quasi type II core/shells have been investigated to improve surface passivation and decrease or even eliminate surface trapping by confining the charge carriers within the core material to prevent their overlap with the surface of the nanocrystals. Type II structures have been utilized to decouple the charge carriers from one another in studies to investigate various fundamental processes in nanocrystals.
Early studies by Klimov and Bawendi investigated a thin shell of ZnS on CdSe nanocrystals which have a type I band alignment confining charge carriers to the CdSe core. These first investigations of ultrafast dynamics with addition of a type I shell indicated that the ZnS shell on CdSe nanocrystals did not alter the coupling of the excited electron and hole when investigating intraband relaxation processes (Klimov, 2000b; Klimov et al., 1999a, 2000). Specifically, both the electron and hole relaxation dynamics did not change significantly and were therefore determined to be insensitive to surface chemistry. However, it was observed that electron trapping at the surface decreased in magnitude by about half (down to ∼7% of total relaxation processes) which was attributed to electronic passivation of the surface as evidenced by suppression of the fast decay component (Klimov et al., 1999b).
Kambhampati et al. later investigated surface effects on intraband carrier cooling in CdSe with ZnS shell and observed no effect on the electron cooling dynamics which confirmed the earlier reports (Cooney et al., 2007). However, in contrast to the earlier reports, they found passivation with two monolayers of ZnS increased hole relaxation from 248 fs to 355 fs. This provided evidence of surface sensitivity for the hot hole thermalization leading to their model of nonadiabatic coupling to surface ligands as the primary mechanism for hole cooling, as discussed previously (see Hole cooling).
Lian et al. utilized anthroquinine, an electron acceptor, to illustrate the ability of the CdSe/ZnS type I band alignment in this core/shell system to successfully confine the charge carriers to the core and decouple the excitonic states from the surface states (Zhu et al., 2010). The rates of charge transfer to the electron acceptor and back-transfer to the nanocrystal decrease exponentially with ZnS shell thickness, indicating that the shell decreases electronic coupling of the excited charge carriers with the surface ligands and serves as a tunneling barrier to charge transfer. Ultimately, the exciton lifetimes of the charge separation and recombination for the thickest shells of ∼2.4 monolayers of ZnS increases to 45 ps and 800 ns, respectively. This is a remarkable increase from 3.4 ps to 0.75 ns for the same processes, respectively, in anthroquinine-adsorbed CdSe nanocrystals without ZnS shells, demonstrating the ability of core/shell systems to control the behavior of charge carriers in nanoheterostructures.
Rosenthal et al. also explored CdSe/ZnSe core/shell structures and found that hole trapping actually increased with size in this system (Kippeny et al., 2008). This is attributed to the type II or quasi type II band alignment between the CdSe core and the ZnSe shell. Specifically, for smaller nanocrystals the band alignment promotes hole trapping to the surface (quasi type II) and/or confines electrons to the CdSe core and holes in the ZnSe shell (type II), in which carrier separation makes the holes more susceptible to surface trap states.
Another CdSe-core based structure explored by Zhang et al. encapsulated a type II CdSe/ZnSe core/shell with an additional ZnS shell, ultimately creating a type I CdSe/ZnSe/ZnS core/shell/shell (Fitzmorris et al., 2012a). Comparing the CdSe core-only, CdSe/ZnSe core/shell, and the CdSe/ZnSe/ZnS core/shell/shell with TCSPC reveals three different types of decay on different time scales. Surface trapping by dangling bonds is attributed to a 0.7 ns decay which is present in CdSe core-only, decreases in magnitude for the CdSe/ZnSe core/shell, and is eliminated entirely in the CdSe/ZnSe/ZnS core/shell/shell. The core/shell structures also introduce crystal lattice dislocations due to lattice mismatch at the core/shell interface which serve as sites for nonradiative recombination. In all samples a slow ∼30 ns process is observed that is attributed to shallow trap state mediated processes.
Beyond CdSe-core based core/shell structures, InP/CdS has also been studied as a type II system (Wu et al., 2013). Notably, in this core/shell structure two rise times are observed for different transitions. The B1 bleach, corresponding to the transition from the CdS valence band edge to the delocalized conduction band 1S electron level, has a rise time of 173 fs and is attributed to cooling of hot electrons to the delocalized 1Se level in the conduction band. The B2 bleach, corresponding to the transition from the 1S hole level in the InP core to the delocalized conduction band 1S electron level, has a rise time of 452 fs and is attributed to the process of hole localization into the 1S in the InP core because the hole is generated in the shell or potentially delocalized throughout the core/shell.
Additional study of InP core-based nanocrystals includes recent work by Rosenthal et al. who studied thick-shell InP/ZnSe core/shell nanocrystals and compared an abrupt core/shell interface to an alloyed interface (Freymeyer et al., 2020). Using both fluorescence upconversion and TCSPC, both samples exhibit sub-200 fs carrier cooling, ∼6 ps hole trapping, ∼26–37 ps electron trapping, and ∼16–18 ns long-lived radiative recombination. Electron trapping is the dominant trapping mechanism. Interestingly, the alloyed interface sample has greater trapping, in particular hole trapping, which is attributed to the additional energy levels near the valence band edge caused by inclusion of In3+.
Alloys
The ultrafast dynamics of homogeneously alloyed nanocrystals closely resemble that of binary semiconductor nanomaterials. Rosenthal et al. studied homogeneously alloyed CdSxSe1-x nanocrystals via ultrafast fluorescence upconversion spectroscopy and compared their dynamics to CdSe nanocrystals (Garrett et al., 2008b). Carrier trapping at the surface is observed, similar to binary CdSe. However, an increase in the composition of sulfur leads to an increase in the surface trapping. This increase in surface trapping is attributed to an increase in the amount of surface trap states, which are directly correlated with the amount of sulfur in the nanocrystals.
Alloy nanocrystal materials have also been explored that are heterogeneous in composition, closely resembling core/shells in both structure and charge carrier behavior. Rosenthal et al. synthesized graded alloy CdSxSe1-x nanocrystals with a CdSe-rich core that gradually transitions to a CdS-rich shell instead of featuring a hard interface between core and shell materials (Keene et al., 2014). The graded alloy structure is able to achieve a core/shell-like structure within a much smaller overall diameter than typical core/shell nanocrystals, which can require thick shells in order to achieve complete shell coverage and passivation of the core material. The ultrafast dynamics were studied as sulfur composition was increased. Hole trapping at the surface, observed as a fast 2–3 ps decay of the radiative recombination, is suppressed and even completely eliminated as sulfur composition increases and the quasi-type II band alignment localizes the excited holes within the CdSe-rich core. Eliminating carrier overlap with the surface within such a small nanocrystal of only ∼4 nm diameter was a notable achievement. Decreased hole trapping also corresponds to an increase in photoluminescence quantum yield for the nanocrystals. Electron trapping of 20–30 ps is observed in all sulfur compositions because the conduction bands of core and shell are closely aligned such that the electron is delocalized throughout the entire structure, maintaining access to the surface trap states.
Gradient alloying was also investigated by Zhang et al. in a type I Cd1-xZnxSe/ZnSe/ZnS alloy core/shell/shell structure (Fitzmorris et al., 2013). On the nanosecond timescale, the dynamics between the Cd1-xZnxSe core, Cd1-xZnxSe/ZnSe core/shell, and Cd1-xZnxSe/ZnSe/ZnS core/shell/shell structures are very similar, showing double exponential decays of ∼1 and ∼9 ns with most of the photoluminescence emitting via the slow ∼9 ns component. Within the alloy core, the alloying process results in a graded structure with a Cd-rich core and Zn-rich shell and on the nanosecond timescale the smaller band gap Cd-rich center funnels charges toward the center of the nanocrystal. This process is attributed to a 7 ps decay observed in TA spectroscopy. Nonradiative recombination through surface trap states in the alloy core are attributed to a 55 ps decay and an 80 ps decay observed in the structures with shell(s) is attributed to nonradiative relaxation through lattice dislocations at the core/shell interface. This is a notable example of alloying being used to decrease lattice mismatch at interfaces between core and shell that introduce trap states.
Nanorods
The ultrafast dynamics of charge carriers in one-dimensional nanorods display many of the same processes as observed in zero-dimensional nanocrystal quantum dots. Early work by El-Sayed et al. compared CdSe nanodots (nanocrystals with diameter of 4.2 nm) to CdSe nanorods of 4.2× 13.5 nm (Mohamed et al., 2001). The rise time of the band gap state is faster in the nanorods (400 fs) in comparison to the nanocrystals (1 ps) as is the decay of the higher energy state (300 fs for the nanorods versus 2 ps for the nanocrystals), indicating a faster cooling process that leads to the rise of the bandgap state in the nanorods. In the nanorods, the decay becomes longer as the energy decreases, 300 fs at high energies extended out to 50 ps at the band gap state. Decay of the band gap in the nanorods (50 ps) is longer than the nanocrystals (two decays: 2 and 36 ps). The differences observed between nanorods and nanocrystals is explained by the decrease in symmetry in the nanorods. The decreased symmetry lifts the degeneracy of the energy levels which then lead to longer times for the charges to cascade down the energy levels to the band edge. In addition, the longer decay of the band gap is explained by less effective surface trapping in the nanorods.
Type II nanorod structures have been investigated in which charge carriers are separated between two different materials including CdSe/PbSe and CdSe/CdTe (Jones et al., 2008; Lee et al., 2017). In CdSe/CdTe nanorods, surface states either trap excited charge carriers on the 44–112 ns timescale (depending on surface chemistry and the degree of passivation of the surface trap states) or the band alignment separates the electron into the CdSe and the hole into the CdTe to form a long-lived charge-separated state with a lifetime on the 0.75–1.82 μs timescale (Jones et al., 2008). Similarly for CdSe/PbSe nanorods, after selective excitation of only the PbSe, the type II band alignment promotes transfer of the electron to the CdSe material while the hole remains in the PbSe (Lee et al., 2017). Exciting at high energy, however, shows a bleach of the CdSe exciton band but not that of the PbSe exciton band even though both materials should be excited. This is attributed to the hot electrons generated in PbSe by the high-energy excitation cooling into trap states before relaxing to the PbSe band edge. Introduction of charge trap states is attributed to the cation exchange synthesis and include surface dangling bonds, interfacial defects, and interior defects. Bleach of the trap-state stricken CdSe/PbSe nanorod recovers completely in 5 ps, whereas CdSe-only nanorods exhibit only ∼20% bleach recovery out to 1 ns. Annealing the nanorods reduces the number of defects and suppresses charge trapping, evidenced by a decrease in the amplitude and rate of the fast carrier trapping processes. Electron transfer from the conduction band of CdSe to the conduction band of TiO2 in CdSe/TiO2 nanorods has also been observed as a fast decay (Fitzmorris et al., 2012b). The fast injection rate from CdSe into TiO2 (1.71 × 1011 1/s) is attributed to the large interfacial area and strong coupling between the two materials.
Just as in nanocrystal-based systems (see Charge transfer), charge transfer from nanorods has also been studied and plays an important role in the excited state of nanorod-based systems. Rosenthal, Macdonald, et al. have investigated CdSe dot in CdS nanorod structures functionalized with an iron-chelated bipyridine-based ligand (a hole acceptor) (La Croix et al., 2017). In CdSe/CdS dot-in-rods with native ligands a biexponential decay is observed with TCSPC, attributed to a small amount of carrier trapping at the surface followed by longer-lived band edge recombination with a lifetime of 23.5 ns. Upon ligand exchange with ammonium-[2,2′-bipyridin]-4-ylcarbamadithioate (DTCBipy) holes are extracted by the ligands from the CdS rod before they can be localized to the CdSe core and a consistent 20.9 ns lifetime of the band edge exciton is observed along with a large quench of the photoluminescence quantum yield. In these samples, the valence band offset is such that the holes are not accessible for transfer from the CdSe cores. However, upon coordination of the DTCBipy ligands with Fe(acac)3 fast hole transfer from the CdS to the molecular ligand occurs in 133 ps while the intrinsic CdSe core recombination has a lifetime of 1.24 ns.
Conclusions and perspectives
The behavior of the excited charge carriers in semiconductor nanomaterials ultimately determines the suitability of a nanostructure for a particular application. Fundamental understanding of the evolution of the excited state and its structure-property relationships enables intelligent design of nanomaterial systems with properties optimized for an intended use, be it light harvesting in photovoltaics or light sources in display or lighting devices. To date, ultrafast spectroscopies have yielded the necessary fundamental understanding of the behavior of the excited state and how it evolves in nanocrystalline semiconductor materials on the ultrafast timescale to begin such design strategies. Throughout the lifetime of the excited state, the excited charge carriers experience a wide range of phenomena including: loss of initial anisotropy, electronic dephasing, exciton spin relaxation, carrier cooling to the band edge, charge transfer to or from adsorbed molecules at the nanocrystal surface, charge separation between materials in heterostructures, carrier trapping at surface states, charge trapping at internal interfacial lattice defects, relaxation back to the ground state, and/or relaxation from trap states. Notable discoveries in this field include the lack of a phonon bottleneck, the importance of surface chemistry, and the ability of core/shell or other complex structures to control the behavior of the excited state.
Future perspectives in the field of ultrafast spectroscopy on semiconductor nanocrystals are summarized below:
-
•expand studies to non-CdSe-based systems and systems of greater complexity and higher dimensions, for example:
- ◦cesium lead halide (CsPbX3)
- ◦doped nanocrystals
- ◦hybrid systems and heteronanostructures
- ◦two-dimensional nanomaterials systems
-
•
femtosecond single particle spectroscopy
-
•
higher sensitive detection to reduce pump power needs
-
•
‘all in one’ box for ultrafast experiments for non-laser specialists
The vast majority of the studies that have yielded fundamental understanding of the excited state in semiconductor nanocrystals have focused on CdSe-based systems, as we have detailed. However, the field has most recently begun to expand and apply ultrafast spectroscopy to non-Cd-based and/or more complex systems. In particular, cesium lead halide (CsPbX3) and other perovskite nanocrystals have recently gained popularity for their enhanced efficiencies in quantum dot photovoltaics and high photoluminescence quantum yields and have garnered great interest in understanding their fundamental excitonic properties (Boehme et al., 2020; Carroll et al., 2021; Dai et al., 2021; Qin et al., 2021; Socie et al., 2021). Doped nanocrystal systems have also sparked interest (Dutta et al., 2018) as well as hybrid systems such as multilayered CdSe nanocrystals bridged with bidentate dithiocarbamate ligands (Virgili et al., 2018). Beyond the zero-dimensional nanocrystals which are the focus of this review, and even one-dimensional nanomaterials such as nanorods which we briefly highlighted, recent work has also begun to explore two-dimensional materials and even composites of two-dimensional materials such as nanoplatelets of CdSe, CsPbX3, and graphene oxide, among other materials (Das et al., 2019; Ghosh et al., 2020).
Finally, the ultrafast techniques used to study the carrier dynamics of semiconductor nanocrystals thus far are ensemble measurements, meaning the signal they measure is the sum of a large population of particles. These ensemble techniques therefore lack the sensitivity to study the carrier dynamics of nanomaterials at the single nanoparticle level. Since there is a large particle-to-particle variability between nanocrystals in any given synthesis, at best these results yield an ‘averaged structure’ picture of the structure-property relationships of semiconductor nanocrystals. Recently, Rosenthal, McBride, Hollingsworth, et al. have demonstrated correlation of single particle TCSPC spectroscopy with electron microscopy characterization of the same nanoparticles (Orfield et al., 2015; Reid et al., 2018). Ultrafast techniques must be developed to monitor the carrier dynamics of a single nanoparticle to allow for correlation to structural characterization of the same particle in order to truly realize the interplay between structure and charge carrier behavior in semiconductor nanocrystals.
Acknowledgments
Author contributions
Conceptualization, S.J.R. and J.D.K; Investigation, J.D.K, N.J.F., J.M.R.,& S.J.R.; Writing – Original Draft, J.D.K, N.J.F., J.M.R., and S.J.R.; Writing – Review & Editing, J.D.K, N.J.F., and J.M.R.; Visualization, J.D.K, N.J.F., and J.M.R.; Supervision, J.D.K.; Project Administration, J.D.K.
Declaration of interests
The authors declare no competing interests.
Contributor Information
Joseph D. Keene, Email: keene_jd@mercer.edu.
Sandra J. Rosenthal, Email: sandra.j.rosenthal@Vanderbilt.Edu.
References
- Alivisatos A.P., Gu W., Larabell C. Quantum dots as cellular probes. Annu. Rev. Biomed. Eng. 2005;7:55–76. doi: 10.1146/annurev.bioeng.7.060804.100432. [DOI] [PubMed] [Google Scholar]
- Bakalova R., Ohba H., Zhelev Z., Ishikawa M., Baba Y. Quantum dots as photosensitizers? Nat. Biotechnol. 2004;22:1360–1361. doi: 10.1038/nbt1104-1360. [DOI] [PubMed] [Google Scholar]
- Bawendi M.G., Carroll P.J., Wilson W.L., Brus L.E. Luminescence properties of CdSe quantum crystallites: resonance between interior and surface localized states. J. Chem. Phys. 1992;96:946–954. doi: 10.1063/1.462114. [DOI] [Google Scholar]
- Blanton S.A., Leheny R.L., Hines M.A., Guyot-Sionnest P. Dielectric dispersion measurements of CdSe nanocrystal Colloids: observation of a permanent dipole moment. Phys. Rev. Lett. 1997;79:865–868. doi: 10.1103/PhysRevLett.79.865. [DOI] [Google Scholar]
- Boehme S.C., Brinck S.T., Maes J., Yazdani N., Zapata F., Chen K., Wood V., Hodgkiss J.M., Hens Z., Geiregat P., Infante I. Phonon-mediated and weakly size-dependent electron and hole cooling in CsPbBr 3nanocrystals revealed by atomistic simulations and ultrafast spectroscopy. Nano Lett. 2020;20:1819–1829. doi: 10.1021/acs.nanolett.9b05051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowers M.J., II, McBride J.R., Garrett M.D., Sammons J.A., Dukes A.D., III, Schreuder M.A., Watt T.L., Lupini A.R., Pennycook S.J., Rosenthal S.J. Structure and ultrafast dynamics of white-light-emitting CdSe nanocrystals. J. Am. Chem. Soc. 2009;131:5730–5731. doi: 10.1021/ja900529h. [DOI] [PubMed] [Google Scholar]
- Bowers M.J., McBride J.R., Rosenthal S.J. White-light emission from magic-sized cadmium selenide nanocrystals. J. Am. Chem. Soc. 2005;127:15378–15379. doi: 10.1021/ja055470d. [DOI] [PubMed] [Google Scholar]
- Brus L.E. Electron–electron and electron-hole interactions in small semiconductor crystallites: the size dependence of the lowest excited electronic state. J. Chem. Phys. 1984;80:4403–4409. doi: 10.1063/1.447218. [DOI] [Google Scholar]
- Burda C., Green T.C., Link S., El-Sayed M.A. Electron shuttling across the interface of CdSe nanoparticles monitored by femtosecond laser spectroscopy. J. Phys. Chem. B. 1999;103:1783–1788. doi: 10.1021/jp9843050. [DOI] [Google Scholar]
- Burda C., Link S., Mohamed M., El-Sayed M. The relaxation pathways of CdSe nanoparticles monitored with femtosecond time-resolution from the visible to the IR: assignment of the transient features by carrier quenching. J. Phys. Chem. B. 2001;105:12286–12292. doi: 10.1021/jp0124589. [DOI] [Google Scholar]
- Burda C., Link S., Mohamed M.B., El-Sayed M. The pump power dependence of the femtosecond relaxation of CdSe nanoparticles observed in the spectral range from visible to infrared. J. Chem. Phys. 2002;116:3828–3833. doi: 10.1063/1.1446851. [DOI] [Google Scholar]
- Carroll G.M., Limpens R., Pach G.F., Zhai Y., Beard M.C., Miller E.M., Neale N.R. Suppressing Auger recombination in multiply excited colloidal silicon nanocrystals with ligand-induced holetraps. J. Phys. Chem. C. 2021;125:2565–2574. doi: 10.1021/acs.jpcc.0c11388. [DOI] [Google Scholar]
- Chen O., Zhao J., Chauhan V.P., Cui J., Wong C., Harris D.K., Wei H., Han H.-S., Fukumura D., Jain R.K., Bawendi M.G. Compact high-quality CdSe–CdS core–shell nanocrystals with narrow emission linewidths and suppressed blinking. Nat. Mater. 2013;12:445–451. doi: 10.1038/nmat3539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho B., Peters W.K., Hill R.J., Courtney T.L., Jonas D.M. Bulklike hot carrier dynamics in lead sulfide quantum dots. Nano Lett. 2010;10:2498–2505. doi: 10.1021/nl1010349. [DOI] [PubMed] [Google Scholar]
- Collini E., Gattuso H., Bolzonello L., Casotto A., Volpato A., Dibenedetto C.N., Fanizza E., Striccoli M., Remacle F. Quantum phenomena in nanomaterials: coherent superpositions of fine structure states in CdSe nanocrystals at room temperature. J. Phys. Chem. C. 2019;123:31286–31293. doi: 10.1021/acs.jpcc.9b11153. [DOI] [Google Scholar]
- Cooney R.R., Sewall S.L., Dias E.A., Sagar D.M., Anderson K.E.H., Kambhampati P. Unified picture of electron and hole relaxation pathways in semiconductor quantum dots. Phys. Rev. B. 2007;75:245311. doi: 10.1103/PhysRevB.75.245311. [DOI] [Google Scholar]
- Dai L., Deng Z., Auras F., Goodwin H., Zhang Z., Walmsley J.C., Bristowe P.D., Deschler F., Greenham N.C. Slow carrier relaxation in tin-based perovskite nanocrystals. Nat. Photonics. 2021;15:696–702. doi: 10.1038/s41566-021-00847-2. [DOI] [Google Scholar]
- Das S., Dutta A., Bera R., Patra A. Ultrafast carrier dynamics in 2D–2D hybrid structures of functionalized GO and CdSe nanoplatelets. Phys. Chem. Chem. Phys. 2019;21:15568–15575. doi: 10.1039/C9CP02823D. [DOI] [PubMed] [Google Scholar]
- Dutta A., Bera R., Ghosh A., Patra A. Ultrafast carrier dynamics of photo-induced Cu-doped CdSe nanocrystals. J. Phys. Chem. C. 2018;122:16992–17000. doi: 10.1021/acs.jpcc.8b05422. [DOI] [Google Scholar]
- Dworak L., Bottin A., Roth S., Trinh P.T., Müllen K., Basché T., Wachtveitl J. Photodynamics at the CdSe quantum dot–perylene diimide interface: unraveling the excitation energy and electron transfer pathways. J. Phys. Chem. C. 2021;125:3277–3284. doi: 10.1021/acs.jpcc.0c09891. [DOI] [Google Scholar]
- Ekimov A.I., Kudryavtsev I.A., Efros A.L., Yazeva T.V., Hache F., Schanne-Klein M.C., Rodina A.V., Ricard D., Flytzanis C. Absorption and intensity-dependent photoluminescence measurements on CdSe quantum dots: assignment of the first electronic transitions. J. Opt. Soc. Am. B. 1993;10:100. doi: 10.1364/JOSAB.10.000100. [DOI] [Google Scholar]
- Fan F., Voznyy O., Sabatini R.P., Bicanic K.T., Adachi M.M., McBride J.R., Reid K.R., Park Y.-S., Li X., Jain A., et al. Continuous-wave lasing in colloidal quantum dot solids enabled by facet-selective epitaxy. Nature. 2017;544:75–79. doi: 10.1038/nature21424. [DOI] [PubMed] [Google Scholar]
- Fitzmorris B.C., Cooper J.K., Edberg J., Gul S., Guo J., Zhang J.Z. Synthesis and structural, optical, and dynamic properties of core/shell/shell CdSe/ZnSe/ZnS quantum dots. J. Phys. Chem. C. 2012;116:25065–25073. doi: 10.1021/jp3092013. [DOI] [Google Scholar]
- Fitzmorris B.C., Larsen G.K., Wheeler D.A., Zhao Y., Zhang J.Z. Ultrafast charge transfer dynamics in polycrystalline CdSe/TiO 2nanorods prepared by oblique angle codeposition. J. Phys. Chem. C. 2012;116:5033–5041. doi: 10.1021/jp2121752. [DOI] [Google Scholar]
- Fitzmorris B.C., Pu Y.-C., Cooper J.K., Lin Y.-F., Hsu Y.-J., Li Y., Zhang J.Z. Optical properties and exciton dynamics of alloyed core/shell/shell Cd 1–x Zn x Se/ZnSe/ZnS quantum dots. ACS Appl. Mater.Inter. 2013;5:2893–2900. doi: 10.1021/am303149r. [DOI] [PubMed] [Google Scholar]
- Frederick M.T., Weiss E.A. Relaxation of exciton confinement in CdSe quantum dots by modification with a conjugated dithiocarbamate ligand. ACS Nano. 2010;4:3195–3200. doi: 10.1021/nn1007435. [DOI] [PubMed] [Google Scholar]
- Freymeyer N.J., Click S.M., Reid K.R., Chisholm M.F., Bradsher C.E., McBride J.R., Rosenthal S.J. Effect of indium alloying on the charge carrier dynamics of thick-shell InP/ZnSe quantum dots. J. Chem. Phys. 2020;152:161104. doi: 10.1063/1.5145189. [DOI] [PubMed] [Google Scholar]
- Garrett M.D., Bowers M.J., McBride J.R., Orndorff R.L., Pennycook S.J., Rosenthal S.J. Band edge dynamics in CdSe nanocrystals observed by ultrafast fluorescence upconversion. J. Phys. Chem. C. 2008;112:436–442. doi: 10.1021/jp7099306. [DOI] [PubMed] [Google Scholar]
- Garrett M.D., Dukes A.D., III, McBride J.R., Smith N.J., Pennycook S.J., Rosenthal S.J. Band edge recombination in CdSe, CdS and CdS x Se 1−xalloy nanocrystals observed by ultrafast fluorescence upconversion: the effect of surface trap states. J. Phys. Chem. C. 2008;112:12736–12746. doi: 10.1021/jp803708r. [DOI] [Google Scholar]
- Ghosh G., Dutta A., Ghosh A., Ghosh S., Patra A. Ultrafast carrier dynamics in 2D CdSe nanoplatelets–CsPbX 3 composites: influence of the halide composition. J. Phys. Chem. C. 2020;124:10252–10260. doi: 10.1021/acs.jpcc.0c03206. [DOI] [Google Scholar]
- Gul S., Cooper J.K., Corrado C., Vollbrecht B., Bridges F., Guo J., Zhang J.Z. Synthesis, optical and structural properties, and charge carrier dynamics of Cu-doped ZnSe nanocrystals. J. Phys. Chem. C. 2011;115:20864–20875. doi: 10.1021/jp2047272. [DOI] [Google Scholar]
- Guyot-Sionnest P., Hines M.A. Intraband transitions in semiconductor nanocrystals. Appl. Phys. Lett. 1998;72:686–688. doi: 10.1063/1.120846. [DOI] [Google Scholar]
- Guyot-Sionnest P., Wehrenberg B., Yu D. Intraband relaxation in CdSe nanocrystals and the strong influence of the surface ligands. J. Chem. Phys. 2005;123:074709. doi: 10.1063/1.2004818. [DOI] [PubMed] [Google Scholar]
- Guzelturk B., Cotts B.L., Jasrasaria D., Philbin J.P., Hanifi D.A., Koscher B.A., Balan A.D., Curling E., Zajac M., Park S., et al. Dynamic lattice distortions driven by surface trapping in semiconductor nanocrystals. Nat. Commun. 2021;12:1860. doi: 10.1038/s41467-021-22116-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanifi D.A., Bronstein N.D., Koscher B.A., Nett Z., Swabeck J.K., Takano K., Schwartzberg A.M., Maserati L., Vandewal K., van de Burgt Y., et al. Redefining near-unity luminescence in quantum dots with photothermal threshold quantum yield. Science. 2019;363:1199–1202. doi: 10.1126/science.aat3803. [DOI] [PubMed] [Google Scholar]
- Hartley C.L., Kessler M.L., Dempsey J.L. Molecular-level insight into semiconductor nanocrystal surfaces. J. Am. Chem. Soc. 2021;143:1251–1266. doi: 10.1021/jacs.0c10658. [DOI] [PubMed] [Google Scholar]
- Hines M.A., Guyot-Sionnest P. Synthesis and characterization of strongly luminescing ZnS-capped CdSe nanocrystals. J. Phys. Chem. 1996;100:468–471. doi: 10.1021/jp9530562. [DOI] [Google Scholar]
- Huxter V.M., Kovalevskij V., Scholes G.D. Dynamics within the exciton fine structure of colloidal CdSe quantum dots. J. Phys. Chem. B. 2005;109:20060–20063. doi: 10.1021/jp0546406. [DOI] [PubMed] [Google Scholar]
- Ithurria S., Dubertret B. Quasi 2D colloidal CdSe platelets with thicknesses controlled at the atomic level. J. Am. Chem. Soc. 2008;130:16504–16505. doi: 10.1021/ja807724e. [DOI] [PubMed] [Google Scholar]
- Johnson J.C., Gerth K.A., Song Q., Murphy J.E., Nozik A.J., Scholes G.D. Ultrafast exciton fine structure relaxation dynamics in lead chalcogenide nanocrystals. Nano Lett. 2008;8:1374–1381. doi: 10.1021/nl080126a. [DOI] [PubMed] [Google Scholar]
- Jones M., Kumar S., Lo S.S., Scholes G.D. Exciton trapping and recombination in type II CdSe/CdTe nanorod heterostructures. J. Phys. Chem. C. 2008;112:5423–5431. doi: 10.1021/jp711009h. [DOI] [Google Scholar]
- Jones M., Lo S.S., Scholes G.D. Signatures of exciton dynamics and carrier trapping in the time-resolved photoluminescence of colloidal CdSe nanocrystals. J. Phys. Chem. C. 2009;113:18632–18642. doi: 10.1021/jp9078772. [DOI] [Google Scholar]
- Kamat P.V. Quantum dot solar cells.The next big thing in photovoltaics. J. Phys. Chem. Lett. 2013;4:908–918. doi: 10.1021/jz400052e. [DOI] [PubMed] [Google Scholar]
- Kamat P.V. Quantum dot solar cells.Semiconductor nanocrystals as light harvesters. J. Phys. Chem. C. 2008;112:18737–18753. doi: 10.1021/jp806791s. [DOI] [Google Scholar]
- Kamat P.V., Dimitrijević N.M. Colloidal semiconductors as photocatalysts for solar energy conversion. Sol. Energy. 1990;44:83–98. doi: 10.1016/0038-092X(90)90070-S. [DOI] [Google Scholar]
- Kambhampati P. Unraveling the structure and dynamics of excitons in semiconductor quantum dots. Acc. Chem. Res. 2011;44:1–13. doi: 10.1021/ar1000428. [DOI] [PubMed] [Google Scholar]
- Kambhampati P. Hot exciton relaxation dynamics in semiconductor quantum dots: radiationless transitions on the nanoscale. J. Phys. Chem. C. 2011;115:22089–22109. doi: 10.1021/jp2058673. [DOI] [Google Scholar]
- Keene J.D., McBride J.R., Orfield N.J., Rosenthal S.J. Elimination of hole–surface overlap in graded CdS x Se 1–xnanocrystals revealed by ultrafast fluorescence upconversion spectroscopy. ACS Nano. 2014;8:10665–10673. doi: 10.1021/nn504235w. [DOI] [PubMed] [Google Scholar]
- Kippeny T., Swafford L.A., Rosenthal S.J. Semiconductor nanocrystals: apowerful visual aid for introducing the particle in a box. J. Chem. Educ. 2002;79:1094. doi: 10.1021/ed079p1094. [DOI] [Google Scholar]
- Kippeny T.C., Bowers M.J., Dukes A.D., McBride J.R., Orndorff R.L., Garrett M.D., Rosenthal S.J. Effects of surface passivation on the exciton dynamics of CdSe nanocrystals as observed by ultrafast fluorescence upconversion spectroscopy. J. Chem. Phys. 2008;128:084713. doi: 10.1063/1.2834692. [DOI] [PubMed] [Google Scholar]
- Klimov V.I. Optical gain and stimulated emission in nanocrystal quantum dots. Science. 2000;290:314–317. doi: 10.1126/science.290.5490.314. [DOI] [PubMed] [Google Scholar]
- Klimov V.I. Optical nonlinearities and ultrafast carrier dynamics in semiconductor nanocrystals. J. Phys. Chem. B. 2000;104:6112–6123. doi: 10.1021/jp9944132. [DOI] [Google Scholar]
- Klimov V.I., Bawendi M.G. Ultrafast carrier dynamics, optical amplification, and lasing in nanocrystal quantum dots. MRS Bull. 2001;26:998–1004. doi: 10.1557/mrs2001.256. [DOI] [Google Scholar]
- Klimov V.I., Bolivar P.H., Kurz H. Ultrafast carrier dynamics in semiconductor quantum dots. Phys. Rev. B. 1996;53:1463–1467. doi: 10.1103/PhysRevB.53.1463. [DOI] [PubMed] [Google Scholar]
- Klimov V.I., McBranch D.W. Femtosecond 1 P -to- 1 S electron relaxation in strongly confined semiconductor nanocrystals. Phys. Rev. Lett. 1998;80:4028–4031. doi: 10.1103/PhysRevLett.80.4028. [DOI] [Google Scholar]
- Klimov V.I., McBranch D.W., Leatherdale C.A., Bawendi M.G. Electron and hole relaxation pathways in semiconductor quantum dots. Phys. Rev. B. 1999;60:13740–13749. doi: 10.1103/PhysRevB.60.13740. [DOI] [Google Scholar]
- Klimov V.I., Mikhailovsky A.A., McBranch D.W., Leatherdale C.A., Bawendi M.G. Mechanisms for intraband energy relaxation in semiconductor quantum dots: the role of electron-hole interactions. Phys. Rev. B. 2000;61:R13349–R13352. doi: 10.1103/PhysRevB.61.R13349. [DOI] [Google Scholar]
- Klimov V.I., Schwarz Ch.J., McBranch D.W., Leatherdale C.A., Bawendi M.G. Ultrafast dynamics of inter- and intraband transitions in semiconductor nanocrystals: implications for quantum-dot lasers. Phys. Rev. B. 1999;60:R2177–R2180. doi: 10.1103/PhysRevB.60.R2177. [DOI] [Google Scholar]
- Knowles K.E., McArthur E.A., Weiss E.A. A multi-timescale map of radiative and nonradiative decay pathways for excitons in CdSe quantum dots. ACS Nano. 2011;5:2026–2035. doi: 10.1021/nn2002689. [DOI] [PubMed] [Google Scholar]
- Konstantatos G., Howard I., Fischer A., Hoogland S., Clifford J., Klem E., Levina L., Sargent E.H. Ultrasensitive solution-cast quantum dot photodetectors. Nature. 2006;442:180–183. doi: 10.1038/nature04855. [DOI] [PubMed] [Google Scholar]
- Kovtun O., Tomlinson I.D., Sakrikar D.S., Chang J.C., Blakely R.D., Rosenthal S.J. Visualization of the cocaine-sensitive dopamine transporter with ligand-conjugated quantum dots. ACS Chem. Neurosci. 2011;2:370–378. doi: 10.1021/cn200032r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krivanek O.L., Dellby N., Murfitt M.F., Chisholm M.F., Pennycook T.J., Suenaga K., Nicolosi V. Gentle STEM: ADF imaging and EELS at low primary energies. Ultramicroscopy. 2010;110:935–945. doi: 10.1016/j.ultramic.2010.02.007. [DOI] [Google Scholar]
- La Croix A.D., O’Hara A., Reid K.R., Orfield N.J., Pantelides S.T., Rosenthal S.J., Macdonald J.E. Design of a hole trapping ligand. Nano Lett. 2017;17:909–914. doi: 10.1021/acs.nanolett.6b04213. [DOI] [PubMed] [Google Scholar]
- Labrador T., Dukovic G. Simultaneous determination of spectral signatures and decay kinetics of excited state species in semiconductor nanocrystals probed by transient absorption spectroscopy. J. Phys. Chem. C. 2020;124:8439–8447. doi: 10.1021/acs.jpcc.0c01701. [DOI] [Google Scholar]
- Lee S., Wang Y., Liu Y., Lee D., Lee K., Lee D.C., Lian T. Exciton dynamics in cation-exchanged CdSe/PbSe nanorods: the role of defects. Chem. Phys. Lett. 2017;683:342–346. doi: 10.1016/j.cplett.2017.04.047. [DOI] [Google Scholar]
- Li Y., Hou X., Dai X., Yao Z., Lv L., Jin Y., Peng X. Stoichiometry-controlled InP-based quantum dots: synthesis, photoluminescence, and electroluminescence. J. Am. Chem. Soc. 2019;141:6448–6452. doi: 10.1021/jacs.8b12908. [DOI] [PubMed] [Google Scholar]
- Logunov S., Green T., Marguet S., El-Sayed M.A. Interfacial carriersdynamics of CdS nanoparticles. J. Phys. Chem. A. 1998;102:5652–5658. doi: 10.1021/jp980387g. [DOI] [Google Scholar]
- Mack T.G., Jethi L., Kambhampati P. Temperature dependence of emission line widths from semiconductor nanocrystals reveals vibronic contributions to line broadening processes. J. Phys. Chem. C. 2017;121:28537–28545. doi: 10.1021/acs.jpcc.7b09903. [DOI] [Google Scholar]
- McArthur E.A., Morris-Cohen A.J., Knowles K.E., Weiss E.A. Charge carrier resolved relaxation of the first excitonic state in CdSe quantum dots probed with near-infrared transient absorption spectroscopy. J. Phys. Chem. B. 2010;114:14514–14520. doi: 10.1021/jp102101f. [DOI] [PubMed] [Google Scholar]
- McBride J., Treadway J., Feldman L.C., Pennycook S.J., Rosenthal S.J. Structural basis for near unity quantum yield core/shell nanostructures. Nano Lett. 2006;6:1496–1501. doi: 10.1021/nl060993k. [DOI] [PubMed] [Google Scholar]
- McBride J.R., Kippeny T.C., Pennycook S.J., Rosenthal S.J. Aberration-corrected Z-contrast scanning transmission electron microscopy of CdSe nanocrystals. Nano Lett. 2004;4:1279–1283. doi: 10.1021/nl049406q. [DOI] [Google Scholar]
- McBride J.R., Pennycook T.J., Pennycook S.J., Rosenthal S.J. The possibility and implications of dynamic nanoparticle surfaces. ACS Nano. 2013;7:8358–8365. doi: 10.1021/nn403478h. [DOI] [PubMed] [Google Scholar]
- McBride J.R., Rosenthal S.J. Real colloidal quantum dot structures revealed by high resolution analytical electron microscopy. J. Chem. Phys. 2019;151:160903. doi: 10.1063/1.5128366. [DOI] [PubMed] [Google Scholar]
- McDonald S.A., Konstantatos G., Zhang S., Cyr P.W., Klem E.J.D., Levina L., Sargent E.H. Solution-processed PbS quantum dot infrared photodetectors and photovoltaics. Nat. Mater. 2005;4:138–142. doi: 10.1038/nmat1299. [DOI] [PubMed] [Google Scholar]
- Micic O.I., Curtis C.J., Jones K.M., Sprague J.R., Nozik A.J. Synthesis and characterization of InPquantum dots. J. Phys. Chem. 1994;98:4966–4969. doi: 10.1021/j100070a004. [DOI] [Google Scholar]
- Micic O.I., Sprague J.R., Curtis C.J., Jones K.M., Machol J.L., Nozik A.J., Giessen H., Fluegel B., Mohs G., Peyghambarian N. Synthesis and characterization of InP, GaP, and GaInP2 quantum dots. J. Phys. Chem. 1995;99:7754–7759. doi: 10.1021/j100019a063. [DOI] [Google Scholar]
- Mittleman D.M., Schoenlein R.W., Shiang J.J., Colvin V.L., Alivisatos A.P., Shank C.V. Quantum size dependence of femtosecond electronic dephasing and vibrational dynamics in CdSe nanocrystals. Phys. Rev. B. 1994;49:14435–14447. doi: 10.1103/PhysRevB.49.14435. [DOI] [PubMed] [Google Scholar]
- Mohamed M.B., Burda C., El-Sayed M.A. Shape dependent ultrafast relaxation dynamics of CdSe nanocrystals: nanorods vs nanodots. Nano Lett. 2001;1:589–593. doi: 10.1021/nl0155835. [DOI] [Google Scholar]
- Mooney J., Krause M.M., Saari J.I., Kambhampati P. Challenge to the deep-trap model of the surface in semiconductor nanocrystals. Phys. Rev. B. 2013;87:081201. doi: 10.1103/PhysRevB.87.081201. [DOI] [Google Scholar]
- Moroz P., Boddy A., Zamkov M. Challenges and prospects of photocatalytic applications utilizing semiconductor nanocrystals. Front. Chem. 2018;6:353. doi: 10.3389/fchem.2018.00353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray C.B., Norris D.J., Bawendi M.G. Synthesis and characterization of nearly monodisperse CdE (E = sulfur, selenium, tellurium) semiconductor nanocrystallites. J. Am. Chem. Soc. 1993;115:8706–8715. doi: 10.1021/ja00072a025. [DOI] [Google Scholar]
- Nellist P.D., Pennycook S.J. Incoherent imaging using dynamically scattered coherent electrons. Ultramicroscopy. 1999;78:111–124. doi: 10.1016/S0304-3991(99)00017-0. [DOI] [Google Scholar]
- Nozik A.J. Quantum dot solar cells. Physica E Low Dimens. Syst. Nanostruct. 2002;14:115–120. doi: 10.1016/S1386-9477(02)00374-0. [DOI] [Google Scholar]
- Okuno T., Lipovskii A.A., Ogawa T., Amagai I., Masumoto Y. Strong confinement of PbSe and PbS quantum dots. J. Lumin. 2000;87:491–493. doi: 10.1016/S0022-2313(99)00220-3. [DOI] [Google Scholar]
- Orfield N.J., McBride J.R., Keene J.D., Davis L.M., Rosenthal S.J. Correlation of atomic structure and photoluminescence of the same quantum dot: pinpointing surface and internal defects that inhibit photoluminescence. ACS Nano. 2015;9:831–839. doi: 10.1021/nn506420w. [DOI] [PubMed] [Google Scholar]
- Pal B.N., Ghosh Y., Brovelli S., Laocharoensuk R., Klimov V., Hollingsworth J.A., Htoon H. ‘Giant’ CdSe/CdS core/shell nanocrystal quantum dots as efficient electroluminescent materials: strong influence of shell thickness on light-emitting diode performance. Nano Lett. 2012;12:331–336. doi: 10.1021/nl203620f. [DOI] [PubMed] [Google Scholar]
- Palato S., Seiler H., McGovern L., Mack T.G., Jethi L., Kambhampati P. Electron dynamics at the surface of semiconductor nanocrystals. J. Phys. Chem. C. 2017;121:26519–26527. doi: 10.1021/acs.jpcc.7b09145. [DOI] [Google Scholar]
- Pandey A., Guyot-Sionnest P. Slow electron cooling in colloidal quantum dots. Science. 2008;322:929–932. doi: 10.1126/science.1159832. [DOI] [PubMed] [Google Scholar]
- Pandey A., Guyot-Sionnest P. Multicarrier recombination in colloidal quantum dots. J. Chem. Phys. 2007;127:111104. doi: 10.1063/1.2786068. [DOI] [PubMed] [Google Scholar]
- Park S., Baranov D., Ryu J., Jonas D. OSA; 2016. The Initial Pump-Probe Polarization Anisotropy of Colloidal PbS Quantum Dots; p. UW4A.23. [DOI] [Google Scholar]
- Peng X., Schlamp M.C., Kadavanich A.V., Alivisatos A.P. Epitaxial growth of highly luminescent CdSe/CdS core/shell nanocrystals with photostability and electronic accessibility. J. Am. Chem. Soc. 1997;119:7019–7029. doi: 10.1021/ja970754m. [DOI] [Google Scholar]
- Peng Z.A., Peng X. Formation of high-quality CdTe, CdSe, and CdS nanocrystals using CdO as precursor. J. Am. Chem. Soc. 2001;123:183–184. doi: 10.1021/ja003633m. [DOI] [PubMed] [Google Scholar]
- Pennycook T.J., McBride J.R., Rosenthal S.J., Pennycook S.J., Pantelides S.T. Dynamic fluctuations in ultrasmall nanocrystals induce white light emission. Nano Lett. 2012;12:3038–3042. doi: 10.1021/nl3008727. [DOI] [PubMed] [Google Scholar]
- Peterson M.D., Cass L.C., Harris R.D., Edme K., Sung K., Weiss E.A. The role of ligands in determining the exciton relaxation dynamics in semiconductor quantum dots. Annu. Rev. Phys. Chem. 2014;65:317–339. doi: 10.1146/annurev-physchem-040513-103649. [DOI] [PubMed] [Google Scholar]
- Qin C., Jiang Z., Zhou Z., Liu Y., Jiang Y. Multiexciton dynamics in CsPbBr 3 nanocrystals: the dependence on pump fluence and temperature. Nanotechnology. 2021;32:455702. doi: 10.1088/1361-6528/ac18d7. [DOI] [PubMed] [Google Scholar]
- Reid K.R., McBride J.R., Freymeyer N.J., Thal L.B., Rosenthal S.J. Chemical structure, ensemble and single-particle spectroscopy of thick-shell InP–ZnSe quantum dots. Nano Lett. 2018;18:709–716. doi: 10.1021/acs.nanolett.7b03703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robel I., Subramanian V., Kuno M., Kamat P.V. Quantum dot solar cells. Harvesting light energy with CdSe nanocrystals molecularly linked to mesoscopic TiO 2 films. J. Am. Chem. Soc. 2006;128:2385–2393. doi: 10.1021/ja056494n. [DOI] [PubMed] [Google Scholar]
- Rosenthal S., Mcbride J., Pennycook S., Feldman L. Synthesis, surface studies, composition and structural characterization of CdSe, core/shell and biologically active nanocrystals. Surf. Sci. Rep. 2007;62:111–157. doi: 10.1016/j.surfrep.2007.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenthal S.J., Chang J.C., Kovtun O., McBride J.R., Tomlinson I.D. Biocompatible quantum dots for biological applications. Chem. Biol. 2011;18:10–24. doi: 10.1016/j.chembiol.2010.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenthal S.J., Tomlinson I., Adkins E.M., Schroeter S., Adams S., Swafford L., McBride J., Wang Y., DeFelice L.J., Blakely R.D. Targeting cell surface receptors with ligand-conjugated nanocrystals. J. Am. Chem. Soc. 2002;124:4586–4594. doi: 10.1021/ja003486s. [DOI] [PubMed] [Google Scholar]
- Rosenthal S.J., Yeh A.T., Alivisatos A.P., Shank C.V. In: Ultrafast Phenomena X. Barbara P.F., Fujimoto J.G., Knox W.H., Zinth W., editors. Springer Berlin Heidelberg; 1996. Size dependent absorption anisotropy measurements of CdSe nanocrystals: symmetry assignments for the lowest electronic states; pp. 431–432. [DOI] [Google Scholar]
- Saari J.I., Dias E.A., Reifsnyder D., Krause M.M., Walsh B.R., Murray C.B., Kambhampati P. Ultrafast electron trapping at the surface of semiconductor nanocrystals: excitonic and biexcitonic processes. J. Phys. Chem. B. 2013;117:4412–4421. doi: 10.1021/jp307668g. [DOI] [PubMed] [Google Scholar]
- Sargent E.H. Colloidal quantum dot solar cells. Nat. Photonics. 2012;6:133–135. doi: 10.1038/nphoton.2012.33. [DOI] [Google Scholar]
- Schlossmacher P., Klenov D.O., Freitag B., von Harrach H.S. Enhanced detection sensitivity with a new windowless XEDS system for AEM based on silicon drift detector technology. Micros. Today. 2010;18:14–20. doi: 10.1017/S1551929510000404. [DOI] [Google Scholar]
- Schoenlein R.W., Mittleman D.M., Shiang J.J., Alivisatos A.P., Shank C.V. Investigation of femtosecond electronic dephasing in CdSe nanocrystals using quantum-beat-suppressed photon echoes. Phys. Rev. Lett. 1993;70:1014–1017. doi: 10.1103/PhysRevLett.70.1014. [DOI] [PubMed] [Google Scholar]
- Schreuder M.A., Gosnell J.D., Smith N.J., Warnement M.R., Weiss S.M., Rosenthal S.J. Encapsulated white-light CdSe nanocrystals as nanophosphors for solid-state lighting. J. Mater. Chem. 2008;18:970. doi: 10.1039/b716803a. [DOI] [Google Scholar]
- Schreuder M.A., Xiao K., Ivanov I.N., Weiss S.M., Rosenthal S.J. White light-emitting diodes based on ultrasmall CdSe nanocrystal electroluminescence. Nano Lett. 2010;10:573–576. doi: 10.1021/nl903515g. [DOI] [PubMed] [Google Scholar]
- Seiler H., Palato S., Sonnichsen C., Baker H., Kambhampati P. Seeing multiexcitons through sample inhomogeneity: band-edge biexciton structure in CdSe nanocrystals revealed by two-dimensional electronic spectroscopy. Nano Lett. 2018;18:2999–3006. doi: 10.1021/acs.nanolett.8b00470. [DOI] [PubMed] [Google Scholar]
- Sewall S.L., Cooney R.R., Anderson K.E.H., Dias E.A., Sagar D.M., Kambhampati P. State-resolved studies of biexcitons and surface trapping dynamics in semiconductor quantum dots. J. Chem. Phys. 2008;129:084701. doi: 10.1063/1.2971181. [DOI] [PubMed] [Google Scholar]
- Shen H., Zhou C., Xu S., Yu C., Wang H., Chen X., Li L.S. Phosphine-free synthesis of Zn1−xCdxSe/ZnSe/ZnSexS1−x/ZnS core/multishell structures with bright and stable blue–green photoluminescence. J. Mater. Chem. 2011;21:6046. doi: 10.1039/c0jm03605f. [DOI] [Google Scholar]
- Shiang J.J., Kadavanich A.V., Grubbs R.K., Alivisatos A.P. Symmetry of annealed wurtzite CdSe nanocrystals: assignment to the C3v point group. J. Phys. Chem. 1995;99:17417–17422. doi: 10.1021/j100048a017. [DOI] [Google Scholar]
- Shirasaki Y., Supran G.J., Bawendi M.G., Bulović V. Emergence of colloidal quantum-dot light-emitting technologies. Nat. Photonics. 2013;7:13–23. doi: 10.1038/nphoton.2012.328. [DOI] [Google Scholar]
- Socie E., Vale B.R.C., Burgos-Caminal A., Moser J. Direct observation of shallow trap states in thermal equilibrium with band-edge excitons in strongly confined CsPbBr 3 perovskite nanoplatelets. Adv. Opt. Mater. 2021;9:2001308. doi: 10.1002/adom.202001308. [DOI] [Google Scholar]
- Soloviev V.N., Eichhöfer A., Fenske D., Banin U. Size-dependent optical spectroscopy of a homologous series of CdSe cluster molecules. J. Am. Chem. Soc. 2001;123:2354–2364. doi: 10.1021/ja003598j. [DOI] [PubMed] [Google Scholar]
- Sukhovatkin V., Hinds S., Brzozowski L., Sargent E.H. Colloidal quantum-dot photodetectors exploiting multiexciton generation. Science. 2009;324:1542–1544. doi: 10.1126/science.1173812. [DOI] [PubMed] [Google Scholar]
- Swafford L.A., Rosenthal S.J. Molecular Nanoelectronics. American Scientific Publishers; 2003. Molecular and nanocrystal based photovoltaics; pp. 263–290. [Google Scholar]
- Swafford L.A., Weigand L.A., Bowers M.J., McBride J.R., Rapaport J.L., Watt T.L., Dixit S.K., Feldman L.C., Rosenthal S.J. Homogeneously alloyed CdS x Se 1 - xnanocrystals: synthesis, characterization, and composition/size-dependent band gap. J. Am. Chem. Soc. 2006;128:12299–12306. doi: 10.1021/ja063939e. [DOI] [PubMed] [Google Scholar]
- Taheri M.M., Elbert K.C., Yang S., Diroll B.T., Park J., Murray C.B., Baxter J.B. Distinguishing electron and holedynamics in functionalized CdSe/CdS core/shell quantum dots using complementary ultrafast spectroscopies and kinetic modeling. J. Phys. Chem. C. 2021;125:31–41. doi: 10.1021/acs.jpcc.0c07037. [DOI] [Google Scholar]
- Talapin D.V., Rogach A.L., Kornowski A., Haase M., Weller H. Highly luminescent monodisperse CdSe and CdSe/ZnS nanocrystals synthesized in a hexadecylamine−trioctylphosphine oxide−trioctylphospine mixture. Nano Lett. 2001;1:207–211. doi: 10.1021/nl0155126. [DOI] [PubMed] [Google Scholar]
- Taylor J., Kippeny T., Rosenthal S.J. Surface stoichiometry of CdSe nanocrystals determined by Rutherford backscattering spectroscopy. J. Cluster Sci. 2001;12:571–582. doi: 10.1023/A:1014246315331. [DOI] [Google Scholar]
- Turner D.B., Hassan Y., Scholes G.D. Exciton superposition states in CdSe nanocrystals measured using broadband two-dimensional electronic spectroscopy. Nano Lett. 2012;12:880–886. doi: 10.1021/nl2039502. [DOI] [PubMed] [Google Scholar]
- Underwood D.F., Kippeny T., Rosenthal S.J. Charge carrier dynamics in CdSe nanocrystals: implications for the use of quantum dots in novel photovoltaics. Eur. Phys. J. D. 2001;16:241–244. doi: 10.1007/s100530170101. [DOI] [Google Scholar]
- Underwood D.F., Kippeny T., Rosenthal S.J. Ultrafast carrier dynamics in CdSe nanocrystals determined by femtosecond fluorescence upconversion spectroscopy. J. Phys. Chem. B. 2001;105:436–443. doi: 10.1021/jp003088b. [DOI] [Google Scholar]
- Virgili T., Calzolari A., Suárez López I., Ruini A., Catellani A., Vercelli B., Tassone F. Hybridized electronic states between CdSe nanoparticles and conjugated organic ligands: a theoretical and ultrafast photo-excited carrier dynamics study. Nano Res. 2018;11:142–150. doi: 10.1007/s12274-017-1613-4. [DOI] [Google Scholar]
- Wehrenberg B.L., Wang C., Guyot-Sionnest P. Interband and intraband optical studies of PbSe colloidal quantum dots. J. Phys. Chem. B. 2002;106:10634–10640. doi: 10.1021/jp021187e. [DOI] [Google Scholar]
- Wheeler D.A., Zhang J.Z. Exciton dynamics in semiconductor nanocrystals. Adv. Mater. 2013;25:2878–2896. doi: 10.1002/adma.201300362. [DOI] [PubMed] [Google Scholar]
- Williams D.B., Carter C.B. Plenum Press; 1996. Transmission Electron Microscopy I, Basics, 2nd Edition. [Google Scholar]
- Won Y.-H., Cho O., Kim T., Chung D.-Y., Kim T., Chung H., Jang H., Lee J., Kim D., Jang E. Highly efficient and stable InP/ZnSe/ZnS quantum dot light-emitting diodes. Nature. 2019;575:634–638. doi: 10.1038/s41586-019-1771-5. [DOI] [PubMed] [Google Scholar]
- Wu K., Song N., Liu Z., Zhu H., Rodríguez-Córdoba W., Lian T. Interfacial charge separation and recombination in InP and quasi-type II InP/CdS core/shell quantum dot-molecular acceptor complexes. J. Phys. Chem. A. 2013;117:7561–7570. doi: 10.1021/jp402425w. [DOI] [PubMed] [Google Scholar]
- Zhu H., Song N., Lian T. Controlling charge separation and recombination rates in CdSe/ZnS type I core−shell quantum dots by shell thicknesses. J. Am. Chem. Soc. 2010;132:15038–15045. doi: 10.1021/ja106710m. [DOI] [PubMed] [Google Scholar]
- Zhu H., Song N., Rodríguez-Córdoba W., Lian T. Wave function engineering for efficient extraction of up to nineteen electrons from one CdSe/CdS quasi-type II quantum dot. J. Am. Chem. Soc. 2012;134:4250–4257. doi: 10.1021/ja210312s. [DOI] [PubMed] [Google Scholar]