

Summary
The esophagus and stomach, joined by a unique transitional zone, contain actively dividing epithelial stem cells required for organ homeostasis. Upon prolonged inflammation, epithelial cells in both organs can undergo a cell fate switch leading to intestinal metaplasia, predisposing to malignancy. Here, we discuss the biology of gastroesophageal stem cells and their role as the cell-of-origin in cancer. We summarize the interactions between the stromal niche and gastroesophageal stem cells in metaplasia and the early expansion of mutated stem cell-derived clones during carcinogenesis. Finally, we review new approaches under development to better study gastroesophageal stem cells and advance the field.
Introduction
The upper GI tract comprises the stratified squamous esophagus and the columnar stomach, joined by the squamous-columnar junction (SCJ) which is lined by a transitional epithelium (Jiang et al., 2017). While the stomach and esophagus belong to the same functional unit, they contain distinct stem and progenitor cell populations needed to sustain rapid cellular turnover. Under injurious conditions, such as bile acid reflux and Helicobacter pylori infection, abnormal differentiation of progenitors leads to a mixture of cells with gastric and intestinal differentiation (intestinal metaplasia, IM) in the SCJ or stomach, respectively, which is associated with an increased cancer risk (Cancer Genome Atlas Research et al., 2017; Giroux and Rustgi, 2017; Hayakawa et al., 2016c; Lavery et al., 2014). Emerging models of metaplasia and cancer have proposed new cell-of-origin and molecular mechanisms. Novel cell populations have also been identified in the upper GI. Here, we discuss new findings and individual stem cell populations and their roles in metaplasia and cancer development. We also elaborate the molecular and cellular mechanism promoting abnormal differentiation in the esophagus and stomach.
The stratified squamous epithelium in the esophagus is maintained by stem/progenitor cells (squamous basal cells, SBCs) located in the bottom layers (Fig. 1A). In contrast, the stomach consists of region-specific columnar epithelia maintained by distinct stem/progenitor cells. The stomach is divided anatomically into three separate regions which have been labeled the antrum, corpus, and cardia (Fig. 1B). The antropyloric glands located in the distal stomach contain primarily mucus secreting cells along with tuft and gastrin-secreting cells (G-cells) (Fig. 1C). Oxyntic glands reside more proximally in the corpus and contain parietal cells secreting gastric acid, chief cells secreting digestive enzymes, tuft cells secreting prostaglandins, along with mucous and endocrine cells (Fig. 1D). Cardiac glands are located most proximally, immediately adjacent to the esophagus, and are similar to antropyloric glands but lack G-cells and contain more abundant tuft cells. All three types of gastric glands contain a narrowed mid-gland region known as the isthmus, where proliferating progenitors supply their progenies bidirectionally (Fig. 1C–D). Notably, the mouse stomach contains additionally a stratified squamous forestomach in the proximal region, representing an extension of the squamous esophagus, where in humans there resides a structure called the fundus or fornix (Fig. 1E).
Figure 1. Anatomy and gland structure of the upper GI tract.
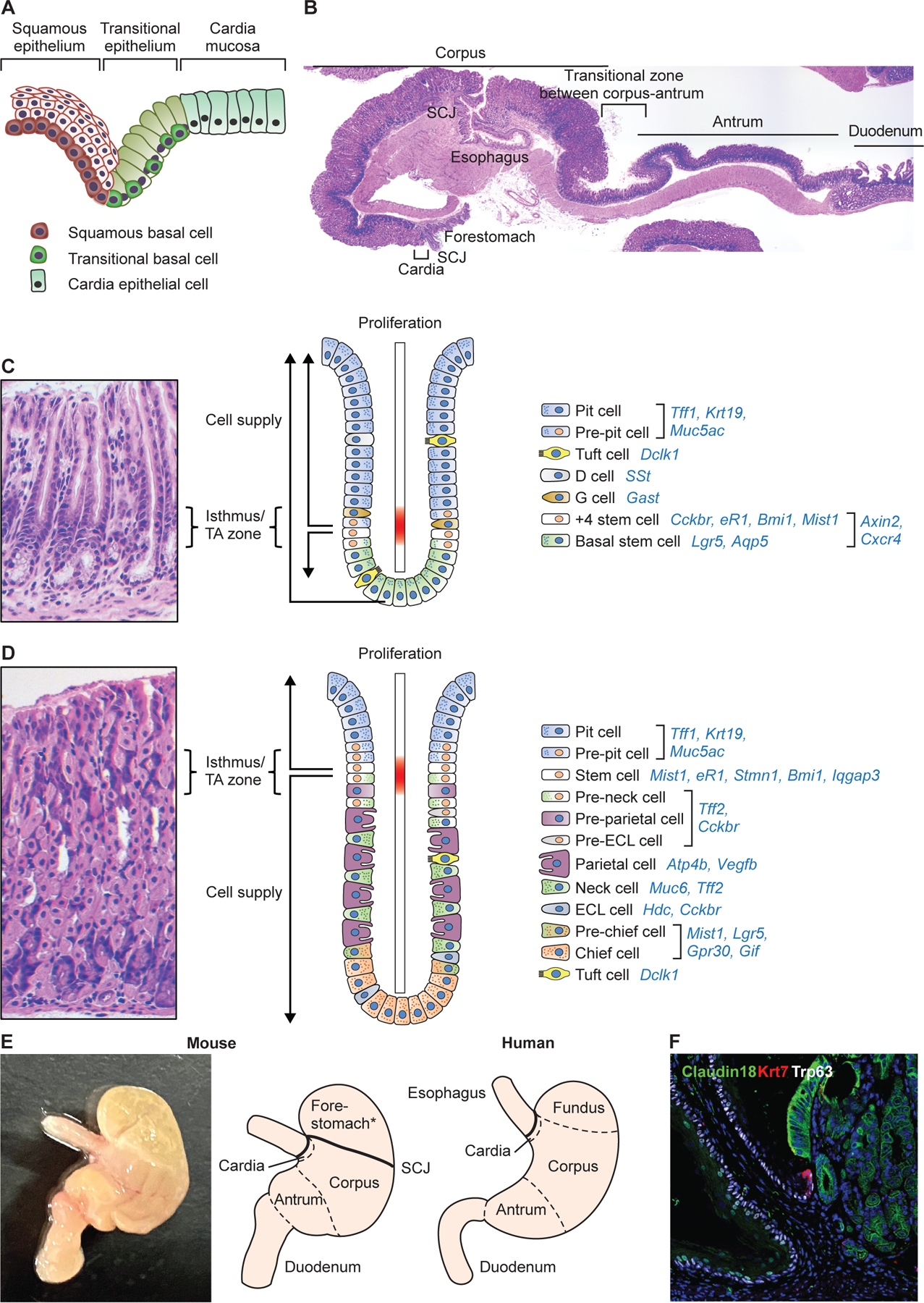
(A) Schematics diagram depicts the transitional zone located between the esophagus and stomach. Note different basal cell populations in the stratified squamous epithelium and transitional zone. (B) H&E images of mouse esophagus~stomach~duodenum. (C) H&E staining and schematic image depicting the structure and cell types with their molecular markers of gastric antral glands. There appears to be two different stem cell populations, +4 isthmus stem cell and basal stem cells. (D) H&E staining and schematic image depicting the structure and cell types with their molecular markers of gastric corpus glands. Stem cells and progenitors reside within the upper isthmus and supply mature cell types bidirectionally. While basal chief cells are thought to be generated via transdifferentiation from neck cells, several recent studies suggested that a subset of chief cells may self-renew and compose independent clones. (E) Gross image and schematic diagram of mouse and human stomach. Note that the mouse stomach has a forestomach that is composed of a stratified squamous epithelium. Cardia contains only a few glands both in mice and human. (F) The transitional epithelium (Krt7+Trp63+Claudin18-) at the squamous-columnar junction.
The SCJ links the fore- and hindstomach in mice, in contrast to linking esophagus and stomach in humans. The transitional epithelium in the SCJ is maintained by transitional basal cells (TBCs) which are distinct from SBCs (Fig. 1F)(Jiang et al., 2017). TBCs have emerged along with gastric cardia stem cells as a possible source for IM and esophageal adenocarcinoma, a malignancy that resembles more closely gastric cancer as opposed to esophageal squamous cell carcinoma (Cancer Genome Atlas Research et al., 2017; Hayakawa et al., 2016c). IM in the SCJ and stomach involves reactivation of developmental signaling, especially with prolonged inflammation (Fig. 2). These signaling pathways (e.g. Wnt, Bmp, Notch) are critical for the morphogenesis of the upper GI tract and setting up esophageal-gastric and gastric-intestinal boundaries, which has been reviewed extensively (Kim and Shivdasani, 2016; Udager et al., 2010; Zhang et al., 2021).
Figure 2. Clonal expansion of metaplastic glands.
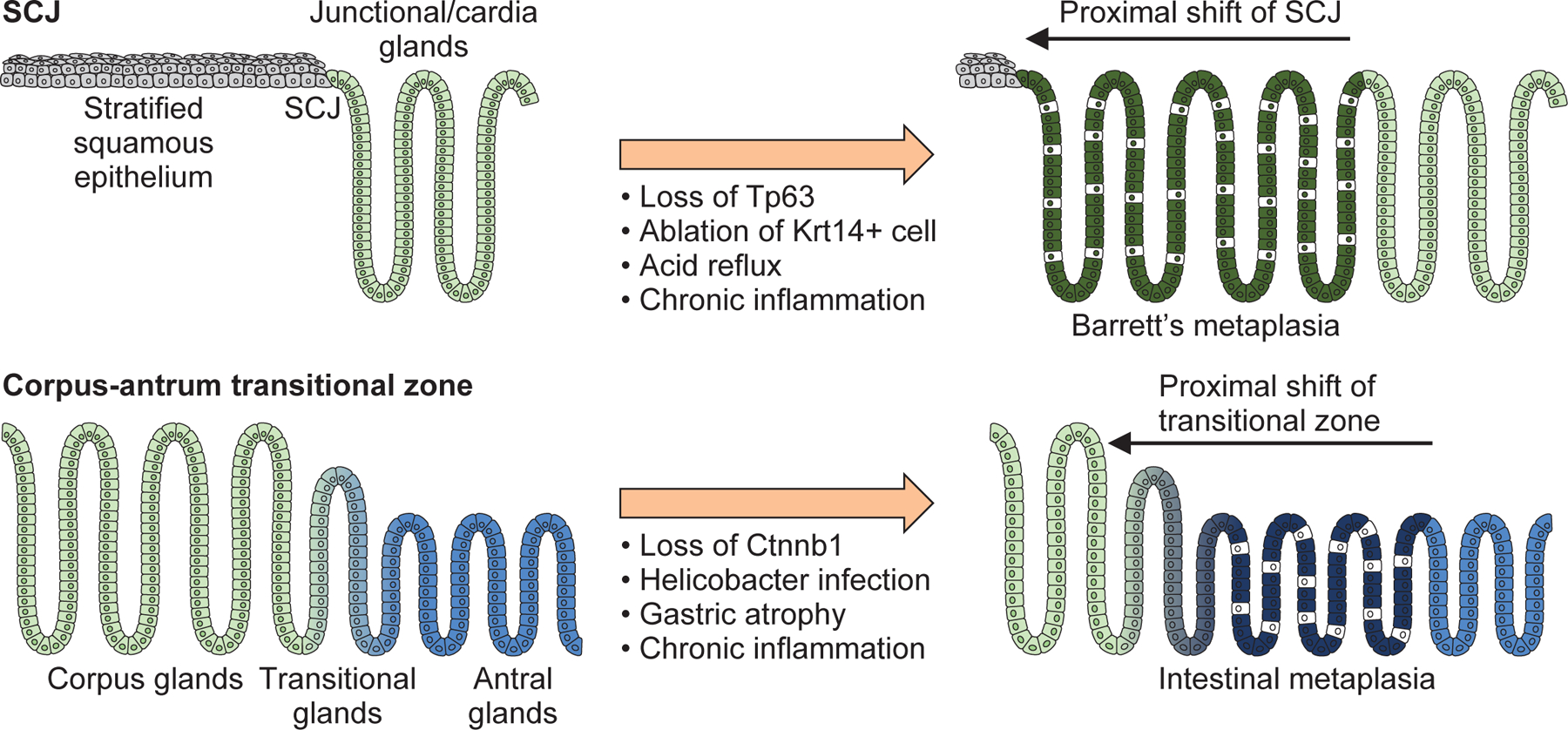
Schematic image depicting the junctional shift at the SCJ and the transitional zone of corpus/antrum glands. Loss of the key transcriptional factors during embryonic stage and chronic inflammation in adult causes similar clonal expansion of metaplastic glands from the distal side.
Chronic inflammation triggers signaling alternations in tissue stem/progenitor cells, in part through secretory factors from nearby stromal and immune cells. Indeed, chronic inflammation secondary to acid reflux or Helicobacter pylori infection is a key trigger of carcinogenesis in the esophagus and stomach (Fig. 2), respectively. It can lead to IM, although the precise relationship of metaplasia to cancer remains uncertain. In mouse models of Barrett’s esophagus (BE, IM of the esophagus) or chronic gastritis, accumulated inflammatory cells and other stroma cell types contribute to stem cell expansion and activation of Wnt, Notch, and MAPK pathways in metaplastic cells via secretion of cytokines and chemokines (Fang et al., 2018; Hayakawa et al., 2015a; Hayakawa et al., 2017; Kunze et al., 2020; Munch et al., 2019; Nienhuser et al., 2020; Quante et al., 2012; Quante et al., 2011; Sakitani et al., 2017; Sigal et al., 2017; Zhao et al., 2014).
Most GI glands are monoclonal, suggesting that normal gastric glands are maintained by long-lived stem cells (Bjerknes and Cheng, 2002; Nomura et al., 1998; Tatematsu et al., 1994; Thompson et al., 1990). Given the monoclonality of metaplastic and dysplastic clones in the inflamed stomach and IM, confirmed through analysis of mitochondrial DNA mutations (Lavery et al., 2014; McDonald et al., 2008; Nicholson et al., 2012), it seems likely that metaplasia and dysplasia also arise from a stem cell origin. Below, we review the biology of stem and progenitor cells of the upper GI tract and their roles in response to injury and the development of metaplasia and carcinoma.
Overview of Gastrointestinal Stem Cells
Early studies of gut stem cells focused on the intestinal crypt (Potten et al., 1978; Potten et al., 2002), and identified a small population of label-retaining cells (LRCs) in the intestinal crypt, situated within the proliferative zone and above the Paneth cells, which appeared to be responsible for cell replacement following radiation-induced injury (Potten et al., 2002). Although LRCs in the upper GI tract have been less characterized, similar cell types may exist in the gastric isthmus and BE (Pan et al., 2013). In 2007, Clevers et al. identified Lgr5-expressing intestinal stem cells residing between Paneth cells at the crypt base, using tamoxifen-inducible lineage-tracing in Lgr5-CreERT mice (Barker et al., 2007). While Lgr5 has remained the major focus of intestinal stem cell research, other groups using similar lineage tracing approaches reported different intestinal stem cell population, expressing a variety of markers, residing at or above the “+4” position (Asfaha et al., 2015; Ayyaz et al., 2019; Barriga et al., 2017; Montgomery et al., 2011; Powell et al., 2012; Takeda et al., 2011), and that appeared to function in the absence of Lgr5+ cells (Tian et al., 2011; Yan et al., 2012) (Fig. 3A).
Figure 3. Gastrointestinal stem cell niche.
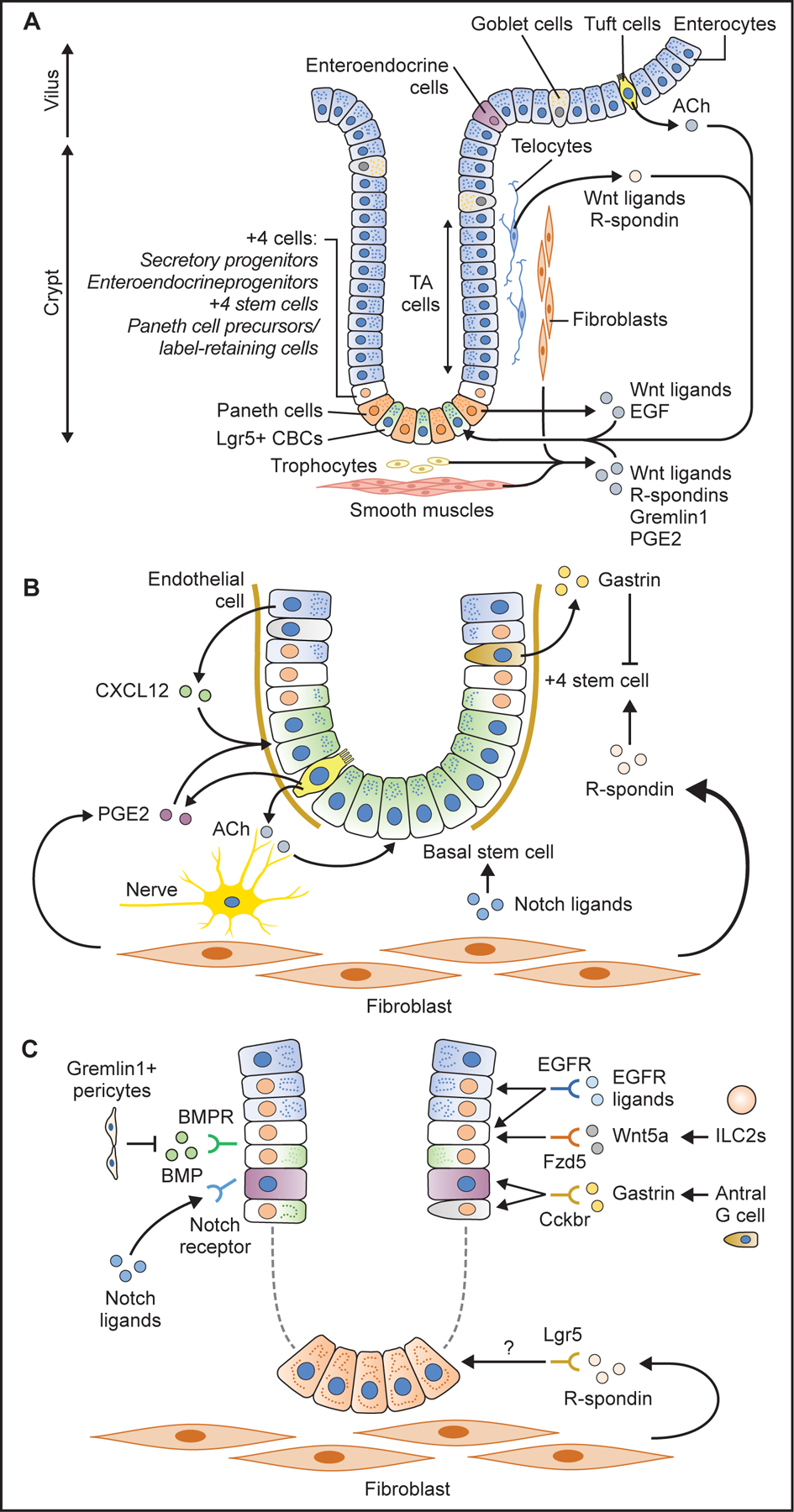
(A) Schema of intestinal stem cells and their niche. Lgr5+ CBCs at the base and +4 stem/progenitor cells are supported by surrounding stromal cells which secrete numerous niche factors such as R-spondins, Wnt ligands, Gremlin1, epidermal growth factor (EGF), prostagrandin E2 (PGE2). Epithelial tuft cells play a role by producing acetylcholine (ACh). (B) Antral stem cell niche factors. R-spondin 3 from stromal myofibroblasts activates +4 stem cells. Gastrin produced from G cells suppress +4 stem cell activity via CCK2R receptor. Notch ligands, ACh from nerves and tuft cells, PGE2 from tuft cells, as well as CXCL12 from endothelial cells promote stem cell expansion and contribute to cancer development. (C) Corpus stem cell niche factors. Isthmus stem cells and progenitors are supported and activated by specific niche factors such as Notch ligands, Wnt5a, gastrin, and EGFR ligands through their corresponding receptors.
GI stem cells are confined to the stem-cell zone in part because of the localized production of essential factors via paracrine secretion by surrounding cells that constitutes the stem cell niche. The stem cell niche containing stromal cells, immune cells, and a subset of epithelial cells assists the maintenance of stem cell longevity, self-renewal and multipotentiality (Farin et al., 2012; Sato et al., 2011). In the intestine, R-spondin, Wnts, and Gremlin1 (a BMP antagonist) are secreted from mesenchymal cells, including telocytes, pericytes, trophocytes, and fibroblasts, all of which maintain stem cell functions and tissue homeostasis (Aoki et al., 2016; Farin et al., 2012; McCarthy et al., 2020; Shoshkes-Carmel et al., 2018; Stzepourginski et al., 2017; Worthley et al., 2015) (Fig. 3A).
One current model of stem cell hierarchy is that intestinal epithelium is normally maintained by actively proliferating crypt basal stem cells that mostly express Lgr5, while in injury states there is dynamic cellular plasticity that contributes to epithelial regeneration through dedifferentiation from reserve progenitor cells residing +4 region into active stem cells (Li and Clevers, 2010). An alternative model is that there are no dedicated reserve progenitors but instead broad plasticity possessed by many post-mitotic cells and short-lived progenitor cells (de Sousa and de Sauvage, 2019). Finally, a more traditional hierarchical model has as yet not been excluded. Nevertheless, while studies of the intestine have provided a useful framework, the stem cell dynamics in the upper GI tract appear to be a bit different. Upper GI stem cells express a unique set of markers and are maintained by distinct niche. As noted below, esophageal and gastric stem cells are dissimilar, and while antral and corpus stem cells are more similar, they are situated differently within the gastric glands. Thus, although several markers (e.g., Lgr5) are shared between the upper GI and intestinal stem cells, one should not assume that they behave equivalently. Furthermore, as discussed later, there are numerous limitations to CreERT-dependent lineage tracing, including the fact that all CreERT drivers are expressed in multiple cell types within GI tissues, making it difficult to distinguish between stem, progenitor and committed cells. Finally, tamoxifen itself has toxic effects in most GI tissues, particularly in the stomach, indicating the need for newer approaches to resolve the many outstanding questions.
Stem cells in the stomach
Early studies involving labeling of proliferating gastric epithelial cells showed that they emerge from the gastric isthmus and supply progenies bidirectionally towards both the top and bottom of the glands, suggesting that stem/ progenitor cells reside primarily within the isthmus (Karam and Leblond, 1993b; Lee and Leblond, 1985b). In particular, in the corpus, a rare undifferentiated, granule-free stem cell was proposed to reside in the isthmus of gastric units in mouse (Karam and Leblond, 1993a; Mills and Shivdasani, 2011). Similarly, stem cells with an undifferentiated, granule-free appearance reside in the isthmus of the antral glands, with clear evidence of bidirectional supply of cells from the stem-cell zone (Lee and Leblond, 1985a). Both isthmus regions also contain immature cells with various types of granules; these were thought to be lineage-committed progenitor cells originally derived from granule-free stem cells.
Antral stem cells
Following the discovery of intestinal Lgr5+ cells, Lgr5+ antral stem cells were identified at gland base using the same Lgr5-CreERT mouse line (Barker et al., 2010). Lgr5-expressing cells at the antral gland base appeared to continuously generate progeny and trace entire antral glands. A multicolor cell labeling study suggested that multiple antral Lgr5+ cells function as active stem cells in each gland, but over time there was a subsequent monoclonal conversion to a single color, interpreted as the outcome of neutral drift (Leushacke et al., 2013). Lgr5 was proposed to label primarily crypt basal stem cells, in the +1 to +3 region, below the proliferative isthmus region of the antrum, contrary to earlier expectations. In this basal stem cell model, proliferative cells in the isthmus were considered transit-amplifying progenitors. However, this concept of transit-amplifying progenitors has been less well defined, and a subset of Lgr5+ cells also reside within the isthmus (Sigal et al., 2017).
Analogous to the intestine, several +4 antral stem cells were subsequently identified in the isthmus of antral glands. These +4 antral stem cells were characterized by the expression of Cckbr, Bhlha15 (also known as Mist1), Bmi1, and eR1, and had a higher proliferation rate than antral Lgr5+ stem cells (Arnold et al., 2011; Hayakawa et al., 2015a; Hayakawa et al., 2015b; Matsuo et al., 2016; Sakitani et al., 2017; Sigal et al., 2017; Yoshioka et al., 2019) (Fig. 1C). In addition, Axin2 (a Wnt target) and a chemokine receptor Cxcr4, are expressed in the lower third of the antral glands, including in the isthmus and gland base (Sakitani et al., 2017; Sigal et al., 2017). The Axin2+ population comprised both Lgr5+ basal stem cells and Lgr5- +4 isthmus stem cells. As expected, Axin2+ cells demonstrated robust long-term lineage tracing ability in Axin2-CreERT mice, and Axin2+Lgr5- cells were able to repopulate entire glands after ablation of the Lgr5+ population. Interestingly, exogenous R-spondin which acts through Lgr5/4 receptors, activates and expands Axin2+Lgr5- isthmus stem cells but not basal Lgr5+ cells (Sigal et al., 2017). These findings suggest that a major target of R-spondin is likely to be the Axin2+Lgr5- isthmus stem cell (Fig. 3B).
Notch signaling also appears to be important for antral stem/progenitor function, as loss of Notch1/2 leads to secretory cell hyperplasia, reduced proliferation and a reduced number of Lgr5+ cells (Gifford et al., 2016). Notch signaling may also be important in the transition to gastric adenocarcinoma (Hibdon et al., 2019). Interestingly, Cckbr+ antral stem cells are Notchlow, and thus are not directly modulated by Notch signaling (Chang et al., 2020).
The means of cell division may also differ between crypt basal stem cells and +4 stem cells. Most Lgr5+ crypt basal stem cells divide symmetrically, with each stem cell giving rise to two stem cells. In this model, the total number of Lgr5+ stem cells is maintained at a stable level via neutral drift, whereby stem cell competition allows for a few dominant stem cells to persist within the niche (Leushacke et al., 2013). In contrast, +4 stem cells likely undergo asymmetric cell division, thereby generating another stem cell and a daughter progenitor cell, the fate of which varies (Guo and Ohlstein, 2015 ). One type of +4 antral stem cell, Cckbr+ cells, undergo predominant asymmetric cell division, expressing Numb and DLL1 and showing long-term label retention (Chang et al., 2020). In contrast, during inflammation and carcinogenesis, the Cckbr+ stem cell population shifts to predominant symmetric cell division, in part due to loss of gastrin-dependent signals, resulting in an expansion of the stem cell pool, driven by the need for tissue regeneration. However, while labeling a relatively small number of antral cells, Cckbr-CreERT is still less than 100% specific for antral stem cells, similar to every other stem cell marker. Nevertheless, earlier studies of LRCs in the intestine supported the concept of asymmetrically-dividing stem cells, with symmetric division (leading to crypt fission) occurring only during regeneration and in carcinogenic states (Booth and Potten, 2000; Potten et al., 2002).
In the gastric antrum, R-spondin 3 is produced in part by myofibroblasts residing near the muscularis mucosae, and contributes to the antral stem cell niche by activating Axin2+ +4 stem cells (Sigal et al., 2017). By contrast, antral crypt basal stem cells expressing Lgr5 or other markers such as Aqp5 are supported and activated by Wnt signaling proteins via, among other targets, the Frizzled-7 receptor (Flanagan et al., 2017; Tan et al., 2020). In addition, gastrin may play a unique role in the antral stem cell niche. Gastrin is secreted from G-cells residing near Cckbr+ stem cells expressing the gastrin receptor. When the gastrin gene is deleted, Cckbr+ stem cells become more proliferative and start to divide symmetrically, suggesting that gastrin maintains the quiescence and/or inhibits antral stem cells (Chang et al., 2020). While NotchlowCckbr+ stem cells are not directly modulated by Notch signaling, Notch activation in the antral glands leads to loss of G-cells, resulting in enhanced proliferation of Cckbr+ stem cells in a gastrin-dependent manner. Given that gastrin deficiency accelerates antral tumorigenesis in mice, antral G-cells appear to be important niche cells, crucial for repressing the stem cell expansion needed for progression to gastric cancer (Hayakawa et al., 2016a; Hayakawa et al., 2015b; Lee et al., 2017).
Furthermore, antral stem cells express various other receptors that mediate responses to stromal niche signals. For example, Lgr5+ cells in the gastric antrum strongly express muscarinic acetylcholine receptors and are regulated by acetylcholine, released from both cholinergic nerves and ChAT-expressing tuft cells (Hayakawa et al., 2017). Moreover, tuft cells produce prostaglandins (Middelhoff et al., 2017; Schutz et al., 2019), which is also secreted by stromal fibroblasts to support intestinal stem cells (Roulis et al., 2020) and presumably gastric stem cells. Both crypt basal and isthmus antral stem cells express the CXCR4 receptor, while its ligand CXCL12 is expressed in vascular endothelial cells adjacent to gastric stem cells (Hayakawa et al., 2015a; Sakitani et al., 2017). Expansion of stem cell niches often precedes the expansion of stem cells during carcinogenesis, which contributes to increase in symmetric cell division and number of stem cells (Chang et al., 2020; Hayakawa et al., 2016b; Sakitani et al., 2017; Sigal et al., 2019).
Corpus stem cells
In contrast to the robust lineage tracing ability of antral Lgr5+ cells, no lineage tracing events were observed initially from corpus Lgr5-expressing cells in Lgr5-CreERT mice (Barker et al., 2010; Nam et al., 2012). Lgr5 does mark basal glandular cells in the corpus, but these represent a large subset of chief cells. Thus, stem cells in the corpus isthmus region are labeled by a distinct set of stem cell markers. Initially, a few corpus cell markers such as Sox2 and Lrig1 were shown to lineage traced corpus glands (Arnold et al., 2011; Powell et al., 2012). However, while they traced isthmal progenitor-like cells, they also marked differentiated cells. Subsequently, a more detailed analysis of corpus stems was performed using Mist1-CreERT mice. Mist1 (Bhlha15) was known to be a marker of gastric chief cells, but was shown by in situ hybridization to mark rare, solitary cells within the corpus isthmus. Consistent with this observation, Mist1-CreERT was expressed in a significant population of corpus granule-free stem cells (Hayakawa et al., 2015a) (Fig. 1D). Mist1+ isthmus cells are relatively slow cycling, dividing on average every 5 days, consistent with the original expectations for mammalian adult stem cells (Malam and Cohn, 2014). Mist1+ stem cells supply daughter cells bidirectionally from the isthmus, generating the entire corpus gland as evidenced by lineage tracing. Thus, Mist1+ isthmus stem cells appear to act as bone fide, quiescent corpus stem cells, contributing to the slow, monoclonal conversion of corpus gland units. However, given the expression of Mist1+ in other cells (mainly zymogenic or chief cells), more work is needed to define the Mist1+ isthmus cell.
After the discovery of Mist1+ stem cells, other isthmus stem and progenitor cell markers were reported, including eR1, Stmn1, Bmi1, Iqgap3 (Arnold et al., 2011; Han et al., 2019; Matsuo et al., 2020; Matsuo et al., 2016; Powell et al., 2012; Yoshioka et al., 2019). Expression of Stmn1 and Iqgap3 was mostly confined to the isthmus and overlapped with the vast majority of Ki67+ cells in this region. These isthmus stem cells give rise to all lineages in the corpus, including surface pit cells, parietal cells, mucous neck cells, and basal chief cells (Matsuo et al., 2020), following a long-term observation (e.g., > 1 year). However, similar to all other corpus isthmus markers to date, based on multi-color fluorescent tracing, Stmn1+ cells likely exist as heterogeneous populations, including long-lived corpus stem cells as well as short-lived proliferating progenitors. Bmi1+ cells in the corpus isthmus are relatively quiescent, but they enter a more active cycling state after epithelial injury. Similarly, isthmal cells expressing Mist1, eR1, or Stmn1 show more rapid lineage tracing following acute epithelial damage or Kras mutation, contributing to the monoclonal conversion of glands (Han et al., 2019; Hayakawa et al., 2015a; Matsuo et al., 2016).
Short-lived, lineage-committed progenitors can be found within the corpus isthmus, characterized by expression of unique markers including Tff2 and Cckbr (Quante et al., 2010; Sheng et al., 2020). Cckbr is robustly expressed in acid-producing parietal cells and histamine-producing enterochromaffin-like (ECL) cells, and mediates acid secretory responses to gastrin. However, Cckbr expression is also seen in the corpus isthmus, and the number and proliferation rate of Cckbr+ isthmal cells are increased by hypergastrinemia (Sheng et al., 2020). Studies with Cckbr-CreERT and Hdc-CreERT mice revealed that while mature ECL cells lack the ability to proliferate or lineage trace gastric glands, Cckbr+ isthmus cells can supply daughter ECL and parietal cells in normal state. Cckbr+ isthmus cells are the source of the expanded ECL cell pool that accumulate with chronic hypergastrinemia (Sheng et al., 2020). Therefore, corpus Cckbr+ cells are distinct from antral Cckbr+ stem cells in terms of their response to gastrin.
Like antral stem cells, corpus stem cells are supported by stem cell niches (Fig. 3C). Wnt, R-spondin, EGF, and noggin are needed for long-term 3D-culture of corpus organoids (Bartfeld et al., 2015; Stange et al., 2013), suggesting that these growth factors are present in the corpus stem cell niche, similar to other parts of the GI tract. In addition, Notch signaling plays a role in the corpus stem cell niche, as Notch activation promotes isthmus proliferation and tumorigenesis (Demitrack et al., 2016). Similarly, activation of the EGF receptor (EGFR) pathway via overexpression of EGFR ligands or induction of mutant Ras/Raf leads to robust isthmal cell proliferation (Kinoshita et al., 2019; Nomura et al., 2005; Okumura et al., 2010; Thiem et al., 2016). Given the specific expression of the EGFR protein in the upper third of the corpus glands (Sheng et al., 2020), EGFR signaling is likely a key pathway that regulates corpus isthmus stem cell function, although the exact cell types producing EGFR ligands remain poorly defined. The noggin/BMP pathway may also be important in corpus homeostasis, as unique stromal cells expressing Gremlin1 reside in close proximity to the corpus isthmus stem cells (Worthley et al., 2015). Since Wnt/R-spondin target genes are not highly expressed in the corpus isthmus, the role of the Wnt/R-spondin pathway at this site remains unclear. In fact, the Wnt/R-spondin target genes Lgr5 and Axin2 are highly enriched at the gland base, as stromal cells near the corpus gland base produce R-spondin (Sigal et al., 2017; Sigal et al., 2019).
Instead of strong dependence on the canonical Wnt pathway, corpus stem cells may be supported and activated by the non-canonical Wnt ligand, Wnt5a. Wnt5a expression is restricted to stromal cells within the isthmus, with type 2 innate lymphoid cells (ILC-2) one of the major sources (Hayakawa et al., 2015a). In regenerative and inflammatory states, Wnt5a+ ILC2s accumulate around the stem-cell zone, responding to IL-33 and CXCL12 secreted from surface pit cells and endothelial cells, respectively, contributing to activation of isthmus stem cells (Hayakawa et al., 2015a; Satoh-Takayama et al., 2020). Frizzled-5, a putative receptor for Wnt5a, is robustly but specifically expressed in gastric isthmus stem/progenitor cells, suggesting that Wnt5a may activate stem cells via this receptor (Nienhuser et al., 2020). Further studies are needed to confirm the role of Wnt5a in modulating corpus progenitors.
Gastric chief cells as potential reserve stem cells
As in the intestine, corpus glands may contain other cell types that have (or can acquire) stem cell function, particularly following severe damage that may diminish normal stem cells. Gastric chief cells have been proposed as the main reserve corpus stem cells following injury, given evidence supporting the potential for dedifferentiation and limited levels of proliferation, and the term “base stem cells” proposed (Han et al., 2019). Chief cells express a number of markers including Lgr5, Mist1, Troy, and GPR30, with GPR30 being the most specific (Hata et al., 2020; Leushacke et al., 2017; Nam et al., 2010; Stange et al., 2013). However, several issues have been raised regarding the interpretation of findings suggesting the conversion of chief cells into stem cells. First, while Mist1-CreERT and Lgr5-CreERT mice label many chief cells, the transgenes are also expressed in cells in the isthmus. Abundant data now indicate that many of the lineage tracing events reported previously are due to isthmus cells rather than chief cells (Hayakawa et al., 2015a; Kinoshita et al., 2018). Second, administration of high dose tamoxifen in combination with CreERT expression may be toxic and produce off-target expression, reducing the specificity of labeling and limiting interpretation of corpus lineage tracing events (Leushacke et al., 2017). Third, in most gastric injury models, stem and progenitor cells in the isthmus region are largely preserved and exhibit more active proliferation within the niche following injury (Han et al., 2019; Hayakawa et al., 2015a; Kinoshita et al., 2018). Therefore, it is important to examine carefully the immediate effects on chief cells and isthmus stem cells in the context of specific models of gastric injury.
Current models suggest that glandular regeneration following injury requires new or existing stem cells. The initial report of Troy-CreERT knockin mice suggested lineage tracing events associated with Troy+ chief cells that emerged when corpus isthmus stem cells were damaged by 5-FU administration (Stange et al., 2013). However, these results need to be interpreted cautiously, as Troy expression is likely not limited to chief cells, but present in other cell type including parietal cells and isthmal progenitors. Moreover, the observed phenotype is strongly dependent on Troy haploinsufficiency in this knockin line. Indeed, lineage tracing from Troy+ chief cells could not be reproduced with BAC transgenic Troy-CreERT mice until one endogenous Troy allele was deleted (Hayakawa et al., 2015a). Furthermore, the emergence of new corpus stem cells at the gland base likely requires a full complement of niche cells needed for corpus stem cell self-renewal. Interestingly, a few days after severe corpus injury, isthmal cell proliferation and the normal bidirectional cellular flow are restored to normal, suggesting that any basal stem cell niche and zymogenic dedifferentiating response to acute injury is likely transient (Kinoshita et al., 2018).
In the vast majority of gastric injury models (including high dose tamoxifen), chief cells have unique cellular kinetics, as they are rapidly depleted following acute injury. Thus, in order to follow the cellular fate of chief cells following gastric injury, it is necessary to employ a tamoxifen-independent genetic tracing system. To this end, a tetracycline-inducible (rtA-tetON) system was developed, whereby Gpr30-rtTA mice were crossed to tetO-Cre; R26-reporter mice, and specific chief cell labeling and tracing was achieved without tamoxifen use (Hata et al., 2020). Interestingly, following high dose tamoxifen, labeled Gpr30-expressing chief cells did not dedifferentiate but instead gradually disappeared. Given that knockout of Gpr30 attenuated the effect by tamoxifen on loss of chief cells, Gpr30, a GPCR form of estrogen receptor expressed specifically by chief cells, may directly bind to tamoxifen and mediate cell-specific effects.
Nevertheless, a working model of two separate progenitor zones (isthmal and basal) within the proximal stomach has been described (Han et al, 2019), and long-term lineage tracing experiments suggest that a subset of chief cells may possess properties of longevity and self-renewal, with the ability to generate clones independent of isthmus-derived lineages (Burclaff et al., 2019; Han et al., 2019). While these recent studies confirm that isthmus-derived clones are the main source of clonal expansion, particularly following mucosal injury, chief cell-derived clones may remain stable at the gland base without ongoing contribution from the isthmus. Thus, in contrast to the expected monoclonality of the vast majority of gastrointestinal glands, and clear evidence of downward differentiation from isthmus progenitors to mucous neck cells to chief cells (Huh et al., 2010; Quante et al., 2010; Ramsey et al., 2007), a basal cell population that escapes monoclonal conversion from isthmus-derived clones may exist.
Stem Cells of the Esophagus and Esophagogastric Junction
Basal stem cells
Basal cells are stem/progenitor cells responsible for the quick turnover of the epithelium, which occurs approximately every 4–6 days in mice and every 11 days in humans (Pan et al., 2013). In the mouse esophagus, proliferating cells are limited to the basal layer of the stratified epithelium, in contrast to the human esophagus where the proliferating cells are enriched in the epilayer or parabasal layers (1–3 layers above the basal layer). Additionally, the basal layers in the human esophagus are more complex, consisting of the papillary basal layer (PBL) and the interpapillary basal layer (IBL) (Seery and Watt, 2000) (Fig. 4A–C). Whether the basal cell is a heterogeneous or homogeneous population has been a topic of intense research and debate for the past 20 years, which we summarize below.
Figure 4. Heterogeneous models of esophageal stem cells.
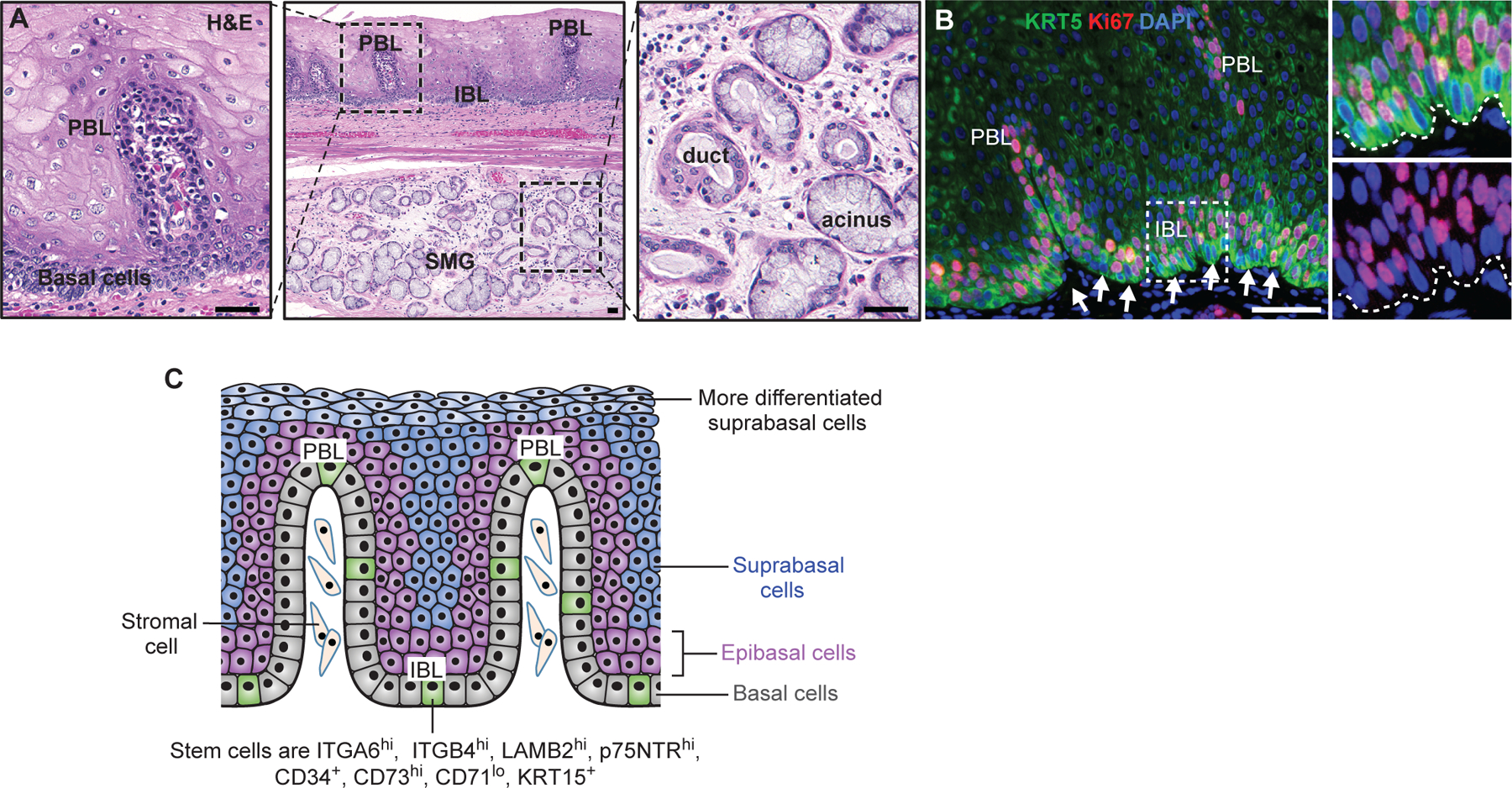
(A) The presence of papilla(e) and submucous gland (SMG) in the human esophagus. (B) Basal cells (Krt5+) are highly proliferative. Note cells at the bottom layer (arrows) are not as proliferative (Ki67+) as epibasal layers. (C) Heterogeneous basal stem cells. Multiple stem cell subpopulations are present in the basal layer which consist of papillary and interpapillary regions. These stem cell subpopulations express different levels of proteins such as integrins and keratins. See text for details. Scale bars: 50µm.
Basal cells are heterogeneous (Fig. 4C).
Basal cells divide approximately four times more frequently in the PBL than in the IBL of human esophagus (Seery and Watt, 2000). Earlier studies suggested differential expression of keratin proteins in the PBL versus IBL. For example, Krt14 and Krt15 transcripts exhibit low and patchy expression pattern in IBL basal cells, in contrast to uniformly high levels in the PBL and epibasal layers, leading to the hypothesis that IBL basal cells are the ‘least differentiated’ cells (Viaene and Baert, 1995). In line with this, IBL basal cells (integrin β1low/β2 laminin chainhigh) were shown to be more clonogenic, forming large colonies when cultured on a 3T3 feeder layer (Seery and Watt, 2000). Basal cells in the mouse esophagus can also be separated into subpopulations. For example, one study demonstrated that basal cells include three subpopulations, integrin α6high/CD71low, integrin α6high/CD71high, integrin α6low/CD71high (Croagh et al., 2007). In another study, mouse basal cells were also found to contain three subpopulations based on the expression levels of integrin α6/β4 and CD73. Among them, the α6/β4high/CD73+ population represents cells exhibiting the highest clonogenic capability in organoid culture (DeWard et al., 2014).
The premise of basal cell heterogeneity was further supported by the Hoechst 33342 dye efflux assay, which identifies a side population that is able to form a stratified squamous epithelium in a 3D culture system (Kalabis et al., 2008). The side population harbors BrdU-labeling retaining cells, rarely-dividing or slowly-cycling cells that reside in the basal compartment. Upon transplantation, the expanded side population integrates into the regenerated epithelium following injury in the esophagus, underscoring that such a population participates in tissue regeneration (Kalabis et al., 2008). It is also noteworthy that TBCs (Krt5+ Krt7+) in the SCJ are distinct from SBCs (Krt5+ Krt7-) in other parts of the esophagus (Fig. 1A)(Jiang et al., 2017).
Lineage tracing of esophageal basal cells was first performed with a transgenic Krt5-CreER mouse line (Jovov et al., 2011). Krt5 is expressed in all esophageal basal cells, and lineage-labelled basal cells demonstrated self-renewal and differentiation capability. More recently, a Krt15-CrePR1 transgenic line which contains the progesterone receptor–fused (PR1-fused) Cre was established to trace the Krt15+ lineage (Giroux et al., 2017). The study showed that 13.6% of basal cells were labeled with five consecutive injections of RU486, which was used to activate genetic labeling. Further comparison of transcripts expressed in Krt15+ vs Krt15- basal cells revealed Wnt signaling-associated genes such as Amer1, Fzd8 and Apcdd1 in the Krt15+ subpopulation. Moreover, Krt15+ basal cells were capable of forming a keratinized stratified squamous epithelium in air-liquid interface culture. Genetic depletion of Krt15+ basal cells led to thinning of the epithelium, accompanied by reduced proliferation in the esophagus of Krt15-CrePR1; R26iDTR mice, confirming the importance of the Krt15+ subpopulation in the maintenance of homeostasis. Krt15+ cells are radioresistant and facilitate tissue regeneration following high-dose irradiation. Krt15+ cells-derived cells organize as clonal units and give rise to all differentiated lineages (Giroux et al., 2017). This is the most compelling in vivo model to date that illuminates the premise that a subset of basal cells has properties of stem cells. In line with these findings, a recent single cell analysis revealed the existence of quiescent COL17A1high; KRT15high stem/progenitor cell population in the basal layer of the human esophagus (Busslinger et al., 2021).
Basal cells are homogenous
Alternatively, it has been suggested that the esophageal epithelium is maintained by a single progenitor cell, similar to models of the epidermis (Clayton et al., 2007; Doupe et al., 2012; Piedrafita et al., 2020). The single-progenitor model proposes that dividing basal cells have an equal probability of generating two basal cells, two differentiated cells or one basal cell and one differentiating daughter cell (Clayton et al., 2007; Doupe et al., 2012). Lineage tracing over one year showed that the clone number decreased due to differentiation, while the size of the remaining clones increased linearly with time. The clone size increased steadily and the size distribution acquired long-term scaling behavior, suggesting a single functionally equivalent population of cells dividing at the same rate (Clayton et al., 2007; Doupe et al., 2010). It is worth pointing out that the single-progenitor model is largely built on the analysis of the mouse esophagus. However, in the human esophagus, the proliferating cells are enriched in the epibasal layers but not in the basal layer (Seery and Watt, 2000) (Fig. 4B), and thus it will be interesting to assess whether the model can be similarly applied to the human esophagus. In addition, the conclusion regarding the absence of slow-cycling or quiescent esophageal stem cells emanates from findings in the HGFP mouse esophagus, although the sensitivity and specificity of HGFP for LRC as defined by Potten(Potten, 1974) is unknown. Notably, in the small intestine, label retention in the HGFP mouse is not necessarily a feature of quiescent or slow-cycling stem cells (Tian et al., 2011).
In summary, epithelial cells in the basal layer clearly contain multiple populations based on molecular profiling. Whether differences in gene expression are stochastic or representative of stem/progenitor cells in different states remains to be determined. In addition, whether similar models can be applied to the human esophagus remains unknown.
Cellular Origin of Metaplasia and Cancer in the Upper GI tract.
Cellular origin of gastric cancers
Studies in mice suggest that mutations in GI stem cells often lead to cancer (Barker, 2014; Barker et al., 2009; Blanpain, 2013; Visvader, 2011; White and Lowry, 2015), while differentiated cells, which are mostly nondividing, tend not to survive for the many decades and cell divisions required to achieve the mutational threshold for malignant transformation (Barker et al., 2009). Recent studies have suggested that a subset of short-lived progenitors or mature cells are able to give rise to cancer following dedifferentiation into long-lived stem-like cells (Hayakawa et al., 2019; Ishibashi et al., 2018; Jones et al., 2019; Westphalen et al., 2014). Nevertheless, stem cells are in many ways the ideal cellular targets for the accumulation of genetic alterations, given their fundamental properties of longevity and self-renewal.
In the mouse gastric antrum, both basal stem cells and +4 stem cells have been shown to act as a source of antral cancers by cell-specific oncogenic mutations (Fig. 5A). Mutations in Apc in antral stem cells cause intramucosal dysplasia or adenoma lesions in the distal stomach (Barker et al., 2010; Sakitani et al., 2017; Sarkar et al., 2016), while the combination of multiple oncogenic drivers such as Apc, p53, Kras, or Smad induces invasive cancers derived from antral stem cells (Chang et al., 2020; Li et al., 2016; Tan et al., 2020). Thus, the induction and accumulation of genetic mutations in antral gastric stem cells is necessary and sufficient for the stepwise development of gastric carcinogenesis.
Figure 5. Stem cells and cellular origin of metaplasia and cancers.
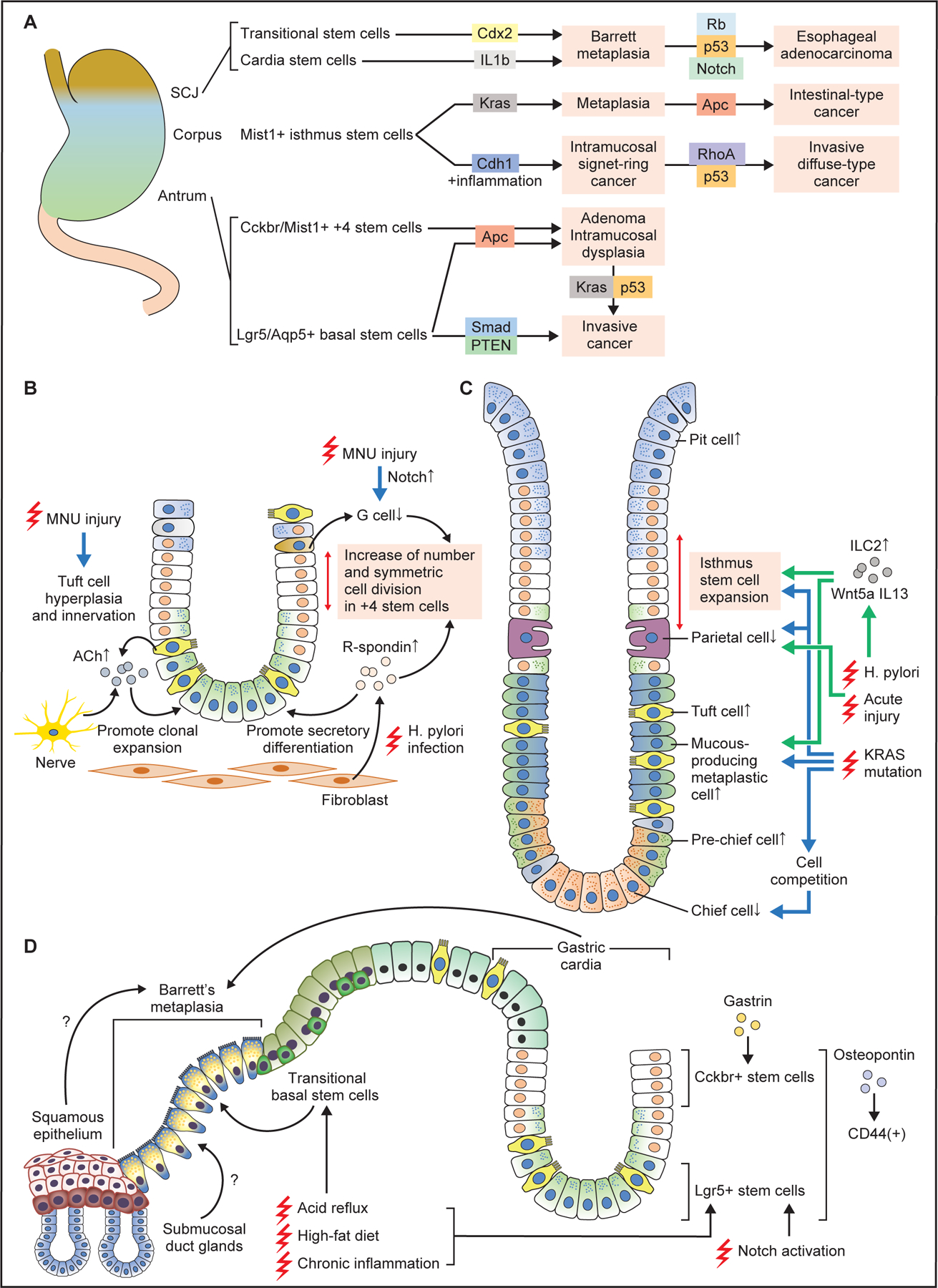
(A) Stem cells in the antrum, corpus, and SCJ give rise to metaplasia and cancer following induction of specific oncogenic mutations. In general, combination of multiple gene mutations causes more advanced, invasive cancers at each site. (B) Changes in antral stem cells and their niche at the early phase of carcinogenesis. Chronic injury by MNU or H. pylori alters stem cell niche, with increase of R-spondin from fibroblasts and acetylcholine (ACh) from nerves and tuft cells, as well as decrease of G cells and gastrin. These contribute to expansion and symmetric cell division of +4 stem cells, and secretory differentiation in Lgr5+ lineage. (C) Cellular changes in the corpus gland during early metaplasia development. Both in the acute and chronic injury models, the numbers of isthmus progenitors, tuft cells, and mucous-producing metaplastic cells are increased, while the numbers of parietal and chief cells are decreased. Wnt5a and IL13 from ILC2s mediate these pathological changes. Induction of Kras mutation in isthmus stem cells causes similar metaplastic changes, while Kras mutation in mature chief cells results in cell competition-dependent loss of chief cells. (D) Schematic image showing Barrett esophagus and carcinogenesis. Stem cells in the gastric cardia glands, including basal Lgr5+ cells and +4 Cckbr+ cells expand and give rise to Barrett metaplasia and cancer in response to chronic injury. High-fat diet and Notch activation promotes the expansion of these cell-derived clones. Transitional basal stem cells also serve as a source of Barrett metaplasia. Esophageal squamous epithelium and submucosal gland ducts might also contribute to Barrett metaplasia development.
In models using gastric carcinogens such as Helicobacter infection and/or MNU, both basal and +4 antral stem cells again appear to act as a cellular source of antral dysplasia or tumors. However, these models require a longer time course to develop tumors, during which time stem cells trace the entire glands, and thus such studies are unrevealing as to the exact cellular origin of tumorigenesis. To understand the cellular origin of clonal expansion, the earliest time points in tumor development must be examined. At a relatively early stage of carcinogenesis, shortly after MNU administration or Helicobacter infection, epithelial damage induces prominent proliferation and cell division in antral stem cells, leading to an increase in stem cell numbers and gland fission (Chang et al., 2020; Hayakawa et al., 2015b; Sigal et al., 2019; Sigal et al., 2015). These changes in the stem cell compartment are mediated and accelerated by changes in the stromal stem cell niche, which is also altered during carcinogenesis (Fig. 5B). For example, acetylcholine-dependent nerve signaling is enhanced during carcinogenesis by increased tuft cells and cholinergic innervation, resulting in clonal expansion of stem cell-derived clones. Furthermore, MNU-induced injury activates the Notch pathway in antral glands, leading to a decrease in gastrin-producing G cells, and further activation of Cckbr+ stem cells (Chang et al., 2020). Similarly, Helicobacter infection increases secretion of R-spondin 3 from stromal fibroblasts, which stimulates expansion of Axin2+ +4 stem cells (Sigal et al., 2017). While expansion of the +4 stem cell population is more evident at initial stage of carcinogenesis, in both models the numbers of basal Lgr5+ stem cells are also later increased (Sigal et al., 2015; Zhao et al., 2014), likely mediated by R-spondin signaling which promotes secretory differentiation (Sigal et al., 2019). Therefore, a detailed analysis at early time points suggests that isthmal stem cells are responsible for epithelial regeneration and clonal gland fission following carcinogenic stimuli, and cancer originates from such stem cell-derived mutated clones.
Similarly, the specific induction of oncogenic mutations in corpus isthmal stem cells has confirmed that they can also act as the origin of cancer. However, corpus stem cells may be somewhat more resistant to malignant transformation compared to antral stem cells, as single gene mutations in corpus stem cells appear insufficient to induce dysplasia or cancer. For example, when the Cdh1 gene is deleted, Mist1+ stem cells undergo anoikis and Cdh1-null Mist1-derived clones gradually disappear. Nevertheless, in the presence of chronic inflammation induced by Helicobacter infection, Cdh1-deleted Mist1-derived clones survive and expand, eventually generating cancers that resemble human signet-ring cell gastric cancers (Hayakawa et al., 2015a). This appears to be due in part to increased Cxcl12 expression and expansion of ILC2 cells, and increased secretion of Wnt5a (Fig. 5A). Additional p53 or Rhoa gene mutations in Cdh1-null Mist1+ stem cells accelerates diffuse-type gastric carcinogenesis (Hayakawa et al., 2015a; Zhang et al., 2019). Moreover, while Apc gene mutation in corpus stem cells is unable to induce cancer at this site, simultaneous mutation in Kras and Apc genes produces rapidly-growing intestinal-type gastric cancer (Hayakawa et al., 2015a). Since both Helicobacter infection and Kras mutation in the stomach leads to gastric atrophy (i.e., loss of parietal cells and chief cells) and metaplasia, such pathological changes may be required for development of corpus cancers derived from isthmus stem cells (Fig. 5C). In particular, since loss of parietal cells triggers expansion of isthmus-derived clones (Han et al., 2019), gastric atrophy, which often precedes cancer development in human cases, may be a critical step for corpus carcinogenesis. Taken together, isthmus stem cells can act as an origin of proximal gastric cancer.
Pseudopyloric and intestinal metaplasia of the gastric corpus
Upper GI cancers are often preceded by precancerous lesions, including metaplasia and dysplasia, raising questions as to the origin of metaplastic lesions and their relationship to the development of cancer. Evidence is emerging that stem cells which acquire mutations (Blokzijl F et al., 2016) can persist and that such mutated stem cells contribute to these precancerous lesions.
In mouse models of Helicobacter infection, cancer development is preceded by gastric atrophy and metaplasia. Metaplasia in the human stomach includes both classical IM as well as pseudopyloric metaplasia or SPEM. In mice, IM rarely develops even after long-term Helicobacter infection, with only occasional goblet-like cells, and thus the origin of IM seen in the human stomach remains uncertain. However, given the monoclonal expansion of IM cells in human stomach, self-renewing, long-lived stem cells are likely the origin of IM.
In addition to Helicobacter infection models, Kras mutation has also been used as an inducer of metaplasia and dysplasia in the mouse proximal stomach (Hayakawa et al., 2015a; Kinoshita et al., 2019; Kinoshita et al., 2018; Leushacke et al., 2017; Matsuo et al., 2020; Matsuo et al., 2016; Okumura et al., 2010). Due to tamoxifen-dependent toxic effects and non-specific expression of most CreERT transgenes, it has been challenging to prove unequivocally the exact cellular source of Kras-induced metaplasia. Recently, studies using the tamoxifen-independent Gpr30-rtTA system verified that Gpr30-expressing chief cells do not dedifferentiate but instead are lost with activation of the Kras mutation (Hata et al., 2020). Loss of basal chief cells triggers compensatory expansion of pre-chief cells, which express both neck and chief cell markers (Fig. 5C). Mechanistically, loss of Kras-mutated chief cells is due to a PDK-dependent cell competition mechanism, whereby mutated cells are extruded by surrounding normal epithelial cells. A similar elimination of Ras-mutated mature cells via cell competition was reported in the intestinal epithelium (Kon et al., 2017). In contrast, induction of a Ras mutation in isthmus stem/progenitors efficiently generates metaplasia and dysplasia in the stomach (Kinoshita et al., 2018; Matsuo et al., 2020). Thus, the effect of a Ras mutation in gastric epithelial cells varies depending on the degree of cellular differentiation. Indeed, in certain post-mitotic epithelial cells, Kras mutation can induce cellular senescence and apoptosis (Cox and Der, 2003; Serrano et al., 1997). Thus, in addition to longevity, cancer-initiating cells also need to be able to stay within the epithelium following acquisition of oncogenic mutations, thereby eluding cell death or extrusion by cell competition. Studies suggest that stem cells likely have such ability and most mature cells do not, although the underlying mechanism remains unclear.
Nevertheless, gastric chief cells have been proposed as a major origin of SPEM in mouse models. SPEM is characterized by abundant expression of markers of mucous neck cells, a precursor of chief cells (Huh et al., 2010; Quante et al., 2010; Ramsey et al., 2007), such as TFF2, MUC6, and GSII, resembling immature chief cells (e.g. GSII+GIF+ cell) that are normally present in corpus glands and express these markers. Experiments using tamoxifen-dependent lineage tracing and nucleic acid analog administration have been considered supportive of a chief cell origin of SPEM (Burclaff et al., 2019; Nam et al., 2010). However, SPEM may also arise from subsets of more immature neck cells with progenitor potential and this remains a subject of some debate. Furthermore, metaplasia and cancer are present in the gastric corpus, cardia and the antrum, while in the latter two regions chief cells are quite scarce.
The precise relevance of SPEM and IM to the development of invasive cancer has not been established. SPEM in acute injury models is quite different histologically and molecularly from SPEM in chronic inflammation models (Bockerstett et al., 2019), although both reflect a regenerative response following tissue damage. However, true SPEM (associated with chronic atrophic gastritis) and IM are markers in the stomach of long-term H. pylori infection, and thus may reflect mutation or epigenetic alterations of gastric stem cells. During regeneration associated with SPEM, cell composition in the corpus glands is dramatically changed, with loss of parietal and chief cells as well as marked expansion of isthmus progenitors and tuft and neck lineages. ILC2s, an important component of corpus niche, are responsible for some of these pathological changes, largely through the secretion of Wnt5a and IL13 (Fig. 5C) (Meyer et al., 2020; Nienhuser et al., 2020). While these metaplastic lesions are associated with an increased risk of upper GI cancers, evidence is lacking that cancer arises directly from metaplastic cells, rather than from undifferentiated stem cells. Indeed, molecules enriched in SPEM lesions such as TFF2 and MUC6 are often tumor-suppressive (Dubeykovskaya et al., 2016; Fox et al., 2007). Thus, it remains uncertain whether SPEM is a true precursor of IM or cancer.
Stem or progenitor cells in the SCJ and cardia give rise to metaplasia and cancer
BE is characterized as the replacement of the distal squamous epithelium by simple columnar cells with features of gastric and intestinal differentiation (McDonald et al., 2015). The origin of BE has been an active subject in recent decades, given its association with EAC which has rapidly increased (Kong et al., 2011; Leedham et al., 2008; Milano et al., 2007; Quante et al., 2012; Sarosi et al., 2008; Wang et al., 2011). Initial in vitro modeling proposed that BE arose from the esophageal squamous epithelium via a process of transdifferentiation (Milano et al., 2007). However, lineage tracing studies have not supported this model. Furthermore, while a recent study showed that ectopic expression in the squamous esophagus of Smoothened, the downstream effector of the Hedgehog pathway, induces the transcription of several columnar markers including Krt8, Muc5ac and Agr2, a simple columnar epithelium was not reported and full transdifferentiation was not achieved (Vercauteren Drubbel et al., 2021). Similarly, esophageal submucosal glands (ESGs, which are not present in the mouse esophagus) have also been considered as a cell of origin for BE (Gonzalez et al., 2016; Owen et al., 2018), given that ESG cells can share the same mitochondria mutations with BE epithelium (Nicholson et al., 2012). But thus far rigorous lineage tracing is lacking for further confirmation. In contrast, recent in vivo studies strongly suggest that the cells-of-origin for BE are stem/progenitor cells in the gastric cardia and the transitional epithelium SCJ connecting the distal esophagus and cardia (Fig. 5D) (Jiang et al., 2017; Lee et al., 2017; Quante et al., 2012).
BE glands contain not only intestinal cell types but also gastric cell types, some of which express gastric mucins such as MUC6, MUC5AC, and TFF2. Thus, the replication of the stem cell organization of pyloric-type gastric glands in BE suggests the notion of the origin of BE in progenitor cells with gastric identify or potential (Lavery et al., 2014). Furthermore, a gastric origin for esophageal cancer was supported by epigenomic studies that revealed that BE and esophageal adenocarcinoma exhibited chromatin features that closely resemble stomach epithelial cells, rather than esophageal tissues (Polak et al., 2015). A more recent study at single cell resolution of open chromatin has shown that BE cells have a mixed, heterogeneous “epigenotype” that comprises distinctive gastric and intestinal states (R. Shivdasani, personal communication).
In L2-IL1b mice overexpressing interleukin-1-beta with chronic carditis and esophagitis, progenitor cells from gastric cardia glands were found to migrate above the SCJ, producing columnar glands within the distal esophagus. Chronic carditis is frequently seen in patients with gastroesophageal reflux disease (Pinto et al., 2019), is associated with IM, and is a predictive of BE development (Leodolter et al., 2012). The origin of these Barrett’s-like glands was confirmed by lineage tracing experiments labeling Lgr5-expressing crypt basal stem cells as well as Cckbr-expressing +4 stem cells in cardia glands (Lee et al., 2017; Quante et al., 2012). Both basal and +4 cardia stem cells express CD44, which are activated by a CD44 ligand, osteopontin (Fig. 5D) (Fu et al., 2020). Development of BE and dysplasia in L2-IL1b mice was accelerated when mice were given an unconjugated bile acid, high-fat diet, or genetic ablation of esophageal squamous cell layer (Lee et al., 2017; Quante et al., 2012). Notch inhibition promotes goblet cell differentiation in the Barrett’s-like epithelium, inhibiting progression to dysplasia, while activation of Notch decreases goblet cells and promotes tumor development (Kunze et al., 2020). Loss of p53 and Rb1 genes in cardia stem cells results in the formation of adenocarcinoma at the SCJ (Fig. 5A) (Fu et al., 2020). In these studies, the presence of intestinal differentiation and abundant goblet-like cells was associated with a reduced risk of early cancer, and the observation has been supported by human studies showing an inverse correlation between goblet cells and cancer risk (Srivastava et al., 2015). Taken together, these findings indicate that progenitor cells in the gastric cardia migrate into the esophagus during chronic inflammation and serve as a source of the cells of origin for BE and adenocarcinoma.
Transitional basal cells (TBCs) have also been shown to generate BE at the SCJ (Jiang et al., 2017). Targeted overexpression of the intestinal gene Cdx2 caused columnar differentiation of TBCs, and the metaplastic epithelial cells expressed gastric and intestinal proteins including AGR2, MUC2 and Villin1. In contrast, ectopic Cdx2 expression failed to drive gastric or intestinal differentiation of squamous basal cells in the esophagus, similar to the observation made in Krt14-Cdx2 mice (Kong et al., 2011). The molecular mechanism by which TBCs acquire gastric and intestinal differentiation capability remains unexplored. Bile acid is a prominent suspect for promoting the pathogenesis of BE in humans. Bile acid reflux induced by jejuno-esophageal anastomosis caused the expansion of TBCs, which subsequently altered their cell fate to become mucus-producing cells (Jiang et al., 2017). Expansion of TBCs was also observed in patients with multilayered columnar epithelium (Jiang et al., 2017), a pathological entity preceding BE (Chen et al., 2008; Glickman et al., 2001). Expansion of TBCs is likely driven by the prolonged bile acid insults and subsequent inflammatory milieu. However, many questions remain. For example, how do TBCs lose some of their identity (e.g. Trp63 expression) while preserving others (e.g. Krt7 expression) during progression towards BE? Lineage tracing showed that TBCs generate Trp63- Krt7+ daughter cells (Jiang et al., 2017). One possible scenario is that these Trp63- Krt7+ daughter cells acquire intestinal differentiation capability and progress towards BE. A similar model has been proposed for the ontogeny of BE-independent EAC originating in the gastric cardia, where Lgr5+ stem cells generate Lgr5- progeny that undergo subsequent transformation upon inactivation of Rb1 and p53 (Fu et al., 2020). Notably, a group of cells termed residual embryonic cells (RECs, Car4+Krt7+Trp63-) have also been considered a possible cell of origin for BE (Wang et al., 2011). These cells are defined as the epithelial cells left behind during development, remaining quiescent in the SCJ in adults. It was proposed that RECs expand into the squamous epithelium upon acid-induced injury, replacing damaged epithelium prior and becoming BE (Wang et al., 2011). Is it possible that these RECs are indeed the progeny of TBCs? Basal cell heterogeneity has been reported in the upper esophagus (Giroux et al., 2017; Kalabis et al., 2008; Seery and Watt, 2000). It is also conceivable that TBCs contain multiple subpopulations, with some able to generate squamous cells while others generate columnar cells. Alternatively, TBCs could be homogenous but multipotent, producing squamous or columnar progeny depending on local needs and the presence or absence of prolonged inflammation.
In summary, lineage tracing evidence confirms that Lgr5+ or Cckbr+ stem cells in the cardia mucosa and TBCs in the SCJ contribute to BE. It is likely that multiple sources of stem/progenitor cells are involved in the initial response to the loss of the squamous epithelium upon acute injury. However, if the repetitive injury persists and chronic inflammation ensues, one of the stem/progenitor populations acquires additional genetic or epigenetic changes, enabling intestinal differentiation capability. Notably, mutation of p53 is relatively common at the early stage of BE pathogenesis (Jenkins et al., 2003; Stachler et al., 2015), which alters genomic stability and may facilitate the acquisition of new genetic mutations.
Basal stem cells as the cell of origin for esophageal squamous cell carcinoma (ESCC)
The first evidence linking basal cells with ESCC initiation comes from the study of Sox2 overexpression with Krt5-CreER mouse line (Liu et al., 2013). SOX2 gene amplification occurs in approximately 30% ESCC (Bass et al., 2009; Liu et al., 2013; Song et al., 2014). However, targeted Sox2 overexpression in the esophagus leads to basal cell hyperplasia but not carcinoma. Invasive squamous cell carcinoma only occurs in the forestomach (Liu et al., 2013), suggesting that the microenvironment plays important roles in the malignant transformation of basal cells. Further analysis revealed that acid-induced inflammation characterized by increased IL-6 and STAT3 activation is essential for tumor initiation (Liu et al., 2013). In a separate study, SCC was also established in the forestomach of Krt5-CreER; LSL-KrasG12D; p53wt/flox mutants upon tamoxifen induction. Interestingly, SCC preferentially occurs at the SCJ when tamoxifen is given at low dose (0.2mg/mouse, i.p.). Similar observations were made in Krt15-CrePR1; LSL-KrasG12D; p53flox/flox mutants upon RU486 injection, suggesting a high susceptibility to neoplasia by basal cells at the SCJ. It is noteworthy that basal cells in the SCJ have high proliferation index compared to basal cells in other regions (Moon et al., 2019).
These findings seem in line with a mathematical model proposing that a higher cell division rate is correlated with a higher incidence of stem cell-originated cancer (Tomasetti and Vogelstein, 2015). It is worth mentioning that prostaglandin-endoperoxide synthase 2 (also known as Cox-2) is a critical mediator of acid-induced tumor formation from basal cells (Moon et al., 2019). Together, these mouse studies reveal that basal cells are the cell of origin for SCC. It seems that common mutation(s) provide proliferation advantages promoting basal cell hyperplasia, and subsequent microenvironmental factors (e.g. bile acid, inflammation) are needed for further transformation of the expanded basal cells.
Limitations of Current Data and Emerging Tools for Studying Upper GI Stem Cells.
Limitations in the previous mouse models and new systems
Much emphasis has been given to findings from inducible Cre lineage tracing, but often these models have been limited by haploinsufficiency of the gene of interest which may alter cell behavior and gene expression (Hayakawa et al., 2015a; Kuo et al., 2016; Westphalen et al., 2014), the use of tamoxifen which induces mucosal and cellular damage in specific organs and cell types (Bohin et al., 2018; Huh et al., 2012; Keeley et al., 2019), and the lack of specificity of many of the Cre drivers. While a number of markers has been described as specific for distinct stem cells, one likely conclusion is that none is perfectly specific and that they may simply overlap with the same stem cell population. In addition, most of the markers have not been confirmed in human tissues. The heterogeneity of marker-positive populations has clearly been underestimated and newer approaches are needed to determine whether there is indeed a homogeneous single progenitor model or a heterogeneous/hierarchical model that defines the upper GI tract. This might include in part the development of better inducible genetic tracing models along with more careful interpretation of such models.
Along this line, doxycycline-dependent tetON/tetOFF system and progesterone-dependent CrePR system have been established used for cell fate mapping studies in the GI tract while avoiding toxic effects by tamoxifen (Hata et al., 2020; Ishibashi et al., 2018; Jiang et al., 2017; Moon et al., 2019). Combination of multiple labeling techniques in one animal, for example, simultaneous activation of Cre/Lox, Dre/Rox, and Frt/Flp systems, would enable us to label marginal populations between two or more markers, or induce stepwise activation of multiple transgenes (He et al., 2017; Schonhuber et al., 2014). Future lineage tracing studies need to better acknowledge the limitation of these previous lineage tracing experiments, and more carefully interpret the specificity of the findings.
Gastric and esophageal organoids derived from PSCs
The in vitro growth of gastric or esophageal organoids (or spheroids) in 3D culture has been a mainstay of stem cell research in the last decade. While the majority of organoid cultures has involved murine tissues, greater attention has been given recently to generating 3D organoid cultures in Matrigel from patient-derived gastric or esophageal tissue, either through primary cultures from surgical specimens or endoscopic biopsies, or through induced pluripotent stem cells (PSC) that are differentiated into gastric or esophageal tissue. A major limitation of studies of human gastro-esophageal organoids has been the limited ability to carry out genetic lineage tracing. In addition, requiring cell proliferation for structure formation, spheroids and organoids assays may not readily detect quiescent stem cells.
Nevertheless, the source of PSC-derived organoids is in theory unlimited, and the associated protocol follows the embryonic gastric developmental stages (McCracken et al., 2017; McCracken et al., 2014). First, PSCs are differentiated by activin A into definitive endoderm, which is then patterned by FGF4 into anterior and foregut endoderm; subsequently, the endodermal cultures are treated with inhibitors of GSK3 (CHIR-99–21) and BMP (noggin). Then, foregut spheroids are directed to the posterior foregut by retinoic acid, while antral organoids containing various endocrine cells (such as G cells) can be obtained by incubating with noggin, RA, and EGF for 4 weeks. Fundic organoid development requires additional GSK3 inhibition, and the addition of a MEK inhibitor and BMP4 is needed to induce differentiation of parietal cells.
The great strength of PSC-derived human gastric organoids (HGOs) (Broda et al., 2019; McCracken et al., 2017) has been the ability to grow gastric epithelial progenitors in conjunction with a stromal niche, and such in vitro models can be used to better understand human gastric stem cells, as well as the critical interactions with niche cell including endothelial, neuronal, immune and mesenchymal cells (Eicher et al., 2021). In addition, HGOs can be used to model disease processes (Bartfeld et al., 2015; McCracken et al., 2014; Sethi et al., 2020). HGOs can also be used for drug screening, transplantation, and in combination with CRISPR-mediated editing, for genetic modeling of diseases including cancer (Nanki et al., 2018).
Esophageal progenitor cells (EPCs) have also been generated by sequential differentiation of hPSCs (Trisno et al., 2018; Zhang et al., 2018). Similar to the generation of gastric epithelium, hPSCs are first differentiated into foregut endodermal progenitor cells. Bmp inhibition and Sox2 expression are critical for the subsequent commitment of progenitor cells towards esophageal instead of gastric or respiratory cell lineage. Indeed this robust reconstitution ability may relate in part to the more fetal-like nature of PSC-derived organoids. This is consistent with the roles of Noggin and Sox2 in the specification of early foregut progenitor cells towards esophageal lineage in mouse embryos (Que et al., 2006; Que et al., 2007). PSCs-derived EPCs generate stratified squamous epithelium in 3D organoid culture and xenograft implanted into the kidney capsule (Zhang et al., 2018). Combined PSC differentiation and mouse genetics allow for the identification of new pathways involved in EPC proliferation and differentiation, such as Notch (Zhang et al., 2018), YAP (Bailey et al., 2019), and Sox2 (Trisno et al., 2018). The PSC differentiation system can also offer insights into the heterogeneity of EPCs when combined with other tools such as single-cell analysis and mathematical modeling. Information gathered through these studies will facilitate the potential use of PSCs-derived EPCs in clinics, producing implantable epithelium.
Single cell analysis
Recently, scRNA-sequencing analysis has been used for examining signaling molecules and transcription factors in foregut development and stem cell homeostasis (Busslinger et al., 2021; Han et al., 2020; Sigal et al., 2019). It has revealed greater heterogeneity in certain stem cell populations, supporting the notion that most stem cell markers are not specific for stem cells. For example, Lgr5+ antral cells contain multiple cell types, including actively proliferating cells, quiescent mucous-producing cells, and several types of differentiated cells (Sigal et al., 2019). Similarly, the Stmn1+ corpus cell population consists not only of actively proliferating cells, but also of multiple lineage-committed progenitors (Han et al., 2019). Integrated analysis of the datasets suggested that stem cell populations in the corpus and antrum are somewhat similar at the transcriptional level, and can be divided into comparable clusters. Consistently, many reported gastric stem cell markers are shared between the antrum and corpus. Antral and corpus gastric stem cells may be somewhat similar, but respond differently to regional stem cell niches by supplying region-specific cell types to the glands.
Spatial gene expression assay on tissue sections has been available for visualizing region- or even cell-specific gene profiles with preserved tissue morphology (Stahl et al., 2016). Moreover, combination of single cell-based gene profiling and lineage tracing has been attempted for mapping transcriptional landscape during cell division and differentiation, by tagging cells with DNA barcodes or CRISPR/Cas9 mutations (Weinreb et al., 2020; Zafar et al., 2020). These technologies would be useful for GI stem and cancer research, and should be a priority for future studies. In any case, scRNA-seq has the potential to analyze cellular hierarchies and assess stemness in a more unbiased manner.
Summary and Perspective
Stem cells play a central role in mucosal homeostasis and are the primary source of metaplasia and cancer in the esophagus and stomach. Both organs share the same embryonic origin while being shaped by distinct region-specific mesenchymal signaling and transcriptional factors. Likewise, esophageal and gastric stem cells in adult tissues are supported by a stromal stem cell niche, and they come together at the transitional zone that lies between the SCJ. Thus, signaling pathways and transcriptional factors appear to diverge during development, and remain continuously but region-specifically activated during organogenesis, homeostasis, and carcinogenesis.
There are still numerous unanswered questions in this field. Recent single cell analysis revealed marked heterogeneity in presumed esophageal and gastric stem/progenitor cell populations. However, understanding such findings will require better methods for sorting individual stem cell subpopulations followed by detailed functional characterization. It needs to be clarified whether there is more than one type of stem cell in each region of the upper GI tract, or whether this is simply a reflection of off-target CreERT expression. While one may wish for the identification of a highly specific stem cell marker, experience from the hematopoietic stem cell field would suggest that this may not be easily forthcoming.
In addition, how metaplasia and cancer develop from stem cells remains largely unknown. It likely involves both genetic and epigenetic changes, and use of single cell analysis - both scRNA-seq and scATAC-seq will facilitate understanding in this regard. Nevertheless, follow-up studies with animal models and 3D organoids are required to further confirm the hypothesis and assess potential corresponding therapeutics. The greater use of human organoids and humanized animal models would enhance the importance and relevance to human disease.
The topic of cellular plasticity remains an open question. While the actual degree of plasticity may be currently overestimated, there are mechanisms in place to restore homeostasis following injury or loss of dedicated stem cells. The plasticity exhibited can be a double-edged sword. On one hand, the plasticity can be explored for facilitating tissue regeneration. On the other hand, the possibility of dedifferentiation or interconversion from progenitors or more differentiated cell types may pose issues when targeting stem cells and/or cancer stem cells for cancer therapy. Nevertheless, a better understanding of the signaling pathways that activate stem cell function, the niche components that generate the signals, and the mechanisms (and limits) for cellular interconversion or dedifferentiation process, are all exciting areas for the future.
Acknowledgements
We appreciate the useful discussions from the NCI Think on the Origins of Gastrointestinal Cancers” on February 14–15, 2019 at NCI Shady Grove Building. We thank to Dr. Rihab Yassin, Ph.D., the organizer of this meeting, and all attendees and presenters, Dr. Jim Goldenring, M.D., Ph.D., AGAF, Vanderbilt University; Dr. Juanita Merchant, M.D., Ph.D., University of Arizona; Dr. Linda Samuelson, Ph.D., University of Michigan; Dr. Kelley Yan, M.D., Ph.D., Columbia University; Dr. Yana Zavros, Ph.D., University of Cincinnati; Dr. Eric Fearon, M.D., University of Michigan; Dr. Simon Leedham, M.D., Oxford University; Dr. Christopher Lengner, Ph.D., University of Pennsylvania; Dr. Linheng Li, Ph.D., Stowers Institute for Medical Research; Dr. Ramesh Shivdasani, M.D., Ph.D., Dana Farber Cancer Institute; Dr. Noah Shroyer, Ph.D., Baylor College of Medicine; Dr. Adam Bass, M.D., Dana Farber Cancer Institute; Dr. J. Alan Diehl, Ph.D., Medical University of South Carolina; Dr. Michael Quante, PD Dr. Med, Technical University of Munich; Dr. Brigid Hogan, Ph.D., FRS, Duke University. We thank for Dr. Andres J. Klein-Szanto (Fox Chase Cancer Center) for esophageal histology.
T.C.W. is supported by the NIH grants R35CA210088 and 5U01DK103155–04, J.Q. by DK100342, DK113144 and DK120650, H.N. by R01DK114436, R01AA026297, P01CA 098101 and U54CA163004, and A.K.R. by P01CA 098101 and U54CA163004. T.C.W., J.Q., H.N. and A.K. R. are also supported by 2P30CA013696.Y.H. is supported by JSPS, Inoue Science Research Award, Takeda Science Foundation Visionary Research Grant, Uehara Memorial Foundation, Naito Foundation, Daiwa-Shoken health foundation, The Waksman Foundation of Japan, Koyanagi Foundation, The Research Foundation for Pharmaceutical Sciences, Yakult Bio-Science Foundation, Tokyo Biochemical Research Foundation, The NOVARTIS Foundation (Japan) for the Promotion of Science, SGH foundation, Daiichi Sankyo Foundation of Life Science, and AMED (PRIME and P-CREATE). Some of the authors’ published work was supported through the Herbert Irving Comprehensive Cancer Center’s shared resources (Confocal and Specialized Microscopy, Flow Cytometry, Genetically Modified Mouse Models, Molecular Pathology and Oncology Precision Therapeutics and Imaging Core).
References:
- Aoki R, Shoshkes-Carmel M, Gao N, Shin S, May CL, Golson ML, Zahm AM, Ray M, Wiser CL, Wright CV, et al. (2016). Foxl1-expressing mesenchymal cells constitute the intestinal stem cell niche. Cell Mol Gastroenterol Hepatol 2, 175–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold K, Sarkar A, Yram MA, Polo JM, Bronson R, Sengupta S, Seandel M, Geijsen N, and Hochedlinger K (2011). Sox2(+) adult stem and progenitor cells are important for tissue regeneration and survival of mice. Cell Stem Cell 9, 317–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asfaha S, Hayakawa Y, Muley A, Stokes S, Graham TA, Ericksen RE, Westphalen CB, von Burstin J, Mastracci TL, Worthley DL, et al. (2015). Krt19(+)/Lgr5(−) Cells Are Radioresistant Cancer-Initiating Stem Cells in the Colon and Intestine. Cell Stem Cell 16, 627–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayyaz A, Kumar S, Sangiorgi B, Ghoshal B, Gosio J, Ouladan S, Fink M, Barutcu S, Trcka D, Shen J, et al. (2019). Single-cell transcriptomes of the regenerating intestine reveal a revival stem cell. Nature 569, 121–125. [DOI] [PubMed] [Google Scholar]
- Bailey DD, Zhang Y, van Soldt BJ, Jiang M, Suresh S, Nakagawa H, Rustgi AK, Aceves SS, Cardoso WV, and Que J (2019). Use of hPSC-derived 3D organoids and mouse genetics to define the roles of YAP in the development of the esophagus. Development 146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker N (2014). Adult intestinal stem cells: critical drivers of epithelial homeostasis and regeneration. Nature reviews Molecular cell biology 15, 19–33. [DOI] [PubMed] [Google Scholar]
- Barker N, Huch M, Kujala P, van de Wetering M, Snippert HJ, van Es JH, Sato T, Stange DE, Begthel H, van den Born M, et al. (2010). Lgr5(+ve) stem cells drive self-renewal in the stomach and build long-lived gastric units in vitro. Cell Stem Cell 6, 25–36. [DOI] [PubMed] [Google Scholar]
- Barker N, Ridgway RA, van Es JH, van de Wetering M, Begthel H, van den Born M, Danenberg E, Clarke AR, Sansom OJ, and Clevers H (2009). Crypt stem cells as the cells-of-origin of intestinal cancer. Nature 457, 608–611. [DOI] [PubMed] [Google Scholar]
- Barker N, van Es JH, Kuipers J, Kujala P, van den Born M, Cozijnsen M, Haegebarth A, Korving J, Begthel H, Peters PJ, et al. (2007). Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature 449, 1003–1007. [DOI] [PubMed] [Google Scholar]
- Barriga FM, Montagni E, Mana M, Mendez-Lago M, Hernando-Momblona X, Sevillano M, Guillaumet-Adkins A, Rodriguez-Esteban G, Buczacki SJA, Gut M, et al. (2017). Mex3a Marks a Slowly Dividing Subpopulation of Lgr5+ Intestinal Stem Cells. Cell Stem Cell 20, 801–816 e807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartfeld S, Bayram T, van de Wetering M, Huch M, Begthel H, Kujala P, Vries R, Peters PJ, and Clevers H (2015). In vitro expansion of human gastric epithelial stem cells and their responses to bacterial infection. Gastroenterology 148, 126–136 e126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bass AJ, Watanabe H, Mermel CH, Yu S, Perner S, Verhaak RG, Kim SY, Wardwell L, Tamayo P, Gat-Viks I, et al. (2009). SOX2 is an amplified lineage-survival oncogene in lung and esophageal squamous cell carcinomas. Nat Genet 41, 1238–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjerknes M, and Cheng H (2002). Multipotential stem cells in adult mouse gastric epithelium. Am J Physiol Gastrointest Liver Physiol 283, G767–777. [DOI] [PubMed] [Google Scholar]
- Blanpain C (2013). Tracing the cellular origin of cancer. Nat Cell Biol 15, 126–134. [DOI] [PubMed] [Google Scholar]
- Blokzijl F, de Ligt J, Jager M, Sasselli V, Roerink S, Sasaki N, Huch M, Boymans S, Kuijk E, Prins P, et al. (2016). Tissue-specific mutation accumulation in human adult stem cells during life. Nature 538, 260–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bockerstett KA, Lewis SA, Wolf KJ, Noto CN, Jackson NM, Ford EL, Ahn TH, and DiPaolo RJ (2019). Single-cell transcriptional analyses of spasmolytic polypeptide-expressing metaplasia arising from acute drug injury and chronic inflammation in the stomach. Gut [DOI] [PMC free article] [PubMed]
- Bohin N, Carlson EA, and Samuelson LC (2018). Genome Toxicity and Impaired Stem Cell Function after Conditional Activation of CreER(T2) in the Intestine. Stem cell reports 11, 1337–1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booth C, and Potten CS (2000). Gut instincts: thoughts on intestinal epithelial stem cells. J Clin Invest 105, 1493–1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broda TR, McCracken KW, and Wells JM (2019). Generation of human antral and fundic gastric organoids from pluripotent stem cells. Nat Protoc 14, 28–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burclaff J, Willet S, Saenz JB, and Mills J (2019). Proliferation and Differentiation of Gastric Mucous Neck and Chief Cells During Homeostasis and Injury-induced Metaplasia. Gastroenterology [DOI] [PMC free article] [PubMed]
- Busslinger GA, Weusten BLA, Bogte A, Begthel H, Brosens LAA, and Clevers H (2021). Human gastrointestinal epithelia of the esophagus, stomach, and duodenum resolved at single-cell resolution. Cell Rep 34, 108819. [DOI] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research, N., Analysis Working Group: Asan, U., Agency, B.C.C., Brigham, Women’s, H., Broad, I., Brown, U., Case Western Reserve, U., Dana-Farber Cancer, I., Duke, U., et al. (2017). Integrated genomic characterization of oesophageal carcinoma. Nature 541, 169–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang W, Wang H, Kim W, Liu Y, Deng H, Liu H, Jiang Z, Niu Z, Sheng W, Napoles OC, et al. (2020). Hormonal Suppression of Stem Cells Inhibits Symmetric Cell Division and Gastric Tumorigenesis. Cell Stem Cell 26, 739–754 e738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Qin R, Liu B, Ma Y, Su Y, Yang CS, Glickman JN, Odze RD, and Shaheen NJ (2008). Multilayered epithelium in a rat model and human Barrett’s esophagus: similar expression patterns of transcription factors and differentiation markers. BMC Gastroenterol 8, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clayton E, Doupe DP, Klein AM, Winton DJ, Simons BD, and Jones PH (2007). A single type of progenitor cell maintains normal epidermis. Nature 446, 185–189. [DOI] [PubMed] [Google Scholar]
- Cox AD, and Der CJ (2003). The dark side of Ras: regulation of apoptosis. Oncogene 22, 8999–9006. [DOI] [PubMed] [Google Scholar]
- Croagh D, Phillips WA, Redvers R, Thomas RJ, and Kaur P (2007). Identification of candidate murine esophageal stem cells using a combination of cell kinetic studies and cell surface markers. Stem Cells 25, 313–318. [DOI] [PubMed] [Google Scholar]
- de Sousa EMF, and de Sauvage FJ (2019). Cellular Plasticity in Intestinal Homeostasis and Disease. Cell Stem Cell 24, 54–64. [DOI] [PubMed] [Google Scholar]
- Demitrack ES, Gifford GB, Keeley TM, Horita N, Todisco A, Turgeon DK, Siebel CW, and Samuelson LC (2016). NOTCH1 and NOTCH2 Regulate Epithelial Cell Proliferation in Mouse and Human Gastric Corpus. Am J Physiol Gastrointest Liver Physiol, ajpgi 00325 02016. [DOI] [PMC free article] [PubMed]
- DeWard AD, Cramer J, and Lagasse E (2014). Cellular heterogeneity in the mouse esophagus implicates the presence of a nonquiescent epithelial stem cell population. Cell Rep 9, 701–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doupe DP, Alcolea MP, Roshan A, Zhang G, Klein AM, Simons BD, and Jones PH (2012). A single progenitor population switches behavior to maintain and repair esophageal epithelium. Science 337, 1091–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doupe DP, Klein AM, Simons BD, and Jones PH (2010). The ordered architecture of murine ear epidermis is maintained by progenitor cells with random fate. Dev Cell 18, 317–323. [DOI] [PubMed] [Google Scholar]
- Dubeykovskaya Z, Si Y, Chen X, Worthley DL, Renz BW, Urbanska AM, Hayakawa Y, Xu T, Westphalen CB, Dubeykovskiy A, et al. (2016). Neural innervation stimulates splenic TFF2 to arrest myeloid cell expansion and cancer. Nature communications 7, 10517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eicher AK, Kechele DO, Berns HM, Sundaram N, Polling HM, Haines L, Helmrath MA, and Wells JM (2021). Engineering complex human gastric tissues from three independent human pluripotent stem cell derived germ layers. Cell Stem Cell (in revision). [DOI] [PMC free article] [PubMed]
- Fang HY, Munch NS, Schottelius M, Ingermann J, Liu H, Schauer M, Stangl S, Multhoff G, Steiger K, Gerngross C, et al. (2018). CXCR4 Is a Potential Target for Diagnostic PET/CT Imaging in Barrett’s Dysplasia and Esophageal Adenocarcinoma. Clin Cancer Res 24, 1048–1061. [DOI] [PubMed] [Google Scholar]
- Farin HF, Van Es JH, and Clevers H (2012). Redundant sources of Wnt regulate intestinal stem cells and promote formation of Paneth cells. Gastroenterology 143, 1518–1529 e1517. [DOI] [PubMed] [Google Scholar]
- Flanagan DJ, Barker N, Nowell C, Clevers H, Ernst M, Phesse TJ, and Vincan E (2017). Loss of the Wnt receptor frizzled 7 in the mouse gastric epithelium is deleterious and triggers rapid repopulation in vivo. Disease models & mechanisms 10, 971–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox JG, Rogers AB, Whary MT, Ge Z, Ohtani M, Jones EK, and Wang TC (2007). Accelerated progression of gastritis to dysplasia in the pyloric antrum of TFF2 −/− C57BL6 x Sv129 Helicobacter pylori-infected mice. Am J Pathol 171, 1520–1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu DJ, Wang L, Chouairi FK, Rose IM, Abetov DA, Miller AD, Yamulla RJ, Schimenti JC, Flesken-Nikitin A, and Nikitin AY (2020). Gastric squamous-columnar junction contains a large pool of cancer-prone immature osteopontin responsive Lgr5(−)CD44(+) cells. Nature communications 11, 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gifford GB, Demitrack ES, Keeley TM, Tam A, La Cunza N, Dedhia PH, Spence JR, Simeone DM, Saotome I, Louvi A, et al. (2016). Notch1 and Notch2 receptors regulate mouse and human gastric antral epithelial cell homoeostasis. Gut [DOI] [PMC free article] [PubMed]
- Giroux V, Lento AA, Islam M, Pitarresi JR, Kharbanda A, Hamilton KE, Whelan KA, Long A, Rhoades B, Tang Q, et al. (2017). Long-lived keratin 15+ esophageal progenitor cells contribute to homeostasis and regeneration. J Clin Invest 127, 2378–2391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giroux V, and Rustgi AK (2017). Metaplasia: tissue injury adaptation and a precursor to the dysplasia-cancer sequence. Nat Rev Cancer 17, 594–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glickman JN, Chen YY, Wang HH, Antonioli DA, and Odze RD (2001). Phenotypic characteristics of a distinctive multilayered epithelium suggests that it is a precursor in the development of Barrett’s esophagus. Am J Surg Pathol 25, 569–578. [DOI] [PubMed] [Google Scholar]
- Gonzalez G, Huang Q, and Mashimo H (2016). Characterization of oncocytes in deep esophageal glands. Dis Esophagus 29, 670–680. [DOI] [PubMed] [Google Scholar]
- Guo Z, and Ohlstein B (2015. ). Stem cell regulation. Bidirectional Notch signaling regulates Drosophila intestinal stem cell multipotency. Science 350, pii: aab0988. doi: 09 10.1126/science.aab0988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han L, Chaturvedi P, Kishimoto K, Koike H, Nasr T, Iwasawa K, Giesbrecht K, Witcher PC, Eicher A, Haines L, et al. (2020). Single cell transcriptomics identifies a signaling network coordinating endoderm and mesoderm diversification during foregut organogenesis. Nature communications 11, 4158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han S, Fink J, Jorg DJ, Lee E, Yum MK, Chatzeli L, Merker SR, Josserand M, Trendafilova T, Andersson-Rolf A, et al. (2019). Defining the Identity and Dynamics of Adult Gastric Isthmus Stem Cells. Cell Stem Cell 25, 342–356 e347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hata M, Kinoshita H, Hayakawa Y, Konishi M, Tsuboi M, Oya Y, Kurokawa K, Hayata Y, Nakagawa H, Tateishi K, et al. (2020). GPR30-Expressing Gastric Chief Cells Do Not Dedifferentiate But Are Eliminated via PDK-Dependent Cell Competition During Development of Metaplasia. Gastroenterology 158, 1650–1666 e1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayakawa Y, Ariyama H, Stancikova J, Sakitani K, Asfaha S, Renz BW, Dubeykovskaya ZA, Shibata W, Wang H, Westphalen CB, et al. (2015a). Mist1 Expressing Gastric Stem Cells Maintain the Normal and Neoplastic Gastric Epithelium and Are Supported by a Perivascular Stem Cell Niche. Cancer Cell 28, 800–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayakawa Y, Chang W, Jin G, and Wang TC (2016a). Gastrin and upper GI cancers. Current opinion in pharmacology 31, 31–37. [DOI] [PubMed] [Google Scholar]
- Hayakawa Y, Jin G, Wang H, Chen X, Westphalen CB, Asfaha S, Renz BW, Ariyama H, Dubeykovskaya ZA, Takemoto Y, et al. (2015b). CCK2R identifies and regulates gastric antral stem cell states and carcinogenesis. Gut 64, 544–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayakawa Y, Sakitani K, Konishi M, Asfaha S, Niikura R, Tomita H, Renz BW, Tailor Y, Macchini M, Middelhoff M, et al. (2016b). Nerve Growth Factor Promotes Gastric Tumorigenesis through Aberrant Cholinergic Signaling. Cancer Cell [DOI] [PMC free article] [PubMed]
- Hayakawa Y, Sakitani K, Konishi M, Asfaha S, Niikura R, Tomita H, Renz BW, Tailor Y, Macchini M, Middelhoff M, et al. (2017). Nerve Growth Factor Promotes Gastric Tumorigenesis through Aberrant Cholinergic Signaling. Cancer Cell 31, 21–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayakawa Y, Sethi N, Sepulveda AR, Bass AJ, and Wang TC (2016c). Oesophageal adenocarcinoma and gastric cancer: should we mind the gap? Nat Rev Cancer 16, 305–318. [DOI] [PubMed] [Google Scholar]
- Hayakawa Y, Tsuboi M, Asfaha S, Kinoshita H, Niikura R, Konishi M, Hata M, Oya Y, Kim W, Middelhoff M, et al. (2019). BHLHA15-Positive Secretory Precursor Cells Can Give Rise to Tumors in Intestine and Colon in Mice. Gastroenterology 156, 1066–1081 e1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He L, Li Y, Li Y, Pu W, Huang X, Tian X, Wang Y, Zhang H, Liu Q, Zhang L, et al. (2017). Enhancing the precision of genetic lineage tracing using dual recombinases. Nat Med 23, 1488–1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hibdon ES, Razumilava N, Keeley TM, Wong G, Solanki S, Shah YM, and Samuelson LC (2019). Notch and mTOR Signaling Pathways Promote Human Gastric Cancer Cell Proliferation. Neoplasia 21, 702–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huh WJ, Esen E, Geahlen JH, Bredemeyer AJ, Lee AH, Shi G, Konieczny SF, Glimcher LH, and Mills JC (2010). XBP1 controls maturation of gastric zymogenic cells by induction of MIST1 and expansion of the rough endoplasmic reticulum. Gastroenterology 139, 2038–2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huh WJ, Khurana SS, Geahlen JH, Kohli K, Waller RA, and Mills JC (2012). Tamoxifen induces rapid, reversible atrophy, and metaplasia in mouse stomach. Gastroenterology 142, 21–24 e27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishibashi F, Shimizu H, Nakata T, Fujii S, Suzuki K, Kawamoto A, Anzai S, Kuno R, Nagata S, Ito G, et al. (2018). Contribution of ATOH1(+) Cells to the Homeostasis, Repair, and Tumorigenesis of the Colonic Epithelium. Stem cell reports 10, 27–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins GJ, Doak SH, Griffiths AP, Tofazzal N, Shah V, Baxter JN, and Parry JM (2003). Early p53 mutations in nondysplastic Barrett’s tissue detected by the restriction site mutation (RSM) methodology. Br J Cancer 88, 1271–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang M, Li H, Zhang Y, Yang Y, Lu R, Liu K, Lin S, Lan X, Wang H, Wu H, et al. (2017). Transitional basal cells at the squamous-columnar junction generate Barrett’s oesophagus. Nature 550, 529–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones JC, Brindley CD, Elder NH, Myers MG Jr., Rajala MW, Dekaney CM, McNamee EN, Frey MR, Shroyer NF, and Dempsey PJ (2019). Cellular Plasticity of Defa4(Cre)-Expressing Paneth Cells in Response to Notch Activation and Intestinal Injury. Cellular and molecular gastroenterology and hepatology 7, 533–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jovov B, Que J, Tobey NA, Djukic Z, Hogan BL, and Orlando RC (2011). Role of E-cadherin in the pathogenesis of gastroesophageal reflux disease. Am J Gastroenterol 106, 1039–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalabis J, Oyama K, Okawa T, Nakagawa H, Michaylira CZ, Stairs DB, Figueiredo JL, Mahmood U, Diehl JA, Herlyn M, et al. (2008). A subpopulation of mouse esophageal basal cells has properties of stem cells with the capacity for self-renewal and lineage specification. J Clin Invest 118, 3860–3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karam SM, and Leblond CP (1993a). Dynamics of epithelial cells in the corpus of the mouse stomach. I. Identification of proliferative cell types and pinpointing of the stem cell. The Anatomical record 236, 259–279. [DOI] [PubMed] [Google Scholar]
- Karam SM, and Leblond CP (1993b). Dynamics of epithelial cells in the corpus of the mouse stomach. V. Behavior of entero-endocrine and caveolated cells: general conclusions on cell kinetics in the oxyntic epithelium. The Anatomical record 236, 333–340. [DOI] [PubMed] [Google Scholar]
- Keeley TM, Horita N, and Samuelson LC (2019). Tamoxifen-Induced Gastric Injury: Effects of Dose and Method of Administration. Cellular and molecular gastroenterology and hepatology 8, 365–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim TH, and Shivdasani RA (2016). Stomach development, stem cells and disease. Development 143, 554–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinoshita H, Hayakawa Y, Konishi M, Hata M, Tsuboi M, Hayata Y, Hikiba Y, Ihara S, Nakagawa H, Ikenoue T, et al. (2019). Three types of metaplasia model through Kras activation, Pten deletion, or Cdh1 deletion in the gastric epithelium. J Pathol 247, 35–47. [DOI] [PubMed] [Google Scholar]
- Kinoshita H, Hayakawa Y, Niu Z, Konishi M, Hata M, Tsuboi M, Hayata Y, Hikiba Y, Ihara S, Nakagawa H, et al. (2018). Mature gastric chief cells are not required for the development of metaplasia. Am J Physiol Gastrointest Liver Physiol [DOI] [PMC free article] [PubMed]
- Kon S, Ishibashi K, Katoh H, Kitamoto S, Shirai T, Tanaka S, Kajita M, Ishikawa S, Yamauchi H, Yako Y, et al. (2017). Cell competition with normal epithelial cells promotes apical extrusion of transformed cells through metabolic changes. Nat Cell Biol 19, 530–541. [DOI] [PubMed] [Google Scholar]
- Kong J, Crissey MA, Funakoshi S, Kreindler JL, and Lynch JP (2011). Ectopic Cdx2 expression in murine esophagus models an intermediate stage in the emergence of Barrett’s esophagus. PLoS One 6, e18280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunze B, Wein F, Fang HY, Anand A, Baumeister T, Strangmann J, Gerland S, Ingermann J, Munch NS, Wiethaler M, et al. (2020). Notch Signaling Mediates Differentiation in Barrett’s Esophagus and Promotes Progression to Adenocarcinoma. Gastroenterology 159, 575–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo TL, Weng CC, Kuo KK, Chen CY, Wu DC, Hung WC, and Cheng KH (2016). APC haploinsufficiency coupled with p53 loss sufficiently induces mucinous cystic neoplasms and invasive pancreatic carcinoma in mice. Oncogene 35, 2223–2234. [DOI] [PubMed] [Google Scholar]
- Lavery DL, Nicholson AM, Poulsom R, Jeffery R, Hussain A, Gay LJ, Jankowski JA, Zeki SS, Barr H, Harrison R, et al. (2014). The stem cell organisation, and the proliferative and gene expression profile of Barrett’s epithelium, replicates pyloric-type gastric glands. Gut 63, 1854–1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee ER, and Leblond CP (1985a). Dynamic histology of the antral epithelium in the mouse stomach: II. Ultrastructure and renewal of isthmal cells. The American journal of anatomy 172, 205–224. [DOI] [PubMed] [Google Scholar]
- Lee ER, and Leblond CP (1985b). Dynamic histology of the antral epithelium in the mouse stomach: IV. Ultrastructure and renewal of gland cells. Am J Anat 172, 241–259. [DOI] [PubMed] [Google Scholar]
- Lee Y, Urbanska AM, Hayakawa Y, Wang H, Au AS, Luna AM, Chang W, Jin G, Bhagat G, Abrams JA, et al. (2017). Gastrin stimulates a cholecystokinin-2-receptor-expressing cardia progenitor cell and promotes progression of Barrett’s-like esophagus. Oncotarget 8, 203–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leedham SJ, Preston SL, McDonald SA, Elia G, Bhandari P, Poller D, Harrison R, Novelli MR, Jankowski JA, and Wright NA (2008). Individual crypt genetic heterogeneity and the origin of metaplastic glandular epithelium in human Barrett’s oesophagus. Gut 57, 1041–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leodolter A, Nocon M, Vieth M, Lind T, Jaspersen D, Richter K, Willich S, Stolte M, Malfertheiner P, and Labenz J (2012). Progression of specialized intestinal metaplasia at the cardia to macroscopically evident Barrett’s esophagus: an entity of concern in the ProGERD study. Scand J Gastroenterol 47, 1429–1435. [DOI] [PubMed] [Google Scholar]
- Leushacke M, Ng A, Galle J, Loeffler M, and Barker N (2013). Lgr5+ Gastric Stem Cells Divide Symmetrically to Effect Epithelial Homeostasis in the Pylorus. Cell Reports 5, 349–356. [DOI] [PubMed] [Google Scholar]
- Leushacke M, Tan SH, Wong A, Swathi Y, Hajamohideen A, Tan LT, Goh J, Wong E, Denil S, Murakami K, et al. (2017). Lgr5-expressing chief cells drive epithelial regeneration and cancer in the oxyntic stomach. Nat Cell Biol [DOI] [PubMed]
- Li L, and Clevers H (2010). Coexistence of quiescent and active adult stem cells in mammals. Science 327, 542–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li XB, Yang G, Zhu L, Tang YL, Zhang C, Ju Z, Yang X, and Teng Y (2016). Gastric Lgr5(+) stem cells are the cellular origin of invasive intestinal-type gastric cancer in mice. Cell research 26, 838–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu K, Jiang M, Lu Y, Chen H, Sun J, Wu S, Ku WY, Nakagawa H, Kita Y, Natsugoe S, et al. (2013). Sox2 cooperates with inflammation-mediated Stat3 activation in the malignant transformation of foregut basal progenitor cells. Cell Stem Cell 12, 304–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malam Z, and Cohn RD (2014). Stem cells on alert: priming quiescent stem cells after remote injury. Cell Stem Cell 15, 7–8. [DOI] [PubMed] [Google Scholar]
- Matsuo J, Douchi D, Myint K, Mon NN, Yamamura A, Kohu K, Heng DL, Chen S, Mawan NA, Nuttonmanit N, et al. (2020). Iqgap3-Ras axis drives stem cell proliferation in the stomach corpus during homoeostasis and repair. Gut [DOI] [PMC free article] [PubMed]
- Matsuo J, Kimura S, Yamamura A, Koh CP, Hossain MZ, Heng DL, Kohu K, Chih-Cheng Voon D, Hiai H, Unno M, et al. (2016). Identification of Stem Cells in the Epithelium of the Stomach Corpus and Antrum of Mice. Gastroenterology [DOI] [PubMed]
- McCarthy N, Manieri E, Storm EE, Saadatpour A, Luoma AM, Kapoor VN, Madha S, Gaynor LT, Cox C, Keerthivasan S, et al. (2020). Distinct Mesenchymal Cell Populations Generate the Essential Intestinal BMP Signaling Gradient. Cell Stem Cell 26, 391–402 e395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCracken KW, Aihara E, Martin B, Crawford CM, Broda T, Treguier J, Zhang X, Shannon JM, Montrose MH, and Wells JM (2017). Wnt/beta-catenin promotes gastric fundus specification in mice and humans. Nature 541, 182–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCracken KW, Cata EM, Crawford CM, Sinagoga KL, Schumacher M, Rockich BE, Tsai YH, Mayhew CN, Spence JR, Zavros Y, et al. (2014). Modelling human development and disease in pluripotent stem-cell-derived gastric organoids. Nature 516, 400–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald SA, Greaves LC, Gutierrez-Gonzalez L, Rodriguez-Justo M, Deheragoda M, Leedham SJ, Taylor RW, Lee CY, Preston SL, Lovell M, et al. (2008). Mechanisms of field cancerization in the human stomach: the expansion and spread of mutated gastric stem cells. Gastroenterology 134, 500–510. [DOI] [PubMed] [Google Scholar]
- McDonald SA, Lavery D, Wright NA, and Jansen M (2015). Barrett oesophagus: lessons on its origins from the lesion itself. Nature reviews Gastroenterology & hepatology 12, 50–60. [DOI] [PubMed] [Google Scholar]
- Meyer AR, Engevik AC, Madorsky T, Belmont E, Stier MT, Norlander AE, Pilkinton MA, McDonnell WJ, Weis JA, Jang B, et al. (2020). Group 2 Innate Lymphoid Cells Coordinate Damage Response in the Stomach. Gastroenterology 159, 2077–2091 e2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Middelhoff M, Westphalen CB, Hayakawa Y, Yan KS, Gershon MD, Wang TC, and Quante M (2017). Dclk1-expressing tuft cells: critical modulators of the intestinal niche? Am J Physiol Gastrointest Liver Physiol 313, G285–G299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milano F, van Baal JW, Buttar NS, Rygiel AM, de Kort F, DeMars CJ, Rosmolen WD, Bergman JJ, J, V.A.M., Wang KK, et al. (2007). Bone morphogenetic protein 4 expressed in esophagitis induces a columnar phenotype in esophageal squamous cells. Gastroenterology 132, 2412–2421. [DOI] [PubMed] [Google Scholar]
- Mills JC, and Shivdasani RA (2011). Gastric epithelial stem cells. Gastroenterology 140, 412–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montgomery RK, Carlone DL, Richmond CA, Farilla L, Kranendonk ME, Henderson DE, Baffour-Awuah NY, Ambruzs DM, Fogli LK, Algra S, et al. (2011). Mouse telomerase reverse transcriptase (mTert) expression marks slowly cycling intestinal stem cells. Proc Natl Acad Sci U S A 108, 179–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon H, Zhu J, Donahue LR, Choi E, and White AC (2019). Krt5(+)/Krt15(+) foregut basal progenitors give rise to cyclooxygenase-2-dependent tumours in response to gastric acid stress. Nat Commun 10, 2225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munch NS, Fang HY, Ingermann J, Maurer HC, Anand A, Kellner V, Sahm V, Wiethaler M, Baumeister T, Wein F, et al. (2019). High-Fat Diet Accelerates Carcinogenesis in a Mouse Model of Barrett’s Esophagus via Interleukin 8 and Alterations to the Gut Microbiome. Gastroenterology 157, 492–506 e492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nam KT, Lee HJ, Sousa JF, Weis VG, O’Neal RL, Finke PE, Romero-Gallo J, Shi G, Mills JC, Peek RM, et al. (2010). Mature chief cells are cryptic progenitors for metaplasia in the stomach. Gastroenterology 139, 2028–2037.e2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nam KT, O’Neal RL, Coffey RJ, Finke PE, Barker N, and Goldenring JR (2012). Spasmolytic polypeptide-expressing metaplasia (SPEM) in the gastric oxyntic mucosa does not arise from Lgr5-expressing cells. Gut 61, 1678–1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nanki K, Toshimitsu K, Takano A, Fujii M, Shimokawa M, Ohta Y, Matano M, Seino T, Nishikori S, Ishikawa K, et al. (2018). Divergent Routes toward Wnt and R-spondin Niche Independency during Human Gastric Carcinogenesis. Cell 174, 856–869 e817. [DOI] [PubMed] [Google Scholar]
- Nicholson AM, Graham TA, Simpson A, Humphries A, Burch N, Rodriguez-Justo M, Novelli M, Harrison R, Wright NA, McDonald SA, et al. (2012). Barrett’s metaplasia glands are clonal, contain multiple stem cells and share a common squamous progenitor. Gut 61, 1380–1389. [DOI] [PubMed] [Google Scholar]
- Nienhuser H, Kim W, Malagola E, Ruan T, Valenti G, Middelhoff M, Bass A, Der CJ, Hayakawa Y, and Wang TC (2020). Mist1+ gastric isthmus stem cells are regulated by Wnt5a and expand in response to injury and inflammation in mice. Gut [DOI] [PMC free article] [PubMed]
- Nomura S, Esumi H, Job C, and Tan SS (1998). Lineage and clonal development of gastric glands. Dev Biol 204, 124–135. [DOI] [PubMed] [Google Scholar]
- Nomura S, Settle SH, Leys CM, Means AL, Peek RM, Leach SD, Wright CV, Coffey RJ, and Goldenring JR (2005). Evidence for repatterning of the gastric fundic epithelium associated with Ménétrier’s disease and TGFalpha overexpression. Gastroenterology 128, 1292–1305. [DOI] [PubMed] [Google Scholar]
- Okumura T, Ericksen RE, Takaishi S, Wang SS, Dubeykovskiy Z, Shibata W, Betz KS, Muthupalani S, Rogers AB, Fox JG, et al. (2010). K-ras mutation targeted to gastric tissue progenitor cells results in chronic inflammation, an altered microenvironment, and progression to intraepithelial neoplasia. Cancer Res 70, 8435–8445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen RP, White MJ, Severson DT, Braden B, Bailey A, Goldin R, Wang LM, Ruiz-Puig C, Maynard ND, Green A, et al. (2018). Single cell RNA-seq reveals profound transcriptional similarity between Barrett’s oesophagus and oesophageal submucosal glands. Nat Commun 9, 4261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Q, Nicholson AM, Barr H, Harrison LA, Wilson GD, Burkert J, Jeffery R, Alison MR, Looijenga L, Lin WR, et al. (2013). Identification of lineage-uncommitted, long-lived, label-retaining cells in healthy human esophagus and stomach, and in metaplastic esophagus. Gastroenterology 144, 761–770. [DOI] [PubMed] [Google Scholar]
- Piedrafita G, Kostiou V, Wabik A, Colom B, Fernandez-Antoran D, Herms A, Murai K, Hall BA, and Jones PH (2020). A single-progenitor model as the unifying paradigm of epidermal and esophageal epithelial maintenance in mice. Nat Commun 11, 1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto D, Plieschnegger W, Schneider NI, Geppert M, Bordel H, Höss GM, Eherer A, Wolf EM, Vieth M, and Langner C (2019). Carditis: a relevant marker of gastroesophageal reflux disease. Data from a prospective central European multicenter study on histological and endoscopic diagnosis of esophagitis (histoGERD trial). Dis Esophagus 32. [DOI] [PubMed] [Google Scholar]
- Polak P, Karlic R, Koren A, Thurman R, Sandstrom R, Lawrence MS, Reynolds A, Rynes E, Vlahovicek K, Stamatoyannopoulos JA, et al. (2015). Cell-of-origin chromatin organization shapes the mutational landscape of cancer. Nature 518, 360–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potten CS (1974). The epidermal proliferative unit: the possible role of the central basal cell. Cell Tissue Kinet 7, 77–88. [DOI] [PubMed] [Google Scholar]
- Potten CS, Hume WJ, Reid P, and Cairns J (1978). The segregation of DNA in epithelial stem cells. Cell 15, 899–906. [DOI] [PubMed] [Google Scholar]
- Potten CS, Owen G, and Booth D (2002). Intestinal stem cells protect their genome by selective segregation of template DNA strands. J Cell Sci 115, 2381–2388. [DOI] [PubMed] [Google Scholar]
- Powell AE, Wang Y, Li Y, Poulin EJ, Means AL, Washington MK, Higginbotham JN, Juchheim A, Prasad N, Levy SE, et al. (2012). The pan-ErbB negative regulator Lrig1 is an intestinal stem cell marker that functions as a tumor suppressor. Cell 149, 146–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quante M, Bhagat G, Abrams JA, Marache F, Good P, Lee MD, Lee Y, Friedman R, Asfaha S, Dubeykovskaya Z, et al. (2012). Bile acid and inflammation activate gastric cardia stem cells in a mouse model of Barrett-like metaplasia. Cancer Cell 21, 36–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quante M, Marrache F, Goldenring JR, and Wang TC (2010). TFF2 mRNA transcript expression marks a gland progenitor cell of the gastric oxyntic mucosa. Gastroenterology 139, 2018–2027 e2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quante M, Tu SP, Tomita H, Gonda T, Wang SS, Takashi S, Baik GH, Shibata W, Diprete B, Betz KS, et al. (2011). Bone marrow-derived myofibroblasts contribute to the mesenchymal stem cell niche and promote tumor growth. Cancer Cell 19, 257–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Que J, Choi M, Ziel JW, Klingensmith J, and Hogan BL (2006). Morphogenesis of the trachea and esophagus: current players and new roles for noggin and Bmps. Differentiation 74, 422–437. [DOI] [PubMed] [Google Scholar]
- Que J, Okubo T, Goldenring JR, Nam KT, Kurotani R, Morrisey EE, Taranova O, Pevny LH, and Hogan BL (2007). Multiple dose-dependent roles for Sox2 in the patterning and differentiation of anterior foregut endoderm. Development 134, 2521–2531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsey VG, Doherty JM, Chen CC, Stappenbeck TS, Konieczny SF, and Mills JC (2007). The maturation of mucus-secreting gastric epithelial progenitors into digestive-enzyme secreting zymogenic cells requires Mist1. Development 134, 211–222. [DOI] [PubMed] [Google Scholar]
- Roulis M, Kaklamanos A, Schernthanner M, Bielecki P, Zhao J, Kaffe E, Frommelt LS, Qu R, Knapp MS, Henriques A, et al. (2020). Paracrine orchestration of intestinal tumorigenesis by a mesenchymal niche. Nature 580, 524–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakitani K, Hayakawa Y, Deng H, Ariyama H, Kinoshita H, Konishi M, Ono S, Suzuki N, Ihara S, Niu Z, et al. (2017). CXCR4-expressing Mist1(+) progenitors in the gastric antrum contribute to gastric cancer development. Oncotarget 8, 111012–111025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar A, Huebner AJ, Sulahian R, Anselmo A, Xu X, Flattery K, Desai N, Sebastian C, Yram MA, Arnold K, et al. (2016). Sox2 Suppresses Gastric Tumorigenesis in Mice. Cell Rep 16, 1929–1941. [DOI] [PubMed] [Google Scholar]
- Sarosi G, Brown G, Jaiswal K, Feagins LA, Lee E, Crook TW, Souza RF, Zou YS, Shay JW, and Spechler SJ (2008). Bone marrow progenitor cells contribute to esophageal regeneration and metaplasia in a rat model of Barrett’s esophagus. Dis Esophagus 21, 43–50. [DOI] [PubMed] [Google Scholar]
- Sato T, van Es JH, Snippert HJ, Stange DE, Vries RG, van den Born M, Barker N, Shroyer NF, van de Wetering M, and Clevers H (2011). Paneth cells constitute the niche for Lgr5 stem cells in intestinal crypts. Nature 469, 415–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh-Takayama N, Kato T, Motomura Y, Kageyama T, Taguchi-Atarashi N, Kinoshita-Daitoku R, Kuroda E, Di Santo JP, Mimuro H, Moro K, et al. (2020). Bacteria-Induced Group 2 Innate Lymphoid Cells in the Stomach Provide Immune Protection through Induction of IgA. Immunity 52, 635–649 e634. [DOI] [PubMed] [Google Scholar]
- Schonhuber N, Seidler B, Schuck K, Veltkamp C, Schachtler C, Zukowska M, Eser S, Feyerabend TB, Paul MC, Eser P, et al. (2014). A next-generation dual-recombinase system for time- and host-specific targeting of pancreatic cancer. Nat Med 20, 1340–1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schutz B, Ruppert AL, Strobel O, Lazarus M, Urade Y, Buchler MW, and Weihe E (2019). Distribution pattern and molecular signature of cholinergic tuft cells in human gastro-intestinal and pancreatic-biliary tract. Scientific reports 9, 17466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seery JP, and Watt FM (2000). Asymmetric stem-cell divisions define the architecture of human oesophageal epithelium. Curr Biol 10, 1447–1450. [DOI] [PubMed] [Google Scholar]
- Serrano M, Lin AW, McCurrach ME, Beach D, and Lowe SW (1997). Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 88, 593–602. [DOI] [PubMed] [Google Scholar]
- Sethi NS, Kikuchi O, Duronio GN, Stachler MD, McFarland JM, Ferrer-Luna R, Zhang Y, Bao C, Bronson R, Patil D, et al. (2020). Early TP53 alterations engage environmental exposures to promote gastric premalignancy in an integrative mouse model. Nat Genet 52, 219–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng W, Malagola E, Nienhuser H, Zhang Z, Kim W, Zamechek L, Sepulveda A, Hata M, Hayakawa Y, Zhao CM, et al. (2020). Hypergastrinemia expands gastric ECL cells through CCK2R+ progenitor cells via ERK activation. Cellular and molecular gastroenterology and hepatology [DOI] [PMC free article] [PubMed]
- Shoshkes-Carmel M, Wang YJ, Wangensteen KJ, Toth B, Kondo A, Massasa EE, Itzkovitz S, and Kaestner KH (2018). Subepithelial telocytes are an important source of Wnts that supports intestinal crypts. Nature 557, 242–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigal M, Logan CY, Kapalczynska M, Mollenkopf HJ, Berger H, Wiedenmann B, Nusse R, Amieva MR, and Meyer TF (2017). Stromal R-spondin orchestrates gastric epithelial stem cells and gland homeostasis. Nature 548, 451–455. [DOI] [PubMed] [Google Scholar]
- Sigal M, Reines MDM, Mullerke S, Fischer C, Kapalczynska M, Berger H, Bakker ERM, Mollenkopf HJ, Rothenberg ME, Wiedenmann B, et al. (2019). R-spondin-3 induces secretory, antimicrobial Lgr5(+) cells in the stomach. Nat Cell Biol 21, 812–823. [DOI] [PubMed] [Google Scholar]
- Sigal M, Rothenberg ME, Logan CY, Lee JY, Honaker RW, Cooper RL, Passarelli B, Camorlinga M, Bouley DM, Alvarez G, et al. (2015). Helicobacter pylori Activates and Expands Lgr5(+) Stem Cells Through Direct Colonization of the Gastric Glands. Gastroenterology 148, 1392–1404 e1321. [DOI] [PubMed] [Google Scholar]
- Song Y, Li L, Ou Y, Gao Z, Li E, Li X, Zhang W, Wang J, Xu L, Zhou Y, et al. (2014). Identification of genomic alterations in oesophageal squamous cell cancer. Nature 509, 91–95. [DOI] [PubMed] [Google Scholar]
- Srivastava A, Golden KL, Sanchez CA, Liu K, Fong PY, Li X, Cowan DS, Rabinovitch PS, Reid BJ, Blount PL, et al. (2015). High Goblet Cell Count Is Inversely Associated with Ploidy Abnormalities and Risk of Adenocarcinoma in Barrett’s Esophagus. PLoS One 10, e0133403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stachler MD, Taylor-Weiner A, Peng S, McKenna A, Agoston AT, Odze RD, Davison JM, Nason KS, Loda M, Leshchiner I, et al. (2015). Paired exome analysis of Barrett’s esophagus and adenocarcinoma. Nat Genet 47, 1047–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stahl PL, Salmen F, Vickovic S, Lundmark A, Navarro JF, Magnusson J, Giacomello S, Asp M, Westholm JO, Huss M, et al. (2016). Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science 353, 78–82. [DOI] [PubMed] [Google Scholar]
- Stange DE, Koo BK, Huch M, Sibbel G, Basak O, Lyubimova A, Kujala P, Bartfeld S, Koster J, Geahlen JH, et al. (2013). Differentiated troy(+) chief cells act as reserve stem cells to generate all lineages of the stomach epithelium. Cell 155, 357–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stzepourginski I, Nigro G, Jacob JM, Dulauroy S, Sansonetti PJ, Eberl G, and Peduto L (2017). CD34+ mesenchymal cells are a major component of the intestinal stem cells niche at homeostasis and after injury. Proc Natl Acad Sci U S A 114, E506–E513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda N, Jain R, LeBoeuf MR, Wang Q, Lu MM, and Epstein JA (2011). Interconversion between intestinal stem cell populations in distinct niches. Science 334, 1420–1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan SH, Swathi Y, Tan S, Goh J, Seishima R, Murakami K, Oshima M, Tsuji T, Phuah P, Tan LT, et al. (2020). AQP5 enriches for stem cells and cancer origins in the distal stomach. Nature 578, 437–443. [DOI] [PubMed] [Google Scholar]
- Tatematsu M, Fukami H, Yamamoto M, Nakanishi H, Masui T, Kusakabe N, and Sakakura T (1994). Clonal analysis of glandular stomach carcinogenesis in C3H/HeN<==>BALB/c chimeric mice treated with N-methyl-N-nitrosourea. Cancer Lett 83, 37–42. [DOI] [PubMed] [Google Scholar]
- Thiem S, Eissmann MF, Elzer J, Jonas A, Putoczki TL, Poh A, Nguyen P, Preaudet A, Flanagan D, Vincan E, et al. (2016). Stomach-Specific Activation of Oncogenic KRAS and STAT3-Dependent Inflammation Cooperatively Promote Gastric Tumorigenesis in a Preclinical Model. Cancer Res 76, 2277–2287. [DOI] [PubMed] [Google Scholar]
- Thompson M, Fleming KA, Evans DJ, Fundele R, Surani MA, and Wright NA (1990). Gastric endocrine cells share a clonal origin with other gut cell lineages. Development 110, 477–481. [DOI] [PubMed] [Google Scholar]
- Tian H, Biehs B, Warming S, Leong KG, Rangell L, Klein OD, and de Sauvage FJ (2011). A reserve stem cell population in small intestine renders Lgr5-positive cells dispensable. Nature 478, 255–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomasetti C, and Vogelstein B (2015). Cancer etiology. Variation in cancer risk among tissues can be explained by the number of stem cell divisions. Science 347, 78–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trisno SL, Philo KED, McCracken KW, Cata EM, Ruiz-Torres S, Rankin SA, Han L, Nasr T, Chaturvedi P, Rothenberg ME, et al. (2018). Esophageal Organoids from Human Pluripotent Stem Cells Delineate Sox2 Functions during Esophageal Specification. Cell Stem Cell 23, 501–515 e507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Udager A, Prakash A, and Gumucio DL (2010). Dividing the tubular gut: generation of organ boundaries at the pylorus. Prog Mol Biol Transl Sci 96, 35–62. [DOI] [PubMed] [Google Scholar]
- Vercauteren Drubbel A, Pirard S, Kin S, Dassy B, Lefort A, Libert F, Nomura S, and Beck B (2021). Reactivation of the Hedgehog pathway in esophageal progenitors turns on an embryonic-like program to initiate columnar metaplasia. Cell Stem Cell [DOI] [PubMed]
- Viaene AI, and Baert JH (1995). Expression of cytokeratin mRNAs in normal human esophageal epithelium. Anat Rec 241, 88–98. [DOI] [PubMed] [Google Scholar]
- Visvader JE (2011). Cells of origin in cancer. Nature 469, 314–322. [DOI] [PubMed] [Google Scholar]
- Wang X, Ouyang H, Yamamoto Y, Kumar PA, Wei TS, Dagher R, Vincent M, Lu X, Bellizzi AM, Ho KY, et al. (2011). Residual embryonic cells as precursors of a Barrett’s-like metaplasia. Cell 145, 1023–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinreb C, Rodriguez-Fraticelli A, Camargo FD, and Klein AM (2020). Lineage tracing on transcriptional landscapes links state to fate during differentiation. Science 367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westphalen CB, Asfaha S, Hayakawa Y, Takemoto Y, Lukin DJ, Nuber AH, Brandtner A, Setlik W, Remotti H, Muley A, et al. (2014). Long-lived intestinal tuft cells serve as colon cancer-initiating cells. J Clin Invest 124, 1283–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White AC, and Lowry WE (2015). Refining the role for adult stem cells as cancer cells of origin. Trends Cell Biol 25, 11–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worthley DL, Churchill M, Compton JT, Tailor Y, Rao M, Si Y, Levin D, Schwartz MG, Uygur A, Hayakawa Y, et al. (2015). Gremlin 1 identifies a skeletal stem cell with bone, cartilage, and reticular stromal potential. Cell 160, 269–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan KS, Chia LA, Li X, Ootani A, Su J, Lee JY, Su N, Luo Y, Heilshorn SC, Amieva MR, et al. (2012). The intestinal stem cell markers Bmi1 and Lgr5 identify two functionally distinct populations. Proc Natl Acad Sci U S A 109, 466–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshioka T, Fukuda A, Araki O, Ogawa S, Hanyu Y, Matsumoto Y, Yamaga Y, Nakanishi Y, Kawada K, Sakai Y, et al. (2019). Bmi1 marks gastric stem cells located in the isthmus in mice. J Pathol 248, 179–190. [DOI] [PubMed] [Google Scholar]
- Zafar H, Lin C, and Bar-Joseph Z (2020). Single-cell lineage tracing by integrating CRISPR-Cas9 mutations with transcriptomic data. Nature communications 11, 3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Schaefer A, Wang Y, Hodge RG, Blake DR, Diehl JN, Papageorge AG, Stachler MD, Liao J, Zhou J, et al. (2019). Gain-of-Function RHOA Mutations Promote Focal Adhesion Kinase Activation and Dependency in Diffuse Gastric Cancer. Cancer discovery [DOI] [PMC free article] [PubMed]
- Zhang Y, Bailey D, Yang P, Kim E, and Que J (2021). The development and stem cells of the esophagus. Development 148, dev193839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Yang Y, Jiang M, Huang SX, Zhang W, Al Alam D, Danopoulos S, Mori M, Chen YW, Balasubramanian R, et al. (2018). 3D Modeling of Esophageal Development using Human PSC-Derived Basal Progenitors Reveals a Critical Role for Notch Signaling. Cell Stem Cell 23, 516–529 e515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao CM, Hayakawa Y, Kodama Y, Muthupalani S, Westphalen CB, Andersen GT, Flatberg A, Johannessen H, Friedman RA, Renz BW, et al. (2014). Denervation suppresses gastric tumorigenesis. Science translational medicine 6, 250ra115. [DOI] [PMC free article] [PubMed] [Google Scholar]