

Graphical abstract
Keywords: Endometrial cancer, ATAC-seq, TFCR, Transcriptional regulation, Diagnostic biomarkers
Abstract
Endometrial cancer (EC) is one of the three fatal tumors of the female reproductive system. Epigenetic alterations have been reported to be important in tumorigenesis, especially the chromatin accessibility changes and transcription factor binding differences. However, the regulatory mechanism underlying epigenetic alterations in EC development remains unclear. Here, we identified and characterized transcription factor binding site clustered regions (TFCRs) by integrating chromatin accessibility and transcription factor binding information. We totally identified 78,820 TFCRs and explored the relationship between TFCRs and regulatory elements, gene expression and mutation. Finally, we constructed a bioinformatic framework to identify candidate oncogenes and screened 13 candidate key genes, which may serve as potential diagnostic markers or therapeutic targets of EC.
1. Introduction
Endometrial cancer (EC) is the sixth most common cause of cancer-related death in women [1], [2]. EC patients in early stage can reach over 90% survival rate after surgical treatment, but some of them may enter the advanced stage before the symptoms and signs appear, and the five-year survival rate is less than 20% [3], [4]. The median survival duration for patients with metastatic or recurrent EC is only 12 to 15 months [5], [6], [7], [8]. Providing histological information by diagnostic curettage under hysteroscopy is the gold standard for early diagnosis. However, hysteroscopy is expensive, invasive and even painful, especially in nulliparous women, bringing a risk of life-threatening uterine perforation and other serious complications [9], [10], [11], [12]. Previous studies demonstrated that patients who have undergone hysteroscopy have a higher incidence of malignant peritoneal cytology at hysterectomy than those who have not [9], [11]. According to the American Cancer Society, there have been 420 thousand new cases of EC diagnosed in 2020 [1], [2], which underscores the urgent need to develop effective strategies for the diagnosis and treatment of EC.
Growing knowledge has confirmed the importance of gene mutations and epigenetics in EC. To date, 16 genomic loci closely associated with EC risk have been identified through genome-wide association studies (GWASs) [13], [14], [15]. Next generation sequencing (NGS) provides a new opportunity to study the clinical significance of somatic mutations in EC [16], [17]. Recent study have demonstrated the importance of the epigenome to cancer initiation and progression [18]. Epigenomic dynamics are governed by reversible covalent modifications such as DNA methylation and histone modifications. Compare with normal endometrium, EC has both hypomethylated and hypermethylated changes and abnormal gene promoter methylation according to global methylation studies [19], [20], [21]. Recent studies indicated that the expression level of histone deacetylase (HDAC) 1/2/3 and SIRT1 are higher in EC than in non-neoplastic endometrium [22], [23]. EZH2, the major enzyme that methylates the H3K27 residue, has been reported to be up regulated in multiple tumors and associated with their aggressiveness [24], [25], [26], [27]. Besides, epigenetic changes lead to alterations of chromatin accessibility and binding of transcription factors (TFs), which collectively result in transcriptional dysregulation [28]. Therefore, exploring gene mutations and epigenetic features in EC is of great significance for understanding the occurrence and development of tumor.
Eukaryotic genome is tightly located in chromatin, and only open chromatin can be targeted by regulatory factors such as TFs. The assay for transposase-accessible chromatin using sequencing (ATAC-seq) makes it possible to quantify chromatin accessibility and thus to assess the gene regulatory landscape in primary human cancers [29]. In our previous research, we developed a method for identifying transcription factor binding site clustered regions (TFCRs) based on chromatin accessibility and TF motif [30]. We found that spatially located adjacent TFCRs act as a whole to regulate gene expression, thereby generating diverse modes of transcriptional regulation [31], and the HOT region where transcription factor binding sites (TFBSs) are most concentrated is significantly enriched in mutation sites related to disease or tumor formation [32]. Based on this finding, we closely matched the ATAC-seq data with the TFCR to facilitate further understanding of the transcriptional regulatory mechanisms of EC. In this study, we first identified TFCRs in EC based on the ATAC-seq data and conducted an in-depth exploration of their nature. Additionally, we proposed a TFCR-based framework for screening candidate oncogenes in EC, which provided new insights into the mechanism underlying development of EC and promoted the discovery of potential diagnostic and therapeutic targets.
2. Results
2.1. Identification of TFCRs in human EC
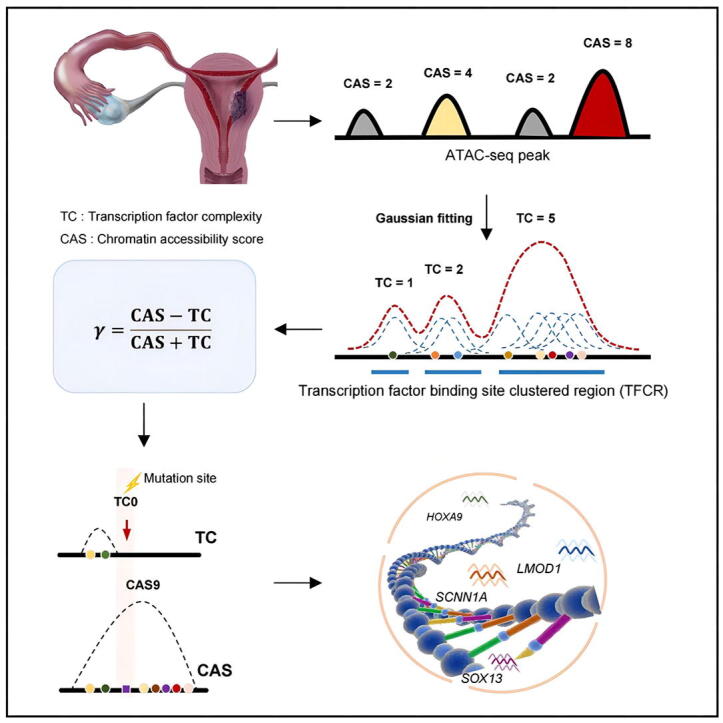
We collected the ATAC-seq data of endometrial cancer from previous study in Science [29]. Based on ATAC-seq data, we performed an improved analysis to identify TFCRs (Fig. 1) [30]. Briefly, we obtained DNA sequence motifs from the TRANSFAC [33], JASPAR [34] and UniPROBE [35] databases and scanned the TFBSs in the open chromatin regions using iFORM [36] (Fig. 1). Then, we performed a Gaussian fitting on all TFBSs and got 78,820 TFBSs clustered regions (TFCRs) (Fig. 1). We next calculated the two features of TFCR, TF complexity (TC) and chromatin accessibility score (CAS) (Fig. 1). TC reflects the number of TFBSs in query TFCR, and CAS reflects the chromatin accessibility level measured by ATAC-seq data of TFCR.
Fig. 1.

Schematic diagram for the identification of TFCRs.
To characterize TFCRs, we compared them with ATAC-seq peaks. The TFCRs covered 95% of the ATAC-seq peaks (Fig. 2A). TFBSs is more enriched on the ATAC-seq peaks overlapping with TFCRs and genes in non-overlapped region are associated with transcription (Fig. 2B, C). The genome-wide coverage rates of TFCRs and ATAC-seq peaks were 2.51% and 1.73%, respectively. The average length of TFCR was 968 bp, and the variance was 300 bp (Table S1). It’s well accepted that tumor results from the accumulated gene mutations and recent studies showed that driver mutations which lead to endometrial cancer are initiated early in life [37], [38], [39]. We obtained EC somatic mutations from the TCGA to compare the mutation rate (ρ, defined as the number of mutation sites listed in the TCGA MAF per kilobase in a specific region of the genome) of the whole genome and different intervals [40]. The mutation rate (ρ) of the whole genome was 0.29 while the mutation rates of ATAC-seq peaks and TFCRs were 0.57 and 0.53, respectively. Furthermore, the mutation rate (ρ) of the TFCR-specific interval (TFCRs without a coincidence interval with ATAC-seq peaks) was 0.51 (Fig. 2D). For EC, both TFCRs and ATAC-seq peaks contain a large number of tumor mutation sites, which indicate that the target genes corresponding to these regulatory elements may play an important role in tumor development.
Fig. 2.

Relationship between TFCRs and ATAC-seq peaks. (A) Pie chart shows the rate of overlap with TFCRs in ATAC-seq peaks. (B) Barplot shows the normalized TFBS distribution for ATAC-seq peaks are overlapped with or without TFCRs. (C) Gene ontology analysis of genes whose ATAC-seq peaks are non-overlapped with TFCRs. (D) Barplot shows the mutation rate (ρ) of the whole genome, TFCRs, ATAC-seq peaks and TFCR-specific peaks. ρ represents the number of TCGA mutation sites per thousand bases in a certain range.
2.2. TFCRs target important cis-regulatory elements
To investigate the relationship between TFCRs and regulatory elements, we compared the proportions of TFCRs or ATAC-seq peaks located in promoters, enhancers, CpG islands and repeat elements. ATAC-seq peaks were divided into 10 groups (from CAS0 to CAS9) according to chromatin accessibility levels. The proportion of ATAC-seq peaks located in promoters first slightly decreased and then gradually increased (Fig. 3A). The proportion from CAS0 to CAS4 decreased from 14.76% to 14.00%, and there was no significant difference in different groups. The proportion reached the highest (62.90%) at CAS9, and the overall increase was 4.99 folds. However, as the TC value increased, the proportion of TFCRs in promoters gradually increased from 7.73% (TC0) to 38.95% (TC9) (Fig. 3A). We next investigated the enhancers, the proportion of TFCRs located near super-enhancers and typical enhancers increased from 3.94% and 12.28% in TC0 to 7.23% and 28.97% in TC9, respectively (Fig. 3B), while the proportion of ATAC-seq peaks located near super-enhancers and typical enhancers increased from 5.16% and 18.52% in CAS0 to 7.6% and 40.32% in CAS9, respectively (Fig. 3B). Mendenhall et al defined HOT regions by studying the chromatin associated protein (CAP) cluster and found that more than 92% of HOT regions can be mapped to candidate promoters or strong enhancer-like states, which suggested that TF clusters might map to enhancers or promoters [41].
Fig. 3.

Genomic characterization of TFCRs and ATAC-seq peaks. (A) Barplot shows the proportion of TFCRs or ATAC-seq peaks located in gene promoters. (B) Stacked barplot shows the proportion of TFCRs or ATAC-seq peaks located in super-enhancers (Red) and typical enhancers (Gray). (C) Barplot shows the proportion of TFCRs or ATAC-seq peaks with CpG islands. (D) Barplot shows the proportion of TFCRs or ATAC-seq peaks on repeats (microsatellite sites). * P < 0.05.
In addition, CpG islands were reported to play an important role in regulating gene expression and gene mutation [42], next we focused on the relationship between TFCRs and CpG islands and found similar results (Fig. 3C). The proportion of TFCRs within CpG islands increased nearly 3folds from TC0 (7.63%) to TC6 (21.19%) (Fig. 3C), while the proportion of ATAC-seq peaks within CpG islands increased only 7.33% (< 2folds) from CAS0 (11.83%) to CAS6 (18.06%) (Fig. 3C). Furthermore, we investigated the proportion of TFCRs located in the promoters, enhancers, and those with CpG islands located near other cis-regulatory elements at the same time. As expected, the proportion at the corresponding regulatory element was observed to increase steadily with the increase of TC value in TFCRs, and similar results were observed at ATAC-seq peaks (fig. S1).
Repeat elements are the same or symmetrical segments in the genome that contain much genetic information and play important role in the gene regulation. Microsatellites (tandem simple sequence repeats, 1–6 bp motif) are abundant across the genome and show high levels of polymorphism [43]. We found that the proportions of TFCRs and ATAC-seq peaks in the microsatellite regions increased as TC and CAS value increased, and the highest proportions in TC9 and CAS9 were 27.33% and 15.09%, respectively (Fig. 3D). In addition, we further analyzed other types of repeats. In general, the proportion of TFCRs on repeats increased with the increase of TC value, while the proportion of ATAC-seq peak on repeat sequence decreased with the increase of CAS value (fig. S2A). On the other hand, interspersed repeats showed a downward trend regardless of TFCRs or ATAC-seq peaks while most tandem repeats showed an upward trend (fig. S2B-G). Taken together, these results confirmed a close relationship between TFCRs and regulatory elements, demonstrating that TFCRs may affect the cis-regulation.
2.3. Relationship between TF complexity and chromatin accessibility in TFCRs
As TC and CAS reflect the two aspects of TFCRs, we next investigated the relationship between TC and CAS in TFCRs. We conducted a correlation analysis and found that the TC was not correlated with CAS (R2 = 0.05) (Fig. 4A), indicating that there may be a large difference between TC and chromatin accessibility score. According to the TC, TFCR was equally divided into 23 bins with 10 as the interval value, and then we investigated the chromatin accessibility level of TFCRs in each bin and found: i) when TC was less than 90, CAS showed a steady upward trend as the TC increased; ii) when TC was between 90 and 110, the upward trend gradually became flat; iii) when TC was greater than 110, CAS showed an unstable decline (Fig. 4B). In other words, there is a significant correlation between TC and CAS when TC is low. However, the number of TFBSs in TFCR may have reached saturation when TC is at a high level, leading TC and CAS to an unbalanced state. Perhaps phase separation is also a contributor to this phenomenon. Many TFs contain intrinsically disordered regions (IDRs), which contain low complexity amino acid sequences that allow weak multivalent interactions with other TFs and cofactors to facilitate liquid–liquid phase separation (LLPS) (Fig. 3D) [44], [45], [46]. Next, we analyzed TFCR with a high imbalance of TC and CAS, and found that cis-regulatory elements preferred to be enriched near TFCRs with high TC or high CAS (fig. S3).
Fig. 4.

Properties of the characteristic parameters TC and CAS in TFCRs. (A) Correlation analysis of the TF complexity and chromatin accessibility score in TFCRs (R2 = 0.05). Boxplot shows (B) the distribution of score in different groups obtained by grouping the TF complexity of TFCRs with an interval value of 10. (C-D) The gene expression levels in different groups in ATAC-seq peaks classified by CAS and TFCRs classified by TC. The upper and lower lines above and below the boxes are the whiskers. (E) Barplot shows the mutation distribution of TFCR with promoter and TFCR without promoter. (F) The line graph shows the mutation rate of TFCRs in gene promoters (CAS: Darkblue; TC: Darkred) classified by TC or CAS. (G-H) Barplots show the proportion of reported cancer driver genes distributed in different classification of TC and CAS.
By exploring the relationship between TC/CAS of TFCRs and gene expression, we found that, similar to ATAC-seq peaks (Fig. 4C), a large number of highly expressed genes were enriched in the TC9 region (Fig. 4D), and CAS was also positively correlated with gene expression (R2 = 0.59) (fig. S4A, S4B).
2.4. TFCRs with high TF complexity or chromatin accessibility score may be the driver of tumorigenesis
Genomic mutations affect the expression of corresponding genes and participate in the occurrence and development of tumors [47]. We next collected the mutation sites from the TCGA to explore the relationship between TFCRs and gene mutation. We found that the overall mutation rate (ρ) in TFCRs did not change significantly between different TC/CAS values (fig. S4C). Gene promoter control the initiation and extent of gene expression (transcription) by binding to TFs [48]. Therefore, mutations in promoter region may be more important for transcriptional regulation. We found that TFCRs with promoter have a higher mutation rate (ρ) compared to those without (Fig. 4E). For TFCRs in promoter region, the mutation rate decreased from TC0 (ρ = 1.69) to TC9 (ρ = 0.76), while it did not change obviously (the range was only 0.36) accompanied with the increase of CAS (Fig. 4F).
Likewise, Moore et al obtained sites information on single base substitution (SBS) driver mutations in normal endometrial gland samples [39]. They suggested that cell clones with these oncogene “driver” mutations usually originate early in life and accumulate to promote EC. We extracted 133 complete sites identified in Moore et al's study and found that 11.28% (15/133) of them located on TFCRs involving RRAS2, ERBB3, ERBB2, FOXA2, and EGFR [39]. Next, we compared previously reported oncogenes with an equal number of randomly selected protein-coding genes and found oncogenes are more likely to fall on TFCRs (fig. S5). At the same time, we examined the human driver genes identified by different research teams and found that most of the cancer driver genes were located on TC9 or CAS9 (Fig. 4G-H) [38], [49], [50], [51], [52], [53], [54], [55], [56].
According to the observations above, TFCRs contain a large number of regions with extremely unbalanced TC and CAS. We found that among TFCRs, the TC0 region had the highest mutation rate and the lowest genome stability (Fig. 4F). Furthermore, we found a higher proportion of known cancer driver genes and enrichment of high expression genes in TC9 and CAS9, suggesting that these regions may be involved in the formation and progression of tumors [49].
2.5. Candidate oncogenes screening framework based on TFCRs
Based on the above, we fused two features of TFCR (TC and CAS), which reflected the information of different dimensions, and constructed a cancer driver gene screening formula to calculate the carcinogenic potential γ of each TFCR (Fig. 5A).
Fig. 5.

Identification of key cancer genes in endometrial cancer. (A) The pipeline used to identify key cancer genes in endometrial cancer based on TFCRs and ATAC-seq peaks. (B) The genome browser shows the positional relationship of the example gene, PRSS8 with TFCRs, ATAC-seq peaks, TCGA mutation sites and transcription factor binding sites in the genome. Each track represents a different component and is distinguished by different colors. The picture below is an enlarged image of the picture above.
The difference between CAS and TC indicated that target TFCR is prone to contain mutations or other complex factors, which is more likely to be associated with the development of cancer. We screened the genes contained in each TFCR with high carcinogenic potential and focused on genes, which distinctly expressed in EC. Among them, genes covered by TFCRs that belong to both TC0 and CAS9 possess the most carcinogenic potential. Finally, we obtained 13 genes which might take part in the development of EC, including SCNN1A, TROAP, UBE2C, TPX2, SOX13, BUB1, HOXA9, MAOA, PRSS8, SCGB2A1, NRP2, LMOD1 and MTMR11 in which BUB1, PRSS8, SCGB2A1, TPX2, TROAP, UBE2C and SCNN1A are up-regulated in EC and others are down-regulated (fig. S6). SCGB2A1 has been reported to be a promising independent prognostic factor in EC and its decreased expression is significantly associated with poor prognostic clinicopathological characteristics [57]. Besides, UBE2C, TPX2 and BUB1 have also been reported to be closely related to proliferation, invasion, differentiation and prognosis of EC [58], [59], [60], [61], [62], [63]. MTMR11 is shown to participate in drug resistance in endometrial cancer cells treated with salinomycin and NPR2 expression increased with the degree of histological differentiation [64], [65]. Proofs that these genes are associated with the development of endometrial cancer suggested that TFCR-based method is indeed effective in screening candidate oncogenes and provided a possibility to explain the mechanism underlying how these genes worked in the development of EC.
2.6. Mutation of PRSS8 in EC
Reports have suggested that PRSS8 functions as a tumor suppressor gene in a variety of tumors [58], [62]. We found two base substitution (G-to-A and A-to-T) located in the binding sites of AHR (aryl hydrocarbon receptor) and ELF1 (E74 like factor 1) of PRSS8 promoter region (chr16:31,131,433–31,135,830), respectively (Fig. 5B). Previous studies have shown that AHR and ELF1 can inhibit cell proliferation and migration [66], [67]. The mutation in the binding sites of AHR and ELF1 may effect PRSS8 to exert its anti-cancer effects in EC.
2.7. Validation of candidate key genes in EC
Except for the genes mentioned above, the role of HOXA9, SCNN1A, TROAP, SOX13, MAOA and LMOD1 have not been validated in EC. HOX genes play important role in reproductive tract development, endometrial cyclic growth and embryo implantation in which HOXA9 has been reported to stimulate cancer-associated fibroblasts and promote ovarian cancer growth. To verify the role of HOXA9 played in EC, we knocked down its expression in human endometrial cell line (HEC-1-A) using siRNA (small interfering RNA) (Fig. 6A), then flow cytometry and transwell assay were used to detect the effect of HOXA9 knockdown on apoptosis, invasion and migration. Decreased expression of HOXA9 enhanced the apoptosis of HEC-1-A cells (Fig. 6B) and the number of cells migrating to the lower membrane in the si-HOXA9 group decreased markedly compared with negative control (Fig. 6C). In addition, HOXA9 knockdown significantly impaired the invasion ability of HEC-1-A cells (Fig. 6C). We detected the migration and invasion ability of HEC-1-A with SCNN1A, TROAP, SOX13, MAOA and LMOD1 knockdown, respectively (fig. S7A). The knockdown of these 5 genes also significantly reduced the migration and invasion of HEC-1-A (fig. S7B, C, D).
Fig. 6.

Effect of HOXA9 knockdown on apoptosis, migration and invasion of HEC-1-A. (A) Effect of HOXA9 knockdown on (B) apoptosis, (C) invasion and migration potential of HEC-1-A, detected by flow cytometry and transwell assay. Data are expressed as the mean ± standard deviation of three independent experiments. * P < 0.05; ** P < 0.01; *** P < 0.001. NC, negative control. (D) TFCR regulation model.
3. Discussion
Tumorigenesis is a multifactorial, multistage, multigenic process involving the accumulation of genetic and epigenetic alterations. Unlike gene mutations and gene deletions, epigenetic modifications are reversible, so they can aid in cancer prevention and treatment by restoring the expression of some key genes in cancers or precancerous lesions. With the application of high-throughput sequencing technology in clinical research, it is possible to conduct a detailed panoramic analysis of the genomes of human cancers. Combined with the richness of diverse, orthogonal data types in the TCGA, the chromatin accessibility landscape in cancer provides a key link between somatic mutations, long-range gene regulation, DNA methylation and gene expression changes that affect cancer prognosis [29]. Transcription factor binding sites are one of the main aspects to study the non-coding regions of the genome. At present, annotations of gene functions focus mainly on the coding regions of genes, while these regions account for only 3% of the whole genome. The functions of noncoding regions, which account for approximately 97% of the whole genome, cannot be ignored. Over the past decade, GWASs have identified a large number of genetic mutations associated with human diseases and traits, most of which are located in noncoding cis-regulatory sequences [68]. Genes are turned on and off in the proper cell types and cell states by TFs acting on DNA regulatory elements that are scattered over the vast noncoding regions of the genome and exert long-range influences [29]. Furthermore, there are more than 1,000 transcription factors in the genome, each corresponding to tens of thousands of TFBSs. TF-TF or TF-genes form an extremely complex regulatory network, which is difficult to study. Instead of considering the regulatory mechanism of a single transcription factor, we take the TF cluster as a whole to explore its regulatory mechanism in tumor formation.
Here, for the first time, we applied the improved TFCR identification method to ATAC-seq data and obtained 78,820 TFCRs in the whole genome. Two features of TFCRs were defined: TC and CAS. TC is a characteristic scalar measuring the ability of a genomic region to bind transcription factors, but the presence of TFBS does not imply a certain binding to the transcription factor. The binding of transcription factors to DNA is affected by some aspects such as epigenetic modification, transcription factor concentration, and the three-dimensional structure of chromatin. CAS is a characteristic scalar to reflect the chromatin accessibility level of the region in the actual sequencing process, which can represent the level of real binding transcription factors to a certain extent. However, sequencing noise, algorithm bias and other factors will also affect its accuracy.
The values of TC and CAS have a strong correlation with genomic location and gene expression. The loci on the genome where TC is imbalanced from CAS are always of special biological significance. Single nucleotide polymorphisms (SNPs), especially in the transcription factor binding sites, may lead to misbinding and loss of transcription factors (Fig. 6D, Table S2), which would influence the function of oncogenes or anti-oncogens and finally lead to carcinogenesis. Neither TC nor CAS can independently characterize the dynamic changes of the genome in human diseases. Combination TC and CAS of TFCRs provide a new perspective for us to explore the mechanism of tumorigenesis and search for more comprehensive disease-related targets. Based on this, we constructed a bioinformatics framework and identified 13 candidate key genes associated with the regulation of TFCRs that may play important roles in endometrial cancer formation, which may provide a direction for tumor therapy targeting transcription factors.
With the enrichment of more comprehensive sequencing data, such as scATAC-seq, Bis-seq, ChIP-seq, and Hi-C data of EC tissues and matched normal tissues, our research will further broaden people's understanding of the mechanism of cancer epigenetic regulation and we can identify cancer key genes more accurately. Our method of screening key cancer genes in EC is not limited to specific types of tumors. It can be widely used in studies on various human tumors and provide a new way to reveal the epigenetic changes of human tumors and predict diagnosis and treatment targets.
4. Materials and Methods
4.1. Datasets
The ATAC-seq data of EC were derived from the work of Corces et al [29]. Raw ATAC-seq data, fastq or aligned BAM files, were downloaded from the NIH Genomic Data Commons portal (https://portal.gdc.cancer.gov/). The somatic mutation data of EC were downloaded from the TCGA database (https://portal.gdc.cancer.gov/repository) [40]. Expression data were obtained from the TCGA database (https://portal.gdc.cancer.gov/repository). We extracted the repeat sequences of the human reference genome GRCH37/hg19 from RepeatMasker (http://www.repeatmasker.org/). For somatic mutation analysis of TFCR, the human reference genome (GRCH38/hg38) was used, and the rest of the analysis used GRCH37/hg19 as reference. The use of the data strictly adhered to the ENCODE Consortium Data Release Policy. The promoter is defined as 2 kb region upstream and downstream of the transcription start site (TSS). The enhancer data in the EC were obtained from the SEdb database (http://www.licpathway.net/sedb/) [69]. CpG islands data were obtained from the UCSC database (https://genome.ucsc.edu/cgi-bin/hgTables).
4.2. Identification of TFBSs
A total of 796 motifs, which corresponded to 542 TFs, were collected from the TRANSFAC [33], JASPAR [34], and UniPROBE [35] databases. We used the genomic sequences under the chromatin open sites in the hg19 genome as input for iFORM [36] to scan for motif instances. The motif instances were combined to generate the TFBSs for each TF.
4.3. Identification of TFCRs in EC
The identification of TFCR has been described in detail by Chen et al [30] . In brief, we regarded each TFBS on the genome wide as a Gaussian distribution with a bandwidth of 300 bp centered on this point and fitted it to form a density distribution curve. The interval corresponding to each peak point of the density curve was identified as the TFBS-clustered region (TFCR). The window for each TFCR was determined by finding the maximum distance (in bp) from the TFCR to a contributing TF and then adding 150 bp. Meanwhile, we filtered out the TFBS with Gaussian signal intensity greater than 0.1 in each TFCR and added them together, and defined the accumulated sum as the complexity of the interval. With this, we examined the overlap of each TFCR and ATAC-seq peaks, and defined the average score of all ATAC-seq peaks with overlapping interval as the chromatin accessibility score, thus reflecting the chromatin accessibility of TFCR for the first time.
4.4. Categorization of TFCRs
To describe TFCRs, we classified them according to their TF complexity and chromatin accessibility score. All TFCRs were sorted according to TF complexity, and then evenly divided into 10 groups, corresponding to TC0 to TC9. The same method was applied to classify TFCRs by the chromatin accessibility score, corresponding to CAS0-CAS9. Therefore, TF complexity (TC) and chromatin accessibility score (CAS) were used as two features of TFCRs to predict the number of TFs could bind and reflect the level of chromatin accessibility.
4.5. Differential expression and cancer survival analyses
We used Gene Expression Profiling Interactive Analysis (GEPIA2) to screen candidate key cancer genes that are significantly differentially expressed between normal and tumor tissues (log2FC ≥ 3) (http://gepia2.cancer-pku.cn/#index). This platform provides a rapid and customized delivery of functionalities based on the TCGA and Genotype-Tissue Expression (GTEx) databases. GEPIA2 was used to evaluate the impact of the key cancer genes identified on survival to determine the prognostic value of their expression.
4.6. Cell culture
Human endometrial cell line (HEC-1-A) was purchased from the Cell Bank of the Chinese Academy of Science (Shanghai, China). Cell lines were cultured in McCoy’s 5A (Sangon Biotech, Shanghai, China) medium supplemented with 10% foetal bovine serum (FBS; Gibco, NY, USA) and 1% antibiotic/antimycotic solution (Sangon Biotech, Shanghai, China) and incubated in a humidified atmosphere with 5% CO2 at 37 °C.
4.7. Gene silencing
HOXA9, SCNN1A and TROAP were knocked down with siRNA designed and synthesized by RiboBio (Guangzhou, China) and scrambled negative control sequences were synthesized (Table S3). For cell transfection, HEC-1-A cells were seeded in 24-well plates and transfected with riboFECTTMCP Transfection Kit containing siRNA of HOXA9, SCNN1A, TROAP or NC (si-NC1), respectively. SOX13, MAOA and LMOD1 were knocked down with siRNA designed and synthesized by GenePharma (Shanghai, China) and scrambled negative control sequences were synthesized (Table S3). Besides, HEC-1-A cells were seeded in 24-well plates and transfected with Lipofectamine™ 2000 containing siRNA of SOX13, MAOA, LMOD1 or NC (si-NC2), respectively. Subsequently, knockdown efficiency was examined by RT-qPCR (quantitative reverse transcription PCR, RT-qPCR) 24 h after transfection.
4.8. RT-qPCR
Total RNA was isolated from cell lines with E.Z.N.A. Total RNA Kit I (Omega, USA) according to the manufacturer’s instructions. RNA was reverse transcribed using ReverTra Ace qPCR RT Master Mix with gDNA Remover (TOYOBO, Japan). Real-time PCR was performed using THUNDERBIRD SYBR® qPCR Mix (TOYOBO, Japan) and primers (Table S4). The results were analyzed with the Step OnePlus Real Time PCR System (Applied Biosystems, USA). GAPDH was used as an endogenous control. The 2-△△Ct method was used to quantify the transcript levels.
4.9. Cell migration and invasion assays
The abilities of cell migration and invasion were measured using transwell assay (Corning Incorporated, Corning, NY, USA) 24 h following transfection. Cells were suspended in 200 μl serum-free medium (1 × 105 cells per well) and seeded in the upper chambers of 24-well plates with an 0.8-μm pore membrane insert. For the cell invasion assay, the upper chamber was pre-coated with 50 μl Matrigel (Corning Incorporated). Then, 700 μl medium containing 10% FBS was added to the bottom of chambers. After incubation at 37 °C for 24 h, cells migrated to the lower side of the membrane were fixed with 4% paraformaldehyde solution and then stained with 0.1% crystal violet for 30 min at room temperature. Then, stained cells were observed and counted under microscope. All experiments performed three times.
4.10. Apoptosis analysis by flow cytometry
Cells were seeded in a six-well plate (5 × 105 cells/well) 24 h following transfection. After 24 h, cells were harvested by centrifugation at 1000 rpm for 5 min and washed with 1 × PBS three times, and then incubated with 5 μl of FITC-conjugated Annexin V and 5 μl of PI for 10 min at room temperature in the dark. The stained cells were detected by the BD FACS Aria II flow cytometer (BD Biosciences, CA, USA). The Annexin V-FITC/PI Apoptosis Detection kit (BD PharmingenTM, BD Biosciences, CA, USA) was used to determine apoptosis of cells.
4.11. Statistical analysis
We used Mann-Whitney U test to verify the significance of TFBSs distribution between TFCRs and ATAC-seq peaks. The Chi-square tests were used to analyze base mutation rates in different genomic regions and the differences between TFCRs or ATAC-seq peaks classified using different features; In the verification part of the experiment, paired two-tailed students were employed to conduct statistical analysis and nominal P values are specified. Differences in all comparisons were considered statistically significant at P values < 0.05.
5. Ethical approval and consent to participate
Not Applicable
6. Consent for publication
All authors have agreed to the publication of this manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (http://www.nsfc.gov.cn; no. 82001520 to XT), the Beijing Nova Program of Science and Technology (https://mis.kw.beijing.gov.cn; no. Z191100001119064 to HC), the National Natural Science Foundation of China (http://www.nsfc.gov.cn; no. 31801112, 61873276 and to 31900488 HC, XB and HL respectively) and the Beijing Natural Science Foundation (http://kw.beijing.gov.cn/; no. 5204040 to HL).
CRediT authorship contribution statement
Xiaohan Tang: Conceptualization, Funding acquisition, Resources, Validation. Junting Wang: Writing – original draft, Formal analysis, Visualization, Writing – review & editing, Data curation. Huan Tao: Investigation, Writing – review & editing. Lin Yuan: Resources, Conceptualization, Writing – original draft. Guifang Du: Data curation, Investigation. Yang Ding: Conceptualization, Formal analysis. Kang Xu: Investigation. Xuemei Bai: Data curation, Investigation, Formal analysis. Yaru Li: Investigation, Writing – review & editing. Yu Sun: Investigation, Data curation. Xin Huang: Investigation, Data curation. Xiushuang Zheng: Validation. Qianqian Li: Validation. Bowen Gong: Validation. Yang Zheng: Data curation. Jingxuan Xu: Investigation. Xiang Xu: Investigation. Zhe Wang: Data curation. Xiaochen Bo: Conceptualization, Funding acquisition, Supervision. Meisong Lu: Conceptualization, Supervision. Hao Li: Conceptualization, Writing – original draft, Writing – review & editing, Software, Funding acquisition, Supervision. Hebing Chen: Conceptualization, Methodology, Writing – original draft, Writing – review & editing, Software, Funding acquisition, Supervision.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
The authors wish to thank Prof. Cheng Li for the critical feedback and discussions.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.csbj.2022.01.014.
Contributor Information
Meisong Lu, Email: lumeisong0417@163.com.
Hao Li, Email: lihao_thu@163.com.
Hebing Chen, Email: chb-1012@163.com.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.Sung H., Ferlay J., Siegel R.L., Laversanne M., Soerjomataram I., Jemal A., et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71(3):209–249. doi: 10.3322/caac.21660. [DOI] [PubMed] [Google Scholar]
- 2.Siegel R.L., Miller K.D., Fuchs H.E., Jemal A. Cancer Statistics, 2021. CA Cancer J Clin. 2021;71(1):7–33. doi: 10.3322/caac.21654. [DOI] [PubMed] [Google Scholar]
- 3.Urick M.E., Bell D.W. Clinical actionability of molecular targets in endometrial cancer. Nat Rev Cancer. 2019;19(9):510–521. doi: 10.1038/s41568-019-0177-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Clarke M.A., Devesa S.S., Harvey S.V., Wentzensen N. Hysterectomy-Corrected Uterine Corpus Cancer Incidence Trends and Differences in Relative Survival Reveal Racial Disparities and Rising Rates of Nonendometrioid Cancers. J Clin Oncol. 2019;37(22):1895–1908. doi: 10.1200/JCO.19.00151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fleming G.F., Brunetto V.L., Cella D., Look K.Y., Reid G.C., Munkarah A.R., et al. Phase III trial of doxorubicin plus cisplatin with or without paclitaxel plus filgrastim in advanced endometrial carcinoma: a Gynecologic Oncology Group Study. J Clin Oncol. 2004;22(11):2159–2166. doi: 10.1200/JCO.2004.07.184. [DOI] [PubMed] [Google Scholar]
- 6.Brooks R.A., Fleming G.F., Lastra R.R., Lee N.K., Moroney J.W., Son C.H., et al. Current recommendations and recent progress in endometrial cancer. CA Cancer J Clin. 2019 doi: 10.3322/caac.21561. [DOI] [PubMed] [Google Scholar]
- 7.Aghajanian C., Filiaci V., Dizon D.S., Carlson J.W., Powell M.A., Secord A.A., et al. A phase II study of frontline paclitaxel/carboplatin/bevacizumab, paclitaxel/carboplatin/temsirolimus, or ixabepilone/carboplatin/bevacizumab in advanced/recurrent endometrial cancer. Gynecol Oncol. 2018;150(2):274–281. doi: 10.1016/j.ygyno.2018.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fader A.N., Roque D.M., Siegel E., Buza N., Hui P., Abdelghany O., et al. Randomized Phase II Trial of Carboplatin-Paclitaxel Compared with Carboplatin-Paclitaxel-Trastuzumab in Advanced (Stage III-IV) or Recurrent Uterine Serous Carcinomas that Overexpress Her2/Neu ( NCT01367002): Updated Overall Survival Analysis. Clin Cancer Res. 2020;26(15):3928–3935. doi: 10.1158/1078-0432.CCR-20-0953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chang Y.-N., Zhang Y., Wang Y.-J., Wang L.-P., Duan H. Effect of hysteroscopy on the peritoneal dissemination of endometrial cancer cells: a meta-analysis. Fertil Steril. 2011;96(4):957–961.e2. doi: 10.1016/j.fertnstert.2011.07.1146. [DOI] [PubMed] [Google Scholar]
- 10.Clark T.J., Voit D., Gupta J.K., Hyde C., Song F., Khan K.S. Accuracy of hysteroscopy in the diagnosis of endometrial cancer and hyperplasia - A systematic quantitative review. Jama-J Am Med Assoc. 2002;288(13):1610. doi: 10.1001/jama.288.13.1610. [DOI] [PubMed] [Google Scholar]
- 11.Morice P., Leary A., Creutzberg C., Abu-Rustum N., Darai E. Endometrial cancer. Lancet. 2016;387(10023):1094–1108. doi: 10.1016/S0140-6736(15)00130-0. [DOI] [PubMed] [Google Scholar]
- 12.Sundar S., Balega J., Crosbie E., Drake A., Edmondson R., Fotopoulou C., et al. BGCS uterine cancer guidelines: Recommendations for practice. Eur J Obstet Gynecol Reprod Biol. 2017;213:71–97. doi: 10.1016/j.ejogrb.2017.04.015. [DOI] [PubMed] [Google Scholar]
- 13.Cheng T.H.T., Thompson D.J., O'Mara T.A., Painter J.N., Glubb D.M., Flach S., et al. Five endometrial cancer risk loci identified through genome-wide association analysis. Nat Genet. 2016;48(6):667–674. doi: 10.1038/ng.3562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.O’Mara T.A., Glubb D.M., Amant F., Annibali D., Ashton K., Attia J., et al. Identification of nine new susceptibility loci for endometrial cancer. Nat Commun. 2018;9(1) doi: 10.1038/s41467-018-05427-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.O'Mara T.A., Glubb D.M., Kho P.F., Thompson D.J., Spurdle A.B. Genome-Wide Association Studies of Endometrial Cancer: Latest Developments and Future Directions. Cancer Epidemiol Biomarkers Prev. 2019;28(7):1095–1102. doi: 10.1158/1055-9965.EPI-18-1031. [DOI] [PubMed] [Google Scholar]
- 16.Chan H.T., Nagayama S., Chin Y.M., Otaki M., Hayashi R., Kiyotani K., et al. Clinical significance of clonal hematopoiesis in the interpretation of blood liquid biopsy. Mol Oncol. 2020;14(8):1719–1730. doi: 10.1002/1878-0261.12727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nair N., Camacho-Vanegas O., Rykunov D., Dashkoff M., Camacho S.C., Schumacher C.A., et al. Genomic Analysis of Uterine Lavage Fluid Detects Early Endometrial Cancers and Reveals a Prevalent Landscape of Driver Mutations in Women without Histopathologic Evidence of Cancer: A Prospective Cross-Sectional Study. PLoS Med. 2016;13(12):e1002206. doi: 10.1371/journal.pmed.1002206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Flavahan W.A., Gaskell E., Bernstein B.E. Epigenetic plasticity and the hallmarks of cancer. Science. 2017;357(6348) doi: 10.1126/science:aal2380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128(4):693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 20.Zhang B.o., Xing XiaoYun, Li J., Lowdon R.F., Zhou Y., Lin N., et al. Comparative DNA methylome analysis of endometrial carcinoma reveals complex and distinct deregulation of cancer promoters and enhancers. BMC Genomics. 2014;15(1) doi: 10.1186/1471-2164-15-868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang Q.-Y., Yi D.-Q., Zhou L.i., Zhang D.-H., Zhou T.-M. Status and significance of CpG island methylator phenotype in endometrial cancer. Gynecol Obstet Invest. 2011;72(3):183–191. doi: 10.1159/000324496. [DOI] [PubMed] [Google Scholar]
- 22.Bartosch C., Monteiro-Reis S., Almeida-Rios D., Vieira R., Castro A., Moutinho M., et al. Assessing sirtuin expression in endometrial carcinoma and non-neoplastic endometrium. Oncotarget. 2016;7(2):1144–1154. doi: 10.18632/oncotarget.6691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Teixeira S.R., Abreu C.M., Parkes L., Davies J., Yao S., Sawhney M.A., et al. Direct monitoring of breast and endometrial cancer cell epigenetic response to DNA methyltransferase and histone deacetylase inhibitors. Biosens Bioelectron. 2019;141:111386. doi: 10.1016/j.bios.2019.111386. [DOI] [PubMed] [Google Scholar]
- 24.Jia N., Li Q., Tao X., Wang J., Hua K., et al. Enhancer of zeste homolog 2 is involved in the proliferation of endometrial carcinoma. Oncol Lett. 2014;8(5):2049–2054. doi: 10.3892/ol.2014.2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jiang F.-Z., He Y.-Y., Wang H.-H., Zhang H.-L., Zhang J., Yan X.-F., et al. Mutant p53 induces EZH2 expression and promotes epithelial-mesenchymal transition by disrupting p68-Drosha complex assembly and attenuating miR-26a processing. Oncotarget. 2015;6(42):44660–44674. doi: 10.18632/oncotarget.6350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Eskander R., Ji T., Huynh B., Wardeh R., Hoang B., et al. Overexpression of enhancer of zeste homolog 2 (EZH2) and focal adhesion kinase (FAK) in high grade endometrial carcinoma. Gynecol Oncol. 2012;127:S1–S34. doi: 10.1016/j.ygyno.2012.07.128. [DOI] [PubMed] [Google Scholar]
- 27.Zhou J., Roh J.-W., Bandyopadhyay S., Chen Z., Munkarah A.R., Hussein Y., et al. Overexpression of enhancer of zeste homolog 2 (EZH2) and focal adhesion kinase (FAK) in high grade endometrial carcinoma. Gynecol Oncol. 2013;128(2):344–348. doi: 10.1016/j.ygyno.2012.07.128. [DOI] [PubMed] [Google Scholar]
- 28.Berger S.L. The complex language of chromatin regulation during transcription. Nature. 2007;447(7143):407–412. doi: 10.1038/nature05915. [DOI] [PubMed] [Google Scholar]
- 29.Corces M.R., Granja J.M., Shams S., Louie B.H., Seoane J.A., Zhou W., et al. The chromatin accessibility landscape of primary human cancers. Science. 2018;362(6413) doi: 10.1126/science:aav1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen H., Li H., Liu F., Zheng X., Wang S., Bo X., et al. An integrative analysis of TFBS-clustered regions reveals new transcriptional regulation models on the accessible chromatin landscape. Sci Rep. 2015;5(1) doi: 10.1038/srep08465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen H, Jiang S, Zhang Z, Li H, Lu Y, et al. (2017) Exploring spatially adjacent TFBS-clustered regions with Hi-C data. Bioinformatics 33: 2611-2614. [DOI] [PMC free article] [PubMed]
- 32.Li H., Chen H., Liu F., Ren C., Wang S., Bo X., et al. Functional annotation of HOT regions in the human genome: implications for human disease and cancer. Sci Rep. 2015;5(1) doi: 10.1038/srep11633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wingender E., Chen X., Hehl R., Karas H., Liebich I., et al. TRANSFAC: an integrated system for gene expression regulation. Nucleic Acids Res. 2000;28:316–319. doi: 10.1093/nar/28.1.316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Portales-Casamar E, Thongjuea S, Kwon AT, Arenillas D, Zhao X, et al. (2010) JASPAR 2010: the greatly expanded open-access database of transcription factor binding profiles. Nucleic Acids Res 38: D105-110. [DOI] [PMC free article] [PubMed]
- 35.Robasky K., Bulyk M.L. UniPROBE, update 2011: expanded content and search tools in the online database of protein-binding microarray data on protein-DNA interactions. Nucleic Acids Res. 2011;39(Database):D124–D128. doi: 10.1093/nar/gkq992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ren C., Chen H., Yang B., Liu F., Ouyang Z., Bo X., et al. iFORM: Incorporating Find Occurrence of Regulatory Motifs. PLoS ONE. 2016;11(12):e0168607. doi: 10.1371/journal.pone.0168607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Turajlic S., Sottoriva A., Graham T., Swanton C. Resolving genetic heterogeneity in cancer. Nat Rev Genet. 2019;20(7):404–416. doi: 10.1038/s41576-019-0114-6. [DOI] [PubMed] [Google Scholar]
- 38.Martincorena I., Raine K.M., Gerstung M., Dawson K.J., Haase K., Van Loo P., et al. Universal Patterns of Selection in Cancer and Somatic Tissues. Cell. 2017;171(5):1029–1041.e21. doi: 10.1016/j.cell.2017.09.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Moore L., Leongamornlert D., Coorens T.H.H., Sanders M.A., Ellis P., Dentro S.C., et al. The mutational landscape of normal human endometrial epithelium. Nature. 2020;580(7805):640–646. doi: 10.1038/s41586-020-2214-z. [DOI] [PubMed] [Google Scholar]
- 40.Levine D.A. Integrated genomic characterization of endometrial carcinoma. Nature. 2013;497(7447):67–73. doi: 10.1038/nature12113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Partridge E.C., Chhetri S.B., Prokop J.W., Ramaker R.C., Jansen C.S., Goh S.-T., et al. Occupancy maps of 208 chromatin-associated proteins in one human cell type. Nature. 2020;583(7818):720–728. doi: 10.1038/s41586-020-2023-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu X.S., Wu H., Ji X., Stelzer Y., Wu X., Czauderna S., et al. Editing DNA Methylation in the Mammalian Genome. Cell. 2016;167(1):233–247.e17. doi: 10.1016/j.cell.2016.08.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pakish J.B., Zhang Q., Chen Z., Liang H., Chisholm G.B., Yuan Y., et al. Immune Microenvironment in Microsatellite-Instable Endometrial Cancers: Hereditary or Sporadic Origin Matters. Clin Cancer Res. 2017;23(15):4473–4481. doi: 10.1158/1078-0432.CCR-16-2655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ahn J.H., Davis E.S., Daugird T.A., Zhao S., Quiroga I.Y., Uryu H., et al. Phase separation drives aberrant chromatin looping and cancer development. Nature. 2021;595(7868):591–595. doi: 10.1038/s41586-021-03662-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chong S., Dugast-Darzacq C., Liu Z., Dong P., Dailey G.M., Cattoglio C., et al. Imaging dynamic and selective low-complexity domain interactions that control gene transcription. Science. 2018;361(6400) doi: 10.1126/science:aar2555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang J., Choi J.-M., Holehouse A.S., Lee H.O., Zhang X., Jahnel M., et al. A Molecular Grammar Governing the Driving Forces for Phase Separation of Prion-like RNA Binding Proteins. Cell. 2018;174(3):688–699.e16. doi: 10.1016/j.cell.2018.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Aitken S.J., Anderson C.J., Connor F., Pich O., Sundaram V., et al. Pervasive lesion segregation shapes cancer genome evolution. Nature. 2019 doi: 10.1038/s41586-020-2435-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cramer P. Organization and regulation of gene transcription. Nature. 2019;573(7772):45–54. doi: 10.1038/s41586-019-1517-4. [DOI] [PubMed] [Google Scholar]
- 49.Martínez-Jiménez F., Muiños F., Sentís I., Deu-Pons J., Reyes-Salazar I., Arnedo-Pac C., et al. A compendium of mutational cancer driver genes. Nat Rev Cancer. 2020;20(10):555–572. doi: 10.1038/s41568-020-0290-x. [DOI] [PubMed] [Google Scholar]
- 50.Kandoth C., McLellan M.D., Vandin F., Ye K., Niu B., Lu C., et al. Mutational landscape and significance across 12 major cancer types. Nature. 2013;502(7471):333–339. doi: 10.1038/nature12634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vogelstein B., Papadopoulos N., Velculescu V.E., Zhou S., Diaz L.A., Kinzler K.W. Cancer genome landscapes. Science. 2013;339(6127):1546–1558. doi: 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lawrence M.S., Stojanov P., Mermel C.H., Robinson J.T., Garraway L.A., Golub T.R., et al. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature. 2014;505(7484):495–501. doi: 10.1038/nature12912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schroeder MP, Rubio-Perez C, Tamborero D, Gonzalez-Perez A, Lopez-Bigas N (2014) OncodriveROLE classifies cancer driver genes in loss of function and activating mode of action. Bioinformatics 30: i549-555. [DOI] [PMC free article] [PubMed]
- 54.Zehir A., Benayed R., Shah R.H., Syed A., Middha S., Kim H.R., et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat Med. 2017;23(6):703–713. doi: 10.1038/nm.4333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sondka Z., Bamford S., Cole C.G., Ward S.A., Dunham I., Forbes S.A. The COSMIC Cancer Gene Census: describing genetic dysfunction across all human cancers. Nat Rev Cancer. 2018;18(11):696–705. doi: 10.1038/s41568-018-0060-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liu S.H., Shen P.C., Chen C.Y., Hsu A.N., Cho Y.C., et al. DriverDBv3: a multi-omics database for cancer driver gene research. Nucleic Acids Res. 2020;48:D863–D870. doi: 10.1093/nar/gkz964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wu L., Ling Z.-H., Wang H., Wang X.-Y., Gui J. Upregulation of SCNN1A Promotes Cell Proliferation, Migration, and Predicts Poor Prognosis in Ovarian Cancer Through Regulating Epithelial-Mesenchymal Transformation. Cancer Biother Radiopharm. 2019;34(10):642–649. doi: 10.1089/cbr.2019.2824. [DOI] [PubMed] [Google Scholar]
- 58.Bao Y., Guo Y., Yang Y., Wei X., Zhang S., Zhang Y., et al. PRSS8 suppresses colorectal carcinogenesis and metastasis. Oncogene. 2019;38(4):497–517. doi: 10.1038/s41388-018-0453-3. [DOI] [PubMed] [Google Scholar]
- 59.Jiang T., Sui D., You D., Yao S., Zhang L., et al. MiR-29a-5p inhibits proliferation and invasion and induces apoptosis in endometrial carcinoma via targeting TPX2. Cell Cycle. 2018;17:1268–1278. doi: 10.1080/15384101.2018.1475829. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 60.Li L., Xu D.-B., Zhao X.-L., Hao T.-Y. Combination analysis of Bub1 and Mad2 expression in endometrial cancer: act as a prognostic factor in endometrial cancer. Arch Gynecol Obstet. 2013;288(1):155–165. doi: 10.1007/s00404-012-2706-7. [DOI] [PubMed] [Google Scholar]
- 61.Liu Y., Zhao R., Chi S., Zhang W., Xiao C., Zhou X., et al. UBE2C Is Upregulated by Estrogen and Promotes Epithelial-Mesenchymal Transition via p53 in Endometrial Cancer. Mol Cancer Res. 2020;18(2):204–215. doi: 10.1158/1541-7786.MCR-19-0561. [DOI] [PubMed] [Google Scholar]
- 62.Zhang L.i., Jia G., Shi B., Ge G., Duan H., Yang Y. PRSS8 is Downregulated and Suppresses Tumour Growth and Metastases in Hepatocellular Carcinoma. Cell Physiol Biochem. 2016;40(3-4):757–769. doi: 10.1159/000453136. [DOI] [PubMed] [Google Scholar]
- 63.Zhou H., Zou X., Li H., Li T., Chen L., et al. Decreased secretoglobin family 2A member 1expression is associated with poor outcomes in endometrial cancer. Oncol Lett. 2020;20:24. doi: 10.3892/ol.2020.11885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Li J., Xu W., Zhu Y. Mammaglobin B may be a prognostic biomarker of uterine corpus endometrial cancer. Oncol Lett. 2020;20:255. doi: 10.3892/ol.2020.12118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Oplawski M., Dziobek K., Grabarek B., Zmarzły N., Dąbruś D., Januszyk P., et al. Expression of NRP-1 and NRP-2 in Endometrial Cancer. Curr Pharm Biotechnol. 2019;20(3):254–260. doi: 10.2174/1389201020666190219121602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bian Y., Li Y., Shrestha G., Wen X., Cai B., Wang K., et al. ITE, an endogenous aryl hydrocarbon receptor ligand, suppresses endometrial cancer cell proliferation and migration. Toxicology. 2019;421:1–8. doi: 10.1016/j.tox.2019.03.017. [DOI] [PubMed] [Google Scholar]
- 67.Ando M., Kawazu M., Ueno T., Koinuma D., Ando K., Koya J., et al. Mutational Landscape and Antiproliferative Functions of ELF Transcription Factors in Human Cancer. Cancer Res. 2016;76(7):1814–1824. doi: 10.1158/0008-5472.CAN-14-3816. [DOI] [PubMed] [Google Scholar]
- 68.Jung I., Schmitt A., Diao Y., Lee A.J., Liu T., Yang D., et al. A compendium of promoter-centered long-range chromatin interactions in the human genome. Nat Genet. 2019;51(10):1442–1449. doi: 10.1038/s41588-019-0494-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jiang Y., Qian F., Bai X., Liu Y., Wang Q., et al. SEdb: a comprehensive human super-enhancer database. Nucl Acids Res. 2019;47:D235–D243. doi: 10.1093/nar/gky1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.