

Summary
Dynamic profiling of changes in gene expression in response to stressors in specific microenvironments without requiring cellular destruction remains challenging. Current methodologies that seek to interrogate gene expression at a molecular level require sampling of cellular transcriptome and therefore lysis of the cell, preventing serial analysis of cellular transcriptome. To address this area of unmet need, we have recently developed a technology allowing transcriptomic analysis over time without cellular destruction. Our method, TRACE-seq (TRanscriptomic Analysis Captured in Extracellular vesicles using sequencing), is characterized by a cell-type specific transgene expression. It provides data on the transcriptome inside extracellular vesicles that provides an accurate representation of stress-responsive cellular transcriptomic changes. Thus, the transcriptome of cells expressing TRACE can be followed over time without destroying the source cell, which is a powerful tool for many fields of fundamental and translational biology research.
Subject areas: Cell biology, Omics, Transcriptomics
Graphical abstract
Highlights
-
•
TRanscriptomic Analysis Captured in Extracellular vesicles using RNA-sequencing
-
•
Excellent correlation of cellular and EV transcriptome using this method
-
•
Accurate profiling of gene expression changes in response to stress signals
Cell biology; Omics; Transcriptomics
Introduction
For several decades, mRNA profiling by microarray and RNA sequencing (Hrdlickova et al., 2017; Ozsolak and Milos, 2011) allowed investigators to push the frontiers of knowledge by providing a better understanding of the architectural complexity that underlies cellular heterogeneity. Despite the development of many techniques with increasingly powerful resolution, a fundamental gap in methodology persists in that the most current techniques are cell destructive (Saadatpour et al., 2015) and are therefore incompatible with studies examining the dynamics of cellular change. Although in situ hybridization in live cells using mRNA probes is feasible (Bao et al., 2009), it is technically challenging to use routinely, not amenable to multiplexing, and cannot provide a longitudinal measure of the whole cell transcriptome.
Substantial effort has been made to profile subpopulations of cells by transcriptomic analysis (Monaco et al., 2019; Corces et al., 2016), focusing on specific cell types (Heiman et al., 2014) or using single cell sequencing methods (Tang et al, 2011; Sandberg, 2014; Saliba et al., 2014). Significant technological advancements have reduced experimental noise while obtaining representative results with low input starting material (Grün and van Oudenaarden, 2015). Recently, development of alternative approaches for time-resolved, longitudinal extraction and quantitative measurement of intracellular mRNA have been reported (Cao et al., 2017), but are limited to in vitro studies and appear unsuitable for routine usage or for in vivo studies. Exploiting the presence of mRNAs in extracellular vesicles (EVs), we aim to develop technology to efficiently and uniformly load cellular mRNAs into EVs in vivo. The noninvasive transcriptome profiling that such a method affords would enable serial, nondestructive, and noninterfering sampling of designated cells simply by collection of physiological fluid.
Here we present a technique, TRACE-seq (TRanscriptomic Analysis Captured in Extracellular vesicles using sequencing), that exploits the cell’s existing gene expression mechanism to secrete mRNAs into EVs across the entire spectrum of relative cellular abundance. TRACE-seq provides a representative sampling of the cytosolic transcriptome allowing monitoring of live cells in physiological or pathological conditions by a serial and nondestructive transcriptomic analysis.
Results
TRACE-seq, an mRNA translation methodology carried by extracellular vesicles
All known cell types secrete EVs and studies demonstrate the presence of RNAs in the cargo of EVs (O'Brien et al., 2020). However, profiling RNA in extracellular vesicles does not always provide an accurate representation of the original cell’s transcriptome. These RNAs consist of a large proportion of fragmented mRNA in addition to multiple small RNA species (Valadi et al., 2007; van Balkom et al., 2015; Pérez-Boza et al., 2018; Wei et al., 2017; Mateescu et al., 2017). This represents challenges in using EV RNA as an accurate proxy for interrogating the cellular/cytosolic transcriptome. We sought to overcome this through the use of a cell-type specific transgene expression which facilitates the packaging of cellular mRNA import into extracellular vesicles, especially Large Extracellular Vesicles (L-EVs), which are used as carriers (Figure 1). A similar technology already exists in a different context, in which researchers used engineered EVs as carriers for protein transportation (Yim et al., 2016). Based on this technology, our methodology comprises two separate components:
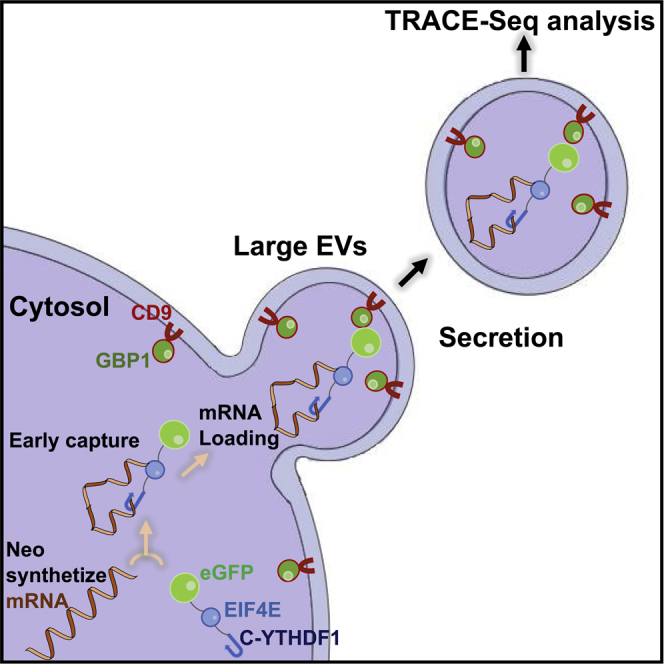
Figure 1.

Overview of the TRACE methodology
(A) Schematic of TRACEseq method. A plasmid corresponding to the two fusion proteins GBP1/CD9 and eGFP/RBP is transfected or transduced to the cells. Both proteins were translated and play their role separately. The RBP “catches” m6A tagged mRNA and by the link of the eGFP/GBP1, brings it into the CD9 coated EVs for an extracellular export.
(B) Confocal image of the transfection of different HEK 293T populations with two constructs: eGFP-C-YTHDF1/GPB1-CD9 (C1) and mCherry-CD9.
(C) NanoFlowCyt of the whole vesicle population secreted by different HEK 293T cells. Untransfected cells (None), cells transfected with the negative control constructs eGFP/GBP1-CD-9 (GFP control), and cells with the TRACE construct eGFP-C-YTHDF1/GPB1-CD9 (C1). For each sample, 5000K events were counted. For the GFP-expressing constructs, there were 731K events (17.5%) that were counted as GFP positive in the GFP control, and 1433K (32%) events for the C1 construct
First, we designed an mRNA “Catcher” which corresponds to a fusion protein composed of a C terminal part of YTHDF1 protein and the fluorescent transmitter enhancer GFP. Recently reported, YTH protein domain recognizes m6A, one of the most abundant internal modifications in eukaryotic mRNA (Wang et al., 2014; Shi et al., 2017; Dominissini et al., 2012). Moreover, the isoform one of the YTH protein domain enhances the translation efficiency and binds to the mRNA close to the nuclear membrane after translocation from the nucleus to the cytosol (Wang et al., 2015; Patil et al., 2018).
Second, we designed another fusion protein which consists of a GFP binding protein 1 (GBP1), with the ability to bind to EGFP while enhancing its fluorescent signal (Tang et al., 2013; Kirchhofer et al., 2010) and the CD9 protein known to be a canonical EV marker (Kowal et al., 2016). Thus, the mRNA “Catcher” EGFP-C-YTHDF1 can trap newly synthesized mRNAs, and then bind to the second fusion protein through the EGFP/GBP1 affinity and transport the mRNA into extracellular vesicles facilitated by tetraspanin CD9 protein, as shown in Figure 1A. To validate part of the construct, we used an additional plasmid as a positive control, mCherry-CD9 (expected to be detected on EVs), and monitored for membrane colocalization signal from our TRACE construct eGFP-C-YTHDF1/GBP1-CD9: C1 (Green) and the positive control plasmid (Red). As expected, we observed this colocalization signal on the cell membrane, as shown in Figure 1B. We then purified L-EVs from the TRACE transfected cells and searched for GFP expressing EVs by High Dimensional Flow Cytometry (also referred to as NanoFlowCyt (Morales-Kastresana et al., 2017)), Figure 1C. A significant number of GFP EVs were detected for both cell groups transfected with the TRACE construct (eGFP-C-YTHDF1/GBP1-CD9: C1) and the control construct (no mRNA catcher: eGFP/GBP1-CD9: GFP control), both of which would be expected to target eGFP to the EVs.
Detection of mRNA inside extracellular vesicles and reverse transcription
A second validation consisted of analyzing the mRNA content of the TRACE transfected cell EVs. To do so, we designed a protocol for a reproducible and robust EV-mRNA profiling (Figure S1), with the generation of a mRNA library based on the SMART-seq2 (Switching Mechanism At 5′ end of RNA Template sequencing) technology (Picelli et al., 2013, 2014). We first evaluated different mRNA catchers (Figure S2). We tested five potential mRNA-binding proteins fused with GFP: the 5′ mRNA cap protein binder eIF4E, the 60S ribosomal protein RPL10A (to mimic the ribosomal immunoprecipitation described in the TRAP-Seq methodology (Rodrigues et al., 2021)) and different isoforms of the YTHDF protein family; the YTHDF2, the C-Term-YTHDF2 (correlated with the mRNA decay pathway) and the C-Term-YTHDF1 (role in mRNA translation). The fused proteins trap mRNA at different binding sites (5′ cap, 5′-UTR,and-3′ UTR) and are precipitated with an anti-GFP antibody. As expected, the YTHDF group and particularly the C-Term-YTHDF1 isoform presented the best mRNA binding capability (ΔCT around three for the five tested genes: GAPDH, β-Actin, LDHA, SDHA, and RPL10A; hence, highest abundance in EVs relative to cellular expression). Based on these results, we subsequently prioritized two constructs for further development with the following design: eGFP-C-YTHDF1/GBP1-CD9 (henceforth referred to as C1), eGFP-EIF4E-C-YTHDF1/GBP1-CD9 (henceforth referred to as 4EC1), which corresponds to the addition of the cap protein EIF4E to the C-term-YTHDF1 for a potential improvement of the mRNA binding and transportation (Figures S3 and S4). The third one, eGFP/GBP1-CD9 (GFP control) was used as a negative control by itself, but in combination with the other two constructs, was important in trafficking the constructs to the membrane. It is important to note that each group of proteins has a specific function (one targets the mRNA and the other brings the complex to the cell membrane), and that each fusion protein is expressed under its own independent promoter. As expected, the presence of the GFP control construct allowed for localization of the protein to the cell membrane (Figure S3).
We hypothesized that premature binding of the mRNA targeted fusion protein (the ‘catcher’) to the EV-targeting fusion protein could direct these complexes into the EV secretion pathway before they could sufficiently ‘capture’ the cellular mRNA. These ‘empty’ complexes could drastically reduce the activity of our TRACE-seq system. The use of two separate promoters (one per fusion protein) would be expected to reduce the potential issue of precocious capture of the eGFP-mRNA catcher by the GBP1-CD9. To confirm this hypothesis, a one-promoter regulated construct: eGFP-C-YTHDF1-T2A-GBP1-CD9 (referred to as T2A) was also cloned and compared with the two-promoter system.
For each of these three constructs and their controls (negative GFP control construct and regular HEK 293T cells), RNA from ∼4 million transfected cells were analyzed by Bioanalyzer. Consistent with prior data that show a predominance of small RNAs in EV cargo, all EV-RNA samples showed a predominant peak corresponding to the vesicular small RNA signal (Figure S4). The T2A construct presented an additional peak at the 28s rRNA region of unclear significance (Figure S5A). To determine if mRNA transcripts were also present in the EVs (but not in sufficient quantity to be detected by the bioanalyzer), RNA was reverse transcribed into cDNA and pre-amplified according to our protocol and analyzed on a High efficiency DNA chip. In transient transfection experiments, although the cDNA signal from the T2A construct did not show any peak corresponding to full length mRNA transcripts (Figure S5B), for the two constructs regulated with distinct CMV promoters, a peak corresponding to full length mRNA was detected (average 1,200nt) and was coincident with the signal from the corresponding cell lysate cDNA signal (Figure 2). These experiments suggested that the generation of the mRNA transcripts for both fusion proteins from separate promoters seems to be a critical step for their anticipated function.
Figure 2.

TRACEseq bioanalyzer analysis from transient expression
Bioanalyzer tracings of cDNA generated from amplified and reverse transcribed RNA isolated from large EVs and their corresponding cell lysates isolated from different transfected HEK 293T populations (triplicate of 4M cells per condition): untransfected cells (None), negative control construct (GFP control), and the two types of TRACE constructs, namely eGFP-C-YTHDF1/GPB1-CD9 (C1) and eGFP-EIF4E-C-YTHDF1/GPB1-CD9 (4EC1).
As opposed to large EVs, it has been observed that the majority of the small EVs (S-EVs) contain fragmented RNA (Pérez-Boza et al., 2018; Batagov and Kurochkin, 2013; Wei et al., 2017). Moreover, the effect of vesicle size on mRNA packaging efficiency demonstrates the better loading capability of L-EVs compared to S-EVs (Wei et al., 2017). We did not focus on these vesicles at first, but for control purposes, we also analyzed S-EVs RNA content. As anticipated, we were not able to generate full-length cDNAs from RNAs isolated from these small EVs, as shown in Figure S6, suggesting that the mRNA brought into the multivesicular bodies during small EVs biogenesis was previously cleaved (Miller and Reese, 2012). In our subsequent experiments we decided to focus all of our design and methodology on large EVs (L-EVs) rather than S-EVs. We repeated the same kind of experiment with a lower number of cells (1 million transfected cells) and observed a similar cDNA signal as before for the two TRACE constructs with proportionally lower amplitude (Figure S7). To confirm that the peak observed on the Bioanalyzer corresponded to mRNAs, qRT-PCR analysis was performed on a small number of genes using one of the constructs (C1) and the two-negative controls (GFP control and untransfected cells). The expression of genes in the cell lysate versus their large EVs is significantly more tightly correlated in the TRACE group (ddCT close to 0), than in the two control groups (ddCT >3 or −3) (Figure S8).
To determine if packaging of mRNA into the large EVs would be more efficient and uniform in stably transfected cell lines, we also generated doxycycline-inducible stable cell lines by lentiviral infection for the two successful constructs (C1 and 4EC1) and the negative control construct (GFP control). L-EVs isolated from the stable cell lines were analyzed and compared to the L-EVs from untransfected HEK293T (Figures 3 and S9). From 10 million cells, a mRNA peak ∼1,300nt was detected in the RNA pico chip for both constructs without detection of mRNA in our negative controls (Figure 3A). Interestingly, regarding the cDNA chip signal, the 4EC1 construct L-EVs group was the only one producing robust and reproducible signals with amplification of full-length cDNA (Figures 3B and S9).
Figure 3.

TRACEseq bioanalyzer validation from stable cell line
(A) Bioanalyzer analysis of large EVs mRNA content extracted from stable cell lines generated with the construct eGFP-C-YTHDF1/GPB1-CD9 (C1), eGFP-EIF4E-C-YTHDF1/GPB1-CD9 (4EC1) the negative control: GFP control, and the regular HEK 293T cells.
(B) Bioanalyzer analysis of cDNA generated from large EVs and their corresponding cDNA generated from cell lysates of the different stable cell lines (Same as above), triplicate of 10M cells per condition.
To next determine if expression of these constructs affected cellular growth, we ran a viability assay and quantified the GFP and GBP1 expression for each of the cell line populations. The presence of GBP1-CD9 expression directed localization of the expressed proteins to the cell membrane in these stable cell lines (Figure S10A); however, prolonged expression of the 4EC1 construct (in the presence of doxycycline) induced a reduction of cell growth (Figure S10B). This justifies the importance of having an inducible system, as also shown in Figure S10B; with no doxycycline induction, growth of the cell line was not affected. Moreover, because we know that the constructs are expressed from two different promoters, the ratio between the two fusion proteins may be 1:1. The 4EC1 group exhibited the closest GFP and GBP1 gene expression level by RT-qPCR, compared to the C1 construct (Figure S10C). The ratio of the two parts of the technology is a critical parameter especially for the protein targeting function and could explain the weaker and inconsistent activity of the C1 construct (Figure S9). Based on these data, we next focused our RNA profiling efforts on transiently transfected cells. For robust proof of concept of the TRACE-seq methodology, we selected samples from the transient transfection batch (Figure 2) for further RNA-seq analysis.
The transcriptome in TRACE-Seq L-EVs is representative of the cellular transcriptome
To further validate the technology, we sequenced the RNA samples from the cell lysates and their corresponding L-EVs from the cells transiently transfected with the C1, 4E-C1, and the GFP control (Figure 2). We focused on a selection of 3603 genes (counts-per-million (CPM) > 0.5 in at least two or more replicates). As shown in the heatmap (Figure 4C), the mRNAs derived from the L-EVs were closer in expression level with the cellular RNAs in the TRACE-seq C1 compared to the GFP control group. In the latter, the majority of the 500 most variable genes are, in terms of level of expression, different compared to cell lysate. Across all RNAs, the L-EVs C1 construct samples are more closely correlated with cellular lysates (Pearson correlation = 0.7; Figure 4C) compared with the correlation between GFP L-EVs and the GFP cell lysate (Pearson correlation = 0.45). Interestingly, DEseq2 analysis (3603 genes) showed that the expression of mRNAs across the transcriptome demonstrate higher correlation for L-EVs C1 construct -C1 Cell lysates construct (with a LogFC between the two groups close to 0 for a large majority of the transcripts, see violin plot in Figure 4C) compared to the L-EVs GFP control-GFP Cell lysate control (which has LogFC values above 1/-1 for half of genes). Accordingly, there are also a far larger number of ‘differentially expressed or present’ transcripts between L-EVs and their corresponding cell lysates in the GFP control group than with the C1 construct (Figure 4D). In the GFP control 62% of genes were significantly differentially expressed in the EVs than in their cell lysates (975 enriched and 595 depleted), compared to 47% of genes in the C1 construct (241 enriched and 264 depleted). Overabundant cellular RNAs are more likely to be present in the control EVs (fisher exact p value = 10−11) compared to their representation in the L-EVs corresponding to the C1 construct (fisher exact p value = 0.49). Together these data suggest that the TRACE-seq constructs lead to a more diverse representation of the cellular transcriptome in L-EVs, with higher correlation of mRNA abundance between the two compartments.
Figure 4.

TRACE DEseq2 analysis from transient expression
DEseq2 analysis of RNAseq data from samples of TRACE eGFP-C-YTHDF1/GPB1-CD9 (C1) construct (duplicate L-EVs and cellular RNAseq): GFP control (duplicate L-EV and cellular RNAseq), and eGFP-EIF4E-C-YTHDF1/GPB1-CD9 (4EC1) construct (duplicate L-EVs and cellular RNAseq) from the transiently transfected cells. Pooled analysis is shown here, for ungrouped results see Figure S11.
(A) Heatmap for the 500 most variable genes across all samples.
(B) Pearson correlation chart from the Limma-voom DEseq2 analysis between each group of samples. Correlations were significant with p < 0.05 noted by asterisks.
(C) All DE genes from the Limma-voom DEseq2 analysis.
(D) Venn diagram from contrast matrix Large EVs-Cell lysates from genes used in Limma-voom DEseq2 (See also Figure S11).
(E) Correlation plot of the Log10 normalized counts of the TRACE 4EC1 construct isolated from L-EVs vs their corresponding Cell lysate
(F) Venn diagram from comparison matrix L-EVs vs Cell lysate for the DEseq 4EC1 construct (selected for genes whose normalized reads are >50).
As our construct preferentially binds to m6A-modified RNAs, we sought to understand whether the correlation between L-EVs and cell lysate would alter if we restricted analysis to m6A YTHDF1 IP isolated genes (RNAs that are expected to bind to YTHDF1 and be targeted to the L-EVs). Our correlations were consistent if we restricted our analysis to genes identified in the POSTAR database (CLIP-RNA-seq on RNAs isolated by co-immuno-pulldown with C-YTHDF1 antibody on HeLa cells) (Hu et al., 2017; Zhu et al., 2019), with the C1 construct showing better correlation than the control construct (Figures S11A, S11B, and S11D). These results show that if we restrict our analysis set of the TRACE-Seq L-EV genes to an m6A YTHDF1 IP gene population, the overall correlation for the TRACE-Seq L-EVs vs whole cell lysate remains consistent.
As noticed in Figure 4, it appears that the TRACE-seq technology transforms the mRNA L-EV cargo content by binding to and importing full length mRNA. Figure S12 shows a clear difference in terms of read coverage across the transcript length (Figure S12A), suggesting that the TRACE construct brings longer mRNA transcripts inside L-EVs with a significant correlation noted between the L-EVs and the cell lysate construct RNAs for the C1 construct. The coverage distribution between the L-EVs and their own cell lysate (Figure S12A) is in closer proximity with C1 Construct than the Control group for transcripts longer than 1000 nucleotides. This is clearly evident in Figure S12B, which shows on a logarithmic scale, the ratio of mRNAs in cell lysates versus L-EVs (L-EVs/Cell lysate). Notably, both C1 construct groups are close to 0, meaning that both L-EVs and cell lysate share a majority of very correlated fragments with the same coverage. In contrast, the ratio of mRNA transcripts in L-EVs versus cells for the control GFP constructs are much more dispersed on this logarithmic scale coverage, suggesting worse correlation between cellular and L-EV transcripts in the control constructs.
Finally, to discriminate between the two constructs C1 and 4EC1, we also decided to sequence transiently transfected samples of the TRACE 4EC1 construct despite the apparently lower signal on the bioanalyzer tracings (Figure 2). As shown in Figure 4E, the Pearson correlation coefficient L-EVs vs their corresponding cell lysate is strongly positive: R = 0.85 and improved compared to the one found for the C1 construct, R = 0.7 (Figure 4B). The Venn diagram generated for the 4EC1 construct (comparison matrix L-EVs vs Cell lysate), Figure 4F. shows a similar trend with 11,002 genes shared between L-EVs and their corresponding cell lysate which presents a very high gene similarity of 76.6%. All these results pooled together tend to qualify the construct 4EC1 as the optimal TRACEseq construct, although our data also suggested that long-term expression of this construct in stably transfected cells may interfere with cell growth pathways.
To assess how our technology compared with other methods to assess nascent RNA transcripts in EVs, we performed iTAG-RNAseq on cellular lysates and their corresponding EVs (Darr et al., 2020), In this methodology, nascent RNAs are labeled with 5-Ethynyl Uridine (5-EU) during transcription and captured by CLICK-iT nascent RNA capture (in cells and EVs), The captured RNAs can be subjected to RNAseq, providing a profile of newly transcribed (nascent) cellular and EV-RNAs (Figure S13). The iTag data batch showed a modest trend of correlation between cellular and EV RNAs. Although 54.1% of the genes were shared between EVs and cells (Figure S13B), we found a relatively modest degree of correlation R = 0.43 (Figure S13C) between cellular and EV-RNA transcripts. These results suggest that our TRACE-seq methodology appears to have significant advantages in sampling cellular RNAs packaged to L-EVs.
Taken together, all these results suggest that TRACE-seq technology can be used to monitor the transcriptome of live cells by sampling the L-EV contents. Our transient transfection data batch demonstrated a higher correlation between the cellular and EV RNAs for the 4EC1 construct compared to the C1, although long term stable expression of the 4EC1 construct appeared to affect cellular growth. Thus, we decided to select the 4EC1 construct to further test how well our technology can assay changes in the cellular transcriptome in response to stress.
Assessment of transcriptomic changes using TRACE-seq in models of cellular stress
We next induced H2O2 and Hypoxic stress to our E4C1 HEK 293T transfected cell line (4EC1 construct) and compared this to non-transfected HEK 293T for 24 h. In order to analyze the oxidative expression pathway, we designed an RTqPCR analysis on genes known to be altered by oxidative stress (TNF-α, TAT, IL-6, PEPCK, GCKR, and HFE). We also included mitochondrial oxidative associated genes (CYP1B1 and CYTC) as the m6A modification in mitochondria is also associated with the YTH protein family member (Koh et al., 2018). As presented in Figure 5, the eight oxidative associated genes tested in the L-EVs from the E4C1 stable cell line are more correlated with their expression in the corresponding cell lysate compared to the untransfected HEK cells in samples from both baseline and H2O2 treated cells (respectively, 4EC1 Construct; H2O2 R:0.976, noH2O2 R: 0.95, and None; H2O2 R:0.90, noH2O2 R: 0.53). As shown in Figures S14A–S14C, the normalized gene expression (treated or untreated cells) appears, as expected, closer between L-EVs and cell lysate in the 4EC1 transfected cells than in untransfected cells. Finally, for these selected transcripts, the change in cellular mRNA transcript abundance induced with oxidative stress (compared to baseline) is well-correlated with their changes in the corresponding L-EVs for the 4EC1 construct (r = 0.91) with far less correlation noted in the control construct (r = 0.59) (Figure 5B).
Figure 5.

H2O2 stress pathway qPCR validation test
Correlation plot obtained from RT-qPCR results generated from L-EVs and Cell lysate mRNA isolated from control cells (None), TRACE construct eGFP-EIFE4-C-YTHDF1/GPB1-CD9 (4EC1) stable cell line on eight genes corresponding to the oxidative stress pathway.
(A) Batch of H2O2 stressed cells and Batch of unstressed cells correlation cell lysates vs L-EVs.
(B) Correlation plots for differentially expressed candidate genes induced by oxidative stress (compared to baseline) in 4EC1 transfected or control cells. Results obtained from triplicate cell populations, 15M cells per condition (See also Figure S14)
Finally, to assess the entire transcriptome in stressed cells, we also analyzed 4EC1 construct in transfected HEK293T cell lines subjected to hypoxia treatment., Isolated RNA from L-EVs and cell lysate were characterized by RNAseq and analyzed it by DEseq (Figure 6). Across the entire transcriptome, the expression level (as assessed by log normalized read counts) were highly correlated between cellular and EV-RNAs both at baseline (Figure 6A, also shown in Figure 4) and with hypoxic stress (Figure 6B) (R = 0.8). This confirms the capability of the 4EC1 TRACE construct to follow the hypoxic stress activation. When looking specifically at differentially expressed genes, the correlation between transcripts that were altered with hypoxic stress (either upregulated or downregulated) was excellent (R = 82, Figure 6C. all DEseq upregulated or downregulated genes). This correlation was even higher for the top 52 DEseq genes with a value of R = 0.92 (Figure 6D), suggesting that TRACE-seq may be used for nondestructive profiling of transcriptomic changes during cellular stress, and could accurately identify the transcripts that were the most altered with stress.
Figure 6.

Hypoxic stress TRACE-seq validation test.DEseq analysis generated from L-EVs, and cell lysate mRNA isolated from eGFP-EIF4E-C-YTHDF1/GPB1-CD9 (4EC1) TRACE construct transfected cells with and without hypoxic stress induction
(A) Correlation plot of the Log10 normalized counts of the TRACE 4EC1 construct isolated at the baseline from L-EVs vs their corresponding Cell lysate.
(B) Correlation plot of the Log10 normalized counts of the TRACE 4EC1 construct isolated with hypoxic stress from L-EVs vs their corresponding Cell lysate.
(C) Correlation plot Log2 Fold Change of the all DE genes under Hypoxic stress of the TRACE-seq isolated L-EVs vs their corresponding Cell lysate.
(D) Correlation plot Log2 Fold Change of the top 52 DE genes under Hypoxic stress of the TRACE-seq isolated L-EVs vs their corresponding Cell lysate, 15M cells per condition. Pearson correlations were obtained using the ‘cor’ function in R package
Discussion
TRACE-seq enables monitoring cells in vitro by collecting media and analyzing the mRNA cargo of L-EVs secreted by the cells. Our methodology brings a representative part of the cellular transcriptome into L-EVs as a useful and cell-sparing method for a large range of analyses including monitoring drug effect, assessing cellular maturation or differentiation, or analyzing cellular response to stress. Traditional transcriptome analysis methodologies require cellular lysis with the exception of the NEX process (Cao et al., 2017) which is designed exclusively for in vitro living cell monitoring (on cell culture plates) and not applicable in vivo. Our study represents the first detailed analysis of a methodology that we developed, that when coupled with appropriate delivery tools (such as viral gene delivery) is fully compatible with in vivo monitoring of the cellular transcriptome.
We evaluated a number of strategies that target cellular mRNAs to the cellular EV secretome, and ultimately settled on an isoform of the YTHDF1 proteins that bind to m6-mRNAs and modify their translation dynamics, based on their ability to ‘trap’ nascent cellular mRNAs. Additionally, addition of the cap protein EIF4E to the C-term-YTHDF1 allowed for further increase in the efficiency of mRNA binding. The second component of our strategy was the use of the GBP1-CD9 fusion protein to bind to the GFP moiety of the mRNA-trapping component to allow for transport to the EVs. Finally, we determined that transcription of these two critical components of our 4EC1 from separate promoters markedly improved the efficiency of mRNA capture and minimized transport of ‘empty’ constructs without mRNA capture to EVs. Our experiments suggested that although both the C1 and the 4EC1 constructs allow for excellent correlation of cellular and L-EV transcriptome, there are certain advantages to each. The C1 construct appears to have slightly higher transient transfection efficiency; however, the 4EC1 construct has better efficiency in the stably transfected cell lines, although prolonged expression of this construct does lead to retardation of cell growth. Interestingly, the 4EC1 construct allows for better efficiency of mRNA capture, likely because of an idealized 1:1 ratio of the two constructs (GBP1-CD9 and 4EC1). For these reasons, we chose to focus on the 4EC1 construct for the cellular stress experiments.
Finally, we have focused on the L-EVs population rather than smaller vesicles, as our mRNA targeting/export constructs allowed for the export of full-length mRNA content to this EV population compared to small EVs. We did not explicitly study the mechanisms of targeting of ‘captured mRNAs’ into L-EVs, and whether biophysical characteristics (such as volume of L-EVs) is correlated with the number/size of mRNAs; such an analysis would be interesting in future studies. Our study also does not allow us to clearly confirm if small EVs transport full-length mRNA or just fragments, but as already exposed in other studies, the YTHDF2 protein interacts with the CCR4-Not1 complex (Du et al., 2016), which may be one explanation why our TRACE fusion proteins are not able to pack full length RNA into CD9 bearing small EVs. When analyzed by RNAseq or RT-PCR of key cellular transcripts, our construct showed high correlation of cellular mRNA transcripts in the L-EV compartment across a range of mRNA abundance thereby allowing for an accurate representation of the cellular transcriptome. Notably, TRACE-seq shows a better efficiency than the i-Tag system (Darr et al., 2020) and would be a simpler and more effective methodology compared to the i-Tag system, which is quite cumbersome and requires specialized transgenic mice for adoption to in vivo analysis.
Our methodology was designed to assess dynamic changes in the cellular transcriptome with stress. In this study we were able to demonstrate that mRNA changes with oxidative stress or hypoxia within the cell were accurately captured by measurement of these changes within L-EVs secreted from these cells. Although the degree of correlation was high across the transcriptome (as shown in Figure 6), it was notably high when comparison was further limited to those transcripts that were most changed with the stress signal (either downregulated or upregulated). Together these data demonstrate that our methodology provides a significant advance to the field and may be adaptable to in vivo systems in the future.
It is important to also point out that introduction of these constructs into cells may affect cellular biology. Notably, long-term expression of our construct (using a lentiviral system) does affect cellular growth, a limitation that could be overcome by using an inducible system. Secondly, our construct may alter the EV biogenesis pathways given that we aim to alter expression of the tetraspanin CD9 (with our GBP/CD9 construct). Although we did not systematically assess this, the full C1 construct led to a larger number of secreted EVs compared to the GBP1-CD9 construct. Although this may be potentially useful when assaying EV content (by increasing the amount of input material), the implications for cellular homeostasis over longer periods of times were not assessed.
We expect TRACE-seq to have broad applicability and serve as a tool for the transcriptomic analysis field to improve our understanding of gene expression and regulation in living cells or tissues. TRACE-seq analysis could be applied to in vivo studies (e.g., for monitoring of transplanted organs transfected with the TRACE-seq inducible constructs) through the analysis of EV-RNAs isolated from the circulation sorted by NanoFACS for GFP positivity (Morales-Kastresana et al., 2019). Finally, TRACE-seq technology offers the possibility of sampling the transcriptome of targeted cells without any major source of disruption at different time points providing a window to dynamic regulation of the transcriptome in response to stressors, and these types of repeated measurements over time domain would be the subject of future studies.
Limitations of the study
TRACEseq allows an easy way of monitoring transcriptome of samples by extracting L-EVs from culture media. However, our method has not yet been adapted for or tested in an in vivo context. Further testing is required to qualify our methodology in vivo and benefit from the maximum technical advantage offered by TRACEseq. The delivery of the constructs to target tissues (e.g., by lentiviral or AAV-mediated viral transfer) needs to be optimized. TRACEseq, as with any methodology to profile EV-RNAs, requires a sufficient quantity of starting material to be effective and discriminant from the background. Finally, as noted above, the presence of the fusion proteins in the cells for longer periods of time may alter cellular biology, including EV biogenesis and would need to be assessed in more detail.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-GFP antibody | Abcam | Cat# ab290; RRID: AB_183734 |
| Bacterial and virus strains | ||
| DH5α | ThermoFisher | Cat# 18258012 |
| pCW57.1 | David Root unpublished | Addgene |
| TRE-eGFP-C-YTHDF1/pCMV-GBP1-CD9 | This paper | N/A |
| TRE-eGFP-EIF4E-C-YTHDF1/pCMV-GBP1-CD9 | This paper | addgene |
| TRE-eGFP/pCMV-GBP1-CD9 | This paper | N/A |
| psPAX2-D64V | Didier trono unpublished | Addgene |
| pMD2.G | Didier trono unpublished | Addgene |
| Critical commercial assays | ||
| Imprint® RNA immunoprecipitation kit | Sigma-Aldrich | Cat# RIP-12RNX |
| Nextera XT DNA kit | Illumina | Cat# FC-131-1096 |
| High sensitivity DNA chip assays | AgilentTechnologies | Cat# 5067-4626 |
| RNA 6000 Pico chip assays | AgilentTechnologies | Cat# 5067-1513 |
| AmpureXP® beads | Beckman Coulter | Cat# A633880 |
| Smarter® Stranded Total RNA-Seq Kit v2 | Takara Bio | Cat# 634413 |
| Streptavidin-conjugated DynabeadsTM | Thermo Fisher | Cat# 65305 |
| Click-iT™ Nascent RNA Capture Kit | Thermo Fisher | Cat# C10365 |
| Deposited data | ||
| RNA-seq C1 vs GFP control data batch NCBI Gene Expression Omnibus | This paper | GSE162425 |
| RNA-seq 4EC1 data batch NCBI Gene Expression Omnibus | This paper | GSE186954 |
| POSTAR database | Hu et al.,2017 | http://postar.ncrnalab.org/ |
| Experimental models: Cell lines | ||
| HEK 293T Cells | ATCC | Cat# CRL-3216 |
| THP-1 | ATCC | Cat# TIB-202 |
| Oligonucleotides | ||
| For primer used see Table S2 | This paper | N/A |
| For oligo for cloning see Table S1 | This paper | N/A |
| Recombinant DNA | ||
| pCAG-GBP1-10gly-Gal4DBD | Tang et al. 2013 | Addgene |
| pCMV-mCherry-CD9-10 | Michael Davidson unpublished | Addgene |
| pCMV-GBP1-CD9 | This paper | N/A |
| pGEx-4T-1-YTHDF1 | Wang et al. 2014 | Addgene |
| pT7-eGFP-HseIF4E | Peter et al. 2015 | Addgene |
| pCMV-eGFP-C-YTHDF1 | This paper | N/A |
| pCMV-eGFP-YTHDF1 | This paper | N/A |
| pCMV-eGFP-RPL10A | This paper | N/A |
| pCMV-eGFP-C-YTHDF2 | This paper | N/A |
| pCMV-eGFP-YTHDF2 | This paper | N/A |
| pCMV-eGFP-C-YTHDF1/pCMV-GBP1-CD9 | This paper | N/A |
| pGBP1-CD9-T2A-GFP-C-YTHDF1 | This paper | N/A |
| pCMV-eGFP-EIF4E-C-YTHDF1/pCMV-GBP1-CD9 | This paper | Addgene |
| pCMV-eGFP/pCMV-GBP1-CD9 | This paper | N/A |
| Software and algorithms | ||
| STAR | Dobin et al., 2013 | https://physiology.med.cornell.edu/faculty/skrabanek/lab/angsd/lecture_notes/STARmanual.pdf |
| FeatureCounts | Liao et al., 2019 | https://rdrr.io/bioc/Rsubread/man/featureCounts.html |
| edgeR | Robinson et al., 2010 | https://bioconductor.org/packages/release/bioc/html/edgeR.html |
| Limma-voom | Ritchie et al., 2015 | https://ucdavis-bioinformatics-training.github.io/2018-June-RNA-Seq-Workshop/thursday/DE.html |
| bedTools | Patwardhan, Mayura et al. unpublished | http://phanstiel-lab.med.unc.edu/bedtoolsr.html |
Resource availability
Lead contact
Further information requests should be directed to the lead contact, Doctor Saumya Das (sdas@mgh.harvard.edu).
Materials availability
Upon publication of the manuscript, the plasmids will be deposited and available from Addgene.
Experimental model and subject details
This study used HEK 293T Cell line, Human Embryonic Kidney cells available from Cat# CRL-3216 ATCC, Manassas, VA, USA. Cells were prepared and cultured as described by ATCC in regular complete culture media: Dulbeccco’s modified Eagle’s media (D-MEM; Gibco Gaithersnurg, MD, USA) supplemented with 10% fetal bovine serum (FBS; Gibco) 1% penicillin/streptomycin (Gibco). Cells were cultured in a 10cm dish (Corning, NY, USA) previously coated with Matrigel (Corning, NY, USA) diluted in F-12 media (F-12 Nutrient Mixture, Gibco Gaithersnurg, MD, USA) and propagated at 37 °C in 5% CO2 as recommended by the manufacturer.
We also used THP-1 cells, human monocyte cells (ATCC, TIB-202) Cells are cultured in T75 flasks in 20 ml complete cell culture medium: Dulbeccco’s modified Eagle’s media (D-MEM; Gibco Gaithersnurg, MD, USA) supplemented with 10% fetal bovine serum (FBS; Gibco) 1% penicillin/streptomycin (Gibco). and propagated at 37 °C in 5% CO2 as recommended by the manufacturer.
Method details
Cells
HEK 293T, Human Embryonic Kidney cells (CRL-3216, ATCC, Manassas, VA, USA), were prepared and cultured as described by ATCC in regular complete culture media: Dulbeccco’s modified Eagle’s media (D-MEM; Gibco Gaithersnurg, MD, USA) supplemented with 10% fetal bovine serum (FBS; Gibco) 1% penicillin/streptomycin (Gibco). Cells were cultured in a 10cm dish (Corning, NY, USA) previously coated with Matrigel (Corning, NY, USA) diluted in F-12 media (F-12 Nutrient Mixture, Gibco Gaithersnurg, MD, USA) as recommended by the manufacturer. During the vesicles collection, a specific media was used and we called it Exo-media: Dulbeccco’s modified Eagle’s media (D-MEM; Gibco Gaithersnurg, MD, USA) supplemented with 10% exosome depleted fetal bovine serum (Exosome-Depleted FBS; Gibco) and 1% penicillin/streptomycin (Gibco). THP-1 cells, human monocyte cells (ATCC, TIB-202) Cells are cultured in T75 flasks in 20 ml complete cell culture medium: Dulbeccco’s modified Eagle’s media (D-MEM; Gibco Gaithersnurg, MD, USA) supplemented with 10% fetal bovine serum (FBS; Gibco) 1% penicillin/streptomycin (Gibco). and propagated at 37 °C in 5% CO2 as recommended by the manufacturer.
Plasmids and cloning strategy
All PCR primers and oligo used for this work are listed in the Tables S1 and S2. The GBP1 fragment was PCR amplified (a flexible linker GGGGGGGGGG on the C-terminal part of the sequence was added) from the pCAG-GBP1-10gly-Gal4DBD plasmid which was a gift from Connie Cepko (Tang et al., 2013) (Addgene, #49438). This fragment was cloned in the pCMV-mCherry-CD9-10 plasmid which was a gift from Michael Davidson (Addgene, #55013) to generated, the pCMV-GBP1-CD9 plasmid. The C terminal part of the YTHDF1 gene was amplified (a flexible linker SGGGGGGGGGG on the N-terminal part of the sequence was added) from pGEx-4T-1-YTHDF1 which was a gift from Chuan He (Wang et al., 2014) (Addgene, #70087) and cloned into the pT7-eGFP-HseIF4E gifted from Elisa Izaurralde (Peter et al., 2015) (Addgene, #79437) and gave the pCMV-eGFP-C-YTHDF1. The two neo-generated plasmids were merged by cloning the pCMV-EGFP-C-YTHDF1 fragment into the pCMV-GBP1-CD9 plasmid. The pCMV-EGFP-C-YTHDF1 fragment was purified using the two digestions site PciI and MluI, and brought into the pCMV-GBP1-CD9 plasmid by digestion of PciI enzyme followed by blunting ends and cloning procedure for both fragments. The final construct obtained was pCMV-eGFP-C-YTHDF1/pCMV-GBP1-CD9. A negative control plasmid was generated by double digestion with XhoI and AclI endonucleases and blunt ligation of the plasmid to remove the C-YTHDF1 sequence and obtain the following plasmid: pCMV-eGFP/pCMV-GBP1-CD9.
Another plasmid was also created. First, the self-cleavable peptide T2A sequence was directly included in the reverse primer for the PCR amplification of the C-term part of the CD9 sequence from the pCMV-GBP1-CD9 with the flanking restriction enzyme sequences BbsI and EcoRI. This C-term CD9-T2A fragment was cloned into the pCMV-GBP1-CD9 plasmid with the same couple of enzymes and had allowed to bring back a full length CD9 sequence without stop codon. The next step was to digest the new plasmid with the enzymes MefI and AfeI to isolate the GBP1-CD9-T2A fragment in order to clone it into the GFP-C-YTHDF1 plasmid with the same couple of digestion enzymes. We generated the plasmid pGBP1-CD9-T2A-GFP-C-YTHDF1. Secondly, we added the EIF4E fragment amplified by PCR from the original pT7-eGFP-HseIF4E plasmid and cloned into the pCMV-eGFP-C-YTHDF1/pCMV-GBP1-CD9 with the digestion enzymes BglII and XhoI to finally obtain the pCMV-eGFP-EIF4E-C-YTHDF1/pCMV-GBP1-CD9 plasmid.
For the lentivirus construct, plasmids and the two endonucleases AfeI and MluI were used to insert the fragments eGFP-C-YTHDF1/pCMV-GBP1-CD9, eGFP-EIF4E-C-YTHDF1/pCMV-GBP1-CD9 and eGFP/pCMV-GBP1-CD9 in the lentivirus backbone. The pCW57.1 plasmid (which was a gift from David Root, Addgene plasmid # 41393) was opened with the two enzymes BmtI and AgeI and a small fragment (annealing of the two complementary ssDNA Afe1/Mlu1 Fragments) was cloned inside with the same enzymes. The new pCW57.1 plasmid containing Afe1 and Mlu1 was ready to receive each construct which were cloned with these two endonucleases. We obtained at the end the inducible tet-on lentivirus plasmid: pCW57.1 TRE-eGFP-C-YTHDF1/pCMV-GBP1-CD9, pCW57.1 TRE-eGFP-EIF4E-C-YTHDF1/pCMV-GBP1-CD9 and pCW57.1 TRE-eGFP/pCMV-GBP1-CD9 for the plasmid control. TRE promoter with tet operators were also obtained from PCR to the pCW57.1 Afe1 Mlu1 plasmid. The generated TRE promoter fragment was cloned in each plasmid construct with NsiI and SgrAI enzymes to remove pCMV promoter and get the GBP1-CD9 fusion protein expression under control of the tet-on induction system. We finally obtained the two following plasmids: pCW57.1 TRE-eGFP-C-YTHDF1/TRE-GBP1-CD9, pCW57.1 TRE-eGFP-EIF4E-C-YTHDF1/TRE-GBP1-CD9 and pCW57.1 TRE-eGFP/TRE-GBP1-CD9 for the plasmid control.
Transfection, transduction and virus production
We develop two ways of delivery which are related to the capability of in vitro /in vivo monitoring. We directly transfected the pCMV-eGFP-C-YTHDF1/pCMV-GBP1-CD9 and pCMV-eGFP/pCMV-GBP1-CD9 plasmids to 1 or 4 million HEK 293T cells in a 10 cm dish with lipofectamine 3000 (Invitrogen, Carlsbad, CA, USA) using the protocol recommended by the manufacturer. A negative control with regular HEK293T (untransfected cells) was also included and treated as samples like the following. After 12 hours of incubation at 37⁰C, all the plates were washed two times with PBS (Gibco Gaithersnurg, MD, USA) and 10mL of fresh exo-media was added complemented with RNAse A (Qiagen, Hilden, Germany) at a final concentration of 30μg/mL. Cells were left in the collection media for 24h at 37⁰C for EVs production.
Regarding the lentivirus, three plasmids were transfected at the same time. First, one of the two constructs: pCW57.1 TRE-eGFP-C-YTHDF1/TRE-GBP1-CD9, pCW57.1 TRE-eGFP-EIF4E-C-YTHDF1/TRE-GBP1-CD9 or the pCW57.1 TRE-eGFP/TRE-GBP1-CD9 for the plasmid control. One of these three plasmids (depending on conditions) was mixed with the psPAX2 (psPAX2 was a gift from Didier Trono (Addgene plasmid # 12260) and the pMD2.G plasmids (pMD2.G was a gift from Didier Trono Addgene plasmid # 12259). All three were transfected in HEK293T in 10 cm dishes with lipofectamine 3000 with the protocol recommended by the manufacturer. Twelve hours after transfection, the media was replaced and collected at 36h and 52 hours after transfection. The collection media was spun at 1,000g for 5 min, the supernatant was filtered with 0.8 μm filters (Millipore Co, Burlington, MA, USA) and let in incubation overnight with PEG-it virus precipitation solution (System Biosciences LLC, Palo Alto, CA, USA). After incubation, the lentivirus solution was spun at 1,500g for 30 min and the virus pellet was resuspended in 1 ml PBS and stored at -80⁰C in 100μL aliquots. A fraction of each lentivirus production was used for a functional virus titration on HEK293T cells and gave us the MOI of 4.2.
Stable cell line
All HEK293T stable cell lines were obtained by infecting with the lentivirus generated from the two following constructs pCW57.1 TRE-eGFP-C-YTHDF1/TRE-GBP1-CD9, pCW57.1 TRE-eGFP-EIF4E-C-YTHDF1/TRE-GBP1-CD9 and pCW57.1 TRE-eGFP/TRE-GBP1-CD9 for the plasmid control. Ten thousand cells where plated in 6 wells dish solution in regular complete media and incubated for 24h at 37⁰C. After incubation, in regards of the MOI of the virus, cell media was removed and replaced by 30μL of lentivirus PBS solution from the -80⁰C stock + 1.960ml of DMEM complete + 10 μg/mL polybrene infection reagent (Sigma-Aldrich, Saint Louis, MO, USA) and incubated for 48h. After the infection time, the media was switched to selection media (regular complete media + 2μg/ml Doxycycline (Fisher Scientific, Hampton, NH, USA) with 2μg/ml puromycin (Fisher Scientific, Hampton, NH, USA)) and incubated for 2 weeks with 2 cell passages with Accutase (STEMCELL Technologies Inc, Vancouver, Canada), the selection media being refreshed every two days. At the last cell passage, the remaining green cells were counted and a subcloning in a 96 well plate was performed in order to dilute the cells down to 1 per well in 100μL of the selection media with puromycin 0.5 μg/ml. After the apparition of cell clusters, the best clone of each stable cell line was selected regarding GFP membranous signal and passaged with Accutase in 6 well dishes with regular complete culture media without doxycycline. At 70% confluency, each stable cell line is ready to use for experiments and a fraction of them was stocked in liquid nitrogen according to our standard procedure (complete media + 10% DMSO) (Sigma-Aldrich, Saint Louis, MO, USA). To reduce cell stress and protect the membranous proteins (especially CD9), all passages were executed with Accutase.
Immunoprecipitation
We generated a batch of 5 plasmids for this experiment: pCMV-eGFP-C-YTHDF1, pCMV-eGFP-YTHDF1, pT7-eGFP-HseIF4E and decided to generate a new plasmid with the C-YTHDF2 sequence directly amplified from the cDNA library of HEK 293T cells. This C-YTHDF2 sequence was cloned inside the pCMV-eGFP-C-YTHDF1 and we removed the C-YTHDF1 sequence to finally get pCMV-eGFP-C-YTHDF2. In the same manner, the RPL10A sequence was amplified from the cDNA library of HEK 293T cells and cloned it into the pCMV-eGFP-C-YTHDF1 plasmid to remove the C-YTHDF1 fragment and obtain the pCMV-eGFP-RPL10A plasmid. Each HEK 293T sample was transfected with one of these plasmids (Invitrogen, Carlsbad, CA, USA) using the protocol recommended by the manufacturer and cultured in a 10cm dish (Corning, NY, USA). After 48 hours of culture, the cells were harvested with a trypsin solution at 0.25% (Gibco, Trypsin 0.25%) for 1min at 37⁰C and the reaction was stopped with regular complete media. Each cell population (10 Million Cells) was spun down at 1500g for 5min at 4⁰C, the residual media was removed, cells were washed with PBS, spun down again and resuspended in 200 μL of PBS. The immunoprecipitation was realized with the Imprint® RNA immunoprecipitation kit (cat# RIP-12RNX, Sigma-Aldrich, Saint Louis, MO, USA) using an anti-GFP antibody (Cat# ab290, Abcam, Cambridge, UK) with the protocol recommended by the manufacturer. The mRNA isolated from each sample was analyzed by RT-PCR for the batch of 5 genes of interest, trapped samples vs whole cell lysate.
EVs purification
L-EVs were directly purified from the supernatant of 1 or 4 million transfected, 10 million HEK 293T cells. For the transfected cells, they were washed two times with PBS after a 12h incubation in the transfection media. Exo-media was complemented with RNAse A at 30μg/mL and cells were incubated for 24h. For the lentivirus cell line, cells were previously treated with Doxycycline (Fisher Scientific, Hampton, NH, USA) at a final concentration of 1μg/ml two times at 24 and 48h before the incubation in the exo-media. Next, cells were washed two times with PBS and incubated in the exo-media complemented with RNAse A at 30μg/mL (to degrade any RNAs not protected within EVs) and 2μg/ml Doxycycline was added and let incubated for 24h. After incubation, the exo-media was collected, and multiple centrifugations were performed. First, a 300g spin at 4⁰C for 10 min to remove cells and big debris was performed. The supernatant was collected and spun a second time at 1500g at 4⁰C for 15 min to eliminate the large majority of the apoptotic bodies. We also filtered all supernatants with 0.8μm filters (Millipore Co, Burlington, MA, USA) and made a final spin at 16.500g, 4⁰C for 30 min. After this spin, the remaining large EVs (L-EVs) pellet was carefully resuspended in PBS supplemented with vanadyl ribonucleoside complex (NEB, Ipswich, MA, USA) at a final concentration of 10 mM to protect any mRNA released from clamped vesicles. To purify small EVs (S-EVs), a last spin of the supernatant was performed at 100000g for 70 min at 4⁰C and the S-EVs pellet was resuspended in 200 μL PBS supplemented with vanadyl ribonucleoside complex. EVs fraction (1/10) are also analyzed and sorted based on their green signal by NanoFlowCyt: Beckman Coulter MoFlo AstriosEQ 4 laser system.
Cell purification
Each aliquot of EVs producer cells is isolated by a treatment of trypsin solution at 0.25% (Gibco, Trypsin 0.25%) for 1min at 37⁰C and the reaction was stopped with regular complete media. A fraction of each cell population (10000 Cells) was spun down at 1500g for 5min at 4⁰C, the residual media was removed, cells were wash with PBS, spin down again and resuspended in 200 μL PBS and ready for the next step of the TRACE protocol.
RNA purification
Right after EVs and cells isolation, the RNA purification protocol was started. Each EV and cell solution was treated with 700μL TRIzol LS Reagent (Fisher Scientific, Hampton, NH, USA), vortexed 5 sec and incubated at 25⁰C for 3 min. A pure solution of 140 μL of Chloroform (Fisher Scientific, Hampton, NH, USA) was added and the tubes were vortexed for 15 sec and incubated at 25⁰C for 5min. Right after the incubation, tubes were spun down at 12,000g for 15 min at 4⁰C. The upper phase of each tube was carefully collected and mixed with 2 vol of 100% pure Ethanol solution (Fisher Scientific, Hampton, NH, USA). Each solution was added to the RNA clean up Qiagen column (RNeasy MinElute Cleanup Kit Qiagen, Hilden, Germany) and spun down at 8,000g for 30 sec. Only for the cell lysates, the column filters were treated with DNAse 1 solution from Qiagen as the protocol recommended by the manufacturer. Columns were then washed two times with a 70% ethanol solution, a volume of 700μL and 500μL for the second one was used. Columns were spun for the first wash at 8,000g for 30 sec and at 8,000g for 1min for the second one. The columns were also dried at 8,000g for 1min and eluted with 10μL of RNAse free water (Qiagen, Hilden, Germany) at 8,000g for 1min after a 1min incubation at room temperature. To attest the presence and quality of the purified RNA, each sample was analyzed on a RNA 6000 Pico chip from Agilent technologies (Agilent Technologies, Santa Clara, CA, USA, cat# 5067-1513) by loading 1μL of each EVs eluent and 1μL of a diluted RNA from cell lysate (usually correspond to 5ng RNA for the lysate).
Reverse transcription and PCR preamplification
The reverse transcription protocol and PCR preamplification were performed according to the Smart-Seq 2 manuscript (Picelli et al., 2014) except for the TSO oligo which were designed and ordered through the Qiagen custom LNA oligonucleotides tool (www.qiagen.com, Qiagen, Hilden, Germany). Volumes were adapted as following: in 0.2mL PCR tube: 9μL form each solution and EVs RNA were mixed with 1μL of 10μM Vnd30T oligo and 1μL of dNTPs mix at 2mM each. 0.5μL of each cell lysate (which corresponds to 30ng) was mixed with 8.5μL RNAse free Water and with 1μL of 10μM Vnd30T oligo and 1μL of dNTPs mix at 2mM each. Samples were annealed in a thermocycler as recommended in the SMART-Seq2 protocol and cDNA reaction mix was performed as the following:
| Component | Volume (μL) | Final concentration |
|---|---|---|
| RNA reaction mix SuperScript II (200 U μL−1) |
11 1 |
– 100 U |
| RNAse Inhibitor (40 U μL−1) | 0.5 | 10 U |
| SuperSript II Buffer (5X) | 4 | 1X |
| DTT (100 mM) | 1 | 5 mM |
| Betaine (5 M) | 2 | 0.5 M |
| MgCl2 (1 M) | 0.1 | 5 mM |
| TSO (100 μM) | 0.2 | 1 μM |
| NRAse free water | 0.2 | – |
| Total volume | 20 | – |
The 9μL cDNA reaction mix was added to each sample to obtain a final reaction volume of 20μL. After mixing the solution gently and spinning down at 700g for 10s at 25⁰C, samples were incubated in the thermocycler as recommended in the Smart-Seq2 protocol.
Right after the RT the PCR mix for the preamplification is prepare as the following:
| Component | Volume (μL) | Final concentration |
|---|---|---|
| First Strand reaction | 20 | – |
| KAPA HiFi HotStart ReadyMix (2X) | 22.5 | 1X |
| IS PCR primers (10 μM) | 0.45 | 0.1 μM |
| NRAse free water | 2.05 | – |
| Total volume | 45 | – |
The PCR run was performed as specified in the Smart-Seq2 protocol. Finally, each sample was cleaned up using Zymo (Zymo research, Irvine, CA, USA) to cut of small fragments and keep up to 300nt. Each sample was loaded in 40 μL of elution buffer. To attest the quality of all pre-amplified cDNA, all samples were analyzed on High sensitivity DNA chip assays from Agilent technologies (Agilent Technologies, Santa Clara, CA, USA, cat# 5067-4626) by loading 1μL of each EVs eluant and 1μL of a diluted cDNA from cell lysate (usually correspond to 5ng DNA for the lysate) and an expected broad peak with an average size of 600-2000 bp would be observed.
Tagmentation reaction and amplification of adapter-ligated fragments
These following steps were made as recommended in the Smart-Seq2 protocol (Picelli et al., 2014) and using the Illumina Nextera XT DNA kit (Illumina, FC-131-1096). As specified in the manufacturer protocol, each sample is normalized on the same volume for all EVs fractions and for the cell lysate preparation, each of them should never exceed 1 ng. 500pg were used, and the same amount of reagent from the Nextera XT DNA sample preparation was used, as the following, in 0.2mL PCR tube:
| Component | Volume (μL) | Final concentration |
|---|---|---|
| Tagment DNA buffer | 10 | 1X |
| Amplicon tagment mix | 5 | – |
| Amplified DNA sample | Variable | – |
| Nuclease free water | Variable | – |
| Total volume | 20 | – |
Samples were incubated in the thermocycler as recommended by the manufacturer. The tagmentation reaction was stopped by adding 5μL of Nextera NT Buffer and incubating for 5min at RT.
To all tagmented samples, the following PCR mix was added:
| Component | Volume (μL) | Final concentration |
|---|---|---|
| Tagmented DNA Sample | 25 | – |
| Nextera PCR master mix | 15 | – |
| Index 1 primers (N7xx) | 5 | – |
| Index 2 primers (N5xx) | 5 | – |
| Total volume | 50 | – |
All samples were incubated in thermocycler and the PCR amplification steps program was performed as recommended by the illumina Nextera® protocol. The amount of PCR cycles during this amplification depends on the starting DNA material originally used as recommended in the Smart-Seq2 protocol (Picelli et al., 2014). As we used 500pg, 10 cycles were performed for this amplification. After this final amplification step, each sample was cleaned up using AmpureXP® beads (Beckman Coulter Co. Brea, CA, USA, cat# A633880).
Library QC and quantification
To attest the cDNA library quality, each sample was analyzed on High sensitivity DNA chip assay from Agilent technologies, 1μL loaded (an expected broad peak with an average size of 300-800pb should be observed) and quantified with the Qubit 2 Fluorometer (Invitrogen, Carlsbad, CA, USA).
Based on the relative molarity of each library, each sample was properly diluted to get a final library solution of 2nM each. At this step, all samples from the Nextera XT® kit were pooled together by following the recommendation from the manufacturer. The pooled library was again checked by Qubit and adjusted if necessary to keep the solution at 2nM.
Cell line in oxidative stress testing
GFP-EIF4E-C-YTHDF1/GBP1-CD9 cell line and regular HEK 293T cells were tested in H2O2 oxidative stress. Plates of 150 mm were made (6 for the TRACE cell line and 6 for the regular HEK 293T cells). Cells were cultured with regular media. When cells reached 15M per well, they were washed two times with PBS and placed in Exo-media with RNAseA at 30μg/mL final concentration + 2μg/ml Doxycycline. Half of each cell population (3 plates of GFP-EIF4E-C-YTHDF1/GBP1-CD9 cell line and 3 plates of None regular cell) was treated with H2O2 peroxide (hydrogen peroxide solution 30% (v/v) from Sigma-Aldrich, Saint Louis, MO, USA) at a final concentration of 50μM. After 24h of culture at 37⁰C, 5% CO2, conditioned media from each plate was collected and processed according to the EVs purification protocol previously described and, in the meantime, each cell lysate was collected as previously described. For each sample, RNA was extracted by phenol/chloroform extraction and RNA clean up Qiagen columns were used to isolate RNA population from each sample. Directly after their isolation, the RNA was reverse transcripted into cDNA and pre-amplified as previously described. The pre amplification step was different as a total number of 25 cycles was performed this time. Samples were purified as usual with zymo kit and samples were eluted into 40μL for qPCR analysis on a batch of 8 specific oxidative genes, 1μL of cDNA per reaction and the 2X SYBRtm Green PCR Master mix (Applied Biosystemstm Foster City, CA, USA). The qPCR was run in triplicate with 10μL per reaction on a QuantStudio 6 384-well formats (Applied Biosystemstm Foster City, CA, USA).
Other GFP-EIF4E-C-YTHDF1/GBP1-CD9 HEK 293T cell line samples were placed with and without hypoxic stress induction (dishes culture in incubator, gaz mixture 2% O2 for 24h) and used to generate a sequencing library via the Smart-seq2 protocol and data were collected according to the previous protocol (see section Primary data processing). Batch of row data were normalized as previously (see section Deferential expression) and a DEseq analysis was generated. This DEseq genes analysis was based on the log2FC (padj <0.05) and the Top 52 DEseq gene selection were obtained based on the log2FC (padj <0.05 & FC in cells larger than 1.5 and in EVs larger than 2).
The capture and sequencing of nascent RNAs in cells and EVs. Nascent RNAs in THP1 cells were labeled with 5-ethynyluridine (EU) and captured using Click-iT™ Nascent RNA Capture Kit (Thermo Fisher, C10365). Briefly, 10 million THP1 cells were plated in 100 mm cell culture dish with R10 medium (RPMI1640 supplemented with 10% FBS and 0.05 mM 2-mercaptoethanol) containing 0.2mM EU. 24 hours later, THP1 cells were washed twice with basal RPMI1640 medium and cultured in fresh EV-depleted R10 medium for additional 24 hours. Then, 0.5 million cells were fixed with 4% PFA for EU incorporation validation using Click-iT™ RNA Alexa Fluor™ 594 Imaging Kit (Thermo Fisher, C10330). Remaining cells and cell culture medium were subjected to cellular and extracellular RNA isolation as previously described (1) . 4 μg cellular and extracellular RNAs were used for Click-iT chemistry reaction to label the incorporated EU with Biotin and cleaned up using Zymo RNA Clean&Concentrator-5 kit (Zymo, R1014). Next, 700 ng Biotinylated cellular and extracellular RNAs were captured using Streptavidin-conjugated DynabeadsTM (Thermo Fisher scientific 65305). The Dynabeads-captured RNAs were then extracted using TRIzol. The long RNA libraries were prepared using Smarter® Stranded Total RNA-Seq Kit v2 - Pico Input (Takara Bio, 634413) and subjected to Illumina NextSeq.
Quantification and statistical analysis
Primary data processing
Data were collected using 50 × 8 × 50 reads on a HiSeq 2500 (paired-end). Reads were aligned to hg19 using STAR (Dobin et al., 2013) and counting reads associated genes were detected with the FeatureCounts module (Liao, Smyth and Shi, 2019).
We proceeded to a filtration to remove low expressed genes with the cpm function from the edgeR package (Robinson, McCarthy and Smyth, 2010). In this dataset, we retain genes if they were expressed at a counts-per-million (CPM) > 0.5 in at least two or more counts.
Differential expression
A normalization was performed on the data set to eliminate composition biases between libraries using the calcNormFactor function. Normalized data were tested for differential expression by using the limma-voom package (Ritchie et al., 2015). Thus, data were voom transformed and differential expression test was made using the lmFit function form the limma package (DESeq2). A differential expression test between TRACE-seq experiments and results from the POSTAR database (Hu et al., 2017; Zhu et al., 2019) (corresponding to the methylated mRNA [m6A targeted] from YTHDF1 IP lysate from Hela cells) was made through the same way of analysis as above.
RNA coverage analysis
Coverage is first computed using the bedTools package. Then a sliding-window approach is used to identify and quantify consistently covered regions of detected transcripts. Briefly, using a window of 50nt, we require an average of at least 10 reads in order to class this window as ‘covered’. Then the fraction of windows across each transcript that satisfy this detection criteria is reported as the fraction covered.
Differential expression for hypoxic stress test
Data were collected according to the previous protocol (see section Primary data processing). Batch of row data were normalized as previously (see section Deferential expression) and a DEseq analysis was generated. This DEseq genes analysis was based on the log2FC (padj <0.05) and the Top 52 DEseq gene selection were obtained based on the log2FC (padj <0.05 & FC in cells larger than 1.5 and in EVs larger than 2). All correlation analyses were done in R package (cor function) to generate Pearson correlations.
Acknowledgments
We thank members from the Benichou and Das laboratories for their expertise and advice on this project, as well as the Beth Israel flow cytometry core for their assistance with the NanoFlowCyt. This work was supported by the French National research agency with the national thesis grant program and the US National Institutes of Health (R35HL150807 to S.D., 5R01HL130391-02 to I.D.).
Author contributions
F.C., G.L., P.S., P.G., and A.P. performed the research experiments. All authors designed experiments and interpreted the data. F.C. wrote the paper. A.P., G.L., S.D., G.B., R.C., and I.D. provided critical reading and editing of the manuscript. S.D. and I.D. provided funding and supervised the planning and analysis of the experiments.
Declaration of interests
Francois Cherbonneau and Aurore Prunevieille are founders of QUIDDITAS Therapeutics which did not play any role in this study. Dr. Das is a founder of Switch Therapeutics and Long QT Therapeutics, which did not play any role in this study, and has consulted for Amgen.
Inclusion and diversity
We worked to ensure diversity in experimental samples through the selection of cell lines. In addition to citing references scientifically relevant for this work, we also actively worked to promote gender balance in our reference list.
Published: February 18, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2022.103806.
Supplemental information
Data and code availability
-
•
RNA-seq data have been deposited at NCBI Gene Expression Omnibus (GEO) and are publicly available as of the date of publication. Accession numbers are listed in the key resources table (GSE162425 and GSE186954).
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Bao G., Rhee W.J., Tsourkas A. Fluorescent probes for live-cell RNA detection. Annu. Rev. Biomed. Eng. 2009;11:25–47. doi: 10.1146/annurev-bioeng-061008-124920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batagov A.O., Kurochkin I.V. Exosomes secreted by human cells transport largely mRNA fragments that are enriched in the 3'-untranslated regions. Biol. Direct. 2013;8:12. doi: 10.1186/1745-6150-8-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Y., Hjort M., Chen H., Birey F., Leal-Ortiz S.A., Han C.M., Santiago J.G., Paşca S.P., Wu J.C., Melosh N.A. Nondestructive nanostraw intracellular sampling for longitudinal cell monitoring. Proc. Natl. Acad. Sci. U S A. 2017;114:E1866–E1874. doi: 10.1073/pnas.1615375114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corces M.R., Buenrostro J.D., Wu B., Greenside P.G., Chan S.M., Koenig J.L., Snyder M.P., Pritchard J.K., Kundaje A., Greenleaf W.J., et al. Lineage-specific and single-cell chromatin accessibility charts human hematopoiesis and leukemia evolution. Nat. Genet. 2016;48:1193–1203. doi: 10.1038/ng.3646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darr J., Tomar A., Lassi M., Gerlini R., Berti L., Hering A., Scheid F., Hrabě de Angelis M., Witting M., Teperino R. iTAG-RNA isolates cell-specific transcriptional responses to environmental stimuli and identifies an RNA-based endocrine Axis. Cell Rep. 2020;30:3183–3194.e4. doi: 10.1016/j.celrep.2020.02.020. [DOI] [PubMed] [Google Scholar]
- Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominissini D., Moshitch-Moshkovitz S., Schwartz S., Salmon-Divon M., Ungar L., Osenberg S., Cesarkas K., Jacob-Hirsch J., Amariglio N., Kupiec M., et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature. 2012;485:201–206. doi: 10.1038/nature11112. [DOI] [PubMed] [Google Scholar]
- Du H., Zhao Y., He J., Zhang Y., Xi H., Liu M., Ma J., Wu L. YTHDF2 destabilizes m(6)A-containing RNA through direct recruitment of the CCR4-NOT deadenylase complex. Nat. Commun. 2016;7:12626. doi: 10.1038/ncomms12626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grün D., van Oudenaarden A. Design and analysis of single-cell sequencing experiments. Cell. 2015;163:799–810. doi: 10.1016/j.cell.2015.10.039. [DOI] [PubMed] [Google Scholar]
- Heiman M., Kulicke R., Fenster R.J., Greengard P., Heintz N. Cell type-specific mRNA purification by translating ribosome affinity purification (TRAP) Nat. Protoc. 2014;9:1282–1291. doi: 10.1038/nprot.2014.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hrdlickova R., Toloue M., Tian B. RNA-Seq methods for transcriptome analysis. Wiley Interdiscip. Rev. RNA. 2017;8:wrna.1364. doi: 10.1002/wrna.1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu B., Yang Y.T., Huang Y., Zhu Y., Lu Z.J. POSTAR: a platform for exploring post-transcriptional regulation coordinated by RNA-binding proteins. Nucleic Acids Res. 2017;45:D104–D114. doi: 10.1093/nar/gkw888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirchhofer A., Helma J., Schmidthals K., Frauer C., Cui S., Karcher A., Pellis M., Muyldermans S., Casas-Delucchi C.S., Cardoso M.C., et al. Modulation of protein properties in living cells using nanobodies. Nat. Struct. Mol. Biol. 2010;17:133–138. doi: 10.1038/nsmb.1727. [DOI] [PubMed] [Google Scholar]
- Koh C.W.Q., Goh Y.T., Toh J.D.W., Neo S.P., Ng S.B., Gunaratne J., Gao Y.G., Quake S.R., Burkholder W.F., Goh W.S.S. Single-nucleotide-resolution sequencing of human N6-methyldeoxyadenosine reveals strand-asymmetric clusters associated with SSBP1 on the mitochondrial genome. Nucleic Acids Res. 2018;46:11659–11670. doi: 10.1093/nar/gky1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowal J., Arras G., Colombo M., Jouve M., Morath J.P., Primdal-Bengtson B., Dingli F., Loew D., Tkach M., Théry C. Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes. Proc. Natl. Acad. Sci. U S A. 2016;113:E968–E977. doi: 10.1073/pnas.1521230113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y., Smyth G.K., Shi W. The R package Rsubread is easier, faster, cheaper and better for alignment and quantification of RNA sequencing reads. Nucleic Acids Res. 2019;47:e47. doi: 10.1093/nar/gkz114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mateescu B., Kowal E.J., van Balkom B.W., Bartel S., Bhattacharyya S.N., Buzás E.I., Buck A.H., de Candia P., Chow F.W., Das S., et al. Obstacles and opportunities in the functional analysis of extracellular vesicle RNA - an ISEV position paper. J. Extracell Vesicles. 2017;6:1286095. doi: 10.1080/20013078.2017.1286095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller J.E., Reese J.C. Ccr4-Not complex: the control freak of eukaryotic cells. Crit. Rev. Biochem. Mol. Biol. 2012;47:315–333. doi: 10.3109/10409238.2012.667214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monaco G., Lee B., Xu W., Mustafah S., Hwang Y.Y., Carré C., Burdin N., Visan L., Ceccarelli M., Poidinger M., et al. RNA-seq signatures normalized by mRNA abundance allow absolute deconvolution of human immune cell types. Cell Rep. 2019;26:1627–1640.e7. doi: 10.1016/j.celrep.2019.01.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales-Kastresana A., Musich T.A., Welsh J.A., Telford W., Demberg T., Wood J.C.S., Bigos M., Ross C.D., Kachynski A., Dean A., et al. High-fidelity detection and sorting of nanoscale vesicles in viral disease and cancer. J. Extracell Vesicles. 2019;8:1597603. doi: 10.1080/20013078.2019.1597603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales-Kastresana A., Telford B., Musich T.A., McKinnon K., Clayborne C., Braig Z., Rosner A., Demberg T., Watson D.C., Karpova T.S., et al. Labeling extracellular vesicles for nanoscale flow cytometry. Sci. Rep. 2017;7:1878. doi: 10.1038/s41598-017-01731-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Brien K., Breyne K., Ughetto S., Laurent L.C., Breakefield X.O. RNA delivery by extracellular vesicles in mammalian cells and its applications. Nat. Rev. Mol. Cell Biol. 2020;21:585–606. doi: 10.1038/s41580-020-0251-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozsolak F., Milos P.M. RNA sequencing: advances, challenges and opportunities. Nat. Rev. Genet. 2011;12:87–98. doi: 10.1038/nrg2934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patil D.P., Pickering B.F., Jaffrey S.R. Reading m6A in the transcriptome:m6A-binding proteins. Trends Cell Biol. 2018;28:113–127. doi: 10.1016/j.tcb.2017.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peter D., Igreja C., Weber R., Wohlbold L., Weiler C., Ebertsch L., Weichenrieder O., Izaurralde E. Molecular architecture of 4E-BP translational inhibitors bound to eIF4E. Mol. Cell. 2015;57:1074–1087. doi: 10.1016/j.molcel.2015.01.017. [DOI] [PubMed] [Google Scholar]
- Picelli S., Björklund Å., Faridani O.R., Sagasser S., Winberg G., Sandberg R. Smart-seq2 for sensitive full-length transcriptome profiling in single cells. Nat. Methods. 2013;10:1096–1098. doi: 10.1038/nmeth.2639. [DOI] [PubMed] [Google Scholar]
- Picelli S., Faridani O.R., Björklund A.K., Winberg G., Sagasser S., Sandberg R. Full-length RNA-seq from single cells using Smart-seq2. Nat. Protoc. 2014;9:171–181. doi: 10.1038/nprot.2014.006. [DOI] [PubMed] [Google Scholar]
- Pérez-Boza J., Lion M., Struman I. Exploring the RNA landscape of endothelial exosomes. RNA. 2018;24:423–435. doi: 10.1261/rna.064352.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie M.E., Phipson B., Wu D., Hu Y., Law C.W., Shi W., Smyth G.K. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson M.D., McCarthy D.J., Smyth G.K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigues D.C., Mufteev M., Ellis J. Quantification of mRNA ribosomal engagement in human neurons using parallel translating ribosome affinity purification (TRAP) and RNA sequencing. STAR Protoc. 2021;2:100229. doi: 10.1016/j.xpro.2020.100229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saadatpour A., Lai S., Guo G., Yuan G.C. Single-cell analysis in cancer genomics. Trends Genet. 2015;31:576–586. doi: 10.1016/j.tig.2015.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saliba A.E., Westermann A.J., Gorski S.A., Vogel J. Single-cell RNA-seq: advances and future challenges. Nucleic Acids Res. 2014;42:8845–8860. doi: 10.1093/nar/gku555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandberg R. Entering the era of single-cell transcriptomics in biology and medicine. Nat. Methods. 2014;11:22–24. doi: 10.1038/nmeth.2764. [DOI] [PubMed] [Google Scholar]
- Shi H., Wang X., Lu Z., Zhao B.S., Ma H., Hsu P.J., Liu C., He C. YTHDF3 facilitates translation and decay of N6-methyladenosine-modified RNA. Cell Res. 2017;27:315–328. doi: 10.1038/cr.2017.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang F., Lao K., Surani M.A. Development and applications of single-cell transcriptome analysis. Nat. Methods. 2011;8:S6–S11. doi: 10.1038/nmeth.1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang J.C., Szikra T., Kozorovitskiy Y., Teixiera M., Sabatini B.L., Roska B., Cepko C.L. A nanobody-based system using fluorescent proteins as scaffolds for cell-specific gene manipulation. Cell. 2013;154:928–939. doi: 10.1016/j.cell.2013.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valadi H., Ekström K., Bossios A., Sjöstrand M., Lee J.J., Lötvall J.O. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 2007;9:654–659. doi: 10.1038/ncb1596. [DOI] [PubMed] [Google Scholar]
- van Balkom B.W., Eisele A.S., Pegtel D.M., Bervoets S., Verhaar M.C. Quantitative and qualitative analysis of small RNAs in human endothelial cells and exosomes provides insights into localized RNA processing, degradation and sorting. J. Extracell Vesicles. 2015;4:26760. doi: 10.3402/jev.v4.26760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X., Lu Z., Gomez A., Hon G.C., Yue Y., Han D., Fu Y., Parisien M., Dai Q., Jia G., et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature. 2014;505:117–120. doi: 10.1038/nature12730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X., Zhao B.S., Roundtree I.A., Lu Z., Han D., Ma H., Weng X., Chen K., Shi H., He C. N(6)-methyladenosine modulates messenger RNA translation efficiency. Cell. 2015;161:1388–1399. doi: 10.1016/j.cell.2015.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Z., Batagov A.O., Schinelli S., Wang J., Wang Y., El Fatimy R., Rabinovsky R., Balaj L., Chen C.C., Hochberg F., et al. Coding and noncoding landscape of extracellular RNA released by human glioma stem cells. Nat. Commun. 2017;8:1145. doi: 10.1038/s41467-017-01196-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yim N., Ryu S.W., Choi K., Lee K.R., Lee S., Choi H., Kim J., Shaker M.R., Sun W., Park J.H., et al. Exosome engineering for efficient intracellular delivery of soluble proteins using optically reversible protein-protein interaction module. Nat. Commun. 2016;7:12277. doi: 10.1038/ncomms12277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y., Xu G., Yang Y.T., Xu Z., Chen X., Shi B., Xie D., Lu Z.J., Wang P. POSTAR2: deciphering the post-transcriptional regulatory logics. Nucleic Acids Res. 2019;47:D203–D211. doi: 10.1093/nar/gky830. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
RNA-seq data have been deposited at NCBI Gene Expression Omnibus (GEO) and are publicly available as of the date of publication. Accession numbers are listed in the key resources table (GSE162425 and GSE186954).
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.